

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disease that causes chronic cognitive dysfunction. Most of the AD cases are late onset, and the apolipoprotein E (APOE) isoform is a key genetic risk factor. The APOE gene has 3 key alleles in humans including APOE2, APOE3, and APOE4. Among them, APOE4 is the most potent genetic risk factor for late-onset AD (LOAD), while APOE2 has a defensive effect. Research data suggest that APOE4 leads to the pathogenesis of AD through various processes such as accelerated beta-amyloid aggregations that raised neurofibrillary tangle formation, cerebrovascular diseases, aggravated neuroinflammation, and synaptic loss. However, the precise mode of actions regarding in what way APOE4 leads to AD pathology remains unclear. Since APOE contributes to several pathological pathways of AD, targeting APOE4 might serve as a promising strategy for the development of novel drugs to combat AD. In this review, we focus on the recent studies about APOE4-targeted therapeutic strategies that have been advanced in animal models and are being prepared for use in humans for the management of AD.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder in which the death of nerve cells causes memory loss and cognitive decline that is serious enough to interfere with daily life [1–4]. Neuropathologically, AD is characterized by the deposition of extracellular beta-amyloid (Aβ) as well as increased intracellular neurofibrillary tangles along with the activation of glia and neuronal death [5–9]. Besides environmental and lifestyle-related risk factors, genetic constituents are also deliberated to raise the risk of progressing AD [10, 11]. Undeniably, research has detected several loci connected with AD, such as both pathogenic and susceptibility genes. AD is categorized into 2 forms according to the age of onset, including early-onset AD (EOAD) that begins in people who are below 65 years old and it is estimated to be about 1–5% of all cases, and late-onset AD (LOAD) that starts in people aged 65 or above 65 and it is projected to be greater than 95% of all cases [12]. There are 3 leading congenital gene mutations, such as presenilin-1 (PSEN1), presenilin-2 (PSEN2), and amyloid precursor protein (APP), which play a central role in raising the generation of Aβ and have been directly related to EOAD [13–15]. On the contrary, LOAD has been detected to be more complicated and is linked to several genes with increased vulnerability [16]. Genetically, the apolipoprotein E4 (APOE4) allele is considered as the paramount determinant for LOAD [17–20].
People who inherit one copy of the APOE4 allele may cause a higher risk of raising AD, and people who possess two copies of the APOE4 allele are at greater risk of progressing AD [21, 22]. The APOE4 gene may also be accompanied by an earlier onset of memory dysfunction and other symptoms in comparison with AD patients who do not have this gene. It is unknown how the APOE4 allele is connected with the risk of AD. Conversely, the APOE4 gene is also linked with a greater number of protein clusters called amyloid plaques, which are found in the AD brain tissue [23, 24]. Furthermore, an aggregation of amyloid plaques is greatly responsible for the death of neurons and the developing symptoms of AD [25, 26]. It has been found that the interaction between herpes simplex virus type 1 (HSV-1) and APOE isoforms indicate a connection between HSV-1 deoxyribonucleic acid (DNA) detection in AD tissues and the existence of the APOE4 allele [27, 28]. In addition, recent investigations have shown a potential relationship between APOE isoform-dependent modifications in tau pathology and neurodegeneration [29]. Therefore, it is evident that people who have the APOE4 allele inherit a greater risk of progressing AD, not the disease itself [30].
Oxidative stress has also been connected with APOE4 in AD patients. ApoE4 is linked to higher oxidative stress as well as diminished antioxidant enzyme activity in the hippocampus of AD patients [31–33]. Oxidative stress markers such as increased oxidized proteins, glycosylated products, elevated levels of lipid peroxidation, formation of aldehydes, alcohols, ketones, free carbonyls, and cholestenone, as well as oxidative modifications in ribonucleic acid (RNA) and mitochondrial and nuclear DNA were observed in postmortem brain tissue and in peripheral systems such as cells and isolated mitochondria from initial phases of AD and APOE4 carriers [34–51]. On the other hand, APOE was shown to act as an antioxidant directly or indirectly against hydrogen peroxide-mediated cytotoxicity in a B12 APOE expressing cell line [52]. According to the study by Hayek et al. [53], the increased levels of peroxidized plasma low-density lipoproteins in APOE-deficient mice were observed. Furthermore, the levels of lipid oxidation were considerably elevated in the frontal cortex of AD patients who were heterozygous or homozygous APOE4 carriers compared to homozygous APOE3 carriers and controls [54]. Although upregulation of catalase activity was completely found in the frontal cortex tissue of homozygous APOE4 carriers, the activities of superoxide dismutase and glutathione concentrations were not as different as those from controls [54].
Hitherto, there are no approved drugs directly targeting APOE4, even though APOE4 was detected about 25 years ago [55–57]. Hence, due to its genetic predominance, APOE isoforms have turned into an auspicious target for better understanding the pathophysiological pathways of AD, identifying patients who are at greater risk for the progression of AD and opening a novel therapeutic approach against AD. Moreover, some clinical researches both in animals and humans have verified that APOE4 remarkably affects the various independent biological pathways in the brain which play a pivotal role in the development of AD [55, 58]. In this review, we emphasize the current studies regarding APOE4-targeted promising therapeutic strategies to combat AD pathogenesis.
2. APOE—Polymorphism and Susceptibility to Alzheimer's Disease
APOE is a chylomicron lipoprotein that is necessary for the transport of lipids and the metabolism of lipoproteins, and it is encoded by the APOE gene which is situated in the long arm of chromosome 19q13.2 [59, 60] (Figure 1). The APOE gene has 3 variants, called APOE2, APOE3, and APOE4, which are present at ~7%, 79%, and ~14%, respectively in the whole populace, and these show dissimilarities in lipid- as well as receptor-binding efficiency. The APOE allelic proteins vary by merely 1 or 2 amino acids including cysteine and arginine at residues 112 and 158, with APOE2 (cys112, cys158), APOE3 (cys112, arg158), and APOE4 (arg112, arg158) [61, 62]. Although the APOE2 gene is associated with type III hyperlipoproteinemia, however, it has a defensive effect against AD. The APOE3 is the most predominant allele and doesn't seem to influence risk. Conversely, the APOE4 gene is connected with a greater risk for AD and coronary artery disease [63, 64]. The morphology of the APOE4 protein reduces the capability of APOE4 to remove the Aβ protein from the brain, causing the development of AD. The existence of one copy of the APOE4 allele raises the AD risk by 3 times, whereas people who inherit two copies of APOE4 alleles are 8 times more likely to progress with AD in comparison with those lacking any APOE4 allele [56]. These data recommend that APOE4 is the highest identified genetic risk factor for AD compared to any other genes so far.
Figure 1.
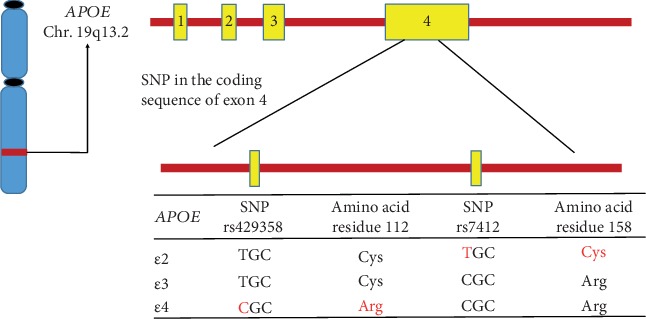
Schematic presentation of the human APOE genotype and APOE polymorphisms. The human APOE gene is situated in the long arm of chromosome 19. In exon 4 of chromosome 19, two nonsynonymous single nucleotide polymorphisms (SNPs) including rs429358 and rs7412 produce 3 main allelic variants (E2, E3, and E4). The resulting APOE2, APOE3, and APOE4 isoforms vary from one another at amino acid residues 112 or 158.
3. APOE-Targeted Treatment for Alzheimer's Disease
It has been suggested that the APOE4 allele considerably alters various biological pathways toward vulnerable conditions for the progression of AD. It is evident that APOE modifies multiple biological pathways by its analogous protein APOE, whereas the key mechanism connecting with APOE4 and neurodegeneration until now remains unclear. Hence, alterations in the APOE allele, along with APOE structures, are fortunate targets for novel drug design and treatment for AD. In this appraisal, we emphasize on the aspects of APOE4 for which therapeutic strategies are being developed (Figure 2). There are several comprehensive studies demonstrating the molecular mechanisms underlying the effects of APOE4 [65–69]. Furthermore, some therapeutic strategies for targeting APOE in AD [70] are being undertaken in the scientific community (Table 1). We emphasized the diverse characteristics of APOE4 via which therapeutic strategies are advanced, and these are discussed below.
Figure 2.
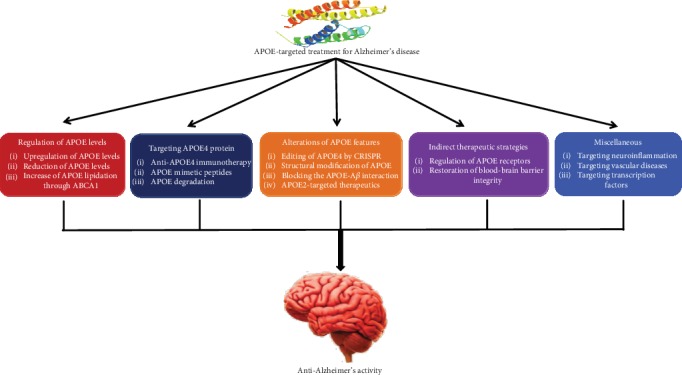
Classification of APOE-targeted treatment strategies for Alzheimer's disease.
Table 1.
Outline of therapeutic agents for targeting APOE4 for Alzheimer's disease.
| Therapeutic strategies | Principle | Studies | References | |
|---|---|---|---|---|
| Animal models | Humans | |||
| RXR, LXR, PPARγ agonists | Increases the lipidation of APOE and promotes Aβ clearance | Mouse | Yes | [75–90, 91, 98] |
| Anti-APOE4 monoclonal antibody | Increases amyloid clearance and decreases APOE-associated toxic effects | Yes | [99–102, 122, 196] | |
| Small peptides comprising the receptor-binding region in APOE | Reduces inflammation and neurotoxicity, increases APOE3-linked protective functions | Mouse | No | [123, 132, 133, 197, 198] |
| Small molecules | Increases Aβ clearance, APOE signaling, and cholesterol transport | Mouse | No | [175, 176] |
| APOE4 structure correctors (GIND25 and PH002) | Interferes with domain-domain interaction in APOE4 thus reducing its toxic effects | No | [145–147] | |
| Viral-mediated APOE2 expression | Enhances APOE-connected neurodefensive effects | Mouse | Yes | [166, 169, 199] |
| Aβ12-28P, Aβ20-29 peptide, small-molecule inhibitors | Increases amyloid clearance | Mouse | No | [153–157] |
| Cyclosporine A | Decreases leakage of blood-derived toxic molecules in APOE4-carrying brain | Mouse | Yes | [179, 200] |
3.1. Regulation of APOE Levels
3.1.1. Upregulation of APOE Levels
Several studies for the hallmarks of AD have revealed contradictory outcomes regarding whether the quantity of APOE in plasma and CSF are decreased in AD patients in comparison with healthy individuals [71–74]. However, many animal studies demonstrated the therapeutic potential of compounds that raise APOE levels in the brain [75–91]. APOE transcription is positively controlled by retinoid X receptors (RXRs) and nuclear receptors, as well as liver X receptors (LXRs) which generate heterodimers [92]. Actually, the oral intake of an RXR agonist including bexarotene raised the levels of APOE in the brain, decreased the accumulation of Aβ, and enhanced cognitive abilities in the amyloid mice model [76]. Furthermore, bexarotene has already been recognized as a drug by the Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma [93–96], and the emerging application of this drug for the treatment of AD has been receiving the attention of numerous researchers for many years. Clinical research recommended that bexarotene considerably reduced cognitive dysfunction in the amyloid model mouse, which expresses human APOE3 and APOE4 [77], as well as returned the APOE4-mediated neuronal and cognitive dysfunctions in mice devoid of amyloid background [75]. In addition, bexarotene is also useful for the restoration of age-dependent synaptic proteins' loss [97]. Conversely, there are contradictory data about the negative effects of bexarotene therapy on amyloid pathology in animal studies [77, 79, 81, 82, 97]. Additionally, bexarotene showed adverse effects such as hepatic failure in mice models [78, 80]. A study by Cummings et al. [98] demonstrated that 4 weeks of bexarotene therapy in the AD human model did not diminish amyloid plaque in the brain as determined by positron emission tomography scans, although the size of the sample was noticeably small. Hence, although an RXR agonist including bexarotene has a promising effect on stopping the pathogenesis of AD, further evaluations as well as dosage optimization are necessary for its emerging therapeutic use for the treatment of AD.
3.1.2. Reduction of APOE Levels
Current studies have claimed that APOE haploinsufficiency in APOE-targeted-replacement (TR) mice weakens the accumulation of Aβ despite APOE genotypes [99, 100]. It is evident that the intraperitoneal injection of an anti-APOE antibody has also been exposed to enhance spatial learning ability by decreasing the quantity of soluble APOE in the brain and slowing the accumulation of Aβ in the brain in an amyloid animal model with no obvious side effects [101, 102]. Although the reduction of APOE levels may be useful for ameliorating the pathology of Aβ; however, this strategy must be justified cautiously for clinical use because the lack of APOE can cause severe hyperlipidemia as reported to take place in an APOE-lacking individual homozygous for an ablative APOE frameshift mutation [103].
APOE4 is widely accepted as an isoform that leads to the pathogenesis of AD by not only gain-of-functional but also loss-of-functional characteristics in comparison with APOE3 [19, 104]. Therefore, these bidirectional properties of APOE4 are necessary to be deliberated upon for advancing favorable APOE-targeted treatments for AD. Furthermore, targeted removal of APOE4 will be a hopeful approach to lessen its toxicity. A study by Luz et al. [105] showed that anti-APOE4 monoclonal antibody (9D11) therapy might prevent the APOE4-induced cognitive damage and the hyperphosphorylation of tau in the brain in APOE4 mice, even though follow-up investigations are mandatory. Moreover, some inactive types of APOE such as APOE fragments and depositions are expected to be detrimental [106–110] where APOE4 possibly has a greater chance of adopting such structural alterations or proteolysis than other polymorphisms [111]. Hence, eliminating APOE depositions but not the original form of functional APOE with the help of immunodepletion can be a different therapeutic approach. Pharmacological strategies to remove APOE fragmentation and depositions might also be considered as potential therapeutic approaches.
3.1.3. Increase of APOE Lipidation through ABCA1
The lipidation condition of APOE considerably affects its activity [112], since the APOE protein (biologically active) is connected with lipids [113, 114]. ATP-binding cassette subfamily A member 1 (ABCA1) is responsible for loading lipid particles on APOE proteins in the brain [115]. The removal of ABCA1 reduces the production of suitably lipidated APOE particles leading to the highly amyloidogenic pathologies in an amyloid mice model [116]. Conversely, ABCA1 overexpression diminishes the accumulation of Aβ in an APOE-dependent way in the amyloid mouse model [117], which recommends a favorable role of ABCA1-induced APOE lipidation for the stopping of the AD pathogenesis (Figure 3). As depicted, those agonists for RXRs and LXRs could lessen the accumulation of Aβ and improve cognition in an amyloid mice model and these processes regulate the transcription of APOE and ABCA1 [77, 84, 91]. Furthermore, lipid transport to cells takes place as APOE is endocytosed through members of the low-density lipoprotein receptor (LDLR) family [118]; endocytosis enhances neurite outgrowth [119], synapse formation [120], and neuronal sprouting [121]. Additionally, recent investigations have found that lipidated APOE plays a pivotal role in demonstrating various functions, such as lipid/cholesterol transport, regeneration of synapse, immune modulation, and clearance of Aβ [85, 102, 122, 123]. Therefore, these examinations advocate the idea that raising the lipidation of APOE may be a useful strategy for the treatment of AD, even though it might be arduous to control the lipidated condition without changing APOE levels.
Figure 3.
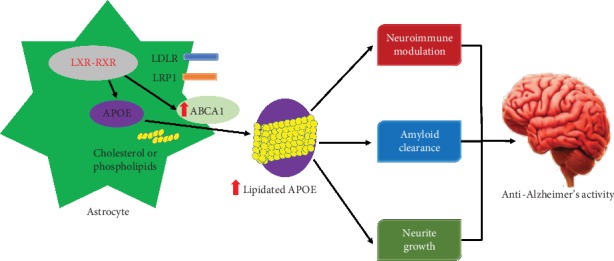
Increasing lipidation of the astrocytic pool of APOE. APOE is principally produced by astrocytes in the brain. Activation of nuclear hormone receptors including LXR and RXR plays an essential role in the expression of APOE and ABCA1. Moreover, ABCA1 is very important for the lipidation of APOE and increasing the expression of ABCA1 contributes to enhanced lipidation of all APOE polymorphisms (indicated by red arrows). It is assumed that enhanced lipidation of APOE will be valuable for several AD-relevant endpoints.
Furthermore, APOE4 is less lipidated than APOE2 and APOE3 both in APOE-TR mice [124, 125] and in humans [126]. Mounting evidence recommends that lipid-driven pathways may contribute to the harmful characteristics of APOE4 [127]. A study by Boehm-Cagan and Michaelson [75] demonstrated that RXR agonists including bexarotene were used in the treatment of APOE4-TR mice resulting in reversed APOE4-mediated neuronal and cognitive deficits apparently by raising APOE4 lipidation. Hence, altering the lipidation condition of APOE4 appears to be a potential therapeutic strategy [128], although the LXR/RXRs-ABCA1 axis activation can possibly raise the levels of APOE4, thus increasing the gain-of-toxic functions of APOE4 [19].
3.2. Targeting APOE4 Protein
3.2.1. Anti-APOE4 Immunotherapy
The fundamental principle of APOE4 immunotherapy is close to that applied in the immunotherapy of tau and Aβ [129], specifically to produce or introduce antibodies against these molecules in the periphery that can neutralize their target (this strategy presumes a toxic effect of APOE4) after their penetration into the brain. In theory, the use of immunotherapy to APOE is encountered by the difficulty that the APOE level in the periphery is about 10-times greater than that in the brain [130], as a result, anti-APOE antibodies must be titrated out in the periphery prior to reaching the brain. Many studies demonstrated that the peripheral use of anti-mouse APOE antibodies in APP transgenic (TR) mice can suppress the amyloid deposition before the beginning of plaque and reduce its deposition after the formation of plaque [101, 102]. Despite the mode of actions involving these pivotal effects of the anti-APOE monoclonal antibodies, these outcomes have an enormous significance and offer a fundamental idea about the reliability of anti-APOE4 immunotherapy as a promising therapeutic strategy. Furthermore, this strategy has now been expanded to APOE3- and APOE4-directed mice using an antibody that reacts particularly with APOE4 [131]. This reveals that repetitive intraperitoneal injection of these antibodies in mice leads to their aggregation in the brain and also in the generation of APOE/IgG complexes, especially in APOE4 mice. Moreover, this was connected with the restoration of cognitive damages in APOE4 mice as well as with the restoration of central synaptic and AD-associated pathological effects of APOE4 [131].
3.2.2. APOE Mimetic Peptides
Using of APOE mimetic peptides represent a further therapeutic strategy against AD. Furthermore, these tiny peptides, which either conform to the receptor-binding APOE domain [132–134] or to a discrete domain of APOE including amphipathic helix domains [134], significantly lessen the neurodegeneration after brain insults [133, 135–138] and defend against tau- and Aβ-mediated pathogenesis in TR mice and analogous model domains [132–134]. The fundamental mechanism of the defensive effects of these tiny peptides might be owing to their anti-inflammatory effects. Conversely, a study of Vitek et al. [133] revealed that these APOE mimetic peptides were defensive after brain insults in both APOE3 and APOE4 mice.
3.2.3. APOE Degradation
APOE4 generates an intermediate molten globule structure, which makes it less stable when compared with APOE3 and its C- and N-terminal interaction as described earlier. Furthermore, this domain interaction in neurons makes APOE4 particularly vulnerable to discrete proteases and responsible for the production of C-terminal neurotoxic portions of APOE4 [67, 139–141]. As stress raises the neuronal APOE generation, it has been anticipated that the raised generation of intraneuronal APOE4 portions under stressful states may play a pivotal role in inducing the pathogenic actions of APOE4 [67, 139–141]. Hence, the neuronal degradation of APOE4 leads to the detection of the proteases as well as the advancement of inhibitors against them demonstrating an additional strategy for neutralizing the actions of APOE4.
3.3. Alterations of APOE Features
3.3.1. Editing of APOE4 by CRISPR
The transformation of the APOE4 gene to either APOE2 or APOE3 and the abolition of the concentration dissimilarity between them would play an important role in solving the crux of the APOE4 problem in spite of the poor understanding of the mechanisms regarding the effects of APOE4. It was quite impossible to enable the exact gene editing before the advancement of the clustered regularly interspaced short palindromic repeats (CRISPR) gene-editing method [142]. Furthermore, this method is specifically the opposite for the APOE gene, in which the coding of DNA for APOE4 varies from that of APOE3, which is considered as the benign gene for AD, by merely 1 nucleotide (explicitly, the 112th position is cysteine in APOE3 and arginine in the case of APOE4). Particularly, the CRISPR method can be used for transforming the APOE4 gene into APOE3 as given in Figure 4. Conversely, it can also be applied in an APOE4-knockout model where APOE3 homozygote mice is transformed by APOE3/APOE4 heterozygote mice, which would be predicted to be defensive when a noxious effect of APOE4 is supposed. Moreover, in a study of Komor et al. [143], CRISPR cell culture research demonstrated the precise transformation of APOE4 to an APOE3 derivative, and this method was also used to silence APOE4 devoid of influencing the expression of APOE3 [144]. The later strategy might therapeutically work against the assumed toxicity related to APOE4. In contrast, effective in vivo use of CRISPR to APOE4 mice has not been established so far. Moreover, it is very essential to remember that the CRISPR method is in its initial stages and data are still promising about probable off-target gene modification and mosaicism, where not all copies of the target gene are edited.
Figure 4.
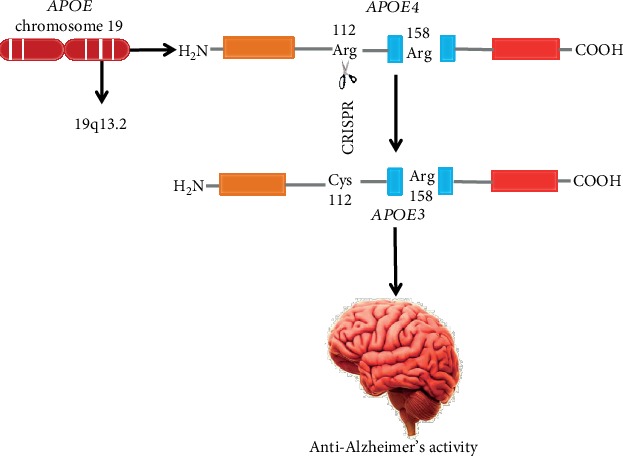
Editing of APOE4 by CRISPR to generate an APOE3-like structure. The APOE4 allele is generally produced by APOE chromosome 19 (19q13.2). The coding of DNA for APOE4 varies from APOE3, which is considered as the benign gene for AD, by only 1 nucleotide (explicitly, the 112th position is cysteine in APOE3 and arginine in the case of APOE4). Particularly, the CRISPR method could be used for converting the APOE4 gene into APOE3.
3.3.2. Structural Modification of APOE
In the N-terminal, the amino acid residues 112 and 158 are the markers of APOE polymorphisms; however, the interaction between residues Glu255 and Arg61 is possibly a further characteristic, which structurally differentiates APOE4 from APOE3. Furthermore, this domain-domain interaction probably produces an aberrant structure in APOE4 that leads to neurotoxicity [145], even though the structure of the complete, native APOE has not been examined until now. In neuronal N2A cells, the expression of APOE4 triggers mitochondrial dysfunction, although this effect is abolished in an APOE4 mutant lacking the interaction of domain (APOE4-R61T) [146]. Moreover, various molecular compounds (PH002 and GIND25) have been accounted to counter harmful actions of APOE4 by obstructing the domain-domain interaction [147] as shown in Figure 5. Hence, altering the pathological conformation of APOE4 including the domain-domain interaction in neurons might be a potential therapeutic strategy for the treatment of AD. Conversely, the effectiveness of these compounds on AD-linked processes has not been determined in vivo so far.
Figure 5.
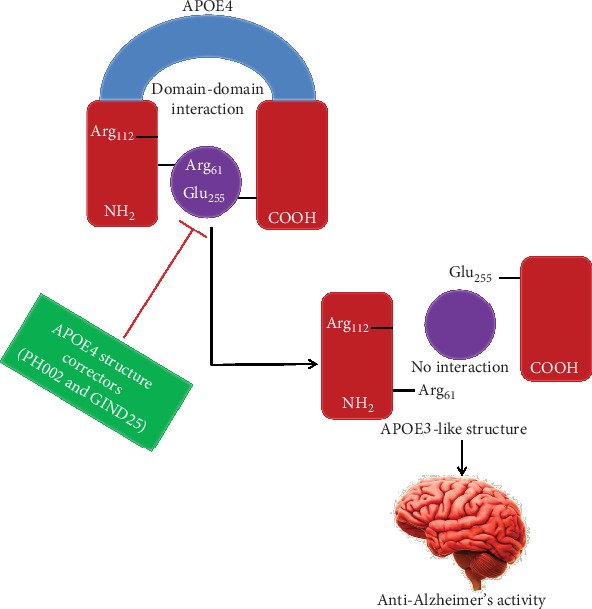
APOE4 structure correctors can disrupt ApoE4 domain-domain interaction. In the N-terminal domain, Arg-61 interacts with Glu-255 in the C-terminal domain in APOE4. APOE4 structure correctors (PH002 and GIND25) that are expected to interact with APOE4 in the region of Arg-61 would interrupt domain-domain interaction and transform APOE4 to an APOE3-like molecule.
On the other hand, AD might be influenced both by the sequence of DNA and by epigenetic profiles in the APOE region. Remarkably, epigenetic mechanisms, including DNA methylation, histone modification, and noncoding RNA can control the expression of the gene whereas the fundamental sequence of DNA remains the same. The epigenome is also influenced both by basic genetic variants and by environmental factors such as environmental pollutants, social environment, and health behaviors [148]. Methylation of cytosine-phosphate-guanine (CpG) dinucleotides is the best understood epigenetic mechanism. It has been recognized that epigenomic patterns of DNA methylation alter with age [149]. In a study by Ma et al. [150], reported that methylation levels at different CpG sites in APOE were considerably connected with age in lymphocytes, and these age-related alterations in DNA methylation were altered by APOE genetic variants. Thus, epigenetic regulation can be a strategy to combat AD pathogenesis.
3.3.3. Blocking the APOE-Aβ Interaction
Although the interaction of APOE with Aβ might be negligible under physiological states [151], Aβ and APOE colocalize in amyloid plaques in AD brains [152]. It is widely accepted that the interaction between Aβ and APOE induces the accumulation of Aβ in human brains [5]. Undeniably, suppression of the interaction of APOE with Aβ by an artificial peptide (Aβ 12-28P, analogous to the APOE binding site on the complete Aβ) played a crucial role in reducing the aggregation of Aβ and intraneuronal deposition of Aβ, and improved memory dysfunctions in amyloid animal models [153, 154]. According to the study of Liu et al. [155] on the 3xTg AD mouse model, tau and Aβ pathology were both sheltered and the interaction of APOE with Aβ was obstructed by Aβ 12-28P, resulting in the diminished accumulation of Aβ and the aggregation of insoluble tau in AD brain. Moreover, the therapy with Aβ 12-28P played an essential role in decreasing the oligomers of Aβ and a load of amyloid plaque also improved neuritic deterioration in the amyloid model mouse with an APOE4-TR or APOE2-TR mouse background [156]. Therefore, suppression of the interaction between Aβ and APOE seems to be favorable for stopping the accumulation of Aβ despite the APOE polymorphisms, although the pharmacological effect of artificial peptides may vary according to the APOE polymorphism being targeted. Additionally, Hao et al. [157] demonstrated that Aβ 20–29 peptides might obstruct the interaction of APOE with Aβ, thus decreasing the fibrillogenesis as well as cytotoxicity of Aβin vitro. Captivatingly, immunotherapy of APOE noticeably reduces the accumulation of Aβ in amyloid mouse models as discussed earlier [101, 102]. Recently, in an in vivo test, using antibodies that accept both human APOE3 and APOE4 and then combining specifically to nonlipidated APOE rather than lipidated APOE result in diminishing the accumulation of Aβ in a TR mice model [158]. In addition, APOE antisense oligonucleotides are also used for the lessening of amyloid plaques [159]. It is evident that APOE3 and APOE4 bind diversely and directly to Aβ [125]; however, a study by Verghese et al. [151] revealed that the interaction of APOE4 with Aβ might be indirect as well as intervened through a third molecule. Therefore, blocking the interaction between APOE4 and Aβ may be a promising therapeutic strategy for the treatment of AD.
3.3.4. APOE2-Targeted Therapeutics
The occurrence of APOE2 in AD patients is about 2-times lower than the general populace, and it is connected with less noticeable brain pathology than that found in non-APOE2 AD subjects [160]. The heterozygosity of APOE2 is also linked with longevity [160] and lessened age-dependent cognitive dysfunction [161–164]. In addition, APOE2 increases the neuroprotective activity against AD via various molecular mechanisms [165]. Consequently, in the case of neurodegenerative disorders that are directly linked with neuronal and synaptic loss, APOE2 is defensive because of its capability to excite the restoration of these pathways. Various investigations recommended that the brain pathological effects of APOE4 in TR mice could be counteracted by the injection (intracerebral) of viral vectors expressing APOE2 [166, 167], recommending a new anti-APOE4 therapeutic strategy [168]. According to the study of Hu et al. [169], APOE4 is hypolipidated compared with APOE3 and APOE2 is hyperlipidated in relation to APOE3. Moreover, it is evident that APOE2 and APOE4 affect the identical pathway such as lipidation of APOE; however, they drive in opposite directions. In contrast, the probability that APOE4 and APOE2 activate through diverse nonoverlapping processes with contrasting physiological outcomes could not be denied.
3.4. Indirect Therapeutic Strategies
3.4.1. Regulation of APOE Receptors
The LDLR family, including LDLR and low-density lipoprotein receptor-related protein 1 (LRP1) [170], plays a central role in mediating the endocytosis of APOE. APOE accelerates the cellular uptake of Aβ via these receptors either by producing the complexes of APOE/Aβ or by inhibiting the Aβ interaction through competition for the binding receptor. Furthermore, diverse mechanisms including APOE isoform-dependent, lipidation-status-dependent, and concentration-dependent mechanisms [112, 170] are responsible for complicating these events. Captivatingly, aggregating data have demonstrated the crucial roles of LDLR as well as LRP1 in the clearance of Aβ in the brain and the metabolism of lipids [171–174]; however, how APOE mediated these phenotypes remains unclear. Therefore, raising LDLR and LRP1 can be considered as a therapeutic strategy to trigger the clearance processes of Aβ in human AD. Undeniably, some compounds such as fluvastatin, caffeine, and rifampicin probably have defensive actions for the management of AD by mounting levels of LRP1 [175, 176]. Moreover, LRP1 and a different APOE receptor including APOE receptor 2 (APOER2) are playing key roles in controlling synaptic activities [170, 177]. On the other hand, a large extracellular matrix glycoprotein, for example, reelin, improves the activity of synaptic glutamate receptors via APOER2, and APOE4 interrupts this process by damaging the recycling process of APOER2 [178]. A study by Gilat-Frenkel et al. [69] showed that the levels of APOER2 in the brain hippocampus are also decreased particularly in APOE4-TR mice. Hence, controlling the levels of APOER2 can serve as an APOE-driven anti-AD treatment, even though additional researches are required.
3.4.2. Restoration of Blood-Brain Barrier Integrity
According to the study of Bell et al. [179], the expression of APOE4 in astrocytes activated the proinflammatory cyclophilin A- (CyA-) nuclear factor κB-matrix metalloproteinase 9 (MMP-9) processes in brain pericytes, thus resulting in the degradation of the tight junction protein and the breakdown of the blood-brain barrier (BBB) in an animal model. Prominently, these pathologies of BBB in APOE4-TR mice were restored by the treatment of cyclosporine A [179]. Age-dependent BBB breakdown as determined by the QAlb index along with raised CyA and active levels of MMP-9 were identified in CSF from cognitively regular APOE4 carriers [179]. Therefore, cyclosporine A may be considered as a therapeutic agent for the treatment of AD, whereas it is still debatable whether APOE4-driven BBB breakdown is adequate to change the worldwide homeostatic capability of BBB leading to the pathogenesis of AD [180, 181].
3.5. Miscellaneous
3.5.1. Targeting Neuroinflammation
Various inflammation-related targets have been recommended, such as microglia, where the current detection of gene expression patterns connected with diverse phases of the activation of microglia presents new targets wherein microglial activation can be controlled [182, 183] and which have been demonstrated to be useful in neurodegeneration-linked models [184, 185]. Furthermore, these advancements and the relationship of APOE4 with raised neuroinflammation recommend that inflammation-linked therapies can be specifically favorable in APOE4 carriers. Conversely, neuroinflammation is a double-edged sword, supposed to be defensive at initial phases and pathological at consequent chronic phases. The use of APOE4- and AD-linked immunotherapeutic approaches is thereby anticipated to be dependent on the inflammatory reaction by which AD patients are treated. In addition, this may differ in diverse brain regions. Therefore, novel hallmarks that detect the phase and brain area of neuroinflammation are required to solve this matter.
3.5.2. Targeting Vascular Diseases
There are some vascular risk factors including atherosclerosis, diabetes, and hypertension that raise the AD risk [186, 187]. Furthermore, APOE4 is connected with the raised risk for atherosclerosis and vascular dementia [188, 189] as well as with damaged vasculature integrity and BBB [189]; therefore, it is suggested that the role of APOE4 in the pathogenesis of AD may be mediated partly by a vascular constituent. Hence, the detection of the molecules by which the AD-linked vascular effects of APOE4 are driven, and which can, therefore, be considered as an AD-APOE4 vascular therapeutic approach, remains presently unclear [186]. Conversely, as significant features of vascular diseases could be treated pharmacologically and by lifestyle changes [190], such strategies are anticipated to lessen the role of vascular and APOE4/vascular pathology to AD.
3.5.3. Targeting Transcription Factors
Generally, the pathological mechanisms of APOE4 are mediated extracellularly or through membrane transport and cytosolic pathways, but it has currently been recommended that APOE4 also goes through nuclear translocation and it combines particularly with high affinity to copious DNA sites [191]. Most of these sites are located in promoter areas, recommending that APOE4 can work as a transcription factor for a lot of diverse genes such as autophagy and growth factor-related genes [192, 193]. A study by Rohn [194] demonstrated that APOE4 is contained in the nucleus and this pathway is associated with a particular proteolytic APOE4 degradation. Furthermore, these investigations and outcomes suggested that APOE4 combines with the genes' promoters involved in diverse pathways that are linked with aging and AD [195] contributing to the exciting recommendation that APOE4 could serve as a transcription factor. There are many queries that remain to be investigated, including in what way APOE escapes the endoplasmic reticulum and is transferred to the nucleus as well as what is the influence of this mechanism compared with other pathological pathways.
4. Conclusions
In this review, we discuss several APOE4-mediated strategies, varying from the APOE gene to the APOE protein and its interacting molecules not only in cellular but also in animal model systems. These investigational strategies have been advanced to act against the pathological effects of APOE4 in the animal model. The role of APOE4 in raising AD risk is multifaceted, linking a wide range of cell types and activities, which needs to be considered for APOE-mediated drug development. Currently, there is no established proof for the human APOE4-mediated therapeutic investigations and it is expected that developments in animal studies will offer auspicious opportunity for the advancement of novel APOE-targeted treatment for AD.
Acknowledgments
This work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-Track Research Funding Program.
Conflicts of Interest
The authors proclaim no conflict of interest.
Authors' Contributions
AAM and MSU conceived the original idea and designed the outlines of the study. AAM, MSU, and MFBB prepared the draft of the manuscript. AAM and MSU prepared the figures for the manuscript. SZ, YB, IJB, MSI, MSS, BM, MSA, and GMA participated in the literature review of the manuscript. MNB-J, SAM, and MMA-D participated in the revision and improved the manuscript. All authors have read and approved the final manuscript.
References
- 1.Uddin M. S., Mamun A. A., Labu Z. K., Hidalgo-Lanussa O., Barreto G. E., Ashraf G. M. Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. Journal of Cellular Physiology. 2018;234(6):8094–8112. doi: 10.1002/jcp.27588. [DOI] [PubMed] [Google Scholar]
- 2.Al Mamun A., Uddin M. S. KDS2010: a potent highly selective and reversible MAO-B inhibitor to abate Alzheimer’s disease. Combinatorial Chemistry & High Throughput Screening. 2020;23 doi: 10.2174/1386207323666200117103144. [DOI] [PubMed] [Google Scholar]
- 3.Uddin M. S., Mamun A. A., Takeda S., Sarwar M. S., Begum M. M. Analyzing the chance of developing dementia among geriatric people: a cross-sectional pilot study in Bangladesh. Psychogeriatrics. 2018;19(2):87–94. doi: 10.1111/psyg.12368. [DOI] [PubMed] [Google Scholar]
- 4.Hossain M. F., Uddin M. S., Uddin G. M. S., et al. Melatonin in Alzheimer’s disease: a latent endogenous regulator of neurogenesis to mitigate Alzheimer’s neuropathology. Molecular Neurobiology. 2019;56(12):8255–8276. doi: 10.1007/s12035-019-01660-3. [DOI] [PubMed] [Google Scholar]
- 5.Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2011;1(1, article a006189) doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uddin M. S., al Mamun A., Kabir M. T., et al. Nootropic and anti-Alzheimer’s actions of medicinal plants: molecular insight into therapeutic potential to alleviate Alzheimer’s neuropathology. Molecular Neurobiology. 2019;56(7):4925–4944. doi: 10.1007/s12035-018-1420-2. [DOI] [PubMed] [Google Scholar]
- 7.Kabir M. T., Abu Sufian M., Uddin M. S., et al. NMDA Receptor Antagonists: Repositioning of Memantine as a Multitargeting Agent for Alzheimer's Therapy. Current Pharmaceutical Design. 2019;25(33):3506–3518. doi: 10.2174/1381612825666191011102444. [DOI] [PubMed] [Google Scholar]
- 8.Uddin M. S., Kabir M. T., Jeandet P., et al. Novel Anti-Alzheimer’s Therapeutic Molecules Targeting Amyloid Precursor Protein Processing. Oxidative Medicine and Cellular Longevity. 2020;2020:19. doi: 10.1155/2020/7039138.7039138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman M. A., Rahman M. R., Zaman T., et al. Emerging potential of naturally occurring autophagy modulators against neurodegeneration. Current Pharmaceutical Design. 2020;26(7):772–779. doi: 10.2174/1381612826666200107142541. [DOI] [PubMed] [Google Scholar]
- 10.Uddin M. S., Mamun A. A., Jakaria M., et al. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Science of The Total Environment. 2020;707, article 135624 doi: 10.1016/j.scitotenv.2019.135624. [DOI] [PubMed] [Google Scholar]
- 11.Uddin M. S., Kabir M. T., Tewari D., Mathew B., Aleya L. Emerging signal regulating potential of small molecule biflavonoids to combat neuropathological insults of Alzheimer's disease. Science of The Total Environment. 2020;700, article 134836 doi: 10.1016/j.scitotenv.2019.134836. [DOI] [PubMed] [Google Scholar]
- 12.Reitz C., Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochemical Pharmacology. 2014;88(4):640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends in Neurosciences. 1997;20(4):154–159. doi: 10.1016/S0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 14.Lanoiselée H. M., Nicolas G., Wallon D., et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Medicine. 2017;14(3, article e1002270) doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al Mamun A., Uddin M. S., Kabir M. T., et al. Exploring the promise of targeting ubiquitin-proteasome system to combat Alzheimer’s disease. Neurotoxicity Research. 2020:1–10. doi: 10.1007/s12640-020-00185-1. [DOI] [PubMed] [Google Scholar]
- 16.Jiang T., Yu J.-T., Tian Y., Tan L. Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Current Alzheimer Research. 2013;10(8):852–867. doi: 10.2174/15672050113109990155. [DOI] [PubMed] [Google Scholar]
- 17.Harold D., Abraham R., Hollingworth P., et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert J.-C., European Alzheimer's Disease Initiative (EADI), Ibrahim-Verbaas C. A., et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature Genetics. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C. C., Kanekiyo T., Xu H., Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature Reviews Neurology. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabir M. T., Uddin M. S., Begum M. M., et al. Cholinesterase inhibitors for Alzheimer’s disease: multitargeting strategy based on anti-Alzheimer’s drugs repositioning. Current Pharmaceutical Design. 2019;25(33):3519–3535. doi: 10.2174/1381612825666191008103141. [DOI] [PubMed] [Google Scholar]
- 21.Montufar S., Calero C., Vinueza R., et al. Association between the APOE ε4 allele and late-onset Alzheimer’s disease in an Ecuadorian mestizo population. International Journal of Alzheimer's Disease. 2017;2017:1–9. doi: 10.1155/2017/1059678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uddin M. S., Tewari D., Al Mamun A., et al. Circadian and sleep dysfunction in Alzheimer’s disease. Ageing Research Reviews. 2020;60, article 101046 doi: 10.1016/J.ARR.2020.101046. [DOI] [PubMed] [Google Scholar]
- 23.Carter D. B. The interaction of Amyloid-β with ApoE. Sub-Cellular Biochemistry. 2005;38:255–272. doi: 10.1007/0-387-23226-5_13. [DOI] [PubMed] [Google Scholar]
- 24.Safieh M., Korczyn A. D., Michaelson D. M. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Medicine. 2019;17(1):p. 64. doi: 10.1186/s12916-019-1299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uddin M. S., Hossain M. F., Al Mamun A., et al. Exploring the multimodal role of phytochemicals in the modulation of cellular signaling pathways to combat age-related neurodegeneration. Science of The Total Environment. 2020;725, article 138313 doi: 10.1016/j.scitotenv.2020.138313. [DOI] [PubMed] [Google Scholar]
- 26.Uddin M. S., Kabir M. T., Rahman M. H., et al. Exploring the multifunctional neuroprotective promise of rasagiline derivatives for multi-dysfunctional Alzheimer’s disease. Current Pharmaceutical Design. 2020;26 doi: 10.2174/1381612826666200406075044. [DOI] [PubMed] [Google Scholar]
- 27.Itzhaki R. F., Lin W. R., Shang D., Wilcock G. K., Faragher B., Jamieson G. A. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. The Lancet. 1997;349(9047):241–244. doi: 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- 28.Linard M., Letenneur L., Garrigue I., Doize A., Dartigues J.-F., Helmer C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer's & Dementia. 2020;16(1):200–208. doi: 10.1002/alz.12008. [DOI] [PubMed] [Google Scholar]
- 29.Koller E. J., Gonzalez de la Cruz E., Weinrich M., et al. Intracerebral expression of AAV-APOE4 is not sufficient to alter Tau burden in two distinct models of tauopathy. Molecular Neurobiology. 2020;57(4):1986–2001. doi: 10.1007/s12035-019-01859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanson A., Craft S., Banks W. The APOE genotype: modification of therapeutic responses in Alzheimer’s disease. Current Pharmaceutical Design. 2014;21(1):114–120. doi: 10.2174/1381612820666141020164222. [DOI] [PubMed] [Google Scholar]
- 31.Ramassamy C., Averill D., Beffert U., et al. Oxidative Insults Are Associated with Apolipoprotein E Genotype in Alzheimer's Disease Brain. Neurobiology of Disease. 2000;7(1):23–37. doi: 10.1006/nbdi.1999.0273. [DOI] [PubMed] [Google Scholar]
- 32.Uddin M. S., Upaganlawar A. B. Oxidative Stress and Antioxidant Defense: Biomedical Value in Health and Diseases. Hauppauge, NY, USA: Nova Science Publishers; 2019. [Google Scholar]
- 33.Uddin M. S., Kabir M. T. Oxidative stress in Alzheimer’s disease: molecular hallmarks of underlying vulnerability. In: Ashraf G., Alexiou A., editors. Biological, Diagnostic and Therapeutic Advances in Alzheimer's Disease. Singapore: Springer; 2019. pp. 91–115. [DOI] [Google Scholar]
- 34.Smith M. A., Rudnicka-Nawrot M., Richey P. L., et al. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. Journal of Neurochemistry. 1995;64(6):2660–2666. doi: 10.1046/j.1471-4159.1995.64062660.x. [DOI] [PubMed] [Google Scholar]
- 35.Butterfield D. A., Di Domenico F., Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2014;1842(9):1693–1706. doi: 10.1016/j.bbadis.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosenko E., Solomadin I., Tikhonova L., Reddy V., Aliev G., Kaminsky Y. Pathogenesis of Alzheimer disease: role of oxidative stress, amyloid-β peptides, systemic ammonia and erythrocyte energy metabolism. CNS & Neurological Disorders Drug Targets. 2014;13(1):112–119. doi: 10.2174/18715273113126660130. [DOI] [PubMed] [Google Scholar]
- 37.Castellani R. J., Moreira P. I., Perry G., Zhu X. The role of iron as a mediator of oxidative stress in Alzheimer disease. BioFactors. 2012;38(2):133–138. doi: 10.1002/biof.1010. [DOI] [PubMed] [Google Scholar]
- 38.Zhu X., Raina A. K., Lee H. G., et al. Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxidants & Redox Signaling. 2003;5(5):571–576. doi: 10.1089/152308603770310220. [DOI] [PubMed] [Google Scholar]
- 39.Markesbery W. R. The role of oxidative stress in Alzheimer disease. Archives of Neurology. 1999;56(12):1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 40.Praticò D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends in Pharmacological Sciences. 2008;29(12):609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Schippling S., Kontush A., Arlt S., et al. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radical Biology & Medicine. 2000;28(3):351–360. doi: 10.1016/s0891-5849(99)00247-6. [DOI] [PubMed] [Google Scholar]
- 42.Smith M. A., Kutty R. K., Richey P. L., et al. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. The American Journal of Pathology. 1994;145(1):42–47. [PMC free article] [PubMed] [Google Scholar]
- 43.Ramassamy C., Krzywkowski P., Averill D., et al. Impact of apoE deficiency on oxidative insults and antioxidant levels in the brain. Molecular Brain Research. 2001;86(1-2):76–83. doi: 10.1016/S0169-328X(00)00268-0. [DOI] [PubMed] [Google Scholar]
- 44.Picklo M. J., Montine T. J., Amarnath V., Neely M. D. Carbonyl Toxicology and Alzheimer's Disease. Toxicology and Applied Pharmacology. 2002;184(3):187–197. doi: 10.1006/taap.2002.9506. [DOI] [PubMed] [Google Scholar]
- 45.Bassett C. N., Montine T. J. Lipoproteins and lipid peroxidation in Alzheimer’s disease. The Journal of Nutrition, Health & Aging. 2003;7:24–29. [PubMed] [Google Scholar]
- 46.Mecocci P., Beal M. F., Cecchetti R., et al. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Molecular and Chemical Neuropathology. 1997;31(1):53–64. doi: 10.1007/BF02815160. [DOI] [PubMed] [Google Scholar]
- 47.Lovell M. A., Markesbery W. R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Research. 2007;35(22):7497–7504. doi: 10.1093/nar/gkm821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lovell M. A., Markesbery W. R. Oxidatively modified RNA in mild cognitive impairment. Neurobiology of Disease. 2008;29(2):169–175. doi: 10.1016/j.nbd.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradley-Whitman M. A., Timmons M. D., Beckett T. L., Murphy M. P., Lynn B. C., Lovell M. A. Nucleic acid oxidation: an early feature of Alzheimer’s disease. Journal of Neurochemistry. 2014;128(2):294–304. doi: 10.1111/jnc.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Migliore L., Fontana I., Trippi F., et al. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiology of Aging. 2005;26(5):567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Sultana R., Mecocci P., Mangialasche F., Cecchetti R., Baglioni M., Butterfield D. A. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this Dementing Disorder. Journal of Alzheimer's Disease. 2011;24(1):77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 52.Miyata M., Smith J. D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and β-amyloid peptides. Nature Genetics. 1996;14(1):55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 53.Hayek T., Oiknine J., Brook J. G., Aviram M. Increased plasma and lipoprotein lipid peroxidation in apo E-deficient mice. Biochemical and Biophysical Research Communications. 1994;201(3):1567–1574. doi: 10.1006/bbrc.1994.1883. [DOI] [PubMed] [Google Scholar]
- 54.Ramassamy C., Averill D., Beffert U., et al. Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein E genotype. Free Radical Biology & Medicine. 1999;27(5-6):544–553. doi: 10.1016/S0891-5849(99)00102-1. [DOI] [PubMed] [Google Scholar]
- 55.Fernandez C. G., Hamby M. E., McReynolds M. L., Ray W. J. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Frontiers in Aging Neuroscience. 2019;11:p. 14. doi: 10.3389/fnagi.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corder E., Saunders A., Strittmatter W., et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 57.Strittmatter W. J., Saunders A. M., Schmechel D., et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim H., Yoo J., Shin J., et al. Modelling APOE ɛ3/4 allele-associated sporadic Alzheimer’s disease in an induced neuron. Brain. 2017;140(8):2193–2209. doi: 10.1093/brain/awx144. [DOI] [PubMed] [Google Scholar]
- 59.Mahley R. W., Rall S. C., Jr. APOLIPOPROTEINE: far more than a lipid transport protein. Annual Review of Genomics and Human Genetics. 2000;1(1):507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 60.Uddin M. S., Kabir M. T. Emerging signal regulating potential of genistein against Alzheimer’s disease: a promising molecule of interest. Frontiers in Cell and Development Biology. 2019;7:1–12. doi: 10.3389/fcell.2019.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hixson J. E., Vernier D. T. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. Journal of Lipid Research. 1990;31(3):545–548. [PubMed] [Google Scholar]
- 62.Uddin M. S., Rhman M. M., Jakaria M., et al. Estrogen signaling in Alzheimer’s disease: molecular insights and therapeutic targets for Alzheimer’s dementia. Molecular Neurobiology. 2020:1–17. doi: 10.1007/s12035-020-01911-8. [DOI] [PubMed] [Google Scholar]
- 63.Chen Q., Reis S. E., Kammerer C. M., et al. APOE polymorphism and angiographic coronary artery disease severity in the Women's Ischemia Syndrome Evaluation (WISE) study. Atherosclerosis. 2003;169(1):159–167. doi: 10.1016/s0021-9150(03)00160-6. [DOI] [PubMed] [Google Scholar]
- 64.Martins C. A. R., Oulhaj A., de Jager C. A., Williams J. H. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology. 2005;65(12):1888–1893. doi: 10.1212/01.wnl.0000188871.74093.12. [DOI] [PubMed] [Google Scholar]
- 65.Sando S. B., Melquist S., Cannon A., et al. APOE ε4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurology. 2008;8(1) doi: 10.1186/1471-2377-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagy Z. S., Esiri M. M., Jobst K. A., et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience. 1995;69(3):757–761. doi: 10.1016/0306-4522(95)00331-C. [DOI] [PubMed] [Google Scholar]
- 67.Harris F. M., Brecht W. J., Xu Q., Mahley R. W., Huang Y. Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. The Journal of Biological Chemistry. 2004;279(43):44795–44801. doi: 10.1074/jbc.M408127200. [DOI] [PubMed] [Google Scholar]
- 68.Bennett R. E., Esparza T. J., Lewis H. A., et al. Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-β immunoreactivity after traumatic brain injury in 3xTG-AD mice. Journal of Neuropathology and Experimental Neurology. 2013;72(5):396–403. doi: 10.1097/NEN.0b013e31828e24ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilat-Frenkel M., Boehm-Cagan A., Liraz O., Xian X., Herz J., Michaelson D. Involvement of the Apoer2 and Lrp1 receptors in mediating the pathological effects of ApoE4 in vivo. Current Alzheimer Research. 2014;11(6):549–557. doi: 10.2174/1567205010666131119232444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uddin M. S., Kabir M. T., Al Mamun A., Abdel-Daim M. M., Barreto G. E., Ashraf G. M. APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Molecular Neurobiology. 2019;56(4):2450–2465. doi: 10.1007/s12035-018-1237-z. [DOI] [PubMed] [Google Scholar]
- 71.Talwar P., Sinha J., Grover S., et al. Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer's disease. Journal of the Neurological Sciences. 2016;360:179–187. doi: 10.1016/j.jns.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 72.Martínez-Morillo E., Hansson O., Atagi Y., et al. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathologica. 2014;127(5):633–643. doi: 10.1007/s00401-014-1266-2. [DOI] [PubMed] [Google Scholar]
- 73.Simon R., Girod M., Fonbonne C., et al. Total ApoE and ApoE4 isoform assays in an Alzheimer’s disease case-control study by targeted mass spectrometry (n = 669): a pilot assay for methionine-containing proteotypic peptides. Molecular & Cellular Proteomics. 2012;11(11):1389–1403. doi: 10.1074/mcp.m112.018861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heywood W. E., Galimberti D., Bliss E., et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Molecular Neurodegeneration. 2015;10(1) doi: 10.1186/s13024-015-0059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boehm-Cagan A., Michaelson D. M. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. The Journal of Neuroscience. 2014;34(21):7293–7301. doi: 10.1523/jneurosci.5198-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cramer P. E., Cirrito J. R., Wesson D. W., et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Price A. R., Xu G., Siemienski Z. B., et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models.”. Science. 2013;340(6135):p. 924–d. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- 78.Tachibana M., Shinohara M., Yamazaki Y., et al. Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Experimental Neurology. 2016;277:1–9. doi: 10.1016/j.expneurol.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.LaClair K. D., Manaye K. F., Lee D. L., et al. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Molecular Neurodegeneration. 2013;8(1):p. 18. doi: 10.1186/1750-1326-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tai L. M., Koster K. P., Luo J., et al. Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. The Journal of Biological Chemistry. 2014;289(44):30538–30555. doi: 10.1074/jbc.M114.600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tesseur I., Lo A. C., Roberfroid A., et al. Comment on "ApoE-Directed therapeutics rapidly Clear β-Amyloid and reverse deficits in AD mouse Models". Science. 2013;340(6135):p. 924–e. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- 82.Veeraraghavalu K., Zhang C., Miller S., et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models.”. Science. 2013;340(6135):p. 924. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- 83.Burns M. P., Vardanian L., Pajoohesh-Ganji A., et al. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. Journal of Neurochemistry. 2006;98(3):792–800. doi: 10.1111/j.1471-4159.2006.03925.x. [DOI] [PubMed] [Google Scholar]
- 84.Donkin J. J., Stukas S., Hirsch-Reinshagen V., et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. The Journal of Biological Chemistry. 2010;285(44):34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koldamova R. P., Lefterov I. M., Staufenbiel M., et al. The liver X receptor ligand T0901317 decreases amyloid β productionin vitroand in a mouse model of alzheimer's disease. The Journal of Biological Chemistry. 2005;280(6):4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 86.Casali B. T., Corona A. W., Mariani M. M., Karlo J. C., Ghosal K., Landreth G. E. Omega-3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2015;35(24):9173–9181. doi: 10.1523/jneurosci.1000-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Riddell D. R., Zhou H., Comery T. A., et al. The LXR agonist TO901317 selectively lowers hippocampal Aβ42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Molecular and Cellular Neurosciences. 2007;34(4):621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 88.Vanmierlo T., Rutten K., Dederen J., et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiology of Aging. 2011;32(7):1262–1272. doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 89.Escribano L., Simón A. M., Gimeno E., et al. Rosiglitazone rescues memory impairment in alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35(7):1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Skerrett R., Pellegrino M. P., Casali B. T., Taraboanta L., Landreth G. E. Combined liver X receptor/peroxisome proliferator-activated receptor γ agonist treatment reduces amyloid β levels and improves behavior in amyloid precursor protein/presenilin 1 mice. The Journal of Biological Chemistry. 2015;290(35):21591–21602. doi: 10.1074/jbc.M115.652008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang Q., Lee C. Y. D., Mandrekar S., et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58(5):681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong C., Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nature Reviews Drug Discovery. 2014;13(6):433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 93.Mehta N., Wayne A. S., Kim Y. H., et al. Bexarotene is active against subcutaneous panniculitis-like T-cell lymphoma in adult and pediatric populations. Clinical Lymphoma, Myeloma & Leukemia. 2012;12(1):20–25. doi: 10.1016/j.clml.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gregoriou S., Rigopoulos D., Stamou C., Nikolaou V., Kontochristopoulos G. Treatment of mycosis fungoides with bexarotene results in remission of diffuse plane xanthomas. Journal of Cutaneous Medicine and Surgery. 2013;17(1):52–54. doi: 10.2310/7750.2012.12022. [DOI] [PubMed] [Google Scholar]
- 95.Scarisbrick J. J., Morris S., Azurdia R., et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. The British Journal of Dermatology. 2013;168(1):192–200. doi: 10.1111/bjd.12042. [DOI] [PubMed] [Google Scholar]
- 96.Väkevä L., Ranki A., Hahtola S. Ten-year experience of bexarotene therapy for cutaneous T-cell lymphoma in Finland. Acta Dermato-Venereologica. 2012;92(3):258–263. doi: 10.2340/00015555-1359. [DOI] [PubMed] [Google Scholar]
- 97.Fitz N. F., Cronican A. A., Lefterov I., Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340(6135):p. 924. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cummings J. L., Zhong K., Kinney J. W., et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimer's Research & Therapy. 2016;8(1):p. 4. doi: 10.1186/s13195-016-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bien-Ly N., Gillespie A. K., Walker D., Yoon S. Y., Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2012;32(14):4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim J., Jiang H., Park S., et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. The Journal of Neuroscience. 2011;31(49):18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liao F., Hori Y., Hudry E., et al. Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. The Journal of Neuroscience. 2014;34(21):7281–7292. doi: 10.1523/JNEUROSCI.0646-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim J., Eltorai A. E. M., Jiang H., et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. The Journal of Experimental Medicine. 2012;209(12):2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mak A. C. Y., Pullinger C. R., Tang L. F., et al. Effects of the absence of apolipoprotein E on lipoproteins, neurocognitive function, and retinal function. JAMA Neurology. 2014;71(10):1228–1236. doi: 10.1001/jamaneurol.2014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nature Reviews Neuroscience. 2009;10(5):333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luz I. L. O., Nakaryakov A., Smorodinsky N. I., Michaelson D. Anti-apoE4 immunotherapy: a novel approach to the treatment of Alzheimer’s disease (AD) Journal of Molecular Neuroscience. 2013;51:S75–S76. [Google Scholar]
- 106.Marques M. A., Owens P. A., Crutcher K. A. Progress toward identification of protease activity involved in proteolysis of apolipoprotein E in human brain. Journal of Molecular Neuroscience. 2004;24(1):073–080. doi: 10.1385/jmn:24:1:073. [DOI] [PubMed] [Google Scholar]
- 107.Tolar M., Marques M. A., Harmony J. A. K., Crutcher K. A. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. The Journal of Neuroscience. 1997;17(15):5678–5686. doi: 10.1523/jneurosci.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chang S., Ma T., Miranda R. D., Balestra M. E., Mahley R. W., Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Elliott D. A., Tsoi K., Holinkova S., et al. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiology of Aging. 2011;32(2):257–271. doi: 10.1016/j.neurobiolaging.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 110.Bien-Ly N., Andrews-Zwilling Y., Xu Q., Bernardo A., Wang C., Huang Y. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Aβ) and acts in concert with Aβ to elicit neuronal and behavioral deficits in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(10):4236–4241. doi: 10.1073/pnas.1018381108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hatters D. M., Zhong N., Rutenber E., Weisgraber K. H. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. Journal of Molecular Biology. 2006;361(5):932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 112.Kanekiyo T., Xu H., Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mahley R. W., Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. Journal of Medicinal Chemistry. 2012;55(21):8997–9008. doi: 10.1021/jm3008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hauser P. S., Narayanaswami V., Ryan R. O. Apolipoprotein E: from lipid transport to neurobiology. Progress in Lipid Research. 2011;50(1):62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wahrle S. E., Jiang H., Parsadanian M., et al. ABCA1 is required for normal central nervous system apoE levels and for lipidation of astrocyte-secreted apoE. The Journal of Biological Chemistry. 2004;279(39):40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- 116.Wahrle S. E., Jiang H., Parsadanian M., et al. Deletion ofAbca1Increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. The Journal of Biological Chemistry. 2005;280(52):43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 117.Wahrle S. E., Jiang H., Parsadanian M., et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. The Journal of Clinical Investigation. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harris-White M. E., Frautschy S. A. Low density lipoprotein receptor-related proteins (LRPs), Alzheimer’s and cognition. Current Drug Targets. CNS and Neurological Disorders. 2005;4(5):469–480. doi: 10.2174/156800705774322102. [DOI] [PubMed] [Google Scholar]
- 119.Nathan B. P., Bellosta S., Sanan D. A., Weisgraber K. H., Mahley R. W., Pitas R. E. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264(5160):850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 120.Mauch D. H., Nägler K., Schumacher S., et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 121.Teter B., Xu P. T., Gilbert J. R., Roses A. D., Galasko D., Cole G. M. Human apolipoprotein E isoform-specific differences in neuronal sprouting in organotypic hippocampal culture. Journal of Neurochemistry. 1999;73(6):2613–2616. doi: 10.1046/j.1471-4159.1999.0732613.x. [DOI] [PubMed] [Google Scholar]
- 122.Michaelson D. M. APOE ε4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimer's & Dementia. 2014;10(6):861–868. doi: 10.1016/j.jalz.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 123.Osei-Hwedieh D. O., Amar M., Sviridov D., Remaley A. T. Apolipoprotein mimetic peptides: mechanisms of action as anti-atherogenic agents. Pharmacology & Therapeutics. 2011;130(1):83–91. doi: 10.1016/j.pharmthera.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Youmans K. L., Tai L. M., Nwabuisi-Heath E., et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. The Journal of Biological Chemistry. 2012;287(50):41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tai L. M., Bilousova T., Jungbauer L., et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. The Journal of Biological Chemistry. 2013;288(8):5914–5926. doi: 10.1074/jbc.M112.442103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hanson A. J., Bayer-Carter J. L., Green P. S., et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurology. 2013;70(8):972–980. doi: 10.1001/jamaneurol.2013.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Buttini M., Akeefe H., Lin C., et al. Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience. 2000;97(2):207–210. doi: 10.1016/S0306-4522(00)00069-5. [DOI] [PubMed] [Google Scholar]
- 128.Tai L. M., Mehra S., Shete V., et al. Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4-induced AD risk. Molecular Neurodegeneration. 2014;9(1):p. 2. doi: 10.1186/1750-1326-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bittar A., Sengupta U., Kayed R. Prospects for strain-specific immunotherapy in Alzheimer's disease and tauopathies. NPJ Vaccines. 2018;3(1):p. 9. doi: 10.1038/s41541-018-0046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mahley R. W. Central nervous system lipoproteins. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(7):1305–1315. doi: 10.1161/atvbaha.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Luz I., Liraz O., Michaelson D. M. An anti-apoE4 specific monoclonal antibody counteracts the pathological effects of apoE4 in vivo. Current Alzheimer Research. 2016;13(8):918–929. doi: 10.2174/1567205013666160404120817. [DOI] [PubMed] [Google Scholar]
- 132.Ghosal K., Stathopoulos A., Thomas D., Phenis D., Vitek M. P., Pimplikar S. W. The apolipoprotein-E-mimetic COG112 protects amyloid precursor protein intracellular domain-overexpressing animals from Alzheimer’s disease-like pathological features. Neurodegenerative Diseases. 2013;12(1):51–58. doi: 10.1159/000341299. [DOI] [PubMed] [Google Scholar]
- 133.Vitek M. P., Christensen D. J., Wilcock D., et al. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegenerative Diseases. 2012;10(1-4):122–126. doi: 10.1159/000334914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang W., Zhu X. HDLmimetic peptides affect apolipoprotein E metabolism: equal supplement or functional enhancer? Journal of Neurochemistry. 2018;147(5):580–583. doi: 10.1111/jnc.14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Laskowitz D. T., Song P., Wang H., et al. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. Journal of Neurotrauma. 2010;27(11):1983–1995. doi: 10.1089/neu.2010.1396. [DOI] [PubMed] [Google Scholar]
- 136.Yao X., Vitek M. P., Remaley A. T., Levine S. J. Apolipoprotein mimetic peptides: a new approach for the treatment of asthma. Frontiers in Pharmacology. 2012;3:p. 37. doi: 10.3389/fphar.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang H., Anderson L. G., Lascola C. D., et al. ApolipoproteinE mimetic peptides improve outcome after focal ischemia. Experimental Neurology. 2013;241:67–74. doi: 10.1016/j.expneurol.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 138.White C. R., Garber D. W., Anantharamaiah G. M. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: a review. Journal of Lipid Research. 2014;55(10):2007–2021. doi: 10.1194/jlr.R051367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brecht W. J., Harris F. M., Chang S., et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. The Journal of Neuroscience. 2004;24(10):2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang Y., Liu X. Q., Wyss-Coray T., Brecht W. J., Sanan D. A., Mahley R. W. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harris F. M., Brecht W. J., Xu Q., et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;100(19):10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Doudna J. A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):p. 1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 143.Komor A. C., Kim Y. B., Packer M. S., Zuris J. A., Liu D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rabinowitz R., Kadair A., Ben-Zur T., Michaelson D., Offen D. ApoE4 allele specific knockout using a synthetic Cas9 variant as a potential gene therapy approach for Alzheimer's disease. Cytotherapy. 2019;21(5, article e7) doi: 10.1016/j.jcyt.2019.04.022. [DOI] [Google Scholar]
- 145.Zhong N., Scearce-Levie K., Ramaswamy G., Weisgraber K. H. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement. 2008;4(3):179–192. doi: 10.1016/j.jalz.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen H.-K., Ji Z.-S., Dodson S. E., et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. The Journal of Biological Chemistry. 2011;286(7):5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brodbeck J., McGuire J., Liu Z., et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. The Journal of Biological Chemistry. 2011;286(19):17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cacabelos R., Torrellas C. Epigenetics of aging and Alzheimer’s disease: implications for pharmacogenomics and drug response. International Journal of Molecular Sciences. 2015;16(12):30483–30543. doi: 10.3390/ijms161226236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Smith J. A., Zagel A. L., Sun Y. V., et al. Epigenomic indicators of age in African Americans. Hereditary Genetics. 2014;3(3) doi: 10.4172/2161-1041.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ma Y., Smith C. E., Lai C. Q., et al. Genetic variants modify the effect of age on APOE methylation in the genetics of lipid lowering drugs and diet network study. Aging Cell. 2015;14(1):49–59. doi: 10.1111/acel.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Verghese P. B., Castellano J. M., Garai K., et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):E1807–E1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Namba Y., Tomonaga M., Kawasaki H., Otomo E., Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Research. 1991;541(1):163–166. doi: 10.1016/0006-8993(91)91092-F. [DOI] [PubMed] [Google Scholar]
- 153.Kuszczyk M. A., Sanchez S., Pankiewicz J., et al. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. The American Journal of Pathology. 2013;182(5):1750–1768. doi: 10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sadowski M. J., Pankiewicz J., Scholtzova H., et al. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(49):18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu S., Breitbart A., Sun Y., et al. Blocking the apolipoprotein E/amyloid β interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid β and tau pathology. Journal of Neurochemistry. 2014;128(4):577–591. doi: 10.1111/jnc.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pankiewicz J. E., Guridi M., Kim J., et al. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathologica Communications. 2014;2(1):p. 75. doi: 10.1186/PREACCEPT-1147957959132865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hao J., Zhang W., Zhang P., et al. Aβ20–29 peptide blocking apoE/Aβ interaction reduces full-length Aβ42/40 fibril formation and cytotoxicity in vitro. Neuropeptides. 2010;44(4):305–313. doi: 10.1016/j.npep.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 158.Liao F., Li A., Xiong M., et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. The Journal of Clinical Investigation. 2018;128(5):2144–2155. doi: 10.1172/JCI96429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huynh T. P. V., Liao F., Francis C. M., et al. Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron. 2017;96(5):1013–1023.e4. doi: 10.1016/j.neuron.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rebeck G. W., Kindy M., LaDu M. J. Apolipoprotein E and Alzheimer’s disease: the protective effects of ApoE2 and E3. Journal of Alzheimer's Disease. 2002;4(3):145–154. doi: 10.3233/JAD-2002-4304. [DOI] [PubMed] [Google Scholar]
- 161.Hyman B. T., Gomez-Isla T., Briggs M., et al. Apolipoprotein E and cognitive change in an elderly population. Annals of Neurology. 1996;40(1):55–66. doi: 10.1002/ana.410400111. [DOI] [PubMed] [Google Scholar]
- 162.Helkala E. L., Koivisto K., Hänninen T., et al. Memory functions in human subjects with different apolipoprotein E phenotypes during a 3-year population-based follow-up study. Neuroscience Letters. 1996;204(3):177–180. doi: 10.1016/0304-3940(96)12348-x. [DOI] [PubMed] [Google Scholar]
- 163.Staehelin H. B., Perrig-Chiello P., Mitrache C., Miserez A. R., Perrig W. J. Apolipoprotein E genotypes and cognitive functions in healthy elderly persons. Acta Neurologica Scandinavica. 1999;100(1):53–60. doi: 10.1111/j.1600-0404.1999.tb00723.x. [DOI] [PubMed] [Google Scholar]
- 164.Wilson R. S., Bienias J. L., Berry-Kravis E., Evans D. A., Bennett D. A. The apolipoprotein E varepsilon2 allele and decline in episodic memory. Journal of Neurology, Neurosurgery, and Psychiatry. 2002;73(6):672–677. doi: 10.1136/jnnp.73.6.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Conejero-Goldberg C., Gomar J. J., Bobes-Bascaran T., et al. APOE2 enhances neuroprotection against Alzheimer's disease through multiple molecular mechanisms. Molecular Psychiatry. 2014;19(11):1243–1250. doi: 10.1038/mp.2013.194. [DOI] [PubMed] [Google Scholar]
- 166.Dodart J.-C., Marr R. A., Koistinaho M., et al. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Suri S., Heise V., Trachtenberg A. J., Mackay C. E. The forgotten APOE allele: A review of the evidence and suggested mechanisms for the protective effect of APOE ɛ2. Neuroscience and Biobehavioral Reviews. 2013;37(10):2878–2886. doi: 10.1016/j.neubiorev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 168.Rosenberg J. B., Kaplitt M. G., de B. P., et al. AAVrh.10-mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer’s disease. Human Gene Therapy. Clinical Development. 2018;29(1):24–47. doi: 10.1089/humc.2017.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hu J., Liu C. C., Chen X. F., Zhang Y. W., Xu H., Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Molecular Neurodegeneration. 2015;10(1):p. 6. doi: 10.1186/s13024-015-0001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Holtzman D. M., Herz J., Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2012;2(3, article a006312) doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kanekiyo T., Zhang J., Liu Q., Liu C. C., Zhang L., Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal Amyloid-β uptake. The Journal of Neuroscience. 2011;31(5):1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu Q., Trotter J., Zhang J., et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. The Journal of Neuroscience. 2010;30(50):17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim J., Castellano J. M., Jiang H., et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron. 2009;64(5):632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kanekiyo T., Cirrito J. R., Liu C.-C., et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. The Journal of Neuroscience. 2013;33(49):19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shinohara M., Sato N., Kurinami H., et al. Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Aβ clearance. The Journal of Biological Chemistry. 2010;285(29):22091–22102. doi: 10.1074/jbc.M110.102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Qosa H., Abuznait A. H., Hill R. A., Kaddoumi A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. Journal of Alzheimer's Disease. 2012;31(1):151–165. doi: 10.3233/JAD-2012-120319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lane-Donovan C., Philips G. T., Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014;83(4):771–787. doi: 10.1016/j.neuron.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen Y., Durakoglugil M. S., Xian X., Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(26):12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bell R. D., Winkler E. A., Singh I., et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alata W., Ye Y., St-Amour I., Vandal M., Calon F. Human apolipoprotein E ε4 expression impairs cerebral vascularization and blood-brain barrier function in mice. Journal of Cerebral Blood Flow and Metabolism. 2014;35(1):86–94. doi: 10.1038/jcbfm.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bien-Ly N., Boswell C. A., Jeet S., et al. Lack of widespread BBB disruption in Alzheimer’s disease models: focus on therapeutic antibodies. Neuron. 2015;88(2):289–297. doi: 10.1016/j.neuron.2015.09.036. [DOI] [PubMed] [Google Scholar]
- 182.Keren-Shaul H., Spinrad A., Weiner A., et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 183.Brown G. C., St George-Hyslop P. H. Deciphering microglial diversity in Alzheimer’s disease. Science. 2017;356(6343):1123–1124. doi: 10.1126/science.aan7893. [DOI] [PubMed] [Google Scholar]
- 184.Wattananit S., Tornero D., Graubardt N., et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. The Journal of Neuroscience. 2016;36(15):4182–4195. doi: 10.1523/JNEUROSCI.4317-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Uddin M. S., Kabir M. T., Mamun A. A. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. International Immunopharmacology. 2020;84, article 106479 doi: 10.1016/j.intimp.2020.106479. [DOI] [PubMed] [Google Scholar]
- 186.Bell R. D. The imbalance of vascular molecules in Alzheimer’s disease. Journal of Alzheimer's Disease. 2012;32(3):699–709. doi: 10.3233/JAD-2012-121060. [DOI] [PubMed] [Google Scholar]
- 187.Altman R., Rutledge J. C. The vascular contribution to Alzheimer’s disease. Clinical Science. 2010;119(10):407–421. doi: 10.1042/CS20100094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rohn T. T. Is apolipoprotein E4 an important risk factor for vascular dementia? International Journal of Clinical and Experimental Pathology. 2014;7(7):3504–3511. [PMC free article] [PubMed] [Google Scholar]
- 189.Mahley R. W., Weisgraber K. H., Huang Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. Journal of Lipid Research. 2009;50:S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Davey D. A. Alzheimer’s disease and vascular dementia: one potentially preventable and modifiable disease? Part II: management, prevention and future perspective. Neurodegenerative Disease Management. 2014;4(3):261–270. doi: 10.2217/nmt.14.14. [DOI] [PubMed] [Google Scholar]
- 191.Theendakara V., Peters-Libeu C. A., Spilman P., Poksay K. S., Bredesen D. E., Rao R. V. Direct transcriptional effects of apolipoprotein E. The Journal of Neuroscience. 2016;36(3):685–700. doi: 10.1523/JNEUROSCI.3562-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Parcon P. A., Balasubramaniam M., Ayyadevara S., et al. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018;14(2):230–242. doi: 10.1016/j.jalz.2017.07.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Urfer-Buchwalder A., Urfer R. Identification of a nuclear respiratory factor 1 recognition motif in the apolipoprotein E variant APOE4 linked to Alzheimer's disease. Scientific Reports. 2017;7(1) doi: 10.1038/srep40668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rohn T. T., Moore Z. D. Nuclear localization of apolipoprotein E4: a new trick for an old protein. International Journal of Neurology and Neurotherapy. 2017;4(2) doi: 10.23937/2378-3001/1410067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Theendakara V., Peters-Libeu C. A., Bredesen D. E., Rao R. V. Transcriptional effects of ApoE4: relevance to Alzheimer’s disease. Molecular Neurobiology. 2018;55(6):5243–5254. doi: 10.1007/s12035-017-0757-2. [DOI] [PubMed] [Google Scholar]
- 196.Budd Haeberlein S., O'Gorman J., Chiao P., et al. Clinical development of aducanumab, an anti-Aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. Journal of Prevention of Alzheimer’s Disease. 2017;4(4):255–263. doi: 10.14283/jpad.2017.39. [DOI] [PubMed] [Google Scholar]
- 197.Minami S. S., Cordova A., Cirrito J. R., et al. ApoE mimetic peptide decreases Aβ production in vitro and in vivo. Molecular Neurodegeneration. 2010;5(1):p. 16. doi: 10.1186/1750-1326-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Handattu S. P., Monroe C. E., Nayyar G., et al. In vivo and in vitro effects of an apolipoprotein E mimetic peptide on amyloid-β pathology. Journal of Alzheimer's Disease. 2013;36(2):335–347. doi: 10.3233/JAD-122377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hudry E., Dashkoff J., Roe A. D., et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Science Translational Medicine. 2013;5(212, article 212ra161) doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kockx M., Guo D. L., Traini M., et al. Cyclosporin A decreases apolipoprotein E secretion from human macrophages via a protein phosphatase 2B-dependent and ATP-binding cassette transporter A1 (ABCA1)-independent pathway. The Journal of Biological Chemistry. 2009;284(36):24144–24154. doi: 10.1074/jbc.M109.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]