

Abstract
Sorafenib is a small, orally-active multikinase inhibitor that is most frequently used for the management of renal cell carcinoma, hepatocellular carcinoma, and radioactive iodine-resistant thyroid carcinoma. However, recent reports have associated sorafenib with hepatotoxicity that can limit its clinical application, although the mechanism of hepatotoxicity is still to be elucidated. Thus, our study was designed to explore the molecular mechanisms underlying sorafenib-induced hepatotoxicity in an in vivo model. Twenty male adult Wistar rats were randomly placed into two groups; the first group received an oral dose of normal saline (vehicle), and the second received sorafenib (30 mg/kg) once daily for twenty-one consecutive days. After twenty-one days, liver tissues and blood samples were used for gene expression, protein expression, and biochemical analysis. Sorafenib treatment resulted in markedly increased levels of alanine aminotransferase and alkaline phosphatase, which indicate the presence of liver damage. Additionally, sorafenib administration induced the inflammatory and oxidative stress marker NF-κB-p65, while antioxidant enzymes were attenuated. Moreover, sorafenib caused upregulation of both gene and protein for the apoptotic markers cleaved Caspase-3, Bax, and Bid, and downregulation of the antiapoptotic protein Bcl-2. In conclusion, our findings suggest that sorafenib administration is associated with hepatotoxicity, which might be due to the activation of oxidative stress and apoptotic pathways.
1. Introduction
Tyrosine kinases (TKs) are key enzymes that play essential roles in a variety of biological activities, especially proliferation, differentiation, and survival of cells. Upon activation by extracellular signaling molecules, TKs activate several signaling pathways that are actively involved in diverse aspects of cellular physiology. Recently, it has been reported that mutations in TKs or overexpression of defective TKs are linked to cancer initiation, progression, and metastasis [1–3]. Because TKs are involved in the process of cancer development, a new class of drugs known as tyrosine kinase inhibitors (TKIs) was developed to block the activation of TKs and the signaling pathways downstream [1]. Currently, TKIs are used to treat a number of types of cancers [2–4].
Drugs that inhibit the activities of multiple TKs are known as multikinase inhibitors (MKIs). One of the most commonly used MKIs is sorafenib (SORA), a small molecule that is orally active and has anticancer and antiangiogenic activities [5, 6]. SORA was designed as a strong candidate for the inactivation of the unregulated Raf/MEK/ERK signaling cascade, which has been reported as a key factor in several cancers and other diseases [7]. SORA has been reported to target B-RAF, C-RAF, platelet-derived growth factor receptor- (PDGFR-) β, vascular endothelial growth factor receptors (VEGFR-1, -2, -3), c-kit, RET, and Fms-like tyrosine kinase-3 (Flt-3). Furthermore, SORA has the ability to hinder cancer growth, progression, metastasis, and angiogenesis, as well as to downregulate mechanisms that prevent cancers from being subject to apoptosis [8, 9]. Accordingly, SORA was approved for the use in renal cell carcinoma (RCC) and in the management of hepatocellular carcinoma (HCC) [10–12].
Although SORA is indicated for the treatment of different types of cancers and has been documented to improve survival in cancer patients, a spectrum of side effects has been reported, including loss of appetite, high blood pressure, diarrhea, hand-foot syndrome, acne, and other health-related problems [13, 14]. Recently, Zhang and his colleagues investigated the possible hepatotoxic effects of 31 FDA-approved TKIs [15]. They reported that SORA was one of three TKIs that were hepatotoxic at their Cmax concentrations and concluded that mitochondrial toxicity may contribute to this hepatotoxicity [15]. An in vitro study has also suggested that SORA-induced apoptosis is realized through reactive oxygen species (ROS) generation, JNK/p38-MAPK activation, and Bax translocation [16]. In addition, it has been shown that SORA treatment induced the activity of NF-κB-p65, which is reported to be induced in response to oxidative stress and inflammation leading to the development of HCC [17–19].
It is possible that SORA-induced hepatotoxicity is mediated through alterations in oxidative stress and apoptosis. However, no report to date has confirmed this; the precise mechanisms underlying SORA-induced hepatotoxicity remain unclear. In one study, Amemiya and his colleague studied the toxic effects of SORA and sunitinib using the mouse model. In their study, they found that 14 days of treatment of sunitinib (26.7 mg/kg), but not SORA (20.6 mg/kg), resulted in hepatotoxicity. Therefore, the current study aimed at examining the association of chronic treatment of SORA with liver toxicity in an in vivo model. Our results confirmed that chronic treatment with SORA induced liver toxicity, which manifested in terms of elevated liver enzymes, elevated oxidative stress markers, and dysregulated antioxidant mechanisms.
2. Methodology
2.1. Animals
Animals used in our study were taken from the animal facility at the College of Pharmacy, King Saud University and maintained in conditions regulated for temperature and humidity (23°C and 12 h. light/dark cycles) with free access to drinking water and standard diet. Animals were housed in clean cages and left to acclimatize without disturbance for 10 days prior to the start of the experiments. The experimental protocols and procedures mentioned in our study were in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals, and it is completely approved and accepted by the local institutional research ethics committee of King Saud University (KSU-SE-18-41).
2.2. Experimental Design and Treatment Protocol
Twenty male adult Wistar rats (weighing between 180 and 200 g) were used in our study and were randomly divided into two groups, with ten rats per group. Animals in group 1 (control) and group 2 (SORA), respectively, received equal doses of normal saline (0.9% NaCl P.O.) and sorafenib (30 mg/kg P.O.) once daily for 21 consecutive days [20]. Body weight was monitored daily during the study and the dose adjusted as needed.
At the end of the study, rats were fasted overnight and anesthetized by i.p. injection of ketamine/xylazine solution (ketamine 100 mg/kg and xylazine 10 mg/kg) [21], after which blood was collected directly from the hearts, and the plasma separated in order to measure liver enzymes and assess liver markers. In addition, liver tissues were harvested and washed immediately with cold phosphate-buffered saline (PBS) and directly kept in liquid nitrogen then stored at -80°C until the time of experiments. Thereafter, frozen liver tissues were used to conduct biochemical, protein expression, and gene expression analyses using commercially available kits according to their protocols.
2.3. Measurement of Plasma Markers
Plasma was obtained from whole blood samples by centrifugation for 5 minutes at 2000 g and 4°C. Then, the levels of cholesterol, triglycerides, high-density lipoprotein (HDL), low-density lipoprotein (LDL), very low-density lipoprotein (VLDL), alanine transaminase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), bilirubin, albumin, and urea were measured using the automated Dimension® RXL MAX Integrated Chemistry System (Siemens, USA).
2.4. Western Blot Analysis
Total proteins were extracted from liver tissue by homogenizing the samples in cold lysis buffer (Thermo Scientific, USA) that was mixed with protease and phosphatase inhibitors (Thermo Scientific, USA). The resulting tissue homogenates were centrifuged, clear supernatants collected, and total proteins quantified using a Direct Detect® spectrometer (EMD Millipore, USA). After that, the protein lysates were mixed with 2x Laemmli buffer (Bio-Rad, USA) that was supplemented with β-mercaptoethanol (βME). Then, protein lysates were heated at 95°C for five minutes. Thereafter, equal amounts of proteins were loaded in the wells of a 12% SDS-PAGE gel, resolved, and transferred to PVDF membranes using the Trans-Blot Turbo Transfer System (Bio-Rad, USA). Membranes were then blocked for one hour with 5% nonfat dry milk in tris-buffered saline (TBS) containing 0.1% Tween-20 at room temperature. After blocking, membranes were separately incubated at 4°C on a rocker with primary antibodies specific to the protein of interest; these were rabbit antisuperoxide dismutase-2 (SOD2) antibody (cat# A1340), anti-B-cell lymphoma-2 (Bcl-2) antibody (cat# A0208), anti-Bcl-2-associated X (Bax) antibody (cat# A12009), anticleaved Caspase-3 antibody (cat# A2156), and mouse anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (cat# AC001). All the primary antibodies were purchased from ABclonal Technology, USA, and used in dilution of 1 : 1000. Subsequently, the membranes were incubated with a suitable HRP-conjugated secondary antibody (Cell signaling technology, USA) for one hour. Finally, bands were developed using a chemiluminescence reagent (Merck Millipore, USA) and visualized using a ChemiDoc MP Imaging System (Bio-Rad, USA). The visualized blots were quantified and analyzed using ImageJ software [22].
2.5. Gene Expression Analysis by Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
From liver tissues, total RNA was isolated using TRIzol ™ reagent (Thermo Scientific, USA) according to the manufacturer's guidance. The purity and concentrations of the isolated RNA were measured using a NanoDrop™ 8000 Spectrophotometer (Thermo Scientific, USA). Thereafter, cDNA was synthesized from the isolated RNA using a TaqMan™ Reverse Transcription kit (Thermo Scientific, USA). Changes in the expression of various genes were quantified by a Quant Studio 6 Flex real-time PCR System (Thermo Fisher Scientific, USA) using SYBR green master mix (Bimake, USA) with β-actin as the reference housekeeping gene. Relative expressions of mRNA were calculated by the ΔΔCt method [23]. Sequences of the forward and reverse primers (IDT, Belgium) that were used in the present study are as follow; Bax (5′-TAGCAAACTGGTGCTCAAGG-3′; 5′-TCTTGGATCCAGACAAGCAG-3′), Bid (5′-CCCACACTGGTGAGACAACT-3′; 5′-TGTCGTTCTCCATGTCCCTA-3′), NF-κB-p65 (5′-CATGCGTTTCCGTTACAAGTGCGA-3′; 5′-TGGGTGCGTCTTAGTGGTATCTGT-3′), Bcl-2 (5′-CATGCGACCTCTGTTTGA-3′; 5′-GTTTCATGGTCCATCCTTG-3′), GPX-1 (5′-AGTTCGGACATCAGGAGAATGGCA-3′; 5′-TCACCATTCACCTCGCACTTCTCA-3′), HO-1 (5′-ACAGGGTGACAGAAGAGGCTAA-3′; 5′-CTGTGAGGGACTCTGGTCTTTG-3′), SOD (5′-TTCGTTTCCTGCGGCGGCTT-3′; 5′-TTCAGCACGCACACGGCCTT-3′), and β-actin (5′-CCAGATCATGTTTGAGACCTTCAA-3′; 5′-GTGGTACGACCAGAGGCATACA-3′).
2.6. Catalase Activity Measurement
Catalase enzyme activity was quantified using an EnzyChrom™ Catalase Assay Kit (cat# ECAT-100, BioAssay Systems, USA) as per the manufacturer's directions. Briefly, liver tissues were homogenized in cold PBS and then centrifuged at high speed for ten minutes. Afterwards, 10 μl of samples was mixed with 90 μl of 50 μM H2O2 to initiate the catalase reaction and incubated at room temperature for 30 minutes. After that, 100 μl of the detection reagent was added to the mixture and incubated for 10 minutes at room temperature. Then, the optical density (ΔOD) was measured at 550 nm.
2.7. Measurement of Glutathione (GSH)
Reduced glutathione (GSH) and oxidized glutathione (GSSG) were measured in liver tissue homogenates using an EnzyChrom™ Glutathione Assay Kit (BioAssay Systems, USA) according to the manufacturer's guidelines. In general, liver tissues were homogenized in cold buffer containing 1 mM EDTA and 50 mM phosphate (pH = 7) in the presence or absence of scavenger to measure GSSG and GSH, respectively. Then, tissue lysates were centrifuged at high speed for five minutes at 4°C and deproteinated using metaphosphoric acid. Afterwards, 200 μl of samples was incubated with 100 μl of detection reagent, which includes assay buffer, NADPH, DNTB, and glutathione reductase (GR). Finally, the optical density differences (ΔOD) were measured at 412 nm.
2.8. NAD+/NADH Measurement
NAD+/NADH was measured in liver tissue homogenates using an EnzyChrom™ NAD+/NADH Assay Kit (BioAssay Systems, USA) according to the manufacturer's procedure. Briefly, liver tissues were homogenized in NAD and NADH extraction buffers to determine NAD and NADH, respectively. Homogenates were heated at 60°C for five minutes, and then incubated with the opposite extraction buffer for neutralization. Next, the samples were centrifuged at 14,000 rpm for five minutes. After that, differences in optical density (ΔΔOD) were measured at 565 nm.
2.9. Statistical Analysis
Results are presented as mean ± SD. All statistical analyses used a two-tailed student's t-test with the level of significance set at P < 0.05. Statistical analyses were achieved using GraphPad Prism 6.01 (CA, USA).
3. Results
3.1. Effect of Sorafenib on Liver Function Tests and Lipid Profile
A number of plasma parameters are used clinically for the analysis of liver functions. To examine whether sorafenib (SORA) administration compromises liver function, we measured several liver function-associated enzymes (ALT, AST, and ALP), plasma proteins (albumin, bilirubin, and urea), and lipid profiles (cholesterol, triglycerides, HDL, LDL, and VLDL). We found that twenty-one days of oral SORA administration at a dose of 30 mg/kg significantly induced ALT (1.5 folds), ALP (1.8 folds), cholesterol (1.2 folds), LDL (1.8 folds), and urea (1.4 folds), whereas levels of AST, bilirubin, triglycerides, HDL, and VLDL were statistically comparable between groups (Figures 1(a)–1(k)). Furthermore, plasma albumin levels were noticeably lower in SORA-treated rats compared to control rats (1.1 folds) (Figure 1(d)). Taken together, these results suggest that SORA treatment is associated with liver damage.
Figure 1.
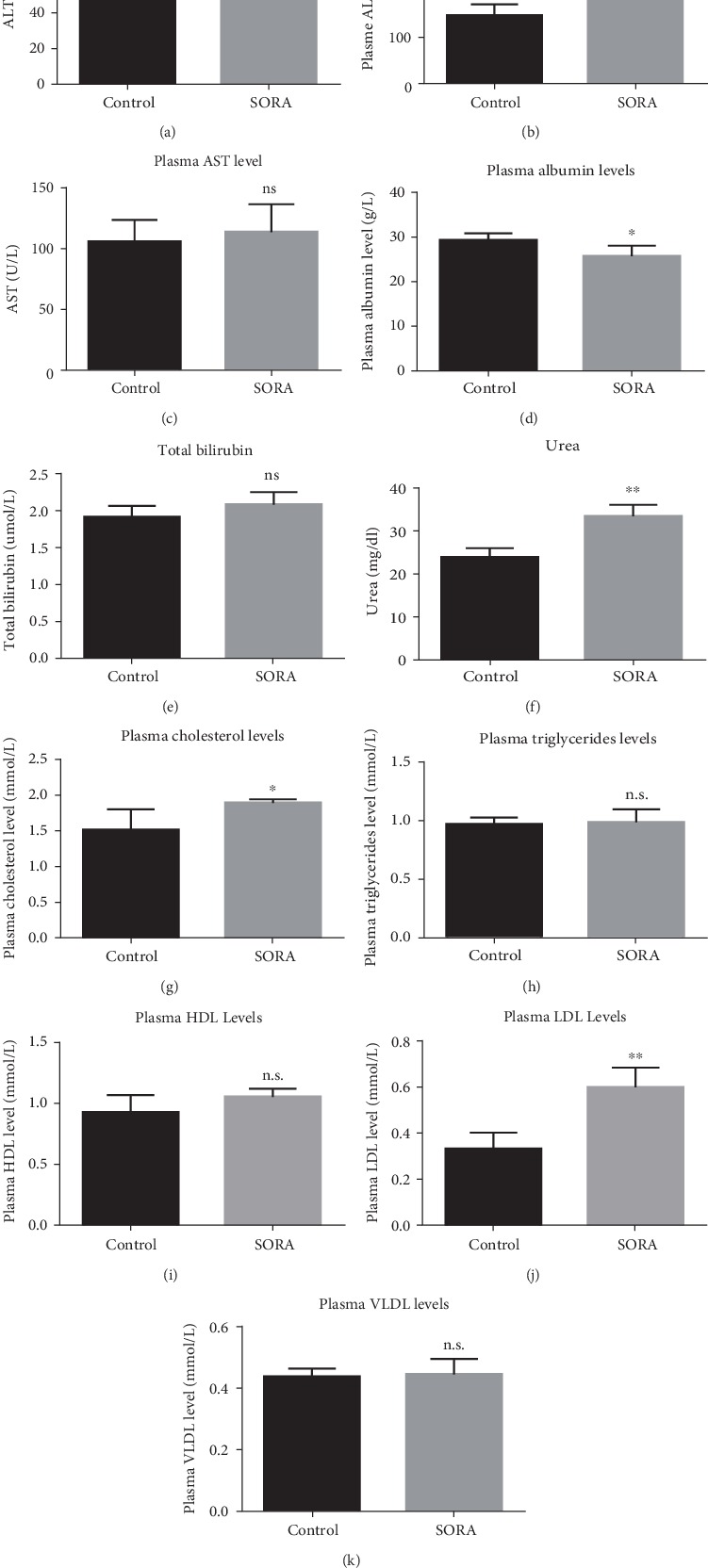
Plasma liver markers. Whole blood samples were used to separate plasma and measure the liver enzymes and liver markers. Data are presented as mean ± SD. Comparison between the control group and the sorafenib group is presented by ∗, where ∗P < 0.05, while ∗∗P < 0.01. n.s. means there were no significant changes (P > 0.05). SORA: sorafenib; ALT: alanine transaminase; ALP: alkaline phosphatase; AST: aspartate aminotransferase; HDL: high-density lipoprotein; LDL: low-density lipoprotein; VLDL: very low-density lipoprotein.
3.2. Sorafenib Activates Oxidative Stress and Apoptotic Pathways
To further investigate the mechanisms underlying SORA-induced hepatotoxicity, we measured the gene expression of multiple genes involved in apoptotic and oxidative stress pathways. Our mRNA analysis revealed that SORA administration considerably induced expression of proapoptotic genes (BAX and BID) (1.4 folds and 1.5 folds, respectively) and produced a noteworthy decrease in the levels of an antiapoptotic gene (Bcl-2) (2 folds) (Figures 2(a), 2(b), and 2(d), respectively). In addition, we detected a considerable increase in the expression of NF-κB-p65 (1.6 folds) in the SORA-treated group (Figure 2(c)). Furthermore, we found that the antioxidant genes GPX-1, HO-1, and SOD2 had considerably reduced expression in SORA-treated rats (1.5 folds, 1.5 folds, and 1.7 folds, respectively) (Figures 2(e)–2(g)). Together, these changes in the expression of proapoptotic and antiapoptotic genes suggest that SORA treatment is associated with liver toxicity.
Figure 2.
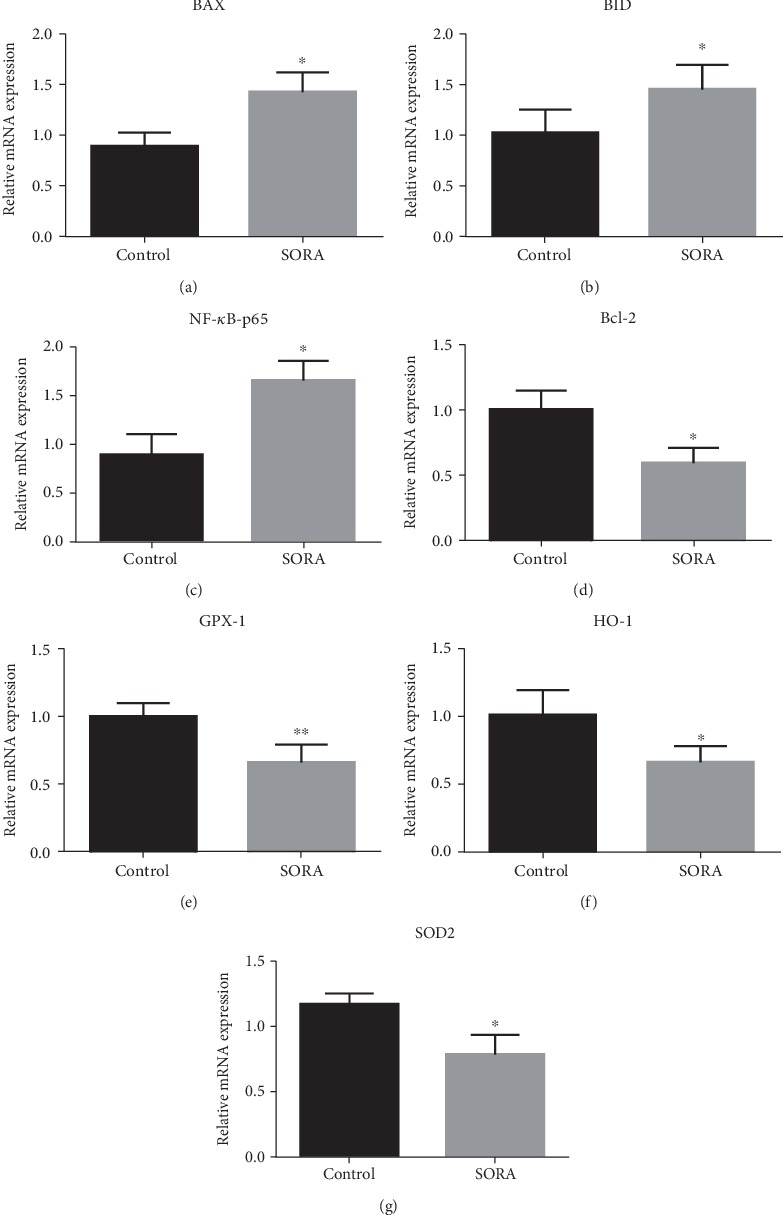
Gene expression analysis. RNA was isolated from the liver of different samples and was used to measure mRNA levels of different genes in each group (a–g) using Quantitative RT-QPCR. Data are presented as mean ± SD. Comparison between the control group and the sorafenib group is presented by ∗, where ∗P < 0.05, while ∗∗P < 0.01. SORA: sorafenib; BAX: Bcl-2 Associated X; BID: BH3 interacting-domain death agonist; NF-κB-p65: Nuclear factor kappa B; Bcl-2: B-cell lymphoma-2; GPX-1: Glutathione peroxidase-1; HO-1: Heme Oxygenase-1; SOD2: superoxide dismutase-2.
3.3. Sorafenib Reduces the Levels of Antioxidant and Antiapoptotic Proteins
To further confirm our findings from gene expression studies, we used western blot analysis to determine levels of the proapoptotic proteins Bax and cleaved Caspase-3, the antiapoptotic protein Bcl-2, and the antioxidant protein SOD2. We found that SORA treatment remarkably induced Bax and cleaved Caspase-3 protein expression relative to the control group (1.6 folds and 1.7 folds, respectively) (Figures 3(a) and 3(b)). Furthermore, we observed considerable decreases in the expression of Bcl-2 and SOD2 proteins in SORA-treated rats (2 folds and 1.5 folds, respectively) (Figures 3(c) and 3(d)). These results further confirm that SORA treatment is associated with liver toxicity, as it activated oxidative stress and apoptotic pathways.
Figure 3.
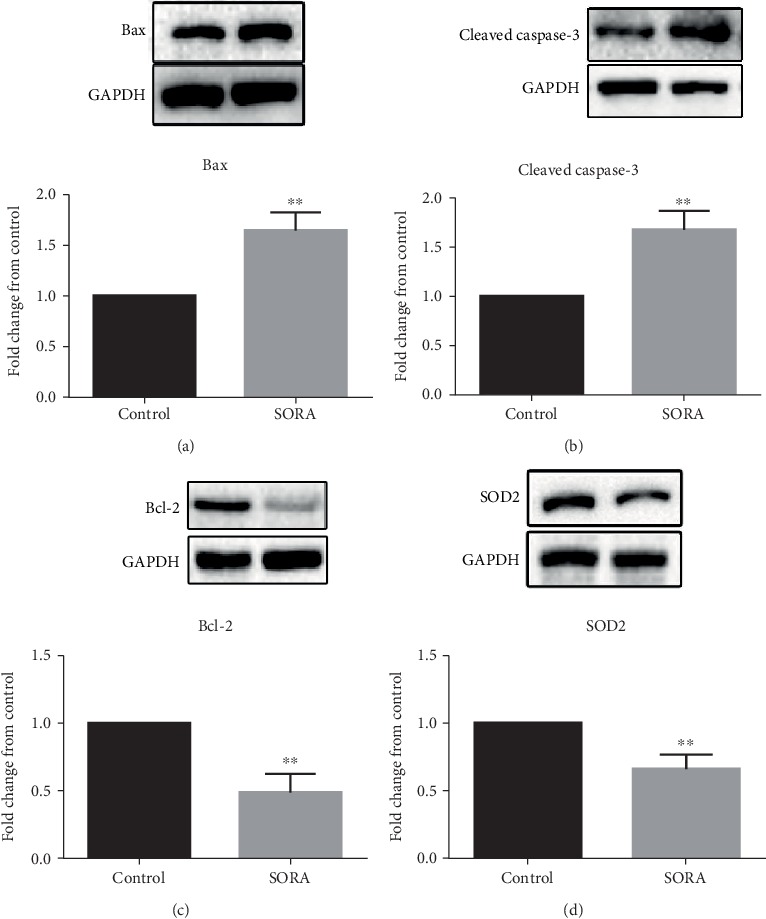
Protein expression analysis. Representative blots analysis of protein levels of (a) Bax, (b) cleaved Caspase-3, (c) Bcl-2, and (d) SOD2. Data are presented as mean ± SD. Where ∗∗P < 0.01. SORA: sorafenib; BAX: Bcl-2 Associated X; Bcl-2: B-cell lymphoma-2; SOD2: superoxide dismutase-2.
3.4. Sorafenib Attenuates the Activity of Antioxidant Enzymes
Several publications have reported that SORA treatment alters the activity of catalase, glutathione (GSH), oxidized glutathione (GSSG), and NAD+ [24–26]. Therefore, to further explore the toxic effects of SORA, we measured the enzyme activity of catalase and GSH along with the NAD+/NADH ratio. As shown in Figure 4, SORA administration significantly diminished the enzyme activity of catalase (5.5 folds) and the NAD+/NADH ratio (2 folds), as well as GSH (1.5 folds) (Figures 4(a)–4(c)). However, we demonstrated a marked increase in the levels of GSSG in SORA treated rats (1.3 folds) (Figure 4(d)). Together, these results support our previous findings that SORA treatment is associated with the induction of hepatotoxicity.
Figure 4.
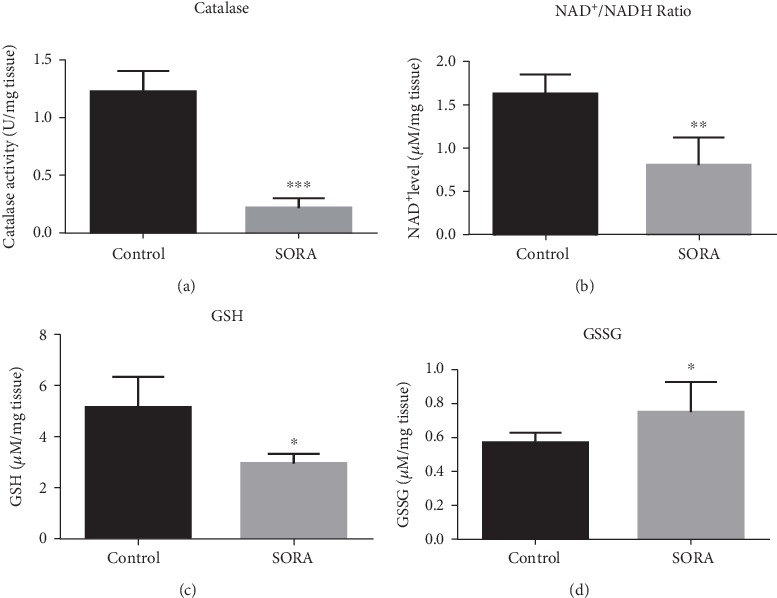
Biochemical assays. Liver tissues homogenates from control and SORA groups were obtained to analyze catalase activity (a), NAD+/NADH ratio (b), GSH (c), and GSSG (d). Data are presented as mean ± SD. Comparison between the control group and the sorafenib group is presented by ∗, where ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. SORA: sorafenib; NAD: Nicotinamide adenine dinucleotide; GSH: glutathione; GSSG: oxidized glutathione.
4. Discussion and Conclusion
In the current study, we studied the molecular mechanisms by which the TKI sorafenib (SORA) induces hepatotoxicity in rats. Our study mainly shows that at a dose of 30 mg/kg, SORA produces liver toxicity as evidenced by the reduction of antioxidant enzyme activities, elevation of hepatic enzymes, activation of the apoptotic pathway, and induction of oxidative stress markers. To the best of our knowledge, this is the first study to report the hepatotoxic effect of SORA using an in vivo model and to investigate the potential mechanisms underlying that toxicity. Future studies are needed to fully elucidate the molecular mechanisms underlying SORA-induced liver toxicity.
Drug-induced hepatotoxicity is a frequent and unpredictable potential risk during the treatment of numerous diseases, including gastrointestinal stromal tumor (GIST) [27, 28]. Sorafenib (SORA) is a well-known multikinase inhibitor that has the potential to inhibit cancer cell growth through its antiangiogenic effects and is used effectively in the treatment of advanced renal cell carcinoma and hepatocellular carcinoma. Several mechanisms by which SORA exerts antioncogenic effects have been reported, including the activation of JNK/p38-MAPK pathways and Bax translocation [13, 14, 16, 29]. Nonetheless, recent evidence has also demonstrated an association of SORA treatment with life-threatening hepatotoxicity in a 57-year-old male patient with GIST [28]. Furthermore, in a study that used rat liver mitochondria as a powerful in vitro model to test 31 approved kinase inhibitors, Zhang and his colleagues reported induction of hepatotoxicity by SORA treatment [15]. Of the 31 tested drugs, only sorafenib, pazopanib, and regorafenib showed mitochondrial toxicity when used at concentrations approximately near the maximum serum concentrations (Cmax). Their data collectively demonstrated three mitochondrial mechanisms that contributed to SORA-induced liver toxicity: mitochondrial swelling, decline in mitochondrial membrane potential, and inhibition of selective respiratory chain complexes. Although their study reported mitochondrial toxicity of SORA in an in vitro model, the exact mechanism through which SORA induces hepatotoxicity in vivo is yet to be considered.
Several reports have suggested alterations in liver enzymes such as ALP, AST, ALT, and urea and dysregulation of blood-associated proteins and lipids as reliable indicators of drug-mediated hepatotoxicity [30–34]. In the current study, SORA treatment significantly induced plasma levels of ALT and ALP, but not AST, while decreasing plasma levels of albumin, consistent with previously published reports [33, 35, 36]. Data from our current study demonstrated that SORA resulted in remarkable changes in cholesterol and LDL levels, and had no significant effects on triglyceride, HDL, or VLDL levels. The dramatic changes strongly indicate the ability of SORA to induce liver toxicity in vivo.
NF-κB is a transcriptional factor whose inactive form is located in the cytoplasm and bound to the inhibitory subunit I-κB. When a toxic agent is administered, reactive oxygen species (ROS) are produced, which further activates the NF-κB pathway; this leads to the induction of apoptosis, which ultimately induces toxicity [37–39]. Although the mechanism of apoptosis involves several factors, it is believed that two protein families are primarily involved, the caspase family (especially caspase-3, 8, and 9) and the Bcl-2 family [39]. The caspase enzymes play essential roles in apoptosis, with caspase-3 being considered the most important as it contributes to several biochemical mechanisms that result in the cleavage of cytosolic and nuclear substances, condensation of chromatin, and DNA damage [40–44]. In order to understand the mechanism underlying SORA-induced hepatotoxicity, we analyzed different oxidative stress markers and the expression of antioxidant, proapoptotic, and antiapoptotic genes. Here, we report that SORA treatment significantly upregulated the expression of NF-κB-p65 and the proapoptotic genes Bax and cleaved Caspase-3, whereas both gene and protein expressions of the antiapoptotic protein Bcl-2 and the antioxidant enzyme SOD2 were attenuated. Our results suggest that SORA induces liver toxicity through the activation of apoptotic pathways, which is in accordance with previously published papers [45–47].
An imbalance between antioxidants and the oxidative system of cells can result in a greater generation of oxidative free radicals, which can be removed from the biological system via enzymatic and nonenzymatic antioxidants [48–50]. Glutathione (GSH) plays a dual role; it can act as a nonenzymatic antioxidant that directly interacts with ROS through its thiol (–SH) group and as a cofactor during the enzymatic detoxification of ROS [51–53]. A reduced cytosolic NAD+/NADH ratio was reported in patients with fatty liver disease (NAFLD) and in animal models [54]. Any approach or treatment that increases cytosolic NAD+/NADH can help to improve NAFLD [54]. In the current study, we measured enzyme activities in order to further understand the mechanism of SORA toxicity. We demonstrated that antioxidant enzymes had diminished activities in SORA-treated animals compared to control animals, which further supports our findings that activation of oxidative stress and apoptotic pathways may contribute to the hepatotoxic effects of SORA. Furthermore, we found a significant decrease in the cytosolic NAD+/NADH ratio in liver homogenates of SORA-treated rats, which further suggests that SORA induces liver damage.
In conclusion, the current study is the first to demonstrate in an in vivo model both the potential hepatotoxic effect of SORA and the possible mechanism underlying this toxic effect. Further studies are required to fully understand the toxicity of SORA.
One of the limitations in the current study is that it does not measure the protein expression of NF-κB-p65 to further validate the proposed mechanism. Furthermore, we acknowledge that measuring other direct oxidative stress markers and mitochondrial injury markers, such as H2O2, NADPH oxidase, and cytochrome c, would further support the proposed mechanism. However, a considerable part of our future study will focus on these issues to further confirm the hepatotoxic effect of SORA.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through the Research group No. RG-1441-451.
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Paech F., Mingard C., Grünig D., Abegg V. F., Bouitbir J., Krähenbühl S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology. 2018;395:34–44. doi: 10.1016/j.tox.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Montor W. R., Salas A., Melo F. H. M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Molecular Cancer. 2018;17(1):p. 55. doi: 10.1186/s12943-018-0792-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segovia-Mendoza M., González-González M. E., Barrera D., Díaz L., García-Becerra R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: preclinical and clinical evidence. American Journal of Cancer Research. 2015;5(9):2531–2561. [PMC free article] [PubMed] [Google Scholar]
- 4.Regad T. Targeting RTK signaling pathways in cancer. Cancers. 2015;7(3):1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin S., Li A., Yi M., Yu S., Zhang M., Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. Journal of Hematology & Oncology. 2019;12(1):p. 27. doi: 10.1186/s13045-019-0718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider T. C., Kapiteijn E., van Wezel T., Smit J. W. A., van der Hoeven J. J. M., Morreau H. (Secondary) solid tumors in thyroid cancer patients treated with the multi-kinase inhibitor sorafenib may present diagnostic challenges. BMC Cancer. 2016;16(1):p. 31. doi: 10.1186/s12885-016-2060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts P. J., Der C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 8.Pitoia F., Jerkovich F. Selective use of sorafenib in the treatment of thyroid cancer. Drug Design, Development and Therapy. 2016;10:1119–1131. doi: 10.2147/dddt.s82972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.European Medicines Agency. Nexavar (sorafenib): summary of product characteristics. United Kingdom: European Medicines Agency; 2014. [Google Scholar]
- 10.Tadmor T., Tallman M. S., Polliack A. Sorafenib – a small molecule with big promise? Leukemia & Lymphoma. 2009;51(2):181–182. doi: 10.3109/10428190903470893. [DOI] [PubMed] [Google Scholar]
- 11.Steeghs N., Nortier J. W., Gelderblom H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Annals of Surgical Oncology. 2007;14(2):942–953. doi: 10.1245/s10434-006-9227-1. [DOI] [PubMed] [Google Scholar]
- 12.Strumberg D. Efficacy of sunitinib and sorafenib in non-clear cell renal cell carcinoma: results from expanded access studies. Journal of Clinical Oncology. 2008;26(20):3469–3471. doi: 10.1200/JCO.2008.17.7410. [DOI] [PubMed] [Google Scholar]
- 13.Parikh N. D., Marshall V. D., Singal A. G., et al. Survival and cost‐effectiveness of sorafenib therapy in advanced hepatocellular carcinoma: an analysis of the SEER–Medicare database. Hepatology. 2017;65(1):122–133. doi: 10.1002/hep.28881. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Gao Z. H., Qu X. J. The adverse effects of sorafenib in patients with advanced cancers. Basic & Clinical Pharmacology & Toxicology. 2015;116(3):216–221. doi: 10.1111/bcpt.12365. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J., Salminen A., Yang X., et al. Effects of 31 FDA approved small-molecule kinase inhibitors on isolated rat liver mitochondria. Archives of Toxicology. 2017;91(8):2921–2938. doi: 10.1007/s00204-016-1918-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park G. B., Choi Y., Kim Y. S., Lee H.-K., Kim D., Hur D. Y. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in Sorafenib-induced apoptosis of EBV-transformed B cells. International Journal of Oncology. 2014;44(3):977–985. doi: 10.3892/ijo.2014.2252. [DOI] [PubMed] [Google Scholar]
- 17.Lo J., Lau E. Y. T., Ching R. H. H., et al. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology. 2015;62(2):534–545. doi: 10.1002/hep.27859. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y., Luo R., Zheng H., et al. Synergistic anti-tumor efficacy of sorafenib and fluvastatin in hepatocellular carcinoma. Oncotarget. 2017;8(14):23265–23276. doi: 10.18632/oncotarget.15575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreck R., Albermann K., Baeuerle P. A. Nuclear factor Kb: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radical Research Communications. 2009;17(4):221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 20.Yan J., Tan C., Gu F., et al. Sorafenib delays recurrence and metastasis after liver transplantation in a rat model of hepatocellular carcinoma with high expression of phosphorylated extracellular signal-regulated kinase. Liver Transplantation. 2013;19(5):507–520. doi: 10.1002/lt.23619. [DOI] [PubMed] [Google Scholar]
- 21.Wellington D., Mikaelian I., Singer L. Comparison of ketamine-xylazine and ketamine-dexmedetomidine anesthesia and intraperitoneal tolerance in rats. Journal of the American Association for Laboratory Animal Science. 2013;52(4):481–487. [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider C. A., Rasband W. S., Eliceiri K. W. NIH image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak K. J., Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Coriat R., Nicco C., Chereau C., et al. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Molecular Cancer Therapeutics. 2012;11(10):2284–2293. doi: 10.1158/1535-7163.MCT-12-0093. [DOI] [PubMed] [Google Scholar]
- 25.Chiou J. F., Tai C. J., Wang Y. H., Liu T. Z., Jen Y. M., Shiau C. Y. Sorafenib induces preferential apoptotic killing of a drug- and radio-resistant Hep G2 cells through a mitochondria-dependent oxidative stress mechanism. Cancer Biology & Therapy. 2014;8(20):1904–1913. doi: 10.4161/cbt.8.20.9436. [DOI] [PubMed] [Google Scholar]
- 26.Garten A., Grohmann T., Kluckova K., Lavery G. G., Kiess W., Penke M. Sorafenib-induced apoptosis in hepatocellular carcinoma is reversed by SIRT1. International Journal of Molecular Sciences. 2019;20, article 4048(16) doi: 10.3390/ijms20164048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An Z., Li C., Lv Y., Li P., Wu C., Liu L. Metabolomics of hydrazine-induced hepatotoxicity in rats for discovering potential biomarkers. Disease Markers. 2018;2018:12. doi: 10.1155/2018/8473161.8473161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murad W., Rabinowitz I., Lee F. C. Sorafenib-induced grade four hepatotoxicity in a patient with recurrent gastrointestinal stromal tumor (GIST): a case report and review of literature. ACG Case Reports Journal. 2014;1(2):115–117. doi: 10.14309/crj.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilhelm S. M., Adnane L., Newell P., Villanueva A., Llovet J. M., Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Molecular Cancer Therapeutics. 2008;7(10):3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rashid S., Ali N., Nafees S., et al. Alleviation of doxorubicin-induced nephrotoxicity and hepatotoxicity by chrysin in Wistar rats. Toxicology Mechanisms and Methods. 2013;23(5):337–345. doi: 10.3109/15376516.2012.759306. [DOI] [PubMed] [Google Scholar]
- 31.Ali N., Rashid S., Nafees S., Hasan S. K., Sultana S. Beneficial effects of Chrysin against methotrexate-induced hepatotoxicity via attenuation of oxidative stress and apoptosis. Molecular and Cellular Biochemistry. 2014;385(1-2):215–223. doi: 10.1007/s11010-013-1830-4. [DOI] [PubMed] [Google Scholar]
- 32.Liu X., Zhang H., Liang J. Blood urea nitrogen is elevated in patients with non-alcoholic fatty liver disease. Hepato-Gastroenterology. 2013;60(122):343–345. [PubMed] [Google Scholar]
- 33.Spinella R., Sawhney R., Jalan R. Albumin in chronic liver disease: structure, functions and therapeutic implications. Hepatology International. 2016;10(1):124–132. doi: 10.1007/s12072-015-9665-6. [DOI] [PubMed] [Google Scholar]
- 34.Radhiga T., Sundaresan A., Viswanathan P., Pugalendi K. V. Effect of protocatechuic acid on lipid profile and DNA damage in D-galactosamine-induced hepatotoxic rats. Journal of Basic and Clinical Physiology and Pharmacology. 2016;27(5):505–514. doi: 10.1515/jbcpp-2015-0135. [DOI] [PubMed] [Google Scholar]
- 35.Raish M., Ahmad A., Alkharfy K. M., et al. Hepatoprotective activity of Lepidium sativum seeds against D-galactosamine/lipopolysaccharide induced hepatotoxicity in animal model. BMC Complementary and Alternative Medicine. 2016;16(1):p. 501. doi: 10.1186/s12906-016-1483-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.AL-Megrin W. A., Alkhuriji A. F., Yousef A. O. S., et al. Antagonistic efficacy of luteolin against lead acetate exposure-associated with hepatotoxicity is mediated via antioxidant, anti-inflammatory, and anti-apoptotic activities. Antioxidants. 2020;9(1):p. 10. doi: 10.3390/antiox9010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abraham E. Nuclear factor–κB and its role in sepsis-associated organ failure. The Journal of Infectious Diseases. 2003;187(s2):S364–S369. doi: 10.1086/374750. [DOI] [PubMed] [Google Scholar]
- 38.Lee C. K., Lee E. Y., Kim Y. G., Mun S. H., Moon H. B., Yoo B. Alpha-lipoic acid inhibits TNF-α induced NF-κB activation through blocking of MEKK1–MKK4–IKK signaling cascades. International Immunopharmacology. 2008;8(2):362–370. doi: 10.1016/j.intimp.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 39.Ilbey Y. O., Ozbek E., Cekmen M., Simsek A., Otunctemur A., Somay A. Protective effect of curcumin in cisplatin-induced oxidative injury in rat testis: mitogen-activated protein kinase and nuclear factor-kappa B signaling pathways. Human Reproduction. 2009;24(7):1717–1725. doi: 10.1093/humrep/dep058. [DOI] [PubMed] [Google Scholar]
- 40.Wyllie A. H. Apoptosis: an overview. British Medical Bulletin. 1997;53(3):451–465. doi: 10.1093/oxfordjournals.bmb.a011623. [DOI] [PubMed] [Google Scholar]
- 41.Zhan Y., van de Water B., Wang Y., Stevens J. L. The roles of caspase-3 and bcl-2 in chemically-induced apoptosis but not necrosis of renal epithelial cells. Oncogene. 1999;18(47):6505–6512. doi: 10.1038/sj.onc.1203060. [DOI] [PubMed] [Google Scholar]
- 42.Salvesen G. S., Dixit V. M. Caspases: intracellular signaling by proteolysis. Cell. 1997;91(4):443–446. doi: 10.1016/S0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 43.Yi X., Yin X. M., Dong Z. Inhibition of bid-induced apoptosis by Bcl-2. tBid insertion, Bax translocation, and Bax/Bak oligomerization suppressed. The Journal of Biological Chemistry. 2003;278(19):16992–16999. doi: 10.1074/jbc.M300039200. [DOI] [PubMed] [Google Scholar]
- 44.Yan H., du J., Chen X., et al. ROS-dependent DNA damage contributes to crizotinib-induced hepatotoxicity via the apoptotic pathway. Toxicology and Applied Pharmacology. 2019;383, article 114768 doi: 10.1016/j.taap.2019.114768. [DOI] [PubMed] [Google Scholar]
- 45.Hamzeh M., Hosseinimehr S. J., Khalatbary A. R., Mohammadi H. R., Dashti A., Amiri F. T. Atorvastatin mitigates cyclophosphamide-induced hepatotoxicity via suppression of oxidative stress and apoptosis in rat model. Research in Pharmaceutical Sciences. 2018;13(5):440–449. doi: 10.4103/1735-5362.236837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arjinajarn P., Chueakula N., Pongchaidecha A., et al. Anthocyanin-rich Riceberry bran extract attenuates gentamicin-induced hepatotoxicity by reducing oxidative stress, inflammation and apoptosis in rats. Biomedicine & Pharmacotherapy. 2017;92:412–420. doi: 10.1016/j.biopha.2017.05.100. [DOI] [PubMed] [Google Scholar]
- 47.Ali N., Rashid S., Nafees S., et al. Protective effect of chlorogenic acid against methotrexate induced oxidative stress, inflammation and apoptosis in rat liver: an experimental approach. Chemico-Biological Interactions. 2017;272:80–91. doi: 10.1016/j.cbi.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Tan B. L., Norhaizan M. E., Liew W. P. P., Sulaiman Rahman H. Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Frontiers in Pharmacology. 2018;9:p. 1162. doi: 10.3389/fphar.2018.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newsholme P., Cruzat V. F., Keane K. N., Carlessi R., de Bittencourt P. I. H., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. The Biochemical Journal. 2016;473(24):4527–4550. doi: 10.1042/BCJ20160503C. [DOI] [PubMed] [Google Scholar]
- 50.Rehman M. U., Ali N., Rashid S., et al. Alleviation of hepatic injury by chrysin in cisplatin administered rats: probable role of oxidative and inflammatory markers. Pharmacological Reports. 2014;66(6):1050–1059. doi: 10.1016/j.pharep.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Pratibha R., Sameer R., Rataboli P. V., Bhiwgade D. A., Dhume C. Y. Enzymatic studies of cisplatin induced oxidative stress in hepatic tissue of rats. European Journal of Pharmacology. 2006;532(3):290–293. doi: 10.1016/j.ejphar.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 52.dos Santos N. A. G., Martins N. M., Curti C., de Lourdes Pires Bianchi M., dos Santos A. C. Dimethylthiourea protects against mitochondrial oxidative damage induced by cisplatin in liver of rats. Chemico-Biological Interactions. 2007;170(3):177–186. doi: 10.1016/j.cbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 53.Liao Y., Lu X., Lu C., Li G., Jin Y., Tang H. Selection of agents for prevention of cisplatin-induced hepatotoxicity. Pharmacological Research. 2008;57(2):125–131. doi: 10.1016/j.phrs.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Masia R., McCarty W. J., Lahmann C., et al. Live cell imaging of cytosolic NADH/NAD+ ratio in hepatocytes and liver slices. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2018;314(1):G97–G108. doi: 10.1152/ajpgi.00093.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.