

Summary
Dysregulation of DNA methylation and mRNA alternative cleavage and polyadenylation (APA) are both prevalent in cancer and have been studied as independent processes. We discovered a DNA methylation-regulated APA mechanism when we compared genome-wide DNA methylation and polyadenylation site usage between DNA methylation-competent and DNA methylation-deficient cells. Here we show that removal of DNA methylation enables CTCF binding and recruitment of the cohesin complex, which in turn, form chromatin loops that promote proximal polyadenylation site usage. In this DNA demethylated context, either deletion of the CTCF binding site or depletion of RAD21 cohesin complex protein can recover distal polyadenylation site usage. Using data from The Cancer Genome Atlas, we authenticated the relationship between DNA methylation and mRNA polyadenylation isoform expression in vivo. This DNA methylation-regulated APA mechanism demonstrates how aberrant DNA methylation impacts transcriptome diversity and highlights the potential sequelae of global DNA methylation inhibition as a cancer treatment.
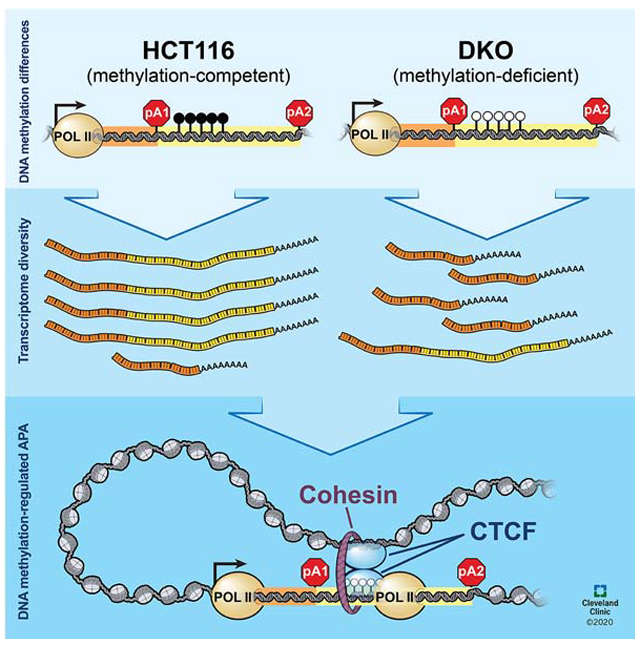
Graphical Abstract
eTOC
The functions of DNA methylation outside of gene promoters are not fully understood. Nanavaty et al. found gene body methylation to modulate transcriptome diversity through alternative cleavage and polyadenylation. In the absence of DNA methylation, CTCF and the cohesin complex orchestrate chromatin loop formation and promote proximal polyadenylation isoform expressions.
Introduction
DNA methylation and alternative cleavage and polyadenylation (APA) are both essential processes in normal mammalian development. DNA methylation, the addition of a methyl group to the 5' position of a cytosine, is a highly conserved epigenetic modification with known roles in genome organization and transcriptional silencing (Zemach and Zilberman, 2010). Deletion of DNA methyltransferases is embryonic lethal in mice (Li et al., 1992; Okano et al., 1999). APA, on the other hand, fine-tunes gene expression in tissue- and cell-specific manners through modulation of mRNA 3′ end maturation (Di Giammartino et al., 2011; Hoque et al., 2013; MacDonald and McMahon, 2010; Wang et al., 2008; Zhang et al., 2005). About 80% of annotated mammalian RNA polymerase II transcripts are known to undergo APA, which can result in either altered protein-coding sequences (intronic and exonic APA) or altered mRNA translational output, stability or localization (3' untranslated region [3' UTR] APA) (Di Giammartino et al., 2011; Gebauer and Hentze, 2004; Sonenberg and Hinnebusch, 2007). Apart from their roles in development, DNA methylation and APA are both dysregulated to drive carcinogenesis. Both lengthening and shortening of mRNA transcripts via APA occur in cancer cells (Morris et al., 2012). Functionally, APA-mediated shortening of 3' UTRs can activate oncogenes by eliminating microRNA (miRNA) binding sites (Mayr and Bartel, 2009) or, in competing endogenous RNAs (ceRNAs), can increase miRNA-mediated degradation of tumor suppressor transcripts (Park et al., 2018). Intronic APA may also promote tumorigenesis by generating truncated proteins (Lee et al., 2018). However, APA regulation is poorly characterized in malignant tissues (Di Giammartino et al., 2011; Neve et al., 2017). At the same time, genome-wide dysregulation of DNA methylation is a hallmark of human cancers, in which much of the functional studies have focused on promoter hypermethylation silencing tumor suppressor genes and promoter hypomethylation activating oncogenes (Baylin and Jones, 2011). Nonetheless, abnormal DNA methylation patterns are not restricted to gene promoters, yet our knowledge of the functions of non-promoter DNA methylation is extremely limited. Thus far, these two dysregulated processes have been studied independently of each other.
Beyond the promoter, gene body methylation positively correlates with gene expression, and increased DNA methylation in this genomic compartment is also pervasive in cancer (Chen and Elnitski, 2019; Yang et al., 2014). Therefore, we hypothesized that genic DNA methylation can impact co-transcriptional processes involved in mRNA transcription and processing and sought to investigate the relationship between aberrant DNA methylation and APA in cancer. Polyadenylation sequencing (poly(A)-seq) and DNA methylation sequencing (MBD-seq) in methylation-competent HCT116 and methylation-deficient DKO cells revealed a significant association between DNA methylation and APA. We further determined that in the absence of DNA methylation, CCCTC-binding factor (CTCF), a methylation-sensitive insulator protein, binds and recruits the cohesin complex to regions downstream of proximal poly(A) sites. This assembly of proteins on the genomic DNA facilitates chromatin looping, analogous to distal enhancer-promoter interactions, and acts as an obstacle to the elongating RNA polymerase II, stalling transcription elongation and promoting proximal poly(A) isoform expression. Additionally, our analysis of RNA-seq and DNA methylation data from The Cancer Genome Atlas (TCGA) corroborated our in vitro observations and provided compelling support for this DNA methylation-regulated APA mechanism in vivo.
Results
Differential DNA methylation correlates with APA
To test if DNA methylation impacts APA, we compared polyadenylation (poly(A)) site usage, using poly(A)-seq, in HCT116 colon cancer cells and a derivative cell line (DKO cells), in which >95% DNA methylation is ablated by genetic deletion of DNA methyltransferases (DNMT) −1 and −3b (Rhee et al., 2002) (Figure S1A; Table S1). We mapped 32,245 poly(A) sites (13,369 genes) in HCT116 and 25,905 poly(A) sites (13,359 genes) in DKO (Tables 1 and S2). In the absence of global APA dysregulation (Figures 1A and S1B), we identified 546 genes undergoing APA between HCT116 and DKO (Tables 1 and S3; Figure S1C). While it has been well documented that the loss of genomic DNA methylation in DKO cells cause widespread promoter demethylation and concurrent gene reactivation, 489 of 546 (90%) candidate genes had comparable total expressions between HCT116 and DKO (Figure 1B). Importantly, the observed APA was independent of changes in the expression of trans-acting factors known to modulate APA (Figures 1C and S1D), and the frequency of hexamers known to precede poly(A) sites in our data was comparable to that in the poly(A) database (Figure 1D). Finally, 412 of 546 (75%) candidate genes preferentially used the proximal poly(A) sites in DKO. To begin elucidating the mechanisms for preferential proximal poly(A) site usage in the absence of DNA methylation, we queried 161 ENCODE transcription factors for potential binding of the DNA sequences between the two most differentially used poly(A) sites in the 412 candidate genes (Figure 1E). Among the top ten most enriched transcription factors, CCCTC-binding factor (CTCF) has well-established, DNA methylation-sensitive binding properties (Phillips and Corces, 2009). This data suggested an association between DNA methylation and APA, which we hypothesized was regulated through CTCF binding in a DNA methylation-sensitive manner.
Table 1.
| HCT116 | DKO | Union | ||||
|---|---|---|---|---|---|---|
| pA sites | Genes | pA sites | Genes | pA sites | Genes | |
| All pA sites | 32,245 | 13,369 | 25,905 | 13,359 | 35,342 | 14,585 |
| Unambig. Genes | 30,112 | 11,614 | ||||
| Genes with more than 1 pA site | 25,420 | 6,922 | ||||
| p.adj < 0.0001 | 2,015 | 1,173 | ||||
| Sites used > 5% in ≥ 1 cell line | 1,819 | 1,106 | ||||
| Site changes > 1.5 fold | 1,163 | 902 | ||||
| > 10% shift in pA site usage | 718 | 546 | ||||
Figure 1: DNA methylation is associated with alternative poly(A) site usage.

a, Poly(A) site usage in HCT116 and DKO cells. n = 4 biological replicates. Red dots, poly(A) sites whose fractional usages are significantly different between HCT116 and DKO; gray dots, poly(A) sites that are not significantly different (NS, non-significant). Numbers in parentheses indicate poly(A) counts in each category. b, Scatter-plot for total gene expression in HCT116 and DKO cells. Red dots, differentially expressed APA candidates between HCT116 and DKO cells; orange dots, non-differentially expressed APA candidates; gray dots, non-APA candidate genes. Numbers in parentheses indicate numbers of genes in each category. c, Expressions of 17 genes known to control APA in duplicate RNA-seq data from HCT116 and DKO. d, Frequencies of known poly(A) signals (AAUAAA and 12 others) in the detected poly(A) sites in HCT116 (replicate 4) and DKO (replicate 4), poly(A) sites in the published Poly(A) database (pAdb2), and a length-matched genomic null set (Null). e, Sequence logos for most enriched ENCODE transcription factors in genomic regions between the proximal and distal poly(A) sites in 412 candidate genes. TCF12 (z-score = 1.7) and BCLX3 (z-score = 0.95) do not have logos in the JASPAR2018 database. See also Figure S1 and Tables S1-S3.
DNA methylation regulates APA via CTCF
We focused on APA candidates with putative CTCF binding sties to interrogate the interaction between DNA methylation, CTCF binding, and APA. Of these genes, we examined novel APA candidates with comparable total expression between HCT116 and DKO. Based on these criteria, we focused on HEAT repeat containing 2 (HEATR2/DNAAF5) and nuclear transcription factor Y subunit alpha (NFYA). In HEATR2, poly(A)-seq analysis showed a 14.6-fold increase in relative usage of the most proximal, intronic poly(A) site in DKO compared to HCT116 (Figure 2A). This increase is at the expense of relative usage of the most distal poly(A) site in the 3' UTR in DKO cells. In NFYA, all four poly(A) sites are in the 3' UTR, and poly(A)-seq detected a 2.6-fold decrease in relative usage of the most distal poly(A) site in DKO compared to HCT116 (Figure 2B). Interestingly, both genes show comparable CTCF binding and DNA methylation between the two cell lines except for the CpG islands (CGIs) between the most increased and decreased poly(A) sites, where we observed enriched CTCF binding and loss of DNA methylation in DKO (Figures 2A and 2B).
Figure 2: DNA methylation regulates APA via CTCF binding.

a, b, Gene model and UCSC genome browser views for HEATR2 (a) and NFYA (b). pA, poly(A) sites; Poly(A), poly(A)-seq; MBD, MBD-seq; CTCF, CTCF ChIP-seq; CGI, annotated CpG islands; BSF, bisulfite sequencing amplicons; boxed regions, differentially methylated CpG islands. c, Bisulfite sequencing of HEATR2 and NFYA in HCT116, DKO, and HCT116 after 72 hours of treatment with DMSO (DMSO) or DAC (DAC). Bars represent percent methylation at individual CpGs. d, ChIP-qPCR for CTCF and POLR2 at HEATR2 and NFYA. Gene models show exons (grey boxes), CpG islands (CGI; green boxes), and poly(A) sites (red lines). ChIP-qPCR amplicons are indicated below the gene model and labeled A-E. n = 3 biological replicates. *, p <0.05 for the difference between DKO and HCT116 or between DAC and DMSO. Data are represented as mean ± SEM. e, qRT-PCR to quantify poly(A) isoform expression at HEATR2 and NFYA in the same cells as in (d). All samples are normalized to HCT116. Data are represented as mean ± SEM. *, p <0.05. f, Northern blot detection of the luciferase reporter gene. The luciferase reporter construct containing NFYA 3′ UTR is shown with red lines indicating poly(A) sites and a blue line denoting the putative CTCF binding site. Mock, no construct; LucNFYA, wildtype NFYA 3′ UTR; LucNFYA*, CTCF-binding site mutant. 28S is a loading control. Normalized luciferase expression for each poly(A) isoform is on the right. See also Figure S2.
We verified the association between DNA methylation and APA by treating HCT116 with the DNA demethylating agent, 5-aza-2′-deoxycytidine (DAC) (Momparler, 2005), which induced substantial decreases in DNA methylation (Figure 2C). In both HEATR2 and NFYA, CTCF was enriched at the differentially methylated CGIs in DKO and DAC-treated HCT116, confirming that CTCF bound these regions in the absence of DNA methylation (Figures 2D, S2A, and S2B). Additionally, we observed an accumulation of RNA Polymerase II (POLR2) near the CTCF binding sites, suggesting that CTCF binding impacted transcriptional dynamics and possibly impeded POLR2 traversal (Figure 2D). The enrichment of CTCF and POLR2 at these locations in DAC-treated cells was not due to global changes in these protein levels (Figure S2C). Isoform-specific qRT-PCR showed that distal poly(A) isoform production decreased 5.8-fold in HEATR2 and 2.5-fold in NFYA in DAC-treated HCT116 cells, comparable to the pattern observed in DKO cells (Figure 2E).
To determine whether CTCF binding was necessary and sufficient for APA regulation, we transfected HCT116 cells with a luciferase reporter construct containing a wild type NFYA 3' UTR (LucNFYA) or a mutant NFYA 3' UTR, lacking the CTCF motif (LucNFYA*; Figure 2F). We expected the LucNFYA* construct to mimic the methylated NFYA allele in terms of poly(A) isoform production due to the loss of CTCF binding. Northern blotting against luciferase showed a 1.44-fold increase in expression of the most distal isoform in the LucNFYA* transfected cells compared to the LucNFYA transfected cells, confirming that CTCF was required for DNA methylation-regulated APA. However, because this was a smaller shift than previously observed at the endogenous locus (Figures 2B and 2E), we postulated that compounding aspects of chromatin structure that could not be recapitulated in a reporter construct may be involved in this APA regulation.
Chromatin loops form at unmethylated CGIs downstream of proximal poly(A) sites
CTCF acts as an anchor point for the cohesin complex to form chromatin loops and topologically associating domains (TAD) (Phillips and Corces, 2009). Therefore, we investigated the possibility that CTCF was cooperating with the cohesin complex in this DNA methylation-regulated APA mechanism. ChIP-seq for RAD21 cohesin complex component (RAD21) and structural maintenance of chromosomes 1 (SMC1) in DKO showed enriched binding of both proteins with CTCF at the unmethylated CGIs in HEATR2 (Figure 3A) and NFYA (Figure 3B) compared to HCT116. Surprisingly, in these same regions, RNA polymerase II phosphorylated at serine 5 of the YSPTSPS repeats (Pol2Ser5p) was also enriched in DKO, but not in HCT116. Pol2Ser5p is part of the transcription initiation complex and is usually found at gene 5′ ends (Bowman and Kelly, 2014). ChIP-seq data for RNA polymerase II phosphorylated at serine 2 (Pol2Ser2p), a transcription elongation complex marker, also showed significant increase in DKO cells in the same regions (Table S4). Hence, the increased POLR2 occupancy near CTCF binding sites (Figures 2D, 3A, and 3B) was likely due to the simultaneous recruitment of a new initiation complex marked by Pol2Ser5p and pausing of elongation complex marked by Pol2Ser2p. Furthermore, histone H3 lysine 27 acetylation (H3K27Ac), a mark associated with enhancer-promoter interactions mediated by the cohesin complex (Creyghton et al., 2010), was increased at the unmethylated CGIs bound by CTCF, RAD21, and SMC1 (Figures 3A and 3B).
Figure 3: DNA methylation-sensitive binding of CTCF and the cohesin complex orchestrate intra-chromosomal looping.

a, b, Gene model and UCSC genome browser views for HEATR2 (a) and NFYA (b). ChIP-seq signals for RAD21, SMC1, RNA Polymerase II phosphorylated at serine 5 (Pol2Ser5p), Total RNA polymerase II (POLR2), and histone H3 acetylated lysine 27 (H3K27Ac) are shown. CGI, annotated CpG islands. c, d, 3c-qPCR analysis of long-distance intra-chromosomal loops at HEATR2 (c) and NFYA (d). Relative contact frequencies between the anchor fragment and distal fragments were normalized to control BAC clones. n = 3 biological replicates. Data are represented as mean ± SEM. *, p <0.05; **, p <0.005. e, f, Visualization and confirmation of 3C-detected contacting genomic fragments for HEATR2 (e) and NFYA (f). Top, UCSC genome browser views of the −30.6 kb region for HEATR2 (e) and the −6.0 kb region for NFYA (f). ChIP-seq signals for CTCF, RAD21, and SMC1 are shown for each fragment. Bottom, Sanger sequencing results showing the ligation between the anchor regions and the distal contact regions in HEATR2 and NFYA. EcoRI, EcoRI restriction enzyme site used in the 3C experiment. See also Figure S3 and Table S4.
The concomitant enrichment of CTCF, cohesin complex proteins, Pol2Ser5p, and H3K27Ac at the unmethylated CGIs compelled us to test if chromatin loops existed at these locations. Using chromosome conformation capture (3C) assay (Hagege et al., 2007), we detected significant interactions between the CGIs (anchor points) and multiple distal genomic sequences at both HEATR2 and NFYA (Figures 3C, 3D and S3). Chromatin loop formation with the anchor points were either significantly increased or only present when the CGIs were unmethylated in DKO cells and DAC-treated HCT116 cells. In particular, the loop contact points located at 30.6 kb upstream of the anchor in HEATR2 (Figure 3E) and at 6.0 kb upstream of the anchor in NFYA (Figure 3F) showed strong binding of CTCF and the cohesin complex proteins. Since CTCF and cohesin complex proteins localized to these distant contact points in both HCT116 and DKO cells, we deduced that the increased contact frequencies between the anchors and these distant sequences were due to unique binding of these proteins at the anchor locations. These long-range interactions were further confirmed by Sanger sequencing of the 3C PCR products. Taken together, these data demonstrated that, when the CGIs downstream of the proximal poly(A) sites were unmethylated, CTCF and the cohesin complex could bind these sequences and mediate intra-chromosomal loop formation and that such chromatin loops mimicked promoter-enhancer interactions, resulting in Pol2Ser5p and H3K27Ac localization to these putative APA control regions.
DNA methylation-regulated APA requires CTCF and the cohesin complex
To directly test the role of CTCF in APA regulation, we deleted the predicted CTCF binding motif within the putative APA control region at the endogenous NFYA locus in HCT116 cells using CRISPR/CAS9. We successfully isolated two independent clones, one with a homozygous 27 bp deletion of the CTCF motif (NFYA−/−ΔCTCF) and the other containing a heterozygous 41 bp deletion of the CTCF motif on one allele (NFYA−/+ΔCTCF), for further characterization (Figure 4A). We treated these CTCF motif-deletion clones with DAC for 72 hours to achieve DNA demethylation, as confirmed by methylation-specific PCR (MSP) and DNMT1 protein depletion (Figures 4B and 4C). Similar to previous experiments, DAC-treated HCT116 cells showed a 2.6-fold decrease in distal poly(A) isoform production (Figure 4D). In contrast to the wildtype HCT116 cells, DAC-treated NFYA−/−ΔCTCF clone did not change its poly(A) isoform expression pattern. In these cells, ChIP-qPCR confirmed loss of CTCF binding and associated RAD21 and Pol2Ser5p co-localization, as would be predicted from the homozygous deletion of CTCF binding site (Figure 4E). Consistent with the genotype, DAC-treated NFYA−/+ΔCTCF showed an intermediate (1.9-fold) decrease in distal poly(A) isoform expression and retained CTCF, RAD21, and Pol2Ser5p enrichment at the APA control region on the remaining wildtype allele (Figures 4D and E). These results confirmed that CTCF binding was necessary for proper recruitment of RAD21 and Pol2Ser5p to the demethylated APA control region to facilitate proximal isoform expression.
Figure 4: DNA methylation-dependent association of CTCF and RAD21 is indispensable for APA regulation.

a, Sequencing results of wildtype (WT) NFYA in HCT116 cells and CRISPR-engineered CTCF motif deletion clones. NFYA−/−ΔCTCF, homozygous deletion clone; NFYA−/+ΔCTCF heterozygous deletion clone; red box, predicted CTCF motif. Genomic coordinates are indicated on top. b, Methylation-specific PCR (MSP) of the APA control region in NFYA in HCT116, DKO, DMSO (DMSO) or DAC (DAC) treated (for 72 hours) HCT116, NFYA−/−ΔCTCF, and NFYA−/+ΔCTCF clones. M, DNA ladder; bp, base pairs. c, Western blot analysis for DNMT1 protein in the same cells as in (b). ACTB is a loading control. MW, molecular weights (kD). d, qRT-PCR to quantify poly(A) isoform expression at NFYA in the same cells as in (b). n = 3 biological replicates for each clone. Samples are normalized to their respective DMSO treatment. Data are represented as mean ± SEM. **, p <0.005. e, ChIP-qPCR for CTCF, RAD21,POLR2, and IgG at NFYA locus. Gene models and ChIP-qPCR amplicons are the same as in Figure 2D. Data are represented as mean ± SEM. *, p <0.05; **, p <0.005. f, Western blot analysis of AIDR-HCT116 treated with DMSO for 72 hours, auxin (3-indoleacetic acid, IAA) for 8 hours (IAA), DAC for 72 hours (DAC), and both DAC and IAA (DAC+IAA). ACTB is a loading control. MW, molecular weights (kD). g, qRT-PCR to quantify poly(A) isoform expression at HEATR2 and NFYA after 4-thiouridine (4sU)-labeling in the same cells as in (f). Total, total RNA from the cells; New transcript, newly transcribed RNA labeled by 4sU; Flowthrough, residual RNA in the wash buffer during 4sU-labeled RNA isolation. n = 2 biological replicates. All samples are normalized to DMSO. Data are represented as mean ± SEM. *, p <0.05; **, p <0.005.
Next, we tested the requirement for the cohesin complex in DNA methylation-regulated APA by leveraging an auxin-inducible, RAD21-degron system in HCT116 cells (AIDR-HCT116) (Natsume et al., 2016). Rapid, auxin-mediated degradation of RAD21 in this cell line led to complete resolution of TADs and chromatin loops (Rao et al., 2017). Therefore, we expected that depletion of RAD21 should recover the decrease in distal poly(A) isoform expression caused by chromatin loop formation at unmethylated APA control regions. In order to accurately and sensitively assess dynamic transcriptional changes in these experiments, we labeled newly transcribed RNA with 4-thiouridine and isolated labeled RNA (New Transcript) from total RNA (Total) for APA isoform-specific qRT-PCR (Radle et al., 2013). When DNA methylation was present, auxin-mediated depletion of RAD21 did not affect poly(A) isoform expression patterns (Figures 4F and 4G). This was consistent with our ChIP-seq and 3C experimental results showing that RAD21 binding and chromatin loops were not present when the APA control regions were fully methylated (Figures 3 and S2). On the other hand, DAC treatment in AIDR-HCT116 cells resulted in DNA demethylation, loss of DNMT1, and decreased distal poly(A) site usage. In this demethylated state, auxin-mediated RAD21 depletion rescued the use of distal poly(A) sites from 17.7% to 53.5% at HEATR2 and from 44.5% to 69.1% at NFYA in the New Transcript fraction (Figure 4G). These results confirmed that RAD21 was required for DNA methylation-regulated APA and supported that the cohesin complex was involved in the regulation.
DNA methylation regulates APA in vivo
Our experimental data at HEATR2 and NFYA suggested a model of DNA methylation-regulated APA, in which CTCF binds to unmethylated APA control regions between two dynamic poly(A) sites and promotes proximal poly(A) site usage by setting up cohesin-mediated chromatin loops that hinder transcription elongation. In contrast, DNA methylation at the APA control regions blocks CTCF binding, thus preventing loop formation and promoting distal poly(A) site usage. We surmised that the coordinated enrichment of CTCF, RAD21, SMC1, H3K27Ac, and Pol2Ser5p at unmethylated APA control regions could serve as signals to determine which of our initial 546 APA candidate genes could be regulated by this mechanism. Therefore, we applied a consensus clustering approach (Monti et al., 2003) on the ChIP-seq data and detected 10 distinct signal clusters based on co-occurring shifts of CTCF, RAD21, SMC1, H3K27Ac, Pol2Ser2p, Pol2Ser5p, and POLR2 enrichment with DNA methylation levels (Figures 5A and S4; Tables S4 and S5). We identified 106 additional genes that may be regulated similarly to HEATR2 and NFYA for future studies.
Figure 5: DNA methylation-regulated APA is evident at additional loci and in TCGA cohorts.

a, ChIP-seq signal characteristics for HCT116 and DKO in clusters 3 and 10 from the integrative consensus clustering. The CpG island of interest in HEATR2 belongs to cluster 10 while that of NFYA belongs to cluster 3. Blue dotted lines, aggregate HCT116 signals; yellow solid lines, aggregate DKO signals. b, Correlation between calculated isoform usage ratio and DNA methylation level at 5 CpG sites in HEATR2. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; Merged, a set containing data from all 11 TCGA cohorts; r2, Pearson correlation coefficient. The number of samples in each cohort is indicated in parentheses. c, Correlation coefficients and p-values across HEATR2 gene for the merged cohort. pA, poly(A) site; PhCon, phastCons100way; CpG, CpG islands. ChIP-seq signals for CTCF, RAD21, H3K27Ac, and Pol2Ser5p in HCT116 and DKO are shown. d, Heatmap of the maximum Pearson correlation coefficients for all 546 APA candidate genes in each of the 11 TCGA cohorts and the Merged set. The 7 best correlations with the highest consistency across cohorts are noted and also shown in Figure S5B. See also Figures S4-S5 and Table S5-S6.
Moreover, we interrogated the effect of DNA methylation on poly(A) site usage in 11 cancer cohorts from The Cancer Genome Atlas (TCGA) and a merged set consisting of data points from all 11 cohorts to identify cohort-specific correlations, as well as correlations across cancer types. We analyzed individual cytosine probes across the entirety of HEATR2 and NFYA. While NFYA showed a statistically significant but modest correlation in 2/11 cohorts, HEATR2 showed a strong negative correlation between DNA methylation and proximal poly(A) site usage for all cytosine probes within chr7: 807,596-809,109 in all cohorts except for kidney renal clear cell carcinoma (Figures 5B and S5A). Importantly, the remarkable correlations in HEATR2 are downstream of the proximal poly(A) site and directly overlap with the differentially methylated APA control region, where the loss of DNA methylation resulted in enrichment of CTCF, the cohesin complex proteins, Pol2Ser5p, and H3K27Ac in vitro (Figure 5C). Additionally, PhastCon scores indicate strong sequence conservation across vertebrates, despite the fact that this region is not part of any annotated exon. Finally, an analysis including all 546 candidate genes revealed 384 genes with a statistically significant correlation in at least one cancer type (Figure 5D; Table S6). Seven genes exhibited correlations across multiple cancer types, and of these, HEATR2 showed the best correlations (Figure S5B). These data provided compelling support for this novel DNA methylation-regulated APA mechanism in vivo.
Discussion
Dysregulation of DNA methylation and APA are pervasive in cancer, and both disruptions alter the cancer transcriptome. These two processes have mostly been studied independently thus far, but here, we discovered a DNA methylation-regulated APA mechanism that can account for the biological function of non-promoter DNA methylation in diversifying the transcriptome through APA. Our data suggested a model, in which DNA methylation at the APA regulatory region between two shifting poly(A) sites can prevent CTCF binding and allow transcription elongation to proceed readily to reach the distal poly(A) site (Figure 6). Thus, DNA methylation here promotes higher usage of the distal poly(A) site and results in higher expression of the distal poly(A) isoform, as seen in HCT116 cells. Conversely, when the DNA at the same regulatory region is unmethylated, CTCF can bind and recruit the cohesin complex. CTCF often associates with the cohesin complex to form 3D chromatin loops that define insulated neighborhoods (Hnisz et al., 2016) or bring together distally positioned enhancers and promoters for transcription activation (Parelho et al., 2008; Ren et al., 2017). A similar structure exists here and is supported by 3C assay results and the degradation of RAD21, which has been shown to eliminate all chromatin loops (Rao et al., 2017). Concomitant with the formation of CTCF/cohesin-mediated loops, we also saw enrichment of Pol2Ser5p, usually found in the transcription initiation complex near gene promoters, and Pol2Ser2p, a marker for active transcription elongation complex, at the demethylated APA control regions. We also observed a significant increase in H3K27Ac. The simultaneous presence of chromatin loops, H3K27Ac, and Pol2Ser5p resembles the chromatin architecture observed for enhancer-promoter interactions, where new RNA polymerase II is recruited. The enrichment of Pol2Ser2p at the same locations suggested pausing of the elongation complex near the proximal poly(A) sites, likely obstructed by the large protein complex consisted of CTCF, cohesin, and Pol2Ser5p. As a consequence, the expression of the proximal poly(A) isoform is increased, as seen in DKO cells.
Figure 6: A model of DNA methylation-regulated APA.

POL II Ser2P, serine 2 phosphorylated POLR2A (transcription elongation complex); POL II Ser5P, serine 5 phosphorylated POLR2A (transcription initiation complex); pA1, proximal poly(A) site; pA2, distal poly(A) site; black lollipops between pA1 and pA2, methylated CpG dinucleotides
The above model of DNA methylation-regulated APA is likely one of several different mechanisms. When considering the interplay between DNA methylation, presence of CGI, and APA in the entire 546 APA candidate gene set, there are in fact several different scenarios, each requiring independent detailed investigation. First, DNA methylation differences occur both within and outside of CGIs, and both types of DNA methylation can impact transcription factor binding. We prioritized for CGIs as these sequences are usually highly conserved for their regulatory potential (Illingworth et al., 2010). Second, changes in DNA methylation can occur upstream from the proximal poly(A) sites, in between the most changed poly(A) sites, downstream of the distal poly(A) sites, or a combination of these. We chose to focus first on DNA methylation changes between the most changed poly(A) sites as this context provides a defined search space for hypothesis generation and testing. Third, we observed both decreased (412 genes) and increased (134 genes) usage of proximal poly(A) sites in the absence of DNA methylation, suggesting that at least two independent mechanisms must exist to support such a dichotomous phenomenon. Finally, it is important to note that DNA methylation-regulated APA may not be restricted to the 546 candidates we uncovered using the HCT116/DKO comparison since these cells do not capture tissue- and cell- type specific DNA methylation patterns that exist. This complexity in genome organization, epigenetic variation, and alternative poly(A) site usage highlights an intersection between epigenetic regulation and transcriptome diversity that warrants further studies (Sweet and Ting, 2016).
Our results have wide-ranging significance in both the basic and applied areas. DAC treatment, an established cancer therapy, induces remission by reactivating genes by promoter demethylation and repressing genes by gene body demethylation (Momparler, 2005; Yang et al., 2014). However, the side effects of global demethylation are challenging to predict without a comprehensive understanding of DNA methylation’s regulatory functions. Gene body DNA methylation has previously been linked to alternative mRNA splicing (Shayevitch et al., 2018; Shukla et al., 2011), intronic antisense transcription (Cowley et al., 2012; Wood et al., 2008), and alternative promoter usage (Maunakea et al., 2010; Nagarajan et al., 2014). Together with our findings here, they emphasize the need to consider the impact of global DNA demethylation not only on what genes are turned on or off but also on what type of transcript variants are expressed. Furthermore, studies of DNA methylation-regulated APA genes may reveal novel therapeutic targets in different types of cancers. For example, it is unclear why many NFYA target genes are upregulated in cancer (Gurtner et al., 2017), and the different poly(A) isoforms controlled by DNA methylation-regulated APA present a new avenue of research as to NFYA’s role in carcinogenesis. In the case of HEATR2, this protein is only known to support ciliary motility (Diggle et al., 2014; Horani et al., 2012; Horani et al., 2018) and has not previously been associated with cancer. Yet, HEATR2 is robustly expressed and shows the strongest inverse correlation between its gene body DNA methylation and proximal poly(A) isoform expression in vivo across 10 different cancer types. The functions of HEATR2 protein and its DNA methylation-regulated poly(A) isoforms in carcinogenesis are also ripe for further investigation. Finally, DNA methylation and APA are not exclusively pathogenic. Rather, they are necessary for normal transcriptome regulation to support gene expression, cell differentiation, and chromosome stability (Jaenisch and Bird, 2003; Mayr, 2017; Tian et al., 2005). While our work focused on DNA methylation-regulated APA in cancer, this mechanism may also operate during development and broadly contribute to the understanding of how the transcriptome and proteome are shaped by non-promoter DNA methylation in healthy and disease contexts.
STAR METHODS
Lead Contact and Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Angela H. Ting (tinga@ccf.org). All unique cells and reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Cell lines and cell culture
HCT116 and DKO (clone #2) cells were cultured in McCoy’s 5A medium with 10% FBS at 37°C and 5% CO2. AIDR-HCT116 cells were cultured in McCoy’s 5A medium with 10% FBS and supplemented with 1 μg/ml puromycin (Life technologies #A1113803), 100 μg/ml geneticin (Gibco #10131-035) and 100 μg/ml hygromycin (Thermo Fisher Scientific #10687010) at 37°C and 5% CO2 (Natsume et al., 2016). HCT116- NFYA−/−ΔCTCF and HCT116- NFYA−/+ΔCTCF cells were cultured in McCoy’s 5A medium with 10% FBS at 37°C and 5% CO2.
Method Details
5-aza-2′-deoxycytidine and Indole-3-acetic acid treatments
Where indicated, HCT116 cells were treated with 1 μM 5-aza-2′-deoxycytidine (DAC; Sigma #A3656) by adding it directly to culture media. AIDR-HCT116 cells were treated with 1 μM DAC for 72 hrs, 500 nM of 3-indoleacetic acid (IAA, Sigma #45533) for 8 hours, or a combination of both DAC and IAA at the above specified concentrations and durations.
Poly(A)-sequencing library preparation
Total cellular RNA was prepared from 4 x 10 cm plates of HCT116 and DKO cells each using AllPrep DNA/RNA/miRNA Universal Kit (Qiagen #80224). Library preparation was similar to previously described (Zagore et al., 2018). Poly(A)+ RNA was selected from 10 μg of total RNA using oligo(dT)25 Dynabeads (Thermo Fisher Scientific #61002) then fragmented in 1X fragmentation buffer at 95°C for 40 minutes. Fragmentation was stopped in cold stop solution and poly(A)+, fragmented RNA precipitated with pellet paint and isopropanol. RNA fragments were separated by 15% PAGE/urea gel electrophoresis, and 50 – 100 nt fragments were excised. The gel was broken up by extrusion through the bottom of a 0.5 mL tube with a hole produced by needle puncture and RNA fragments eluted by shaking in 0.5 mL of diffusion buffer at 37°C for 45 minutes. Gel remnants were removed by centrifugation through Spin-X columns, and RNA was precipitated with pellet paint and isopropanol. Four biological replicates of each cell line were processed for sequencing.
Bioinformatic detection of processing regions
FASTQ files containing the raw sequence reads from poly(A)-seq were de-barcoded by allowing up to 2 mismatches between the first 6 bases and one of 4 barcodes. Reads with more than 2 mismatches from a barcode used in the experiment were discarded. The next 6 bases were designed to contain a random hexamer for de-duplication of PCR duplicates. This sequence was removed from the read and annotated into the read name. Next, sequenced poly(A) tails were removed from the reads by detecting the first instance of 4 consecutive adenosine bases with no more than 1 non-A base in the following 6 base pairs. The remaining sequences were aligned to the hg19 genome build using bowtie 2.3,3,3,1(Langmead and Salzberg, 2012). Uniquely mapping alignments were retained by requiring a mapping quality (MAPQ) score > 20. PCR duplicates were removed by collapsing all alignments with identical random hexamer annotations and the same alignment position into a single alignment. Reads due to predicted internal priming of genomic A were removed by filtering all alignments with at least 7 of 10 or 6 consecutive downstream A bases. Processing regions (PRs), which are used interchangeably with poly(A) sites, were computed by merging all regions with a coverage of at least 10 reads in one experiment within 10 bp of each other into contiguous genomic ranges. Transcription units (TUs) were generated using ENSEMBL genes with biotypes Protein Coding and Long-Noncoding. All processing regions were overlapped and assigned to the gene body or 5 kb downstream flanking region of individual TUs. Only TUs were all assigned PRs were unique (did not overlap with the gene body or flank of any other TUs) were considered for statistical testing of APA.
Statistical testing of APA
Read depths for each PR were joined into a matrix containing the depth at each PR across all experiments. Because changes in PR usage within TUs are analogous analytically to changes in exon usage with TUs, we employed DEXSeq (Anders et al., 2012) to test statistically for changes in PR usage independent of total gene expression changes. TUs with more than one joined PR that was expressed in at least one condition (mean depth ≥ 10 reads) were tested using DEXSeq’s testForDEU() function. P-values were adjusted for multiple testing using the Benjamini-Hochberg procedure. Fractional usages were computed for each PR as the depth of a given PR divided by the sum of the depths of all PRs in the TU. Changes in PR depth between conditions were considered significant when the DEXSeq adjusted p-value was < 1x10−4, the fractional usages between conditions differed by more than 0.1, and the fold change between the fractional usages between conditions was greater than 1.5.
Enrichment testing for transcription factors
For the 412 genes that preferentially use the proximal poly(A) site in DKO, the genomic regions between the most increased and most decreased poly(A) processing regions were interrogated for the enrichment of known binding sites in the ENCODE ChIP-seq database (a union of binding sites across all cell lines tested). A per-bp rate of overlap was computed for each factor. A background set was generated by sampling, without replacement, 10,000 genomic regions between pairs of PRs in genes containing more than one PR, and the per-bp overlap rate was computed for this background set. Enrichment was determined by calculating the Z-scores for each factor against the background set. Transcription factors predicted to bind less than 10% of candidate genes were filtered out prior to a ranking by Z-scores.
Chromatin Immunoprecipitation (ChIP)
Cells were crosslinked in 1% formaldehyde for 10 minutes at RT. Formaldehyde crosslinking was quenched in 125 mM glycine for 5 minutes at RT. Cells were then washed twice with cold PBS, scraped into PBS, centrifuged at 500g for 2 minutes, and lysed by passing 10 times through a 20G needle in lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, Roche Protease Inhibitor Cocktail). Nuclei were collected by centrifugation at 400g for 5 minutes and re-suspended in RIPA buffer. Chromatin from 8 million cell equivalents in 130 μL RIPA was fragmented using Covaris. Chromatin and antibodies conjugated to magnetic beads were incubated with rotation overnight at 4°C. Unbound chromatin was washed 3 times with LiCl wash buffer and 1 time with TE and eluted in elution buffer (1% SDS, 0.1 M NaHCO3). Reverse crosslinking was performed by incubating the eluate at 65°C overnight. DNA was purified by isopropanol precipitation and re-suspended in H2O.
ChIP-sequencing (ChIP-seq)
Cultured cells were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300-500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were re-suspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield. An aliquot of chromatin (30 μg) was precleared with G (for POLR2 Active Motif #91151, Pol2Ser2p Active Motif #61084, and Pol2Ser5p Active Motif #61086) or A (for CTCF Active Motif #61311, RAD21 Santa Cruz #sc98784, SMC1 Bethyl #A300-005A, and H3K27Ac Active Motif #39133) agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 4 μg of antibody against each protein. Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation. Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single-end).
Differential ChIP-seq signal detection
ChIP-seq data were processed using the ENCODE transcription factor and histone ChIP-seq processing pipeline (https://github.com/kundajelab/chipseq_pipeline) using “--se --species hg19 --peak-caller macs2 --type $type”, where “--type” was “TF” for all transcript factors and “histone” for H3K27Ac, Pol2Ser2p, Pol2Ser5p, and POLR2. Peaks from the ENCODE pipeline output for HCT116 and DKO cells were compared using MANorm (Shao et al., 2012) using a window length of either 500 bp (CTCF, SMC1, and RAD21) or 1,000 bp (H3K27Ac, POLR2, Pol2Ser5p, Pol2Ser2p, and POLR2). Log2-fold change (M-Value) and normalized binding depth were reported and used for downstream analysis.
Integrative clustering of multiple differential ChIP-seq signals
Differential ChIP-seq signals and DNA methylation signals for HCT116 and DKO (Serre et al., 2010) that overlap with the genomic sequences between the most increased and most decreased poly(A) processing regions ± 500 bp were extracted for subsequent analysis. Briefly, data from methyl-CpG binding (MBD) sequencing were aligned to hg19 using bowtie 2.3.4.1 and processed for peak calling using MACS2 with the option “-f BAM -g hs -B --broad”. We segmented the genomic regions based on signals from the 8 features such that the mean depth for a given factor is equivalent within a segment. Segments 10 bp or shorter were filtered out, leaving a matrix of 8,013 distinct genomic segments by 8 features filled with a normalized log2-fold change (M-Value from MANorm output) for CTCF, SMC1, RAD21, H3K27Ac, Pol2Ser2p, Pol2Ser5p, and POLR2 and raw mean depths from two cell lines MBD-seq. Next, the genomic segments in this feature count matrix were clustered using a non-negative matrix factorization (NMF)-based (Kim and Park, 2008) consensus clustering method at K = 2 to K = 15. In each NMF clustering experiment, 80% of genomic segments were randomly sampled. Results from a total of 100 clustering runs were used for consensus membership analysis. Based on the delta value curves and the consistency of cluster membership, K = 10 was determined to be optimal. A unique cluster ID was assigned to each genomic segment in the feature count matrix. The distance between two clusters was measured as the average distance between each point in one cluster to every other point in the other cluster. Finally, to visualize the ChIP-seq and DNA methylation signal density distributions for HCT116 and DKO in the different clusters, ggplot2::geom_density() was used to plot the mean read depths (normalized read depth from MANorm and raw mean depth for MBD-seq) corresponding to each genomic segment in each cell line. The difference between two distributions is quantified by Mann–Whitney-Wilcoxon nonparametric test (Table S5).
Western blotting
Cells were lysed in 10 mM Tris pH=7.5, 3% SDS by pipetting then centrifugation through a Qiashredder column (Qiagen #79656). Protein concentrations were determined by BCA assay (Thermo Fisher Scientific #23225), then lysates were resolved on SDS-PAGE in NuPAGE reducing sample buffer (Thermo Fisher Scientific #NP0001) using the Novex system at 120 V for 150 minutes. Proteins were transferred to activated PVDF membrane using a 1X transfer buffer (NuPAGE transfer buffer, Thermo Fisher Scientific #NP0006-1) with 10% methanol at 30V for 3 hours at 4°C. Membranes were then blocked in 10% non-fat dairy milk in 1X TTBS (25 mM Tris-HCl, 155 mM NaCl, 0.1% Tween 20) overnight at 4°C. Membranes were incubated at RT with primary antibody in 5% blocking buffer for 2 hours at the following concentrations: ACTB (Sigma Aldrich #A3584; 1:10,000), CTCF (Cell Signaling #2899S; 1:1,000), DNMT1 (Sigma Aldrich #D4692; 1:1,000), HEATR2 (Proteintech #24578-1-AP; 1:600), NFYA (Abcam #Ab139402; 1:1,000), RAD21 (Abcam #Ab992; 1:1,000), total POLR2 (Active Motif #39497; 1:2,000), and NUDT21 (Abcam #ab183660; 1:1,000). Membranes were incubated at room temperature (RT) for 1 hour with secondary antibody at 1:5,000 (ACTB, CTCF, DNMT1, HEATR2, NFYA, RAD21, and NUDT21) or 1:25,000 (total POLR2) in 5% blocking buffer. Washes consisted of 3x10 minute gentle shaking in 1x TBS 0.1% Tween 20 at RT. Membranes were treated with either ECL or ECL Plus, then exposed to film.
Bisulfite Sequencing and MSP
DNA was bisulfite converted using the EZ DNA methylation-gold kit (Zymo #D5006). Loci of interest were amplified using the primers listed in Table S7 under “Bisulfite sequencing”. Amplicons were resolved on 0.8% agarose gel by gel electrophoresis and purified using the Qiaquick Gel Extraction Kit (Qiagen #28706). Purified PCR product was cloned into the Topo TA Vector and transformed into OneShot TopTen Chemically Competent E. Coli Cells (Thermo Fisher Scientific #K457540). Alleles were analyzed using BISMA after Sanger sequencing (Rohde et al., 2010). Methylation-specific PCR (MSP) primers are listed in Table S7, and PCR products were resolved on a 1.2% agarose gel by gel electrophoresis.
Quantification of poly(A) isoform expression by qRT-PCR
Total RNA was extracted by TRIzol (Ambion #15596026). Oligo-dT(16) primer was used to convert 1 μg of total RNA into cDNA using Superscript III reverse transcriptase (Thermo Fisher #18080093), and 40 ng of cDNA was used for each qPCR reaction with QuantiTect SYBR green reagent (Qiagen #204143). PCR primers for specific poly(A) isoforms are listed in Table S7. Relative isoform expression for HEATR2 was calculated as distal isoform level divided by proximal isoform level, and for NFYA, as distal isoform level divided by total isoform level. Results were calculated from biological triplicates that were assayed by technical duplicate PCR reactions, and statistical significance was determined by Student’s t-test.
Cloning
Plasmids for making anti-sense RNA probes for northern blot were generated by cloning PCR products downstream of T7 RNA polymerase. Briefly, PCR products were generated using the primers from Table S7 under “Northern probe (for cloning constructs for in vitro transcription)”, and PCR products were gel purified, then cloned into pSC-A-amp/kan according to manufacturer’s instructions (Strataclone PCR Cloning Kit; Agilent #240205). Reporter constructs were generated by cloning NFYA 3' UTR genomic sequence downstream of Renilla luciferase in pGL4.74 (Promega) by Gibson assembly (NEB #E5510S).
Northern blotting
100 μg total RNA was poly(A) selected using Dynabeads Oligo(dT)25 (Thermo Fisher Scientific #61002) following the manufacturer’s instructions. Eluted RNA was rebound to the same beads in order to limit non-specific rRNA and finally eluted into 1X Northern loading dye (50% formamide, 2% formaldehyde, 1X MOPS, 40 μg/mL Ethidium Bromide) at 80°C for 2 minutes. Samples were heated to 65°C for 10 minutes, then loaded onto 1% agarose-formaldehyde gels and run for 4 hours at 100 V. Gels were treated with 0.05 M NaOH/1.5 M NaCl for 30 minutes, 0.5 M T ris pH=7.4/1.5 M NaCl for 20 minutes, followed by 20X SSC for 45 minutes, to enhance the transfer of large RNAs, then transferred to positively charged nylon membrane (GE) using overnight capillary transfer. Membranes were washed in 0.1% SDS/0.1X SSC for 1 hour then pre-hybridized for 4 hours in 5X SSC, 5X Denhardt’s solution, 50% formamide, 1% SDS, and 100 μg/ml salmon sperm DNA at 60°C. Riboprobes were generated from constructs linearized by EcoRV using the MAXIscript T7 Transcription kit (Thermo Fisher Scientific #AM1312) and incubated with membranes at 60°C overnight. Blots were washed in 0.1% SDS/2X SSC twice for 5 minutes followed by 0.1% SDS/0.1X SSC for 1 hour at 65 °C before exposure to autoradiography cassettes.
Chromosome Conformation Capture (3C)
3C experiments were performed as described previously (Hagege et al., 2007). In brief, a total of 107 cells were harvested with trypsin, washed, and re-suspended in 9.5 ml PBS. The samples were cross-linked in 1% formaldehyde for 10 minutes at room temperature with rotation. Nuclei were extracted by incubating cells on ice for 10 minutes using 5 mL cold lysis buffer (10 mM Tris-HCl, pH 7.5; 10 mM NaCl; 5mM MgCl2; 0.1 mM EGTA; 1X complete protease inhibitor; Roche #11836145001). Pelleted nuclei were re-suspended in 0.5 mL 1.2x restriction enzyme buffer with 0.3% SDS and incubated for 1 hour at 37°C while shaking at 900 rpm, followed by the addition of Triton X-100 to 2% and incubated for 1 hour at 37°C while shaking. 400 U of EcoRI (NEB #R0101L) was added, and cells were incubated overnight at 37°C with shaking. SDS was added to 1.6%, and samples were incubated at 65°C for 20 minutes with shaking. Ligation of the digested DNA was carried out by adding 6.125 ml of 1.15x ligation buffer (10X: 660 mM Tris-HCl, pH 7.5; 50 mM DTT, 50 mM MgCl2, 10 mM ATP) and adding Triton X-100 to a final concentration of 1%, followed by incubation for 1 hour at 37°C with gentle shaking. Samples were incubated with 100 U of T4 DNA ligase for 4 hours at 16°C followed by 30 minutes at room temperature. Samples were then de-crosslinked overnight at 65°C with 300 μg proteinase K. RNase A was added to a final amount of 300 μg and samples were incubated for 30 minutes at 37°C. DNA was purified using phenol/chloroform extraction and re-suspended in 150 μl of 10 mM Tris pH 7.5. DNA was quantified by SYBR green real-time quantitative PCR on the Roche LightCycler® 96 on 50X diluted 3C DNA and serial dilutions of reference DNA of known concentration using internal primer sets that do not amplify across EcoRI cut sites.
The 3C control template was prepared by obtaining DNA from a single BAC clone spanning the region of interest (CTD-2028A10 for HEATR2 and RP11-439P13 for NFYA from BACPAC resources at Children’s Hospital Oakland Research Institute, https://bacpac.chori.org) followed by digestion with EcoRI. The digested DNA was purified using phenol/chloroform extraction, ligated, and subsequently purified as before. For the control template and 3C template, EcoRI restriction enzyme digestion efficiency was confirmed to be > 80% efficient at each genomic fragments in HCT116 and DKO cells. 25ng of 3C DNA was used per reaction for quantification of the ligated fragments using real-time quantitative PCR on the Roche LightCycler® 96. Standard curves were generated using serial dilutions of the 3C control template spanning 25 ng DNA concentration and run in parallel with 3C experimental samples. Interaction frequencies were calculated accordingly using the slope and intercept of the standard curve generated from the 3C control template for each primer set used (interaction frequency = 10(Ct-b)/a where b=intercept, a=slope, and Ct=Ct of the 3C DNA). Subsequent values were normalized to an internal loading control using primer sets that do not amplify across EcoRI cut sites as well as to a positive control using primer sets detecting a CTCF interaction that was common and robust across both HCT116 and DKO cell lines and unaffected by DNA methylation. All data points were an average of three independent 3C experiments with the qPCR performed in duplicate. The primers used in qPCR are listed in Table S7.
CTCF motif deletion in HCT116 cells
Guide RNAs (gRNAs) to target CTCF motif in the NFYA 3' UTR sequence (chr6:41,068,752-41,068,886) were designed using crispr.mit.edu. Potential guide RNAs were screened for their specificity to the target region using BLAST, and the two guide RNAs used in the experiment are listed in Table S7. The D10A mutation was generated on the lentiCRISPR v2 (Addgene plasmid #52961) backbone, which converts the nuclease to a nickase (Jinek et al., 2012). Annealed gRNAs were cloned into lentiCRISPR v2_D10A plasmid using the BbsI restriction enzyme (NEB #R0539S). A total of 10 μg of plasmids/gRNA were transfected into 70% confluent HCT116 cells using lipofectamine-2000 (Thermo #11668019) following the manufacturer’s protocol. Cells were diluted in 1:20 ratio at 48hrs post-transfection into media containing 1 μg/mL of puromycin (Life technologies #A1113803). Single clones were picked and screened for the deletion of CTCF motifs by Sanger sequencing.
Metabolic labeling of RNA for extraction of de novo transcript
AIDR-HCT116 cells were treated with 1 μM DAC for 72 hrs, 500 nM of 3-indoleacetic acid (IAA, Sigma #45533) for 8 hours, or a combination of both DAC and IAA at the above specified concentrations and durations. 2 hours before harvesting the cells, 100 μM of 4-thiouridine (4sU; Sigma #13957-31-8) was supplemented in the media to allow incorporation into newly transcribed mRNA. Total RNA was extracted by TRIzol (Ambion #15596026), and a fraction was saved as “Total” fraction. 4sU-labeled RNA were isolated as previously described (Radle et al., 2013). Briefly, 100 μg of total RNA was biotinylated using 2 mg/ml of EZ-link HPDP-Biotin (Thermo Fisher Scientific #A35390), extracted using phenol/chloroform three times in phase-lock tubes, and precipitated with ethanol. The biotinylated RNA, which consisted of 4sU-labeled RNA, was captured using Dynabeads™ Myone Streptavidin C1 (Thermo Fisher Scientific #65001). Unbound fraction was collected as “Flowthrough” fraction, and bound biotinylated RNA was eluted with DTT as “New transcript” fraction. 1 μg of RNA from each fraction was converted to cDNA for poly(A) isoform expression quantification.
Correlation analysis of The Cancer Genome Atlas dataset
Matched mRNA-seq BAM files and Infinium HumanMethylation 450k BeadChip array data for 5,284 patients across 11 cancer types from TCGA were used for correlation analysis between poly(A) isoform usage and DNA methylation. In order to infer poly(A) isoform usage for a gene, we manually defined a genomic region unique of an isoform of interest (region “A”) and another shared by all isoforms (region “B) for all 546 APA candidate genes (Table S6). Average coverage was extracted from BAM files for both “A” and “B” regions, and the ratio A/B was calculated as the poly(A) isoform usage for each gene. The analysis was restricted to genes deemed to be confidently expressed in each sample (within the top 8,000 genes ranked by the FPKM-UQ value already available from National Cancer Institute Genomic Data Commons). Cases in which the mean depth in region “A” is greater than in region “B” were excluded from further analysis. Normalized DNA methylation beta (β) values were obtained using the beta mixture quantile dilation (BMIQ) method. Only samples with valid poly(A) isoform usage ratios and β values were included in the correlation analysis. Individual β values for DNA methylation probes within ± 5 kb of each TU were plotted against the isoform usage ratio for the same TU within each cancer cohort or across all cohorts (merged set). Pearson correlation with Benjamini-Hochberg adjusted p-values was computed to quantify the dependence between DNA methylation and isoform usage.
Quantification and Statistical Analysis
Statistical analysis
All statistical tests and numbers of biological replicates are listed in the figure legends. To compare statistical significance between mean values of biological replicates in ChIP-qPCR and qRT-PCR, two-way ANOVA tests were used. To compare statistical significance between mean valuates of biological replicates in 3C assays, two-tailed unpaired t-tests were used. All statistical tests were performed with GraphPad Prism 7 (GraphPad Software).
Data and code availability
Raw and processed sequencing data have been deposited in the Gene Expression Omnibus under accession numbers GSE86178 (poly(A)-seq) and GSE131606 (ChIP-seq). The analysis code is available at https://github.com/hwanglab/apa_atingLab2019.
Supplementary Material
Table S7: PCR primers (Related to Star Methods).
Table S2: Poly(A) sites in HCT116 and DKO cells (Related to Figure 1).
Table S3: APA candidates in HCT116 vs. DKO comparison (Related to Figure 1).
Table S5: NMF-based consensus clustering of dynamic ChIP-seq signals (Related to Figure 5).
Table S6: Correlation between isoform usage and DNA methylation in TCGA datasets (Related to Figure 5).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ACTB antibody produced in rabbit | Sigma-Aldrich | Cat# AV40173, RRID:AB_1844540 |
| CTCF Antibody | Cell Signaling Technology | Cat# 2899, RRID:AB_2086794 |
| Anti-DNMT1 antibody produced in rabbit | Sigma-Aldrich | Cat# D4692, RRID:AB_262096 |
| Anti-HEATR2 antibody produced in rabbit | Proteintech | Cat# 24578-1-AP RRID: AB_2827668 |
| Anti-NFYA antibody produced in rabbit | Abcam | Cat# ab139402 RRID: AB_2827669 |
| Anti-RAD21 antibody produced in rabbit | Abcam | Cat# ab996 RRID: AB_2176601 |
| RNA pol II antibody | Active motif | Cat# 39097, RRID:AB_2732926 |
| Anti-NUDT21 antibody | Abcam | Cat# 183660 RRID: AB_2827670 |
| Bacterial and Virus Strains | ||
| CTD-2028A10_BAC clone | BACPAC_CHORI | N/A |
| RP11-439P13_BAC clone | BACPAC_CHORI | N/A |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-Aza-2′-deoxycytidine | Sigma Aldrich | Cat# A3656 RRID: Not Available |
| 3-indoleacetic acid | Sigma Aldrich | Cat# 45533 RRID: Not Available |
| 4-Thiouridine | Sigma Aldrich | Cat# 13957-31-8 |
| EZ-link HPDP-Biotin | Thermo Fisher Scientific | Cat# A35390 |
| oligo(dT)25 Dynabeads | Thermo Fisher Scientific | Cat# 61002 |
| Critical Commercial Assays | ||
| Gibson Assembly | NEB | Cat# E5510S |
| Topo TA Cloning Kit for Sequencing with One Shot Top10 Chemically Competent E. Coli | Thermofisher | Cat# K457540 |
| AllPrep DNA/RNA Mini Kit | Qiagen | Cat# 80204 |
| Deposited Data | ||
| Raw and analyzed data (Poly(A)-seq) | This paper | GEO: GSE86178 |
| Raw and analyzed data (ChIP-Seq) | This paper | GEO: GSE131606 |
| Raw western blots and Agarose gel images | This paper, Mendeley data | http://dx.doi.org/10.17632/6tstd7xkg8.1 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL247 |
| DKO (clone #2) | Laboratory of Dr. Bert Vogelstein | Rhee et al., 2002 |
| HCT116-mAID-RAD21 | Laboratory of Dr. Masatao T. Kanemaki | Natsume et al., 2016 |
| HCT116- NFYA−/−ΔCTCF | This paper | N/A |
| HCT116- NFYA−/+ΔCTCF | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| Primers for Bisulfite sequencing, methylation specific PCR, ChIP-qPCR, Northern probe, qRTPCR, 3C-qPCR, & CRISPR gRNA, see Table S7 | This paper | N/A |
| Recombinant DNA | ||
| pSC-A-amp/kan | Strataclone PCR Cloning Kit; Agilent | Cat# 240205 |
| lentiCRISPR_V2_D10A | This paper | N/A |
| pGL4.74 | Promega | Cat# E692A |
| Software and Algorithms | ||
| GraphPad Prism 7 for windows for statistical analysis | GraphPad Software | https://www.graphpad.com/ |
| DEXSeq | Anders et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| ChIP-seq data processing | ENCODE pipeline | https://github.com/kundajelab/chipseq_pipeline |
| MANorm | Shao et al., 2012 | http://bcb.dfci.harvard.edu/~gcyuan/MAnorm/MAnorm.htm |
| bowtie | Langmead and Salzberg., 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Other | ||
| Analysis codes | This paper | https://github.com/hwanglab/apa_atingLab2019. |
Highlights.
DNA methylation regulates mRNA alternative cleavage and polyadenylation.
CTCF binds unmethylated CpG islands downstream of proximal poly(A) sites.
CTCF subsequently recruits cohesin complex to form chromatin loops.
Chromatin loops promote usage of proximal poly(A) sites in vitro and in vivo.
Acknowledgment
The authors acknowledge TCGA and the ENCODE Consortium for generating data used in this study. AIDR-HCT116 cells were a generous gift from Dr. Kanemaki. We also thank LRI Computing Services for data and computing resource management and David R. Schumick in the Center for Medical Art and Photography for model illustration. This work is supported by the VeloSano cancer research pilot award, the Developmental Research Program within P50-CA150964, and R01 CA230033 to A.H.T.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Reyes A, and Huber W (2012). Detecting differential usage of exons from RNA-seq data. Genome Res 22, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, and Jones PA (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman EA, and Kelly WG (2014). RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: A tail of two kinases. Nucleus 5, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, and Elnitski L (2019). Aberrant DNA methylation defines isoform usage in cancer, with functional implications. PLoS Comput Biol 15, e1007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley M, Wood AJ, Bohm S, Schulz R, and Oakey RJ (2012). Epigenetic control of alternative mRNA processing at the imprinted Herc3/Nap1l5 locus. Nucleic Acids Res 40, 8917–8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, and Manley JL (2011). Mechanisms and consequences of alternative polyadenylation. Mol Cell 43, 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle CP, Moore DJ, Mali G, zur Lage P, Ait-Lounis A, Schmidts M, Shoemark A, Garcia Munoz A, Halachev MR, Gautier P et al. (2014). HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet 10, e1004577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, and Hentze MW (2004). Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 5, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner A, Manni I, and Piaggio G (2017). NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim Biophys Acta Gene Regul Mech 1860, 604–616. [DOI] [PubMed] [Google Scholar]
- Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, and Forne T (2007). Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc 2, 1722–1733. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016). Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, and Tian B (2013). Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing. Nat Methods 10, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, Thornton KC, Giacalone JC, Albee AJ, Wilson KS, et al. (2012). Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet 91, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Ustione A, Huang T, Firth AL, Pan J, Gunsten SP, Haspel JA, Piston DW, and Brody SL (2018). Establishment of the early cilia preassembly protein complex during motile ciliogenesis. Proc Natl Acad Sci U S A 115, E1221–E1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, and Bird AP (2010). Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, and Bird A (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl, 245–254. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Park H (2008). Toward Faster Nonnegative Matrix Factorization: A New Algorithm and Comparisons. Icdm 2008: Eighth Ieee International Conference on Data Mining, Proceedings, 353–362. [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, and Mayr C (2018). Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 561, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, and Jaenisch R (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. [DOI] [PubMed] [Google Scholar]
- MacDonald CC, and McMahon KW (2010). Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley Interdiscip Rev RNA 1, 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C (2017). Regulation by 3'-Untranslated Regions. Annu Rev Genet 51, 171–194. [DOI] [PubMed] [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momparler RL (2005). Pharmacology of 5-Aza-2'-deoxycytidine (decitabine). Semin Hematol 42, S9–16. [DOI] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, and Golub T (2003). Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Machine Learning 52, 91–118. [Google Scholar]
- Morris AR, Bos A, Diosdado B, Rooijers K, Elkon R, Bolijn AS, Carvalho B, Meijer GA, and Agami R (2012). Alternative cleavage and polyadenylation during colorectal cancer development. Clin Cancer Res 18, 5256–5266. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Zhang B, Bell RJ, Johnson BE, Olshen AB, Sundaram V, Li D, Graham AE, Diaz A, Fouse SD, et al. (2014). Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Res 24, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Neve J, Patel R, Wang Z, Louey A, and Furger AM (2017). Cleavage and polyadenylation: Ending the message expands gene regulation. RNA Biol 14, 865–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, and Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. (2008). Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433. [DOI] [PubMed] [Google Scholar]
- Park HJ, Ji P, Kim S, Xia Z, Rodriguez B, Li L, Su J, Chen K, Masamha CP, Baillat D, et al. (2018). 3' UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat Genet 50, 783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, and Corces VG (2009). CTCF: master weaver of the genome. Cell 137, 1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radle B, Rutkowski AJ, Ruzsics Z, Friedel CC, Koszinowski UH, and Dolken L (2013). Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, Larson DR, and Zhao K (2017). CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell 67, 1049–1058 e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. (2002). DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556. [DOI] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Reinhardt R, and Jeltsch A (2010). BISMA--fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC bioinformatics 11, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Lee BH, and Ting AH (2010). MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res 38, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayevitch R, Askayo D, Keydar I, and Ast G (2018). The importance of DNA methylation of exons on alternative splicing. RNA 24, 1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, and Oberdoerffer S (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2007). New modes of translational control in development, behavior, and disease. Mol Cell 28, 721–729. [DOI] [PubMed] [Google Scholar]
- Sweet TJ, and Ting AH (2016). WOMEN IN CANCER THEMATIC REVIEW: Diverse functions of DNA methylation: implications for prostate cancer and beyond. Endocr Relat Cancer 23, T169–T178. [DOI] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, and Lutz CS (2005). A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Schulz R, Woodfine K, Koltowska K, Beechey CV, Peters J, Bourc'his D, and Oakey RJ (2008). Regulation of alternative polyadenylation by genomic imprinting. Genes Dev 22, 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, and Liang G (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagore LL, Sweet TJ, Hannigan MM, Weyn-Vanhentenryck SM, Jobava R, Hatzoglou M, Zhang C, and Licatalosi DD (2018). DAZL Regulates Germ Cell Survival through a Network of PolyA-Proximal mRNA Interactions. Cell Rep 25, 1225–1240 e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, and Zilberman D (2010). Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol 20, R780–785. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lee JY, and Tian B (2005). Biased alternative polyadenylation in human tissues. Genome Biol 6, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S7: PCR primers (Related to Star Methods).
Table S2: Poly(A) sites in HCT116 and DKO cells (Related to Figure 1).
Table S3: APA candidates in HCT116 vs. DKO comparison (Related to Figure 1).
Table S5: NMF-based consensus clustering of dynamic ChIP-seq signals (Related to Figure 5).
Table S6: Correlation between isoform usage and DNA methylation in TCGA datasets (Related to Figure 5).
Data Availability Statement
Raw and processed sequencing data have been deposited in the Gene Expression Omnibus under accession numbers GSE86178 (poly(A)-seq) and GSE131606 (ChIP-seq). The analysis code is available at https://github.com/hwanglab/apa_atingLab2019.