

Abstract
Background
Tissue oxidative stress, sympathetic activation and nutrient sensing signals are closely related to adult hypertension of fetal origin, although their interactions in hypertension programming remain unclear. Based on a maternal high-fructose diet (HFD) model of programmed hypertension, we tested the hypothesis that dysfunction of AMP-activated protein kinase (AMPK)-regulated angiotensin type 1 receptor (AT1R) expression and sirtuin1 (SIRT1)-dependent mitochondrial biogenesis contribute to tissue oxidative stress and sympathoexcitation in programmed hypertension of young offspring.
Methods
Pregnant female rats were randomly assigned to receive normal diet (ND) or HFD (60% fructose) chow during pregnancy and lactation. Both ND and HFD offspring returned to ND chow after weaning, and blood pressure (BP) was monitored from age 6 to 12 weeks. At age of 8 weeks, ND and HFD offspring received oral administration of simvastatin or metformin; or brain microinfusion of losartan. BP was monitored under conscious condition by the tail-cuff method. Nutrient sensing molecules, AT1R, subunits of NADPH oxidase, mitochondrial biogenesis markers in rostral ventrolateral medulla (RVLM) were measured by Western blot analyses. RVLM oxidative stress was measured by fluorescent probe dihydroethidium and lipid peroxidation by malondialdehyde assay. Mitochondrial DNA copy number was determined by quantitative real-time polymerase chain reaction.
Results
Increased systolic BP, plasma norepinephrine level and sympathetic vasomotor activity were exhibited by young HFD offspring. Reactive oxygen species (ROS) level was also elevated in RVLM where sympathetic premotor neurons reside, alongside augmented protein expressions of AT1R and pg91phox subunit of NADPH oxidase, decrease in superoxide dismutase 2; and suppression of transcription factors for mitochondrial biogenesis, peroxisome proliferator-activated receptor γ co-activator α (PGC-1α) and mitochondrial transcription factor A (TFAM). Maternal HFD also attenuated AMPK phosphorylation and protein expression of SIRT1 in RVLM of young offspring. Oral administration of a HMG-CoA reductase inhibitor, simvastatin, or an AMPK activator, metformin, to young HFD offspring reversed maternal HFD-programmed increase in AT1R and decreases in SIRT1, PGC-1α and TFAM; alleviated ROS production in RVLM, and attenuated sympathoexcitation and hypertension.
Conclusion
Dysfunction of AMPK-regulated AT1R expression and SIRT1-mediated mitochondrial biogenesis may contribute to tissue oxidative stress in RVLM, which in turn primes increases of sympathetic vasomotor activity and BP in young offspring programmed by excessive maternal fructose consumption.
Keywords: Maternal high fructose, AMP-activated protein kinase, Sirtuins, Angiotensin type 1 receptor, Rostral ventrolateral medulla, Programmed hypertension
Background
Emerging evidence from human and animal studies indicates that the increased risk for a number of cardio-metabolic diseases, including hypertension, diabetes, heart failure, myocardial infarction and chronic kidney disease, may have their origins from birth [1, 2]. According to the concept of developmental original of adult health and disease (DOHaD) [3], susceptibility to the development of those non-communicable diseases in adult life could be primed by exposures such as malnutrition, prenatal hypoxia, hyperglycemia, toxin and medication during fetal life [4]. In particular, elevated blood pressure (BP), a major determinant of morbidity and mortality burden to cardio-metabolic risk [5], can be programmed by maternal overnutrition [6, 7]. In a rodent model of DOHaD, we [8, 9] and others [10, 11] demonstrated that maternal exposure to a high fructose diet (HFD) during gestation and/or lactation programs the development of hypertension in adult offspring. Both vascular and renal dysfunctions have been proposed as the culprits [12, 13], with enhanced reactive oxygen species (ROS) and reduced nitric oxide (NO) availability as the major protagonists [14, 15].
Programmed hypertension of fetal origin is associated with a significant activation of the sympathetic nervous system (SNS) [15, 16]. In the rostral ventrolateral medulla (RVLM), where the sympathetic premotor cells reside [17], the bulk of evidence supports the argument that tissue oxidative [18] and nitrosative [18, 19] stress play pivotal roles in central mechanisms of sympathoexcitation associated with hypertension. At the molecular level, angiotensin type 1 receptor (AT1R)-dependent activation of the nicotinamide adenine dinucleotide diphosphate oxidase (NADPH oxidase) signaling [18–20], suppression of endogenous antioxidants [18, 19, 21], impairment of mitochondrial biogenesis [18, 19, 22] and changes in protein expressions of NO synthase (NOS) isoforms [19, 23] have all been reported to contribute to the pathogenesis of hypertension via sympathoexcitation.
AMP-activated protein kinase (AMPK), a nutrient sensing molecule known for its function to ‘fuel gauge’ cellular energy status [24], plays a key role in the regulation of whole-body energy balance [25]. For example, metabolic disturbances in the liver programmed by fetal malnutrition are mediated, at least in part, by the suppression of AMPK phosphorylation at the Thr172 residue [26]. More recently, an interplay between oxidative stress and nutrient sensing signals, including AMPK and sirtuin (SIRT), has been stipulated to be a major component in cardio-metabolic programming of fetal origin [27], although detailed underlying mechanisms of this interplay is wanting.
The present study was undertaken to interrogate whether maternal HFD contributes to the programming of sympathoexcitation and hypertension in adult life by altering the tissue ROS and/or NO homeostasis in RVLM of young offspring. We also delineated the molecular mechanisms underpinning the programmed dysfunctions in RVLM of HFD offspring. Our data revealed that maternal HFD induces tissue oxidative stress in RVLM that primes an increase in sympathetic vasomotor activity and BP in young offspring. We further identified that AMPK-regulated increase in AT1R expression and SIRT1-mediated dysfunction of mitochondrial biogenesis as potential mediators for the enhanced ROS production in RVLM of HFD offspring.
Methods
Animals
Virgin female (n = 24) and male (n = 12) normotensive Sprague-Dawley (SD) rats at age of 10 weeks purchased from BioLASCO (Taipei, Taiwan) were used for breeding. Animals were allowed to acclimatize in a temperature- (22 ± 1 °C), humidity- (55 ± 5%) and light- (12:12 light-dark cycle, light on from 08:00) controlled room in an AAALAC-International accredited animal facility for at least 14 days before the experiments. All experiments were approved by our institutional animal care and use committee (2,012,102,701 and 2,015,122,217), and were carried out in accordance to the guidelines for animal experimentation endorsed by that committee and conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Measurement of blood pressure
We routinely measured systolic blood pressure (SBP) at 14:00–16:00 in rats under conscious condition using the noninvasive tail-cuff method based on electrosphygmomanometry (MK-2000; Muromachi Kikai Co., Tokyo, Japan). Offspring to dams fed maternal normal diet (ND) or 60% high fructose diet (HFD) chow were handled repeatedly and allowed to adapt to the restraint chamber for at least 3 days before the commencement of actual measurements. BP determinations were considered valid only when five consecutive readings were recorded from rats under resting condition and the values did not differ by more than 5 mmHg; the mean of the five readings was then recorded as the measured value. SBP was measured in ND and HFD offspring at age of 6, 8, 10 and 12 weeks; in some offspring until age of 18 weeks. We have previously validated [28, 29] and again in pilot experiment of the current study that BP obtained by tail-cuff plethysmography was comparable to those measured by radiotelemetry.
Evaluation of sympathetic vasomotor tone
Animals were anesthetized with isoflurane (5% for induction and 2% for maintenance) via an anesthesia mask for cannulation of femoral artery. Satisfactory anesthesia was maintained by the absence of withdrawal reflex to hind paw pinch. Animals were allowed to breathe spontaneously with room air, and body temperature was maintained at 37 °C by a heating pad. The arterial pressure was monitored from the femoral artery cannula for 30 min between 14:00 and 16:00. The pressure signal was analyzed by an arterial blood pressure analyzer (Notocord, Le Pecq, France) to obtain systemic arterial pressure (SAP). Continuous on-line, real-time spectral analysis of SAP signals based on fast Fourier transform was used to detect temporal fluctuations in the low frequency (LF, 0.25–0.8 Hz) component of SBP signals. Power density of LF band was used as our experimental index to reflect sympathetic vasomotor tone [30, 31].
Measurement of plasma norepinephrine concentration
Plasma norepinephrine (NE) level was measured by the o-phthaldehyde (OPA) method using high performance liquid chromatography with fluorescence detection [32]. In brief, 500 μL ice cold trichloroacetic acid was added to 500 μL plasma sample and mixed by vortexing for 20 min, followed by centrifugation at 15,000×g for 20 min at room temperature. The supernatant was filtered through a 0.22 μm syringe filter (Chroma Technology Corp., Bellows Falls, VT), mixed with 4 fold methanol and centrifuged at 12,000×g for 15 min at room temperature. The supernatant was filtered again and kept at − 80 °C until analysis.
The sample or standard NE solution (at concentrations of 1–50 mM) was mixed with the derivatization reagent OPA solution at 4:1 (V/V) and placed in dark for 15 min. Each 20 μL aliquot was injected into the HPLC system using a Rheodyne (model 7125, Merck KGaA, Darmstadt, Germany) injector. The HPLC system (Hitachi CM5000, Hitachi Corp., Tokyo, Japan) is comprised of a 5110 syringe pump system, a 5210 autosampler and a 5440 FL spectrofluorimetric detector. Chromatographic separation was achieved on a ZORBAX SB-C18 column (4.6 mm × 250 mm, 5 μm; Agilent Technologies, Taipei, Taiwan). The column temperature was maintained at 30 °C and the flow rate was 1 mL/min. Methanol and acetate buffer (20 mM, pH 3.5, with 1 mM Na2EDTA) with a ratio of 5:4 was used as the mobile phase, and the samples were eluted within 20 min. The fluorescence signal was detected with excitation and emission wavelengths of 340 nm and 450 nm, respectively. Retention time for NE was 2.2–2.6 min. The concentration of NE was computed by comparing the area under curve of each sample against standard solutions of known concentrations. Each sample was analyzed in triplicates and the results are shown as the mean of the three values.
Measurement of blood biochemistry
All measurements were carried out in ND and HFD offspring at age of 6, 9 and 12 weeks, and each assay was performed in triplicates. The animals were fasted for at least 15 h before blood samples were collected from a punch at the tip of the tail. Glucose levels were analyzed using a glucose oxidase kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. For plasma insulin measurement, 200 μL blood sample was centrifuged at 2000×g for 15 min. Fasting plasma insulin was analyzed by means of an ELISA kit (Mercodia, Uppsala, Sweden). Samples were first interacted with plate-coated mouse monoclonal anti-insulin, followed by reacting with peroxidase-conjugated anti-insulin antibodies. The bound conjugate was detected by reaction with 3,3′,5,5′-tetramethylbenzidine and read by a spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA) at 450 nm after the reaction was stopped by adding stop solution. The detected insulin and glucose concentrations were then used to calculate the ‘homeostasis model assessment’ (HOMA) indices of insulin resistance (IR) based on formulas reported previously [33].
The concentration of fasting plasma triglycerides was detected by a triglycerides assay kit (Randox, Antrim, UK). Ten μl plasma from fasting animals was incubated with the reaction mixture at 25 °C for 20 min. Triglyceride concentration was detected by a spectrophotometer (Thermo Fisher Scientific Inc.) at 570 nm. The concentration of fasting plasma leptin was detected using a rat leptin quantikine ELISA kit (R&D Systems, Minneapolis, MN). In brief, after 10 fold dilution into calibrator diluent, 50 μl solution was used to react with equal volume of acetic acid (2.5 N) and urea (10 M) according to the protocols provided by the manufacturer. The level of plasma leptin was analyzed using a microplate spectrophotometer (ThermoScientific, Chantilly, VA) at 540 nm.
Implantation of osmotic minipump
Animals were anesthetized with pentobarbital sodium (50 mg/kg, IP) for implantation of osmotic minipump according to previously reported procedures [30, 34]. Briefly, a midline dorsal neck incision was made and the underneath muscle layers were dissected to expose the dura mater between the foramen magnum and C1 lamina. This was followed by perforation of dura with a 22-gauge steel needle and insertion of a PE-10 catheter (Clay Adams, Sparks, MD) into the cisterna magna. Patency of the implantation was assured by drainage of cerebrospinal fluid (CSF) from the outer end of catheter. The catheter was then sealed to the dura with tissue glue and the incision was closed with layered sutures. The outer end of the catheter was connected to a micro-osmotic minipump (Alzet 1007D; Durect Co., Cupertino, CA), which was placed under the skin in the neck region. Animals received procaine penicillin (1000 IU, IM) injection postoperatively, and only animals that showed progressive weight gain after the operation were used in subsequent experiments. Control infusion of artificial CSF (aCSF) served as the volume and vehicle control. The composition of aCSF was (mM): NaCl 117, NaHCO3 25, Glucose 11, KCl 4.7, CaCl2 2.5, MgCl2 1.2 and NaH2PO4.
RVLM tissue collection
At age of 10 or 12 weeks, ND and HFD offspring were deeply anesthetized with an overdose of pentobarbital sodium (100 mg/kg, IP), followed by intracardial infusion with warm normal saline. The skull was opened and brain stem was rapidly removed and immediately frozen on ice. The medulla oblongata covering rostral ventrolateral medulla (RVLM) was blocked between 0.5 and 1.5 mm rostral to the obex, based on the atlas of Watson and Paxinos [35], using a rodent brain matrix (World Precision Instruments, Sarasota, FL). Both sides of the ventrolateral medulla covering RVLM (approximately 1.5- to 2.5-mm lateral to the midline and medial to the spinal trigeminal tract) were collected by micropunches with a 1-mm inner diameter stainless-steel burr [30, 31, 34]. Medullary tissues collected were stored at − 80 °C for subsequent protein analysis.
Total protein preparation
RVLM tissue samples were homogenized in ice-cold lysis buffer by a Dounce grinder with a tight pestle [30, 31, 34]. A cocktail of protease inhibitors (Sigma-Aldrich, St. Louis, USA) was included in the isolation buffer to prevent protein degradation. Solubilized proteins were centrifuged at 20,000×g at 4 °C for 15 min, supernatant was collected and total protein was quantified by the Bradford assay with a protein assay kit (Bio-Rad, Hercules, CA).
Western blot analysis
Total protein extracted from RVLM samples was used to determine the expression levels of AT1R, gp91phox, p67phox or p47phox subunits of NADPH oxidase, isoforms of superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase, nitric oxide synthase (NOS) isoforms, phosphorylated and total AMPK (p-AMPK and t-AMPK), sirtuin 1 (SIRT1), SIRT3, peroxisome proliferator-activated receptor gamma co-activator α (PGC-1α), mitochondrial transcription factor A (TFAM) or leptin by Western blot analysis. Protein samples were subject to 8–12% SDS-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride transfer membranes (Immobilon-P membrane; Millipore, Bedford, MA) for 1.5 h at 4 °C, using a Bio-Rad miniprotein-III wet transfer unit (Bio-Rad). The transfer membranes were then incubated with a blocking solution (5% nonfat dried milk dissolved in Tris-buffered saline-Tween buffer (pH 7.6, 10 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature [34, 36].
The primary antisera used included goat polyclonal, rabbit polyclonal or monoclonal, or mouse monoclonal antiserum against AT1R (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), gp91phox (1:5000; BD Biosciences, Sparks, MD), p67phox or p47phox (1:5000; Santa Cruz Biotechnology), manganese SOD (SOD1, 1:6000; Stressgen, Ann Arbor, MI), copper/zinc SOD (SOD2, 1:3000; Stressgen), extracellular SOD (SOD3, 1:5000; Stressgen), catalase (1:4000; Stressgen), GPx1 or GPx 3 (1:5000; BD Biosciences), NOS1, NOS2 or NOS3 (1:2000; BD Biosciences), t-AMPKα or p-AMPKα at Thr172 (1:1000; Cell Signaling, Danvers, MA), SIRT1 or SIRT3 (1:1000; Proteintech, Rosemont, IL), PGC-1α (1:1000; Santa Cruz Biotechnology), TFAM (1:1000; Abcam, Rockville, MA), leptin (1:1000; Abcam) or GAPDH (1:10,000; Merck). Membranes were washed three times with TBS-t buffer, followed by the secondary antibodies (1:10000; Jackson ImmunoResearch, West Grove, PA) for 1 h. This was followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Jackson ImmunoReserach). Specific antibody-antigen complex was detected using an enhanced chemiluminescence Western blot detection system (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). The amount of detected proteins was quantified by ImageJ software (NIH, Bethesda, MD), and was expressed as the ratio to loading control (GAPDH).
Measurement of ROS in RVLM
To measure the reactive oxygen species (ROS) in RVLM tissues, total protein extracted was reacted with the oxidation-sensitive fluorescent probe dihydroethidium (DHE, 1 μM; Invitrogen) [36]. RVLM tissues were homogenized in 20 mM sodium phosphate buffer (pH 7.4; containing 0.01 mM EDTA) by a glass-to-glass homogenizer. The homogenate was subjected to low speed centrifugation at 1000×g for 10 min at 4 °C. The supernatant was collected immediately for ROS measurement by reacting the lysate with DHE at 37 °C for 15 min under protection from light. The fluorescence signal was measured in a microplate reader (FluorStar; Biodirect, Inc., Taunton, MA). Specificity for superoxide was determined by the addition of a membrane-permeable superoxide dismutase SOD (350 U/mL) into the incubation medium. Protein sample extracted from RVLM of individual ND or HFD offspring was analyzed in triplicates and the results are shown as the mean of the three values.
Measurement of lipid peroxidation in RVLM
Levels of lipid peroxidation were measured by a malondialdehyde (MDA) assay kit (Biovision, Milpitas, CA), following the protocol provided by the manufacturer. In brief, RVLM tissue was homogenized with MDA lysis buffer on ice to remove precipitated protein. The supernatant was then reacted with thiobarbituric acid. The level of cellular MDA was determined using a microplate spectrophotometer (ThermoScientific), with colorimetric absorbance read at 532 nm. Protein sample extracted from RVLM of individual ND or HFD offspring was analyzed in triplicates and the results are shown as the mean of the three values.
Measurement of tissue NOx level
RVLM tissues were homogenized in lysis buffer. After centrifugation in 20,000×g at 4 °C for 15 min, the supernatant was deproteinized using a Centricon-30 filtrator (Microcon YM-30, Bedford, MA) and stored at − 80 °C until further processing. The level of total nitrate and nitrite (NOx) was determined with the purge system of a NO analyzer (Sievers NOA 280™, Boulder, CO) based on chemiluminescence reaction [36]. All assays were performed in triplicate and the results are shown as the mean of the three values.
Determination of mitochondrial DNA copy number
Total RNA was extracted from RVLM tissue using TRIzol (Invitrogen, Carlsbad, CA) and cDNA was reversely transcribed from RNA (200 ng) using cDNA synthesis kit (EP0442, ThermoScientific). Mitochondrial DNA copy number was determined by the ratio of cDNA amplified from mtDNA-encoded NADH dehydrogenase subunit 1 (ND1) to nucleus-encoded 18S RNA genes. Primers for the ND1 probe correspond to nucleotides 389–408 (forward) and 572–592 (reverse; PCR product of 200 base pairs) of the rat mitochondrial genome (Chromosome MT - NC_001665.2). Primers for the 18S probe correspond to nucleotides 681–702 (forward) and 864–884 (reverse; PCR product of 200 base pairs) of the rat nuclear genome (Chromosome 14 - NC_005113.3). The primer sequences are: ND1, Forward (5′-3′) TCGGAGCCCTACGAGCCGTT /Reverse (5′-3′) AGGGAGCTCGATTTGTTTCTG; 18 s rRNA, Forward (5′-3′) TAGTTGGATCTTGGGAGCGGG /Reverse (5′-3′) CCGCGGTCCTATTCCATTATT.
Quantitative real-time polymerase chain reaction (qPCR) was performed by a Roche LightCycler 480 (Roche Applied Science, Mannheim, Germany) apparatus with LightCycler 480 SYBR Green I Master kit (Roche Applied Science). DNA sample (10 ng) from RVLM was mixed with 10 μL LightCycler 480 SYBR Green I Master Mix that contained forward and reverse primers (at a final concentration of 0.4 μM) in a final volume of 20 μl. The qPCR reactions were conducted as follows: initiation at 50 °C for 2 min, 95 °C for 1 min, 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 20 s, extension at 72 °C for 15 s, and finally holding at 4 °C. The value of the threshold cycle number (Ct) of the 18 s rRNA gene and the ND1 gene was determined for each individual qPCR reaction. ΔCt = [Ct (ND1) - Ct (18S)] represents the relative abundance. The results are expressed as the copy number of mtDNA/sample by 2-ΔCt [22]. Each measurement was performed in triplicate and the results are shown as the mean of the three values.
Measurement of tissue ATP level
RVLM tissues were homogenized in a protein extraction solution (Pierce, Rockford, IL), followed by centrifugation at 10,000×g for 10 min. The supernatant was subject to determination of ATP concentration by an ATP colorimetric assay kit (Biovisoin) [22]. The ATP level was detected using a microplate reader (ThermoScientific) and was normalized to protein concentration of the sample. Each measurement was performed in triplicate and the results are shown as the mean of the three values.
Double immunofluorescence staining
At age of 12 weeks, ND and HFD offspring were perfused transcardially with warm saline under deep sodium pentobarbital anesthesia (100 mg/kg, IP), and the brain stem was removed and post-fixed overnight in 4% paraformaldehyde, followed by 30% sucrose solution for at least 5 days. 30-μm coronal sections of the rostral medulla oblongata were cut using a cryostat (Leica, Nussloch, Germany). The sections were rinsed for 30 min in PBS. After pre-absorption in gelatin (0.375%), normal horse serum (3%) and triton-X 100 (0.2%) in PBS, the sections were incubated with a rabbit polyclonal antibody against AT1R (1:250; Santa Cruz Biotechnology), mouse monoclonal antibody against gp91phox (1:250; BD Biosciences), rabbit polyclonal antibody against SOD2 (1:250; Biovision) or mouse monoclonal or rabbit polyclonal antibody against NeuN (1:2000; Millipore) at 4 °C overnight and then rinsed 3 times in PBS. After incubation in Alex 594 conjugated anti-mouse or rabbit IgG (1:1000; Molecular Probes), or Alex 488 conjugated anti-rabbit or mouse IgG (1:1000; Molecular Probes, Eugene, OR) for 1 h, the sections were rinsed 3 times in MilliQ distilled water. Sections were mounted and observed under a confocol microscope (FluoView FV10i; Olympus, Tokyo, Japan) [34, 37].
Experimental design
One male rat was housed with two females until mating was confirmed by the observation of vaginal plug. Pregnant female rats were randomly assigned to receive normal diet (ND, 46% complex carbohydrate, 3.4 Kcal/g; Harlan Laboratories, Madison, WI) or HFD (60% fructose, 3.6 Kcal/g; TD.89247; Harlan Laboratories) chow during the entire period of pregnancy and lactation. Both food and water were provided ad libitum. Clinical observations indicate that men are more prone to hypertension at a younger age [38]. Accordingly, only male offspring from litters culled to sizes of eight pups after birth were used in subsequent experiments.
After weaning (3 weeks after birth), both ND and HFD offspring returned to ND chow, and body weight as well as food intake of offspring were measured and recorded once per week until age of 12 weeks. Metabolic indices were measured at 6, 9 and 12 weeks of age and BP was monitored weekly from age of 6 to 12 weeks (in some offspring to 18 weeks). At age of 8 weeks, ND and HFD offspring were randomly separated into groups to receive either oral administration via gavage of a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, simvastatin (5 mg·kg− 1·day− 1) or a AMPK activator, metformin (400 mg·kg− 1·day− 1); or microinfusion into the cistern magna of an AT1R antagonist, losartan (3 μg·μL− 1·h− 1). All treatments were applied for 4 weeks. Dosage of the compounds was adopted or modified from previous studies [28, 39] or determined in pilot experiments. During outcome assessments, treatment groups and assignments were masked from the experimenters.
Drugs used
Drugs used include a HMG-CoA reductase inhibitor, simvastatin (5 mg·kg− 1·day− 1; Sigma-Aldrich), an AMPK activator, metformin (400 mg·kg− 1·day− 1; Sigma-Aldrich) or an AT1R antagonist, losartan (3 μg·μL− 1·h− 1; Sigma-Aldrich). Simvastatin and metformin were administered to the ND or HFD offspring via gastric gavage using a blunt-ended needle, whereas losartan was microinfused into the cistern magna via the implanted osmotic minipump. Normal saline served as volume and vehicle control for simvastatin and metformin, and artificial CFS for losartan.
Statistical analysis
Data are expressed as means ± SEM. All statistical analyses were performed by the Graphpad Prism analysis software (La Jolla, CA). One-way or two-way ANOVA with repeated measures was used, as appropriate, to assess group means; to be followed by Tukey’s or Newman-Keuls multiple-range test for post hoc assessment of individual means. P value < 0.05 was considered statistically significant.
Results
Young offspring to maternal HFD exposure exhibit elevated blood pressure, augmented sympathetic vasomotor tone and higher circulatory norepinephrine level
In comparison with ND group, baseline SBP (Fig. 1a), sympathetic vasomotor activity, as reflected by the power density of the LF component of SBP signals (Fig. 1b), and circulatory NE level (Fig. 1c) were significantly increased in male, young offspring exposed to maternal HFD that became significant at age of 10 weeks. The elevated SBP (HFD vs. ND: 165.4 ± 5.7 vs. 141.9 ± 3.9 mmHg, P < 0.05, n = 8) was maintained in adult HFD offspring at age of 18 weeks. Maternal HFD also programmed the development of metabolic syndrome, characterized by increases in fasting plasma insulin, glucose, triglyceride and leptin levels, as well as HOMA index (Table S1). In addition, expression of leptin in RVLM (Fig. 1d) was also augmented.
Fig. 1.

Cardio-metabolic parameters and redox signals at RVLM in young offspring exposed to maternal HFD or ND. Dams were exposed to a 60% HFD or ND during gestation and lactation. Both HFD and ND offspring returned to ND immediate after weaning (at age of 3 weeks). a SBP measured using tail-cuff plethysmography under conscious condition; b sympathetic vasomotor activity represented by power density of low frequency component of SBP signals; c plasma NE determined by high performance liquid chromatography; d protein expression of leptin in RVLM assessed by Western blot; e tissue level of ROS measured by dihydroethidium signal; f MDA determined by colorimetric spectrophotometry; and g NOx evaluated based on chemiluminescence reaction in RVLM. Analysis was performed on tissues collected bilaterally from individual RVLM. All measurements were made in ND (n = 6–12) and HFD (n = 6–12) offspring at age of 6, 8, 10, 11 and/or 12 weeks. Data are presented as mean ± SEM. *P < 0.05 versus ND group at comparable age in post hoc Tukey’s multiple range analysis. There is no significant difference between data shown in G in one-way ANOVA
Maternal HFD leads to oxidative stress in RVLM of young offspring
In tissues collected from RVLM at 10 or 12 weeks of age, offspring to HFD dams showed greater levels of ROS (Fig. 1e) and MDA (Fig. 1f), but not NO (Fig. 1g). These changes were associated with increases in protein expression of AT1R (Fig. 2a) or gp91phox (Fig. 2b) and a decrease of SOD2 (Fig. 2c) in RVLM tissues and neurons (Figure S1) examined at 10 (data not shown) or 12 weeks. All these changes were alleviated by oral administration of simvastatin (5 mg·kg− 1·day− 1) to the offspring commencing at 8 weeks of age. The same treatment also mitigated ROS accumulation (Fig. 2e) and the increased leptin expression (Fig. 2d) in RVLM, as well as the increases in fasting plasma triglyceride and leptin (Table S2) at age of 12 weeks. On the other hand, no apparent change in the expression of p47phox, p67phox, SOD1, SOD3, catalase, GPx, or NOS isoforms (Figure S2) was detected in the RVLM of HFD offspring at comparable ages.
Fig. 2.

Expressions of AT1R, molecules involved in redox homeostasis and leptin in RVLM of young offspring exposed to maternal HFD or ND. Representative gels and densitometric analysis of results from Western blot showing changes in protein expression of (a) AT1R, (b) gp91phox (c) SOD2, and (d) leptin or (e) ROS level in RVLM of ND (n = 6–10) or HFD (n = 6–10) offspring, alone or with additional treatment with a HMG-CoA reductase inhibitor, simvasatatin (5 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM. *P < 0.05 versus ND group, #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Maternal HFD impairs mitochondrial biogenesis in RVLM of young offspring
Compared to ND offspring, the expression of PGC-1α (Fig. 3a) and TFAM (Fig. 3b), two key molecules in transcription and replication of mitochondrial DNA [40], together with mitochondrial DNA copy number (Fig. 3c) were decreased in RVLM of HFD offspring at 10- (data not shown) and 12-week old. These changes are accompanied by a significant decrease in tissue content of ATP (Fig. 3d). Oral administration of simvastatin (5 mg·kg− 1·day− 1) at age of 8 weeks for 4 weeks significantly restored the protein expression of PGC-1α and TFAM, and maintained tissue ATP content in RVLM of HFD offspring at 12 weeks of age.
Fig. 3.

Mitochondrial biogenesis in RVLM of young offspring exposed to maternal HFD or ND. Representative gels (insets) and densitometric analysis of results from Western blot showing changes in protein expression of (a) PGC-1α (n = 10 per group) and (b) TFAM (n = 8 per group), two key molecules in transcription and replication of mitochondrial DNA; (c) mitochondrial DNA copy number (n = 10 per group); and (d) tissue content of ATP (n = 6 per group) in RVLM of ND or HFD offspring, alone or with additional treatment with simvastatin (5 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, are presented as mean ± SEM. *P < 0.05 versus ND group #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Maternal HFD suppresses AMPK activation and SIRT expression in RVLM of young offspring
RVLM tissue collected from HFD offspring at 10 (data not shown) or 12 weeks of age showed reduced expression of phosphorylated AMPK at the Thr172 residue (p-AMPK) (Fig. 4a) and SIRT1 (Fig. 4c), but not total AMPK (t-AMPK) (Fig. 4b) or SIRT3 (Fig. 4d), along with a decreased p-AMPK/t-AMPK ratio (Fig. 4e); all of which were partially protected by oral administration of simvastatin (5 mg·kg− 1·day− 1) to the offspring at 8 weeks of age.
Fig. 4.

AMPK/SIRT signals in RVLM of young offspring exposed to maternal HFD or ND. Representative gels and densitometric analysis of results from Western blot showing changes in protein expression of (a) p-AMPK, (b) t-AMPK, (c) SIRT1 and (d) SIRT3, as well as (e) ratio between p-AMPK/t-AMPK in RVLM of ND or HFD offspring, alone or with additional treatment with simvasatatin (5 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM (n = 10 in all groups). *P < 0.05 versus ND group #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Activation of AMPK retards AT1R upregulation and restores the impaired mitochondrial biogenesis in RVLM of young HFD offspring
Oral administration of an AMPK activator, metformin (400 mg·kg− 1·day− 1), for 4 weeks at age of 8 weeks significantly retarded the increased AT1R (Fig. 5a), restored the decreased SIRT1 (Fig. 5b), PGC-1α (Fig. 5c) or TFAM (Fig. 5d) expression and maintained mitochondrial DNA copy number (Fig. 5e) in RVLM of offspring exposed to maternal HFD. The same treatment also significantly ameliorated the heightened ROS production in RVLM of HFD offspring (Fig. 5f).
Fig. 5.

AMPK activation on the expressions of AT1R and SIRT1, mitochondrial biogenesis and ROS level in RVLM of young offspring exposed to maternal HFD or ND. Representative gels and densitometric analysis of results from Western blot showing changes in protein expression of (a) AT1R; (b) SIRT1, (c) PGC-1α, or (d) TFAM; (e) mitochondrial DNA copy number; and (f) ROS level in RVLM of ND or HFD offspring, alone or with additional treatment with an AMPK activator, metformin (400 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM (n = 8 in all groups). *P < 0.05 versus ND group and #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Protection against RVLM oxidative stress lessens increase in SBP in young HFD offspring
Infusion into the cistern magna of an AT1R antagonist, losartan (3 μg·μL− 1·h− 1), to young offspring from age of 8 weeks for 4 weeks effectively diminished the augmented ROS production (Fig. 6a), as well as the increase in SBP (Fig. 6b) and power density of LF component of SBP signal (Fig. 6c) manifested after exposure to maternal HFD. The same treatment also reversed the increased AT1R (Fig. 6d) and gp91phox (Fig. 6e), as well as the reduced SOD 2 (Fig. 6f) expression in RVLM of HFD offspring. Losartan, on the other hand, exerted no effect on protein expressions of p-AMPK, SIRT1 or p-AMPK/t-AMPK ratio, all of which were suppressed in the HFD offspring (Figure S3). Fasting plasma insulin, glucose, triglyceride or leptin (Table S2) levels measured at age of 12 weeks was not affected by intracisternal infusion of losartan.
Fig. 6.

Effects of AT1R antagonist on blood pressure, sympathetic vasomotor activity, ROS level and expressions of AT1R, gp91phox or SOD2 protein in RVLM of young offspring exposed to maternal HFD or ND. a tissue ROS level; b SBP; c sympathetic vasomotor activity; and representative gels and densitometric analysis of results from Western blot showing changes in protein expression of (d) AT1R, (e) gp91phox, or (f) SOD2 in RVLM of ND or HFD offspring, alone or with additional treatment with an AT1R antagonist, losartan (3 μg·μL− 1·h− 1), microinfused via a minipump into the cistern magna at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM (n = 6 in all groups). *P < 0.05 versus ND group and #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Protection against increase of SBP in young adult HFD offspring by simvastatin and metformin
Oral administration of simvastatin (5 mg·kg− 1·day− 1) or metformin (400 mg·kg− 1·day− 1) from 8 weeks of age effectively reduced the increase in SBP and the heightened sympathetic vasomotor activity (Fig. 7a) in young HFD offspring at age of 12 weeks. The same treatments also lessened the programmed hypertension (Fig. 7b) measured at 18 weeks of age.
Fig. 7.
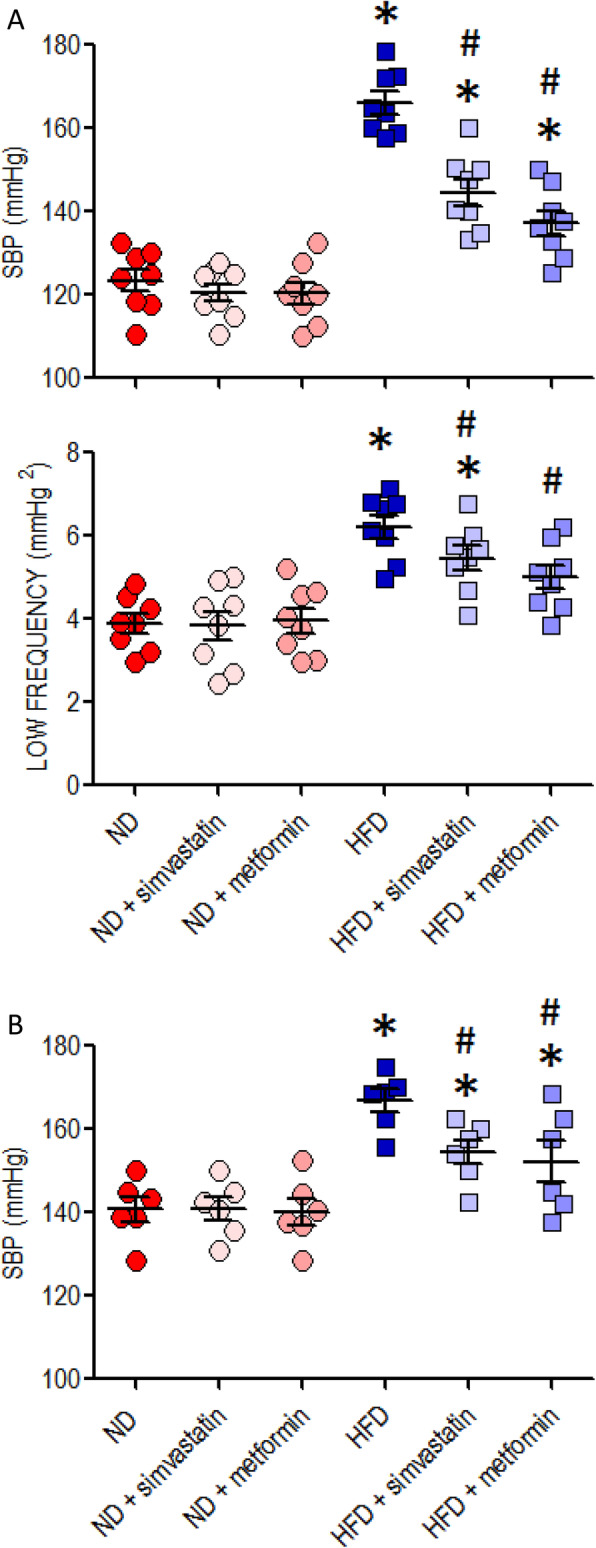
Reprogramming hypertension in young HFD offspring by simvastatin and metformin. Changes in (a) SBP and sympathetic vasomotor activity at age of 12 weeks or (b) SBP at age of 18 weeks in ND or HFD offspring following 4-week treatment with simvastatin (5 mg·kg− 1·day− 1) or metformin (400 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Data are presented as mean ± SEM (n = 6–8 per group). *P < 0.05 versus ND group and #P < 0.05 versus HFD group in post hoc Newman-Keuls multiple-range test
Discussion
The most significant findings of the present study are: (1) maternal exposure to HFD during gestation and lactation increases tissue oxidative stress in RVLM, priming the increases in sympathetic vasomotor activity and SBP in young offspring; (2) the programmed tissue oxidative stress in RVLM of HFD offspring are attributed to the increase in AMPK-regulated AT1R expression and impairment of SIRT1-associated mitochondrial biogenesis; (3) the perturbed redox homeostasis in RVLM of young HFD offspring could be significantly restored by oral administration of simvastatin or metformin; (4) the same treatments reprogram the hemodynamic dysfunctions observed in young HFD offspring, and lessens the programmed hypertension in adult life. Together, these data offer new documentation in support of oxidative stress in RVLM in fetal programming of hypertension in adult offspring that are exposed to maternal HFD, and provide novel insights into the roles of AT1R and mitochondria biogenesis, via the dysregulation of AMPK/SIRT1 signaling, in this process.
Hypertension is a major risk factor for cardio-vascular disease, the leading cause of morbidity and mortality worldwide. Emerging evidence from both epidemiological and animal studies indicates that the pathological process of hypertension in adult life may have its origin at pre- and neonatal stages. This concept of fetal programming of hypertension was first postulated by Barker [3] based on the seminal observations of an inverse relationship between infant birth weight and SBP in adult humans. Numerous experimental studies that mimic intrauterine nutritional insults have thereafter been utilized to investigate the etiology of programmed hypertension. Among the multiple mechanisms proposed, activation of the SNS represents a key pathophysiological process [15, 16]. In line with this postulation, we found in the present study that the increase in SBP of young offspring exposed to maternal HFD was accompanied by a parallel increase in plasma NE level, suggesting an activation of peripheral SNS. The observed augmentation of sympathetic vasomotor activity further indicated the involvement of brain machinery in the sympathoexcitation of HFD offspring.
Both oxidative stress and nitrosative stress in RVLM, where the sympathetic premotor neurons reside, have been demonstrated to mediate sympathoexcitation and promote hypertension [18, 19, 23]. Based on a maternal HFD model of programmed hypertension, our results revealed that oxidative rather than nitrosative stress in RVLM may account for the increase in sympathetic vasomotor activity observed in young offspring. We found that the tissue level of ROS and its distribution in RVLM neurons were enhanced; whereas no apparent change in NO level or expression of NOS isoforms was detected in HFD offspring. At the molecular level, the elevated ROS level in RVLM was attributed by an increase in gp91phox subunit of the NADPH oxidase and downregulation of the antioxidant SOD2, suppression of PGC-1α and TFAM expression, together with decrease in mitochondrial DNA copy number, events known to underpin sympathoexcitation in animal models of hypertension [18–23]. It is likely that activation of AT1R plays a pivotal role in priming tissue oxidative stress in RVLM of HFD offspring. Augmented expression of the receptor protein was induced by maternal HFD. More importantly, inhibition of AT1R action by losartan reversed the induced molecular changes and ameliorated ROS accumulation in RVLM of HFD offspring. Deficiencies in NO production, eNOS expression, NOS coupling and/or NO-cGMP signaling in the kidney [8, 14, 41] and vasculature [42] have been reported as potential underlying mechanisms for fetal programming of hypertension. Whether nitrosative stress in RVLM occurs in HFD offspring at an age earlier or later than that studied in the present study is not immediately clear and awaits further interrogation.
The brain renin-angiotensin system is an important element of hypertension programming during fetal life. Not only the AT1R expression in the brain areas involved in cardiovascular regulation is increased in fetus to antenatal nutrient deprivation, intracerebroventricular injection of AT1R antagonist significantly reduces the increase in mean arterial BP of the offspring [43], although the underlying regulatory mechanism is unknown. In addition to being a key regulator in whole-body energy metabolism [24, 25], AMPK, an evolutionarily conserved serine/threonine kinase, exhibits pluripotent cellular functions. In cultured cardiomyocytes, activation of AMPK reduces angiotensin II (Ang II)-induced AT1R upregulation and protects the cells from Ang II-induced hypertrophy [44]. AMPK is expressed in various tissues, including the brain, although there is currently no study that elucidates the relationship between AMPK and AT1R in neural tissues. Interestingly, we found in the present study an inverse relationship between AT1R expression and AMPK phosphorylation in RVLM of HFD offspring. That the induced increase in AT1R expression was significantly inhibited by treatment with an AMPK activator, metformin, further suggests that AT1R upregulation could be consequential to the blunted AMPK signals programmed by maternal HFD. This relationship between AMPK and AT1R is further supported by the observations in which AT1R blockade exerts negligible effect on the suppressed protein expressions of p-AMPK and SIRT1 in RVLM of HFD offspring. Compromised AMPK signals were recently demonstrated to be associated with intolerance to cardiac insult in offspring of gestational diabetes mellitus [45]. Phosphorylated AMPK was postulated to reduce AT1R levels through down-regulation of genes encoding the receptor via the PGC-1α/PPARγ pathway [46]. It is noteworthy that AMPK exhibits multiple phosphorylation sites on its catalytic subunit α, including the phosphorylation of AMPKα1 on Thr172 and AMPKα1/α2 on Ser471/Ser491 [47]. Depending on the residues and/or sites of phosphorylation, as well as its cellular location, AMPK may exert different cellular actions. In this regard, AMPKα2 gene knockout significantly exacerbates the degree of transverse aortic constriction-induced left ventricle hypertrophy and dysfunction, which are not affected by AMPKα1 gene knockout [48]. At the same time, the metformin-promoted cardioprotection against ischemia/reperfusion injury is mediated via activation of both cytosolic AMPKα1 and nuclear AMPKα2 [49]. Therefore, a full understanding in the roles of AMPK in RVLM on programmed hypertension in HFD offspring remains to be unraveled.
Upon activation at the Thr172 residue, AMPK increases NAD+/NADH ratio, which in turn stimulates the NAD+-dependent deacetylases sirtuins [50], a family of mammalian class III histone deacetylases with implicated functions in health span and longevity [51]. SIRT1 is the most studied sirtuins and may exert cellular protective actions by promoting mitochondrial biogenesis and triggering the turnover of damaged mitochondria [52]. The present study provides novel evidence to suggest that dysregulated SIRT1-dependent mitochondrial biogenesis may play an active role in oxidative stress programmed by maternal HFD exposure in RVLM. As the master regulator of mitochondrial biogenesis, PGC-1α drives transcription and replication of mitochondrial DNA via activation of transcription factors on the promoter region of TFAM [53]. SIRT1, on the other hand, is known to deacetylate PGC-1α to increase its transcriptional activity [54]. It thus is intriguing that we found the expression of SIRT1 protein was significantly decreased, alongside reduced PGC-1α and TFAM expressions and mitochondrial DNA copy number in RVLM of HFD offspring. The expression of SIRT3, another major mitochondrial sirtuin [55], on the other hand, was not affected by the same maternal exposure. We further found that activation of AMPK with metformin mitigated the impaired mitochondrial biogenesis, suggesting that the action of SIRT1 is downstream to AMPK in the signaling cascade. Accordingly, in the presence of metformin, AMPK-induced SIRT1 expression may prevent the reduced expression of PGC-1α and TFAM primed by maternal HFD exposure, resulting in the maintenance of mitochondrial biogenesis and redox homeostasis in RVLM of HFD offspring. These findings confer a novel role for the dysfunctional AMPK/SIRT1 signaling in fetal programming of oxidative stress in RVLM from maternal HFD insult. Dysregulated AMPK-PGC-1α signal has recently been reported to be engaged in adult hypertension programmed by prenatal NO deficiency plus postnatal high-fat diet exposure [56]. In addition to biogenesis impairment, mitochondrial ROS generated from curtail in bioenergetics [19, 21, 30, 31] and activation of cyclophilin D (a regulatory subunit of the mitochondrial permeability transition pore) [57] also contribute to hypertension development. Moreover, it is interesting to note that cyclophilin D interacts with heat shock protein 90 [58] Bcl-2, p53 [59] and PPARα [60] in mediating AMPK activation. As such, additional studies are required to further explore the relationship of AMPK activation, mitochondrial dysfunction and ROS production in developmental programming of hypertension.
An intriguing finding in the present study is that both the increased AT1R expression and impaired mitochondrial biogenesis in RVLM of HFD offspring were ameliorated after 4 weeks of oral administration of simvastatin, a widely used statin in the treatment of cardio-metabolic diseases because of its lipid-lowering effect. These observations suggest that ROS accumulation in RVLM could be primed by peripheral dyslipidemia developed in young offspring to maternal HFD. In this regard, we found both plasma and RVLM leptin levels were increased in young HFD offspring. Moreover, oral simvastatin treatment abolished the higher leptin expression in RVLM, pointing to a permissive role of dyslipidemia in brain uptake of leptin. Leptin has been shown to be transported across the blood-brain barrier to the brain in mice fed high fat diet [61], and the increase in brain leptin level inhibits AMPK phosphorylation [62]. In addition, leptin receptors are colocalized with AT1AR in brain tissue [63]. Central infusion of leptin increases AT1R mRNA expression in forebrain cardiovascular structures [64]. The mechanisms underpinning the effect of leptin on AMPK phosphorylation and AT1R expression in RVLM of HFD offspring, however, require further elucidation. In addition, despite that simvastatin treatment ameliorates the augmented leptin expression in RVLM and that high level leptin inhibits AMPK phosphorylation [62], there is no evidence from the current study for an active role of simvastatin-regulated AMPK-SIRT1 signal in RVLM on dyslipidemia and hyperleptinemia manifested in HFD offspring.
Another salient finding of the present study is the functional impact of the anomalous AMPK-regulated AT1R expression and SIRT1-dependent mitochondrial biogenesis on the programming of elevated BP in young HFD offspring. We found that metformin, at treatment regimen that restored the AMPK-regulated AT1R overexpression and SIRT1-dependent impairment of mitochondrial biogenesis in RVLM, could significantly abolish sympathoexcitation and increase of SBP in young HFD offspring. Similar responses were evoked by intracisternal infusion of losartan; further emphasizing the importance of AT1R of RVLM in fetal programming of hypertension in HFD offspring. The reprogramming of SBP in young HFD offspring after oral administration of simvastatin was interpreted to imply a sympathoexcitatory role of leptin in RVLM. In this regard, RVLM neurons express functional leptin receptors and can respond to leptin by increasing renal sympathetic activity and mean arterial BP [65].
There are a few cautionary notes in conjunction with the present study. First, while animal studies have identified the adverse effects of maternal fructose consumption on fetus and disease risk in offspring, only a small number of human studies have shown an association between excessive fructose consumption with poor pregnancy outcome [66]. As such, the translational implication of the present study remains to be realized. Second, the susceptibility to develop cardio-metabolic dysfunctions in offspring to maternal intrauterine insults could be sex dependent with a higher prevalence in male [67, 68], limiting the use of proposed reprogramming interventions to treat female offspring. Third, the beneficial effects of the studied compounds could be mediated via mechanisms not deciphered in the present study. For example, since AMPK is also a key regulator of PGC-1α [69], metformin may exert its beneficial effect on tissue oxidative stress directly via activation of PGC-1α. In addition, cardiovascular protective action of simvastatin is related to its pleiotropic effect such as inhibition of Rho kinase and increasing NO in the RVLM [70]. Finally, with the exception of losartan, all studied compounds were given via oral administration; therefore, site of action (central versus peripheral) and the proposed signaling cascades of their protective effects await further confirmation. To this end, simvastatin is known to penetrate the blood-brain barrier [71], and brain infusion of simvastatin protects against heart failure through normalization of sympathetic outflow from RVLM [70].
Conclusions
In conclusion, the present study provides novel evidence to suggest that anomalous AMPK-regulated AT1R expression and SIRT1-mediated mitochondrial biogenesis may contribute to tissue oxidative stress in RVLM, which in turn primes the increases in sympathetic vasomotor activity and BP in young offspring exposed to maternal HFD exposure (Fig. 8). Furthermore, the impaired nutrient sensing signaling in RVLM may be initiated by hyperleptinemia in young offspring to maternal HFD exposure.
Fig. 8.

Schematic depiction of the contribution of AMPK/SIRT1 signaling in RVLM to oxidative stress-associated programming of hypertension in young offspring exposed to maternal high fructose. Circulatory dyslipidemia programmed by maternal HFD exposure during gestation and lactation increases leptin level, which may in turn increase AT1R expression and inhibit AMPK phosphorylation in RVLM. AT1R overexpression leads to tissue oxidative stress via an increase in gp91phox and decrease in SOD2 expression. Suppressed AMPK activation results in inhibition of SIRT1 expression and its downstream signals, PGC-1α and TFAM, followed by tissue oxidative stress through the reduction in mitochondrial biogenesis. Suppressed AMPK activation also augments AT1R expression that promotes further oxidative stress. Accumulated ROS in RVLM contributes to the programmed hypertension in young HFD offspring via sympathoexcitation. The hypertension programming in HFD offspring could be protected by treatments with simvastatin to reduce dyslipidemia and hyperleptinemia; metformin to activate the AMPK/SIRT1 signals, and losartan to antagonize AT1R activation in RVLM. Arrows indicate activation; bar-headed lines indicate inhibition. Solid line denotes data from the present study or existing literature; dotted line denotes potential connections that require further documentation
Perspective
The burden of hypertension is an increasingly pressing matter because of the rising prevalence of higher BP in adolescence and its tracking into adult hypertension. Determining its pathogenesis to gain insights into its current and emerging management, particularly at young age, is therefore crucial and timely. Herein, in support of the DOHaD concept, our study highlights the transgenerational detrimental influence of maternal HFD on the pathogenesis of hypertension in young offspring. Our results suggest that programmed hypertension could be developed via common (e.g., activation of the SNS) or specific (e.g., dysfunction in AMPK/SIRT signaling) underlying mechanisms. Therapeutic remedies against programmed hypertension and cardio-metabolic disease should therefore be based on the underlying disease etiology rather than the disease phenotypes. Our observations that treatment with simvastatin or metformin to young offspring (at age of 8 weeks) protects against hypertension manifested in adult life (at age of 18 weeks) further emphasize a window of opportunity to treat adult cardio-metabolic disease of developmental origin at young age. Future studies examining the long-term beneficial effect on adult cardiovascular diseases of prenatal or perinatal treatment are crucial. It is also important to recommend dietary guidelines on fructose intake during pregnancy and lactation.
Supplementary information
Additional file 1: Table S1. Metabolic indices of offspring to maternal ND or HFD exposure. Table S2. Effect of simvastatin or losartan on metabolic indices of young ND or HFD offspring.
Additional file 2: Figure S1. Representative laser-scanning confocal microscopic images showing the distribution of (A) AT1R, (B) gp91pphox or (C) SOD2 (green fluorescence) in cells that were stained positively for a neuronal marker, neuron-specific nuclear protein (NeuN) (red fluorescence) in RVLM of ND or HFD offspring at age of 12 weeks. Scale bar: 50 μm. Figure S2. Representative gels (insets) and densitometric analysis of results from Western blot analysis showing changes in protein expression of (A) p47phox or p67phox of the NADPH oxidase, (B) SOD1 or SOD3, (C) catalase or GPx, or (D) NOS1–3 in RVLM of ND (n = 6–10) or HFD (n = 6–10) offspring, alone or with additional treatment with a HMG-CoA inhibitor, simvastatin (5 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM. No significance difference among all groups in one-way ANOVA. Figure S3. Representative gels (insets) and densitometric analysis of results from Western blot analysis showing changes in protein expression of (A) p-AMPK and (B) SIRT1, as well as (C) ratio between p-AMPK/t-AMPK in RVLM of ND or HFD offspring, alone or with additional treatment with an AT1R antagonist, losartan (3 μg·μL− 1·h− 1), microinfused via a minipump into the cistern magna at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM (n = 10 in all groups). *P < 0.05 versus ND group in post hoc Newman-Keuls multiple-range test. Group data of p-AMPK and SIRT1 in ND and HFD offspring from Fig. 4 are adopted for statistical comparison.
Acknowledgments
Not applicable.
Abbreviations
- AMPK
AMP-activated protein kinase
- AT1R
Angiotensin type 1 receptor
- BP
Blood pressure
- CSF
Cerebrospinal fluid
- DHE
Dihydroethidium
- DOHaD
Developmental original of adult health and disease
- GPx
Glutathione peroxidase
- HFD
High fructose diet
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- HOMA
Homeostasis model assessment
- IR
Insulin resistance
- LF
Low frequency
- MDA
Malondialdehyde
- NADPH oxidase
Nicotinamide adenine dinucleotide diphosphate oxidase
- ND
Normal diet
- ND1
NADH dehydrogenase subunit 1
- NE
Norepinephrine
- NO
Nitric oxide
- NOS
NO synthase
- NOx
Nitrate and nitrite
- OPA
O-phthaldehyde
- PGC-1α
Peroxisome proliferator-activated receptor gamma co-activator α
- qPCR
Quantitative real-time polymerase chain reaction
- ROS
Reactive oxygen species
- RVLM
Rostral ventrolateral medulla
- SAP
Systemic arterial pressure
- SBP
Systolic blood pressure
- SD
Sprague-Dawley
- SIRT
Sirtuin
- SNS
Sympathetic nervous system
- SOD
Superoxide dismutase
- TFAM
Mitochondrial transcription factor A
Authors’ contributions
YMC, KLHW and JYHC participated in the design and coordination of the study, and contributed to writing and preparation of the manuscript. PCT, YMC, and KLHW participated in performed experiments and analyzed data. YLT, SL, and WCL helped to edit the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by research grants from the Chang Gung Medical Foundation to J.Y.H. Chan (CMRPG8C0053, CMRPG8F0031–33 and OMRPG8G0011). Y.M. Chao was supported by a Postdoctoral Fellowship (MOST107–2811-B-182A-501) from the Ministry of Science and Technology, Taiwan.
Availability of data and materials
Analytic methods, and study materials will be made available on publication of this research article. The data will be available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yung-Mei Chao and Kay L. H. Wu contributed equally to this work.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12929-020-00660-z.
References
- 1.Kereliuk Stephanie, Brawerman Gabriel, Dolinsky Vernon. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. International Journal of Molecular Sciences. 2017;18(7):1451. doi: 10.3390/ijms18071451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costa-Silva JH, Simoes-Alves AC, Fernandes MP. Developmental origins of cardiometabolic diseases: role of the maternal diet. Front Physiol. 2016;7:504. doi: 10.3389/fphys.2016.00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 4.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherrer U, Sartori C. Insulin as a vascular and sympathoexcitatory hormone: implications for blood pressure regulation, insulin sensitivity, and cardiovascular morbidity. Circulation. 1997;96:4104–4113. doi: 10.1161/01.cir.96.11.4104. [DOI] [PubMed] [Google Scholar]
- 6.Ha V, Sievenpiper JL, de Souza RJ, Chiavaroli L, Wang DD, Cozma AI, Mirrahimi A, Yu ME, Carleton AJ, Dibuono M, Jenkins AL, Leiter LA, Wolever TM, Beyene J, Kendall CW, Jenkins DJ. Effect of fructose on blood pressure: a systematic review and meta-analysis of controlled feeding trials. Hypertension. 2012;59:787–795. doi: 10.1161/HYPERTENSIONAHA.111.182311. [DOI] [PubMed] [Google Scholar]
- 7.Samuelsson AS, Mullier A, Maicas N, Oosterhuis NR, Eun Bae S, Novoselova TV, Chan LF, Pombo JM, Taylor PD, Joles JA, Coen CW, Balthasar N, Poston L. Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proc Natl Acad Sci U S A. 2016;113:12298–12303. doi: 10.1073/pnas.1607464113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tain YL, Leu S, Wu KLH, Lee WC, Chan JYH. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: roles of nitric oxide and arachidonic acid metabolites. J Pineal Res. 2014;57:80–89. doi: 10.1111/jpi.12145. [DOI] [PubMed] [Google Scholar]
- 9.Tain YL, Lee WC, Wu KLH, Leu S, Chan JYH. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J Nutr Biochem. 2016;38:86–92. doi: 10.1016/j.jnutbio.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Saad AF, Dickerson J, Kechichian TB, Yin H, Gamble P, Salazar A, Patrikeev I, Motamedi M, Saade GR, Costantine MM. High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am J Obstet Gynecol. 2016;215:e371–e376. doi: 10.1016/j.ajog.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 11.Seong HY, Cho HM, Kim M, Kim I. Maternal high-fructose intake induces multigenerational activation of the renin-angiotensin-aldosterone system. Hypertension. 2019;74:518–525. doi: 10.1161/HYPERTENSIONAHA.119.12941. [DOI] [PubMed] [Google Scholar]
- 12.Szostak-Wegierek D. Intrauterine nutrition: long-term consequences for vascular health. Int J Women's Health. 2014;6:647–656. doi: 10.2147/IJWH.S48751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tain YL, Wu KL, Lee WC, Leu S, Chan JYH. Maternal fructose-intake-induced renal programming in adult male offspring. J Nutr Biochem. 2015;26:642–650. doi: 10.1016/j.jnutbio.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Tain You-Lin, Hsu Chien-Ning. Targeting on Asymmetric Dimethylarginine-Related Nitric Oxide-Reactive Oxygen Species Imbalance to Reprogram the Development of Hypertension. International Journal of Molecular Sciences. 2016;17(12):2020. doi: 10.3390/ijms17122020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morton JS, Cooke CL, Davidge ST. In utero origins of hypertension: mechanisms and targets for therapy. Physiol Rev. 2016;96:549–603. doi: 10.1152/physrev.00015.2015. [DOI] [PubMed] [Google Scholar]
- 16.Dasinger JH, Davis GK, Newsome AD, Alexander BT. Developmental programming of hypertension: physiological mechanisms. Hypertension. 2016;68:826–831. doi: 10.1161/HYPERTENSIONAHA.116.06603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross CA, Ruggiero DA, Joh TH, Park DH, Reis DJ. Rostral ventrolateral medulla: selective projections to the thoracic autonomic cell column from the region containing C1 adrenaline neurons. J Comp Neurol. 1984;228:168–185. doi: 10.1002/cne.902280204. [DOI] [PubMed] [Google Scholar]
- 18.Hirooka Y. Oxidative stress in the cardiovascular center has a pivotal role in the sympathetic activation in hypertension. Hypertens Res. 2011;34:407–412. doi: 10.1038/hr.2011.14. [DOI] [PubMed] [Google Scholar]
- 19.Chan SHH, Chan JYH. Brain stem NOS and ROS in neural mechanisms of hypertension. Antioxid Redox Signal. 2014;20:146–163. doi: 10.1089/ars.2013.5230. [DOI] [PubMed] [Google Scholar]
- 20.Chan SHH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JYH. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- 21.Chan SHH, Tai MH, Li CY, Chan JYH. Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radic Biol Med. 2006;40:2028–2039. doi: 10.1016/j.freeradbiomed.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Wu KLH, Wu CW, Chao YM, Hung CY, Chan JYH. Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic Biol Med. 2016;97:58–74. doi: 10.1016/j.freeradbiomed.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Chan JYH, Chan SHH. Differential impacts of brain stem oxidative stress and nitrosative stress on sympathetic vasomotor tone. Pharmacol Ther. 2019;201:120–136. doi: 10.1016/j.pharmthera.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez M, Nogueiras R, Tena-Sempere M, Dieguez C. Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol. 2016;12:421–432. doi: 10.1038/nrendo.2016.67. [DOI] [PubMed] [Google Scholar]
- 26.Philp LK, Muhlhausler BS, Janovska A, Wittert GA, Duffield JA, McMillen IC. Maternal overnutrition suppresses the phosphorylation of 5′-AMP-activated protein kinase in liver, but not skeletal muscle, in the fetal and neonatal sheep. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1982–R1990. doi: 10.1152/ajpregu.90492.2008. [DOI] [PubMed] [Google Scholar]
- 27.Tain You-Lin, Hsu Chien-Ning. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. International Journal of Molecular Sciences. 2018;19(6):1744. doi: 10.3390/ijms19061744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tain YL, Wu KLH, Lee WC, Leu S, Chan JYH. Prenatal metformin therapy attenuates hypertension of developmental origin in male adult offspring exposed to maternal high-fructose and post-weaning high-fat diets. Int J Mol Sci. 2018;19:1066. doi: 10.3390/ijms19041066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu KLH, SHH C, JYH C. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflam. 2012;9:212. doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan SHH, Wu KLH, Chang AYW, Tai MH, Chan JYH. Oxidative impairment of mitochondrial electron tranposrt chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertensin. Hypertension. 2009;53:217–227. doi: 10.1161/HYPERTENSIONAHA.108.116905. [DOI] [PubMed] [Google Scholar]
- 31.Chan SHH, Wu KLH, Kung PSS, Chan JYH. Oral intake of rosiglitazone promotes a central antihypertensive effect via upregulation of peroxisome proliferator-activated receptor-γ and alleviation of oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertension. 2010;55:1444–1453. doi: 10.1161/HYPERTENSIONAHA.109.149146. [DOI] [PubMed] [Google Scholar]
- 32.Gamache P, Ryan E, Svendsen C, Murayama K, Acworth IN. Simultaneous measurement of monoamines, metabolites and amino acids in brain tissue and microdialysis perfusates. J Chromatogr. 1993;614:213–220. doi: 10.1016/0378-4347(93)80311-q. [DOI] [PubMed] [Google Scholar]
- 33.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;8:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 34.Chan SHH, Wu CWJ, Chang AYW, Hsu KS, Chan JYH. Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: significance in superoxide homeostasis and neural regulation of arterial pressure. Circ Res. 2010;107:1127–1139. doi: 10.1161/CIRCRESAHA.110.225573. [DOI] [PubMed] [Google Scholar]
- 35.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 7. New York: American Press; 2013. [DOI] [PubMed] [Google Scholar]
- 36.Wu KLH, Chao YM, Tsay SJ, Chen CH, Chan SHH, Dovinova I, Chan JYH. Role of nitric oxide synthase uncoupling at rostral ventrolateral medulla in redox-sensitive hypertension associated with metabolic syndrome. Hypertension. 2014;64:815–824. doi: 10.1161/HYPERTENSIONAHA.114.03777. [DOI] [PubMed] [Google Scholar]
- 37.Chao YM, Lai MD, Chan JYH. Redox-sensitive endoplasmic reticulum stress and autophagy at rostral ventrolateral medulla contribute to hypertension in spontaneously hypertensive rats. Hypertension. 2013;61:1270–1280. doi: 10.1161/HYPERTENSIONAHA.111.00469. [DOI] [PubMed] [Google Scholar]
- 38.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension. 2001;37:1199–1208. doi: 10.1161/01.hyp.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 39.Wong MJ, Kantores C, Ivanovska J, Jain A, Jankov RP. Simvastatin prevents and reverses chronic pulmonary hypertension in newborn rats via pleiotropic inhibition of RhoA signaling. Am J Physiol Lung Cell Mol Physiol. 2016;311:L985–L999. doi: 10.1152/ajplung.00345.2016. [DOI] [PubMed] [Google Scholar]
- 40.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 41.Hsu CN, Tain YL. Regulation of nitric oxide production in the developmental programming of hypertension and kidney disease. Int J Mol Sci. 2019;20:681. doi: 10.3390/ijms20030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang ZQ, Wang JL, Luo H, Chen C, Pei F, Cai Y, Yang XL, Wang N, Fu JJ, Zu ZC, Zhou L, Zeng CY. Prenatal lipopolysaccharide exposure causes mesenteric vascular dysfunction through the nitric oxide and cyclic guanosine monophosphate pathway in offspring. Free Radic Biol Med. 2015;86:322–330. doi: 10.1016/j.freeradbiomed.2015.05.040. [DOI] [PubMed] [Google Scholar]
- 43.Pladys P, Lahaie I, Cambonie G, Thibault G, Lê NL, Abran D, Nuyt AM. Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr Res. 2004;55:1042–1049. doi: 10.1203/01.PDR.0000127012.37315.36. [DOI] [PubMed] [Google Scholar]
- 44.Hernandez JS, Barreto-Torres G, Kuznetsov AV, Khuchua Z, Javadov S. Crosstalk between AMPK activation and angiotensin II-induced hypertrophy in cardiomyocytes: the role of mitochondria. J Cell Mol Med. 2014;18:709–720. doi: 10.1111/jcmm.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo X, Zou H, Xu P, Wen L, Stanley JL, Jiang X, Han TL, Olson D, Peng C, Zhang C, Xiao X, Tong C, Qi H, Baker PN. Blunted cardiac AMPK response is associated with susceptibility to ischemia/reperfusion in male offspring of gestational diabetic rats. Cell Physiol Biochem. 2019;52:1103–1116. doi: 10.33594/000000075. [DOI] [PubMed] [Google Scholar]
- 46.Na L, Chu X, Jiang S, Li C, Li G, He Y, Liu Y, Li Y, Sun C. Vinegar decreases blood pressure by down-regulating AT1R expression via the AMPK/PGC-1α/PPARγ pathway in spontaneously hypertensive rats. Eur J Nutr. 2016;55:1245–1253. doi: 10.1007/s00394-015-0937-7. [DOI] [PubMed] [Google Scholar]
- 47.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X, Lu Z, Fassett J, Zhang P, Hu X, Liu X, Kwak D, Li J, Zhu G, Tao Y, Hou M, Wang H, Guo H, Viollet B, McFalls EO, Bache RJ, Chen Y. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase α2. Hypertension. 2014;63:723–728. doi: 10.1161/HYPERTENSIONAHA.113.02619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Yang Z, Zheng G, Yu L, Yin Y, Mu N, Ma H. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKα1 and nuclear AMPKα2 pathways. Life Sci. 2019;225:64–71. doi: 10.1016/j.lfs.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Cantó C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 52.Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39:87–95. doi: 10.14348/molcells.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu ZD, Puigserver P, Andersson U, Zhang CY, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 54.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through sirt1/pgc-1α. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radic Biol Med. 2016;100:164–174. doi: 10.1016/j.freeradbiomed.2016.04.197. [DOI] [PubMed] [Google Scholar]
- 56.Chen HE, Lin YJ, Lin IC, Yu HR, Sheen JM, Tsai CC, Huang LT, Tain YL. Resveratrol prevents combined prenatal NG-nitro-L-arginine-methyl ester (L-NAME) treatment plus postnatal high-fat diet induced programmed hypertension in adult rat offspring: interplay between nutrient-sensing signals, oxidative stress and gut microbiota. J Nutr Biochem. 2019;70:28–37. doi: 10.1016/j.jnutbio.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 57.Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, Dikalov SI. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67:1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G, Kranias EG. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A. 2015;112:E6466–E6475. doi: 10.1073/pnas.1508760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen MB, Jiang Q, Liu YY, Zhang Y, He BS, Wei MX, Lu JW, Ji Y, Lu PH. C6 ceramide dramatically increases vincristine sensitivity both in vivo and in vitro, involving AMP-activated protein kinase-p53 signaling. Carcinogenesis. 2015;36:1061–1070. doi: 10.1093/carcin/bgv094. [DOI] [PubMed] [Google Scholar]
- 60.Barreto-Torres G, Hernandez JS, Jang S, Rodríguez-Muñoz AR, Torres-Ramos CA, Basnakian AG, Javadov S. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARα-cyclophilin D interaction in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2015;308:H749–H758. doi: 10.1152/ajpheart.00414.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleinert M, Kotzbeck P, Altendorfer-Kroath T, Birngruber T, Tschöp MH, Clemmensen C. Time-resolved hypothalamic open flow micro-perfusion reveals normal leptin transport across the blood-brain barrier in leptin resistant mice. Mol Metab. 2018;13:77–82. doi: 10.1016/j.molmet.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, Strynadka K, Chohnan S, Smith WW, Tamashiro KL, Ladenheim EE, Ronnett GV, Tu Y, Birnbaum MJ, Lopaschuk GD, Moran TH. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A. 2007;104:17358–17363. doi: 10.1073/pnas.0708385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Claflin KE, Sandgren JA, Lambertz AM, Weidemann BJ, Littlejohn NK, Burnett CM, Pearson NA, Morgan DA, Gibson-Corley KN, Rahmouni K, Grobe JL. Angiotensin AT1A receptors on leptin receptor-expressing cells control resting metabolism. J Clin Invest. 2017;127:1414–1424. doi: 10.1172/JCI88641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xue B, Yu Y, Zhang Z, Guo F, Beltz TG, Thunhorst RL, Felder RB, Johnson AK. Leptin mediates high-fat diet sensitization of angiotensin II-elicited hypertension by upregulating the brain renin-angiotensin system and inflammation. Hypertension. 2016;67:970–976. doi: 10.1161/HYPERTENSIONAHA.115.06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barnes MJ, McDougal DH. Leptin into the rostral ventral lateral medulla (RVLM) augments renal sympathetic nerve activity and blood pressure. Front Neurosci. 2014;8:232. doi: 10.3389/fnins.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Regnault TR, Gentili S, Sarr O, Toop CR, Sloboda DM. Fructose, pregnancy and later life impacts. Clin Exp Pharmacol Physiol. 2013;40:824–837. doi: 10.1111/1440-1681.12162. [DOI] [PubMed] [Google Scholar]
- 67.de Sá FG, de Queiroz DB, Ramos-Alves FE, Santos-Rocha J, da Silva OA, Moreira HS, Leal GA, da Rocha MA, Duarte GP, Xavier FE. Hyperglycaemia in pregnant rats causes sex-related vascular dysfunction in adult offspring: role of cyclooxygenase-2. Exp Physiol. 2017;102:1019–1036. doi: 10.1113/EP086132. [DOI] [PubMed] [Google Scholar]
- 68.Hsu CN, Lai WT, Lin YJ, Tain YL. Postnatal high-fat diet sex-specifically exacerbates prenatal dexamethasone-induced hypertension: mass spectrometry-based quantitative proteomic approach. J Nutr Biochem. 2018;57:268–275. doi: 10.1016/j.jnutbio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 69.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao L, Wang W, Zucker IH. Simvastatin inhibits central sympathetic outflow in heart failure by a nitric-oxide synthase mechanism. J Pharmacol Exp Ther. 2008;326:278–285. doi: 10.1124/jpet.107.136028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saheki A, Terasaki T, Tamai I, Tsuji A. In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme a (HMG-CoA) reductase inhibitors. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Metabolic indices of offspring to maternal ND or HFD exposure. Table S2. Effect of simvastatin or losartan on metabolic indices of young ND or HFD offspring.
Additional file 2: Figure S1. Representative laser-scanning confocal microscopic images showing the distribution of (A) AT1R, (B) gp91pphox or (C) SOD2 (green fluorescence) in cells that were stained positively for a neuronal marker, neuron-specific nuclear protein (NeuN) (red fluorescence) in RVLM of ND or HFD offspring at age of 12 weeks. Scale bar: 50 μm. Figure S2. Representative gels (insets) and densitometric analysis of results from Western blot analysis showing changes in protein expression of (A) p47phox or p67phox of the NADPH oxidase, (B) SOD1 or SOD3, (C) catalase or GPx, or (D) NOS1–3 in RVLM of ND (n = 6–10) or HFD (n = 6–10) offspring, alone or with additional treatment with a HMG-CoA inhibitor, simvastatin (5 mg·kg− 1·day− 1), administered via gastric gavage at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM. No significance difference among all groups in one-way ANOVA. Figure S3. Representative gels (insets) and densitometric analysis of results from Western blot analysis showing changes in protein expression of (A) p-AMPK and (B) SIRT1, as well as (C) ratio between p-AMPK/t-AMPK in RVLM of ND or HFD offspring, alone or with additional treatment with an AT1R antagonist, losartan (3 μg·μL− 1·h− 1), microinfused via a minipump into the cistern magna at age of 8 weeks for 4 weeks. Analysis was performed on tissues collected bilaterally from individual RVLM at age of 12 weeks. Data on protein expression were normalized to the average ND control value, which is set to 1.0, and are presented as mean ± SEM (n = 10 in all groups). *P < 0.05 versus ND group in post hoc Newman-Keuls multiple-range test. Group data of p-AMPK and SIRT1 in ND and HFD offspring from Fig. 4 are adopted for statistical comparison.
Data Availability Statement
Analytic methods, and study materials will be made available on publication of this research article. The data will be available from the corresponding author on reasonable request.