

Early diagnosis of Turner syndrome enhances care, but in routine practice, even within larger referral centers, diagnosis is delayed. Our study examines the utility of an electronic health record (EHR) algorithm in identifying patients at high risk for TS. Six percent of those identified had missed diagnoses of TS.
Turner syndrome results from a complete or partial loss of the X chromosome and affects 25–50 per 100,000 females (1). TS is relatively common in females with unexplained short stature, but the heterogenous phenotype may result in a delayed diagnosis if classic features (e.g. lymphedema and webbed neck) are not present at birth. Only 20–40% of affected individuals are diagnosed in the newborn period with the remaining cases caught during late childhood with abnormal growth or in adolescence with lack of pubertal development (2–4). Savendhahl et al evaluated the age of diagnosis in 81 girls with TS, of which about half were diagnosed in infancy. If not diagnosed in infancy, the mean age of diagnosis was 8.8 ± 5 years (delay in diagnosis averaged 7.7 ± 5.4 years) at a mean height of −2.9 standard deviations (SD) (5). Studies have shown that combined auxological measures (an absolute height SD < −2 and a distance to target height of > 2 SD) provide the best predictive value for pathological short stature, and in patients >3 years old the distance to target height is the most important criterion (4, 6, 7). Careful growth monitoring with attention to such measures is paramount to a timely diagnosis of TS, especially in cases of mosaicism where short stature may be the only obvious sign. Furthermore, with early detection and initiation of growth hormone (GH) therapy, most girls with TS can reach an adult height within the normal range (1).
In addition to improving growth outcomes, prompt detection of TS can be lifesaving, as some patients have unsuspected cardiovascular anomalies and are at increased risk for aortic dilation, dissection, and rupture (8). Health related quality of life measures also improve when treatments such as hormone replacement therapy for primary ovarian failure are initiated within an age-appropriate timeframe, allowing for pubertal induction and progression in congruence with peers (1). Furthermore, early recognition of cognitive and psychosocial deficits can allow for tailored interventions that help promote optimal educational attainment and adaptive skills.
Sophisticated health information technology (HIT) systems have the ability to enhance care with the centralization of biochemical, auxologic, and historical data that can be harnessed for real time phenotype curation—identifying and alerting physicians to groups that fit criteria for potential pathology. Such clinical decision support systems with point of care reminders have been shown to cause small to modest improvements in provider behavior (9) and may be the key to earlier recognition of pathology, such as abnormal growth patterns in females who are at risk for TS. Thus, we hypothesize that a best practice alert guided by an algorithm based on both absolute height SD and a deflection from mid-parental height (MPH) will successfully identify patients at risk for TS, promote appropriate referrals, and lead to more timely diagnostic evaluation.
METHODS:
This study assesses the ability of an algorithm-driven EHR search to identify potentially missed cases of TS within a cohort of females with idiopathic short stature (ISS). This study was approved by the Institutional Review Board (IRB) at Cincinnati Children’s Hospital Medical Center (CCHMC). Consent was obtained from all subjects.
Clinical data of ISS females seen in the Endocrinology Clinic at CCHMC from 2012–2017 was extracted from the hospital’s common EHR system, Epic, and uploaded into a research database for analysis. The algorithm identified patients with a height SD < −2, BMI > 5th percentile, and absence of chronic illness, as evidenced by ICD-10 codes manually reviewed by the study team. We further selected for patients that had height data available on both parents, were ≥ 1 SD below MPH, and had not yet received genetic testing. The MPH was defined as the mean height standard deviation score (HSDS) of both parents. Additionally, we queried the EHR to gather a positive TS control group.
Microarray analysis was performed using the HumanCoreExome kit on the 34 available ISS samples and five TS controls (as a quality control measure). PLINK data files were manually reviewed by our team’s cytogeneticist. Graphic representations of B allele frequency and log R ratio for each patient’s genotype were utilized to identify copy number variation and copy neutral structural variants as well as their intensities (level of mosaicism).
Statistical analysis was done with CRAN R version 3.5.1. A Wilcoxon rank-sum test was used to compare the ISS and TS control groups, as outlined in the results, given that data was not normally distributed. A P value of ≤.05 was considered as statistically significant.
RESULTS:
The EHR search identified 216 ISS females and 76 matched TS controls. The ISS cohort was plotted against TS controls by HSDS (x-axis) and distance to MPH (y-axis) (Figure). By definition, all patients with ISS had a HSDS below −2. Of the children presenting with a HSDS below −2, a distance to MPH of more than 1 SD encompassed all known TS controls. Therefore, we selected a cut-off of > 1 SD from MPH for our algorithm to maximize the sensitivity for detecting missed cases of TS in our study population. 189 patients were more than 1 SD from their target height, of which 117 (62%) had previously normal karyotypes and 72 (38%) never had genetic testing. Comparison of the 2 ISS groups, those previously karyotyped versus non-karyotyped, and the non-karyotyped patients with samples available for microarray analysis are presented in the Table. The patients that previously underwent karyotype analysis were significantly shorter with a greater MPH deflection (Table). Of the 34 non-karyotyped patients studied, their age at presentation and height measures were representative of the remaining non-karyotyped group (Table).
Figure.
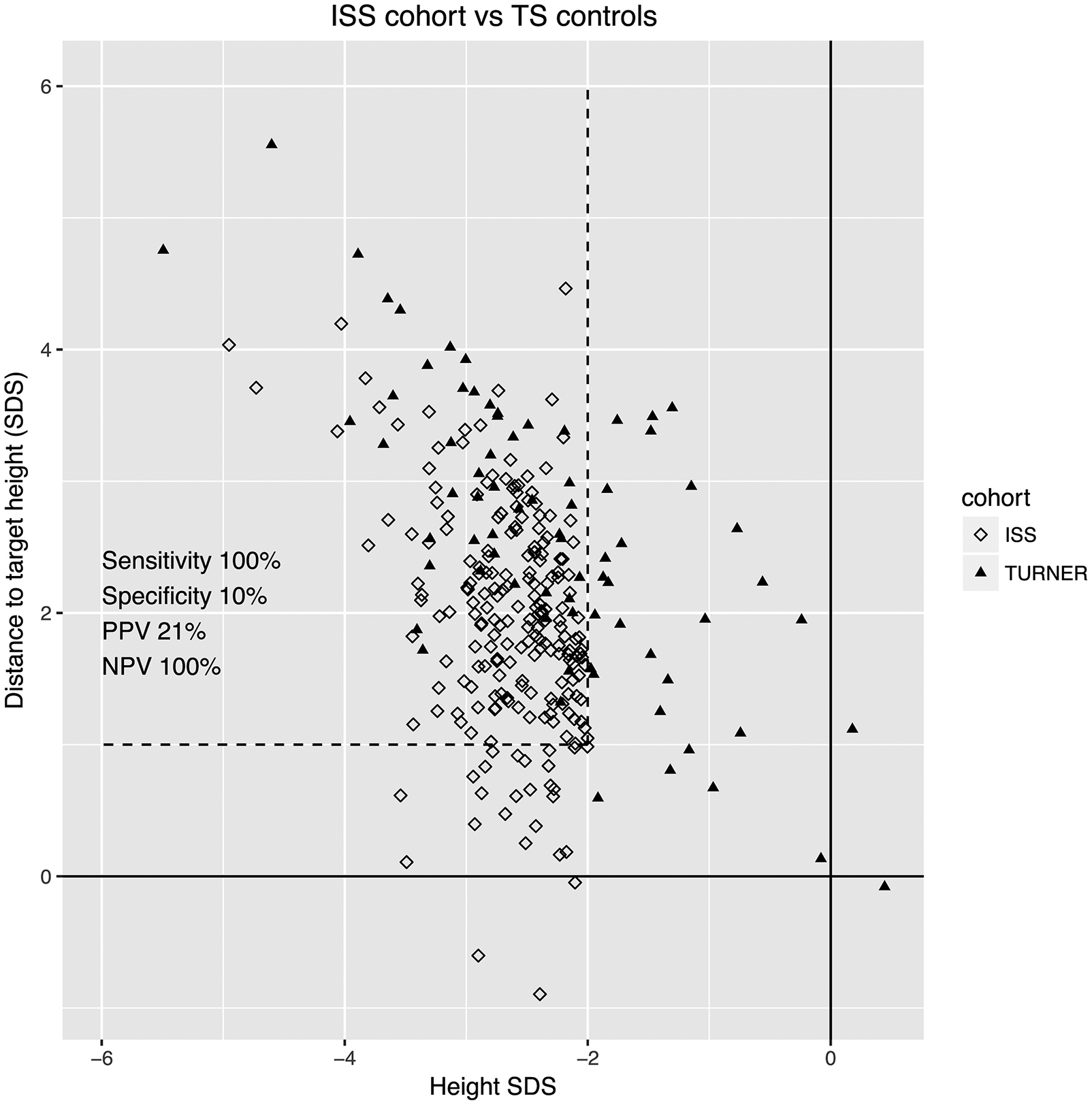
On the x-axis is the patient’s HSDS based on the Centers for Disease Control and Prevention criteria. On the y-axis is the difference between the MPH SDS and the patient’s HSDS (distance to target height). Filled triangles represent Turner syndrome controls and unfilled diamonds represent female patients with ISS. Cutoffs for height SDS −2 and distance to target height of <1 SD are shown. NPV, Negative predictive value; PPV, positive predictive value.
Table 1.
Baseline demographics for ISS females with and without prior karyotype testing, and those without prior karyotype testing included in our study group. Listed are age, height standard deviation score (SDS), and deficit from mid-parental height (MPH), which is the difference between the patient’s height SDS and their MPH SDS. P-values comparing the three groups are listed, with a p-value <0.05 considered statistically significant.
| Karyotype (n=117) | No karyotype (n=72) | No karyotype study population (n=34) | P-value (karyotype vs no karyotype) | P-value (no karyotype vs no karyotype study population) | |
|---|---|---|---|---|---|
| Age (years) | |||||
| Mean | 10.5 | 10.9 | 10.5 | 0.488 | 0.586 |
| Range | 3.3 to 18.5 | 3.2 to 18.6 | 3.2 to 18.6 | ||
| Height SDS | |||||
| Mean | −2.72 | −2.58 | −2.45 | 0.022 | 0.349 |
| Range | −4.98 to −2.0 | −4.95 to −2.02 | −3.21 to −2.02 | ||
| Deficit from MPH (SDS) | |||||
| Mean | −2.28 | −1.99 | −1.88 | 0.002 | 0.652 |
| Range | −4.1 to −1.0 | −4.46 to −0.67 | −4.46 to −0.67 |
Microarray analysis generated data for 32 ISS samples (two samples had insufficient quantity of DNA) and five TS controls. The microarray analysis successfully detected all TS controls, two new cases of TS (6%), and one patient with a copy number variant causing short stature (3%). Both TS patients had mosaicism (45,X[10]/46,XY[10] and 45,X[12]/46,X,r(X)(p22.3q28)[8]) and the other had a large genomic duplication and deletion (2.7 Mb deletion from chromosome 1 (1q25.3–>1q31.1) and 2.1 Mb duplication from 22q11.21).
The patient with 45,X/46,XY karyotype was initially evaluated by an endocrinologist at 9 years, 1 month for short stature (height −2.77 SD, deflection from MPH −2.42 SD, weight −1.66 SD, BMI 0 SD). Workup included normal renal, liver, and thyroid function and a negative celiac screen. Insulin-like growth factor-I (IGF-I) was approximately −2 SD and IGF binding protein-3 (IGFBP-3) was 0 SD. Her bone age was delayed at 7 years, 4 months or −2.5 SD based on Brush Foundation data. Given her reassuring laboratory results, aside from a borderline low IGF-I that was felt to be reflective of her bone age delay, prepubertal stage, and nutritional status, the etiology of her short stature was attributed to poor nutrition as a result of ADHD medication. No genetic testing was performed and follow-up with endocrinology was requested in six months, after which the patient was lost to follow-up and then picked up by our study algorithm. Three and a half years after her initial evaluation, she returned to the endocrine clinic with continued growth failure despite appropriate interval weight gain (height −3.1 SD, weight −0.66 SD, BMI +0.99 SD) and a confirmatory clinical karyotype demonstrated TS mosaicism.
The patient with 45,X/46,X,r(X)(p22.3q28) karyotype was initially seen in the endocrine clinic at 12 years, 1 month for short stature (height −2.1 SD, deflection from MPH −1 SD, weight +0.1 SD, BMI +1.29 SD). She had a history of normal pubertal progression with onset of menses at 11 years, 8 months and an examination without any stigmata of TS. Her prior laboratory results showed normal thyroid function. Her bone age was read as 13 years, 6 months at a chronologic age of 12 years, 1 month. Given a predicted adult height significantly below her genetic potential (142.2 cm versus MPH of 156 cm), the endocrinologist asked the patient to return for a karyotype to evaluate for TS. The patient never returned for testing and was picked up through our study algorithm 1.25 years later and found to have TS mosaicism.
The patient with the deletion/duplication syndrome was initially seen in clinic at 8 years, 9 months for failure to thrive (height −2.80 SD, deflection from MPH −2.04 SD, weight −2.96 SD, BMI −1.62 SD). Other conditions included vesicoureteral reflux and ADHD. Initial evaluation demonstrated a normal GH and thyroid hormone axis, negative celiac screen, normal kidney function, and lack of anemia. Her bone age was read as 7 years, 10 months at a chronologic age of 8 years, 9 months with a predicted adult height of 150 cm. Poor growth was thought to be nutritionally mediated and correlated with initiation of stimulant medication, so genetic testing was not pursued. She was subsequently lost to follow-up and identified through our algorithm two years later. Review of the literature demonstrates that deletions in this area of chromosome 1 are associated with growth retardation, cognitive delays, genitourinary anomalies, and other syndromic features (10). Duplication of chromosome 22q11.21 has highly variable manifestations, including poor growth (11).
DISCUSSION:
In this study, we used an algorithm-based search of the EHR to identify girls with undiagnosed TS from a cohort of girls with ISS. We found that 6% of girls had undiagnosed TS (and one patient had a copy number variant associated with short stature). Furthermore, only 62% of ISS girls had a prior karyotype. Those previously karyotyped were significantly shorter with a larger deflection from their MPH, suggesting that clinicians were falsely reassured by the milder presentation of patients in whom a karyotype was not performed.
Evidence based guidelines encourage screening for pathologic causes of short stature in patients with a HSDS < −2 and a distance to target height of > 2 SD (6). Furthermore, the TS consensus statement recommends karyotype analysis for evaluation of TS in all females with isolated ISS (1). Our study demonstrates that gaps between recommended practice guidelines and provided care continue to exist, as 38% of ISS females had no prior karyotype and one missed case of TS had a distance to MPH of −2.42 SD. Furthermore, this study underscores the importance of screening all ISS females for TS, as the deflection from MPH can be more subtle in cases of mosaicism, as evidenced by one of the missed cases of TS where distance to MPH was −1 SD. In order to improve clinical suspicion and screening for TS, a method that is feasible, reliable, and reproducible within a busy clinic setting is key. There are many approaches to changing physician behavior such as educational interventions and audits with feedback of performance, but perhaps one of the more practical and cost-effective modalities uses clinical decision support systems built into the already widely adopted EHR (9). Automated reminders elicited by algorithms programmed into the EHR can prompt physicians at the point-of-care to recall information they already know, but may overlook in a busy setting and apply this to their clinical decision making. A Cochrane systematic review concluded that the use of such EHR reminders resulted in minor to modest improvements in process adherence, laboratory ordering, and clinical outcomes (9). Therefore, implementation of a best practice alert that accounts for specific growth measures, including an absolute HSDS and deflection from MPH, can help to reliably ascertain patients with abnormal growth patterns that may warrant further investigation. This alert could be implemented in any EHR system that collects the patient and parents’ heights as discrete data elements. These decision support systems would also be of significant value in the primary care setting, as there is a known bias in growth referrals toward males that may overlook serious pathology in females. A retrospective study by Grimberg et al demonstrated a higher prevalence of males than females referred for evaluation of growth disorders (182 boys vs 96 girls, p<0.001). These males were more often of normal height (72% of males vs 48% of females, p<0.0001) or short but healthy, whereas females had a higher rate of underlying organic disease impacting their growth (41% of females vs 15% of males) (12).
This study adds to the existing concerns regarding delays in diagnosis of TS. Six percent of ISS females with a height more than 1 SD below their MPH and available DNA had undiagnosed TS mosaicism. In these cases, the ages at diagnoses were 12 years, 9 months and 13 years, 4 months, missing the window of opportunity for significant height gains and delaying the necessary surveillance for associated comorbidities until their teenage years. Our study was performed at a large academic center with a specialized TS clinic on a referred population; therefore, additional investigation is needed to examine the yield of our algorithm when applied to the general population and in other practice settings. One could postulate that there may be more missed cases of TS in other settings, especially mild mosaic phenotypes and those without obvious short stature due to tall parents, as general clinic settings may overlook the importance of parental height measurements in a child’s growth assessment. Furthermore, our algorithm was applied to an ISS population with HSDS < −2 and, as shown in the Figure, there are several cases of TS with an absolute HSDS > −2 that would have been missed by this cut off. Thus, when considering the generalizability of this algorithm outside of ISS cohorts, we would need to explore other absolute height cutoffs in order to maximize both sensitivity and specificity for detection of TS.
The EHR algorithm was effective at identifying patients at risk for TS who merit genetic testing, and also detecting patients with copy number variants associated with short stature. Of the ISS females ≥ 1 SD below their MPH percentile, 38% never had a karyotype done and on further investigation two new cases of TS mosaicism (6%) were found. Although not all patients had DNA available for microarray analysis, if we assume that all other cases were negative, the rate of undiagnosed TS would still approach 3%. Extrapolating this data to a larger scale suggests that the diagnosis of TS may be delayed for many females with ISS, even after evaluation in endocrine clinics. This reinforces that genetic testing, either a clinical karyotype or microarray, is necessary for all ISS females to evaluate for TS. Furthermore, we recommend the implementation of additional tools in the clinic workflow, such as best practice alerts based on specific growth measures, to increase clinical suspicion and testing for TS.
Acknowledgments
Ssupported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD093622 [to A.D.]). P.B. has received research support from Novo Nordisk, Ipsen, Opko, and Sandoz, and consulting fees from Novo Nordisk and Sandoz. A.D. has received consulting fees from Novo Nordisk, Sandoz, Pfizer, Ipsen, Ascendis, and OPKO Biologics. The other authors declare no conflicts of interest. have nothing to disclose.
ABBREVIATIONS:
- EHR
Electronic health record
- ISS
Idiopathic short stature
- SD
Standard deviation
- MPH
Mid-parental height
- BMI
Body mass index
- HIT
Health information technology
- HSDS
Height standard deviation score
- IGF-I
Insulin-like growth factor-I
- IGFBP-3
Insulin-like growth factor binding protein-3
- GH
Growth hormone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Portions of this study were presented at the ENDO Society Meeting Poster Session, March 25, 2019, New Orleans, Lousiana.
REFERENCES:
- 1.Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177:G1–G70. [DOI] [PubMed] [Google Scholar]
- 2.Massa G, Verlinde F, De Schepper J, Thomas M, Bourguignon JP, Craen M, et al. Trends in age at diagnosis of Turner syndrome. Arch Dis Child. 2005;90:267–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apperley L, Das U, Ramakrishnan R, Dharmaraj P, Blair J, Didi M, et al. Mode of clinical presentation and delayed diagnosis of Turner syndrome: a single Centre UK study. Int J Pediatr Endocrinol. 2018;2018:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Buuren S, van Dommelen P, Zandwijken GR, Grote FK, Wit JM, Verkerk PH. Towards evidence based referral criteria for growth monitoring. Arch Dis Child. 2004;89:336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savendahl L, Davenport ML. Delayed diagnoses of Turner’s syndrome: proposed guidelines for change. J Pediatr. 2000;137:455–9. [DOI] [PubMed] [Google Scholar]
- 6.Grote FK, van Dommelen P, Oostdijk W, de Muinck Keizer-Schrama SM, Verkerk PH, Wit JM, et al. Developing evidence-based guidelines for referral for short stature. Arch Dis Child. 2008;93:212–7. [DOI] [PubMed] [Google Scholar]
- 7.Ouarezki Y, Cizmecioglu FM, Mansour C, Jones JH, Gault EJ, Mason A, et al. Measured parental height in Turner syndrome-a valuable but underused diagnostic tool. Eur J Pediatr. 2018;177:171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sperling M Pediatric endocrinology. 2014.
- 9.Shojania KG, Jennings A, Mayhew A, Ramsay CR, Eccles MP, Grimshaw J. The effects of on-screen, point of care computer reminders on processes and outcomes of care. Cochrane Database Syst Rev. 2009:CD001096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu P, Wang Y, Meng LL, Qin L, Ma DY, Yi L, et al. 1q25.2-q31.3 Deletion in a female with mental retardation, clinodactyly, minor facial anomalies but no growth retardation. Mol Cytogenet. 2013;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guy C, Wang X, Lu X, Lu J, Li S. Two patients with small chromosome 22q11.21 alterations and central nervous system abnormalities. Mol Cytogenet. 2015;8:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimberg A, Kutikov JK, Cucchiara AJ. Sex differences in patients referred for evaluation of poor growth. J Pediatr. 2005;146:212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]