

Abstract
Protein functions can be predicted based on their three-dimensional structures. However, many multidomain proteins have unstable structures, making it difficult to determine the whole structure in biological experiments. Additionally, multidomain proteins are often decomposed and identified based on their domains, with the structure of each domain often found in public databases. Recent studies have advanced structure prediction methods of multidomain proteins through computational analysis. In existing methods, proteins that serve as templates are used for three-dimensional structure prediction. However, when no protein template is available, the accuracy of the prediction is decreased. This study was conducted to predict the structures of multidomain proteins without the need for whole structure templates.
We improved structure prediction methods by performing rigid-body docking from the structure of each domain and reranking a structure closer to the correct structure to have a higher value. In the proposed method, the score for the domain-domain interaction obtained without a structural template of the multidomain protein and score for the three-dimensional structure obtained during docking calculation were newly incorporated into the score function. We successfully predicted the structures of 50 of 55 multidomain proteins examined in the test dataset.
Interaction residue pair information of the protein-protein complex interface contributes to domain reorganizations even when a structural template for a multidomain protein cannot be obtained. This approach may be useful for predicting the structures of multidomain proteins with important biochemical functions.
Keywords: multidomain protein, protein tertiary structure prediction, interaction residue pair, rigid-body docking, conformations reranking
Significance
We have developed a novel multidomain protein structure prediction method named PINE without the need for whole multidomain structure templates. The score for the domain-domain interaction obtained without a structural template of the multidomain protein and score for the three-dimensional structure obtained during protein docking calculation were newly incorporated into the score function. We successfully predicted the structures of 50 of 55 multidomain proteins examined in the test dataset. Interaction residue pair information of the protein-protein complex interface contributes to domain reorganizations even when a structural template for a multidomain protein cannot be obtained.
More than half of prokaryote and eukaryote genes produce multidomain proteins with multiple partial structures known as protein domains [1,2]. Each protein domain is folded into a tight and stable structure that is conserved among different multidomain proteins [3].
Because protein functions differ depending on their three-dimensional (3D) structures, determining these structures is very important [4]. Typically, the 3D structure of a protein is determined by X-ray crystallography analysis, nuclear magnetic resonance analysis, or electron microscopy. However, determining structures experimentally is difficult because multidomain proteins are generally difficult to crystallize [5,6]. Even if the entire 3D structure has not been elucidated, structure information for protein domains is collected in public databases such as the Protein Data Bank (PDB) [7]. Therefore, experimental costs can be reduced by computationally predicting the whole structure based on the structure of each domain. Structure prediction methods include ab initio methods [8–10], template-based methods [11–13], and template-free methods [14–16].
Ab initio methods require high levels of computational resources to construct an entire protein structure, and thus it is difficult to predict the structures of all multidomain proteins using these methods [9]. Template-based methods require homologous templates whose 3D structural information is known; when such proteins are not available or only low sequence similarity proteins are available, prediction accuracy may be low [11]. Template-free methods are based on the property that protein domains are conserved even in multidomain proteins and can be used to predict structures by rigid-body docking with the 3D structure of protein domains. This method is superior to other methods in that the entire multidomain protein structure can be predicted from the 3D structure of protein domains stored in the PDB. Hirako, S. et al. [16] used individual domains of tertiary structures in two-domain proteins and structural docking tools for protein complexes to generate structural models for two-domain protein conformational prediction. They developed a scoring method for selecting an appropriate association model between two domains. However, the scoring method requires information on the structure of a homologous template multidomain protein and cannot be used when the template structures do not exist. The number of protein tertiary structures that can function as templates is continually increasing [7]. A template structure may be sufficient for protein complex structure prediction [17]; still this approach may not be effective in multidomain proteins [11].
In this study, we developed a new scoring method, PINE for selecting a structural model of a two-domain protein even when template structures cannot be obtained. This method estimates the pattern of interaction between domains using dimeric protein-protein interaction (PPI) residue pair information. As a result, the association model of the two domains can be selected with high accuracy when a template structure cannot be obtained.
Materials and Methods
Problem setting
First, we obtained the structure of a protein with two domains from PDB. Next, we separated each domain at the linker region and predicted the original structure from these two domains. The structural model was generated using a rigid-body docking tool, and reranking (scoring) was performed for the generated structural model using a scoring function. By reranking, a structure close to the original structure was predicted to have a higher rank. A structure whose root mean square deviation (RMSD) with the original structure was within 10 Å (acceptable structure) in the top 10 positions was defined as a successful prediction (Fig. 1).
Figure 1.
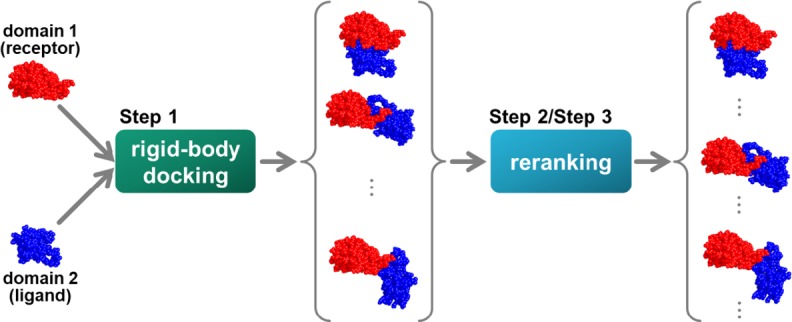
Problem setting. Step 1: We obtained a two-domain protein from the PDB and divided each domain according to the definition. Next, we generated a structural model using a rigid-body docking tool with two input domain structures. Step 2: We calculated some scores for each structural model. Step 3: We evaluated and reranked each model with a score function using some scores. As a result, the prediction was considered as successful when there was at least one acceptable structure within the top 10 solutions.
Division of multidomain proteins into domains was performed according to the definition of SCOP [18]. However, the region between domains, known as the linker, is not always clearly defined. Therefore, we defined the linker region as the region between the last residue of the secondary structure of the N-terminal domain and first residue of the secondary structure of the C-terminal domain. The region without secondary structure between the domains was considered as the linker. DSSP [19] was used to determine whether secondary structures were formed for each residue. Additionally, the linker region was excluded during docking calculation and during model evaluation.
Related work: DINE
DINE is a method of reranking (scoring) the results of rigid-body docking of each domain structure for two-domain proteins. First, 2,000 domain-domain docking poses are generated by rigid-body docking using ZDOCK [20], after which scoring is performed by calculating the linear sum of the binding energy score Szrank, inter-domain distance score Sete, and domain interface score Sint. Finally, the DINE score is obtained from these scores as a linear weighted sum as follows.
Binding energy score
The binding energy score (Szrank) is a value calculated by ZRANK [21], a protein complex scoring tool. The score zr obtained by ZRANK is based on van der Waals energy, electrostatic interaction energy, and desolvation energy [22,23]; a smaller value is preferable [24]. Szrank is calculated with the following equation using zr, where zrmax and zrmin are respectively the maximum and minimum zr values for the generated poses.
Domain interface score
The domain interface score (Sint) is a value of the interface of the binding pose estimated from the homology template structure. Protein domains with 30% or more sequence homology may have the same spatial arrangement and interaction surface within multidomain proteins [25,26]. Therefore, previous studies were conducted to improve prediction accuracy by predicting the interaction surface using the known structure of a multidomain protein. The KIP method [16] was used to predict interaction surfaces and obtain Sint. The KIP method uses a database in which multidomain proteins whose entire structures and interaction residues are known and are clustered by homology. The authors searched for proteins homologous to the query multidomain protein in the database and predicted interacting residues. Sint is the ratio of this predicted interaction residue to the interaction residue of each structural model.
Inter-domain distance score
The inter-domain distance score (Sete) is calculated from the statistics of the residue length of the linker region and inter-domain distance. Sete is calculated using the following equation
where L is the number of residues in the linker region, de is the distance between domains of the structural model, μe(L) and σe(L) are the mean and standard deviation of the distance between domains when the number of linker residues is L, respectively; μe(L) and σe(L) are based on the statistics from 1,657 multidomain proteins [16].
Proposed method: PINE
The DINE Sint function cannot be calculated when no template proteins are available. DINE uses structural information of homologous multidomain proteins as query proteins to calculate Sint. Therefore, in this study, we propose a new scoring method named PINE. PINE solved this problem by using a heterodimeric interaction residue pair score Sppi, which is a new scoring term for predicting the interaction surface, without using a protein as a homologous template, and docking score Sdock, calculated during rigid-body docking. Figure 2 shows the flow of the proposed PINE method.
Figure 2.

a. Outline of the proposed method. Step 1: Obtain domains as in the existing method and generate 10,800 structural models for each multidomain protein with the rigid-body docking tool MEGADOCK. Docking score is calculated at the time of this docking calculation. Step 2: Calculate the proposed score for each structural model. Step 3: Rerank the structural model with the score function PINE using each score and predict the structure. b. Calculation of the interaction amino acid residue score Sppi. The PPI matrix P was generated from protein complex structures in advance and the domain contact matrix C was generated from a predicted two-domain protein structure. Next, Sppi of the predicted structure was calculated with P and C. This score predicts interactions without requiring a template for the overall structure of the multidomain protein.
Model generation
Generation of the structural model was performed using MEGADOCK 4.0.2 [27,28]. The top three structural models of the docking score were output for each rotation angle, and the number of rotation angles was 3,600. Therefore, 3×3,600=10,800 poses were used for structural prediction of one multidomain protein (the option -t 3 -N 10800 was used). Among the domains of multidomain proteins, we considered the domain with a large number of residues as the receptor (argument of -R option) and that with few residues as the ligand (argument of -L option). Moreover, domain-domain docking was performed by excluding the linker region.
Sppi and Sdock
Rather than using the score based on the interaction surface prediction in the score function of the existing method, Sint, two new terms were used. The interaction amino acid residue score (Sppi) derived from the PPI and the docking score (Sdock) derived from the docking calculation with MEGADOCK are new terms in the proposed score function of PINE. Szrank and Sete are the same as those used in the previous study [16]. To calculate Szrank, hydrogen must be added to the structural model, which was performed using reduce ver. 3.23 [29]. The score function is defined as follows:
Interaction amino acid residue score
The interaction amino acid residue score is a term that focuses on the combination of interacting residues. The domain-domain interaction interface is predicted from the pair of amino acid residues involved in the PPI. This eliminates the need to know the entire structure of the multidomain protein homologous to the prediction of the interaction surface. In this study, if the distance between Cα atoms in two domains was within 8 Å, the residue pair was defined as interacting. Proteins are often multimers, and the PDB catalogs conformations of multimers. Therefore, among the dimers stored in the PDB, the number of amino acid residue pairs present in interacting positions was counted using 12,532 complex structures excluding redundancy based on UniProtID. As a result, a 20×20 upper triangular matrix (PPI matrix) P=(pij) indicating the number of interacting amino acid residues pairs was obtained. However, for each element in the PPI matrix, the number of interaction residue pairs counted was the value obtained by min-max normalization of the minimum value to 0 and maximum value to 1 as follows:
In addition, the number of amino acid residue pairs interacting between domains was counted from each domain docking structure model generated, and the 20×20 upper triangular matrix (domain contact matrix) C=(cij) was determined. For these two matrices, the interaction amino acid residue score Sppi of each model was calculated by the following formulas:
which has a minimum value of 0 and maximum value of 1.
Docking score
Model generation was performed using MEGADOCK ver. 4.0.2 [27,28]. Sdock is the docking score calculated for each of the 10,800 structural models to be generated and was min-max normalized, so that the score ranges between 0 and 1.
Dataset
For consistency purposes, we used the same training and test datasets that were used by Hirako, S. et al. [16]. The training set contained 62 non-redundant two-domain proteins originally proposed by Wollacott, A. M. et al. [8] excluding the 14 proteins defined as single-domain proteins in SCOP [18]. The test set contained 55 non-redundant two-domain proteins used by Cheng, T. M. K. et al. [14]. Parameter optimization of the score function weights, wzrank, wete, wppi, and wdock, was conducted using the training dataset, and evaluation of PINE was performed using the test dataset. These training and test datasets are shown in Tables 1 and 2, respectively.
Table 1.
Detail and prediction result of Wollacott dataset (training set, 62 proteins). (values in parentheses are RMSD (Å))
| PDB IDa | Domain 1 | Domain 2 | Linker length | Linker region | Best rank of acceptable docking pose | |
|---|---|---|---|---|---|---|
| Initial weight | Optimized weight | |||||
| 1A62A | 1–44 | 49–125 | 4 | 45–48 | 4 (1.2) | 2 (1.2) |
| 1A6QA | 2–290 | 299–368 | 8 | 291–298 | 1 (0.8) | 1 (0.8) |
| 1A79C | 9–81 | 85–179 | 3 | 82–84 | 1 (1.1) | 1 (1.1) |
| 1A8DA | 1–243 | 266–452 | 22 | 244–265 | 1 (1.6) | 1 (1.6) |
| 1A8LA | 1–116 | 123–226 | 6 | 117–122 | 1 (0.8) | 1 (1.4) |
| 1AMMA† | 1–80 | 90–174 | 9 | 81–89 | 6 (0.9) | 3 (0.9) |
| 1AOAA | 12–244 | 261–375 | 16 | 245–260 | 2 (1.8) | 1 (1.8) |
| 1AVAA | 1–343 | 352–403 | 8 | 344–351 | 1 (1.6) | 1 (1.6) |
| 1B63A | 2–215 | 218–331 | 2 | 216–217 | 1 (1.2) | 1 (1.3) |
| 1BAGA† | 1–343 | 361–425 | 19 | 342–360 | 1 (1.3) | 1 (1.1) |
| 1BG6A | 4–185 | 189–359 | 3 | 186–188 | 1 (1.7) | 1 (1.3) |
| 1BI5A | 1–232 | 237–389 | 4 | 233–236 | 1 (1.4) | 1 (1.4) |
| 1BKBA | 4–73 | 77–139 | 3 | 74–76 | 52 (7.7) | 10 (4.1) |
| 1BU6O | 3–252 | 260–499 | 7 | 253–259 | 1 (1.6) | 1 (1.6) |
| 1C2AA | 4–56 | 72–123 | 15 | 57–71 | 8965 (8.6) | 7455 (8.6) |
| 1CA1A† | 1–245 | 259–370 | 13 | 246–258 | 3 (1.2) | 2 (1.2) |
| 1CDYA | 1–96 | 99–178 | 2 | 97–98 | 4 (1.8) | 5 (1.8) |
| 1CJXB | 4–143 | 156–356 | 12 | 144–155 | 1 (1.6) | 1 (1.6) |
| 1CLCA† | 35–130 | 132–575 | 11 | 129–131 | 1 (1.8) | 1 (1.7) |
| 1CLIB | 1021–1168 | 1180–1345 | 11 | 1169–1179 | 1 (1.5) | 1 (1.5) |
| 1CRZA | 7–136 | 145–409 | 8 | 137–144 | 1183 (9.9) | 806 (9.9) |
| 1CTUA | 1–148 | 157–294 | 8 | 149–156 | 1 (2.4) | 1 (1.7) |
| 1CVRA | 1–339 | 356–432 | 16 | 340–355 | 1 (5.9) | 1 (5.9) |
| 1CX4A | 130–264 | 267–412 | 2 | 265–266 | 1 (1.8) | 1 (1.8) |
| 1D09B | 1–95 | 102–153 | 6 | 96–101 | 56 (4.3) | 173 (4.3) |
| 1D5RA | 14–184 | 192–351 | 7 | 185–191 | 1 (4.3) | 1 (4.3) |
| 1DZFA | 5–140 | 152–215 | 11 | 141–151 | 717 (7.9) | 520 (7.8) |
| 1EGAB | 4–171 | 190–296 | 18 | 172–189 | 78 (1.9) | 21 (1.9) |
| 1EOVA | 71–198 | 208–557 | 9 | 199–207 | 281 (9.7) | 440 (9.7) |
| 1EUDA | 1–128 | 134–306 | 5 | 129–133 | 1 (1.6) | 1 (1.4) |
| 1F1ZA | 8–167 | 170–267 | 2 | 168–169 | 1 (1.6) | 1 (1.6) |
| 1F3AA | 1–77 | 85–221 | 7 | 78–84 | 1 (1.4) | 1 (1.4) |
| 1F5NA | 7–276 | 290–583 | 13 | 277–289 | 1 (1.3) | 1 (1.3) |
| 1FMTA | 1–200 | 209–314 | 8 | 201–208 | 1441 (8.4) | 422 (1.1) |
| 1FTSA | 201–280 | 294–495 | 13 | 281–293 | 1 (1.1) | 1 (1.1) |
| 1GCYA | 1–355 | 362–418 | 6 | 356–361 | 1 (1.6) | 1 (1.6) |
| 1GV1B | 1–141 | 144–299 | 2 | 142–143 | 1 (1.3) | 1 (1.3) |
| 1HANA | 2–118 | 140–289 | 21 | 119–139 | 1 (1.4) | 1 (1.4) |
| 1HYEA | 1–144 | 149–313 | 4 | 145–148 | 1 (1.7) | 1 (1.7) |
| 1I8DB† | 1–86 | 105–206 | 18 | 87–104 | 891 (0.9) | 2189 (0.9) |
| 1J8MF | 3–83 | 99–297 | 15 | 84–98 | 1 (1.4) | 1 (1.4) |
| 1JAKA | 8–147 | 153–506 | 5 | 148–152 | 1 (1.7) | 1 (1.7) |
| 1JGTB | 2–206 | 219–508 | 12 | 207–218 | 1 (1.3) | 1 (1.3) |
| 1JPMA | 1–122 | 129–359 | 6 | 123–128 | 1 (1.3) | 1 (2.1) |
| 1JPNA | 1–85 | 99–296 | 13 | 86–98 | 1 (1.4) | 1 (1.4) |
| 1K0MA | 6–87 | 101–240 | 13 | 88–100 | 1 (1.4) | 1 (1.3) |
| 1KBWC | 13–155 | 165–314 | 9 | 156–164 | 1 (1.6) | 1 (1.6) |
| 1KNYA | 1–124 | 128–253 | 3 | 125–127 | 3 (1.8) | 8 (1.8) |
| 1KS9A | 1–165 | 169–291 | 3 | 166–168 | 1 (1.0) | 1 (1.3) |
| 1LAMA | 1–158 | 161–484 | 2 | 159–160 | 1 (1.2) | 1 (1.2) |
| 1LBUA | 1–76 | 88–213 | 11 | 77–87 | 1 (1.5) | 1 (1.5) |
| 1MGTA | 1–83 | 91–169 | 7 | 84–90 | 1 (1.5) | 1 (1.5) |
| 1NKRA | 6–99 | 109–200 | 9 | 100–108 | 398 (7.7) | 388 (9.0) |
| 1PGSA† | 4–135 | 150–314 | 14 | 136–149 | 1 (1.3) | 1 (1.3) |
| 1PIIA | 1–253 | 258–452 | 4 | 254–257 | 1 (1.9) | 1 (1.9) |
| 1QCSA | 0–83 | 87–201 | 3 | 84–86 | 3 (2.5) | 1 (8.1) |
| 1QFJC | 1–92 | 105–232 | 12 | 93–104 | 1 (1.0) | 1 (1.0) |
| 1QH4B | 2–99 | 112–381 | 12 | 100–111 | 1 (1.2) | 1 (1.2) |
| 1RHSA | 1–135 | 158–293 | 22 | 136–157 | 1 (1.4) | 1 (1.2) |
| 1SMDA | 1–401 | 406–496 | 4 | 402–405 | 1 (1.1) | 1 (1.1) |
| 1TF4B | 1–444 | 463–605 | 18 | 445–462 | 1880 (1.7) | 60 (2.6) |
| 2REBA | 3–266 | 270–328 | 3 | 267–269 | 1 (1.3) | 1 (1.3) |
a The first 4-letters are PDB ID and the 5th letter is chain ID. † It is also included in Cheng dataset (Table 2).
Table 2.
Details and prediction result of Cheng dataset (test set, 55 proteins) (values in parentheses are RMSD (Å))
| PDB IDa | Domain 1 | Domain 2 | Linker length | Linker region | Best rank of acceptable docking pose by PINE |
|---|---|---|---|---|---|
| 1A8PA | 2–92 | 109–258 | 16 | 93–108 | 1 (1.1) |
| 1AH5A | 3–216 | 226–313 | 9 | 217–225 | 1 (1.5) |
| 1AMMA† | 1–80 | 90–174 | 9 | 81–89 | 3 (0.9) |
| 1AORB | 1–204 | 218–605 | 13 | 205–217 | 1 (1.1) |
| 1AQHA | 1–348 | 362–448 | 13 | 349–361 | 1 (1.1) |
| 1AR4A | 1–80 | 94–201 | 13 | 81–93 | 1 (1.1) |
| 1AW7A | 1–89 | 98–194 | 8 | 90–97 | 1 (1.4) |
| 1AW9A | 2–76 | 95–217 | 18 | 77–94 | 1 (2.0) |
| 1B06A | 3–89 | 103–210 | 13 | 90–102 | 1 (1.7) |
| 1B25A | 1–202 | 216–619 | 13 | 203–215 | 1 (1.6) |
| 1B8PA | 3–156 | 161–329 | 4 | 157–160 | 1 (1.3) |
| 1BAGA† | 1–341 | 361–425 | 19 | 342–360 | 1 (1.1) |
| 1BAYA | 1–73 | 85–209 | 11 | 74–84 | 1 (0.8) |
| 1BIKA | 25–76 | 95–134 | 18 | 77–94 | 1 (1.2) |
| 1CA1A† | 1–245 | 259–370 | 13 | 246–258 | 2 (1.2) |
| 1CHMB | 2–155 | 166–402 | 10 | 156–165 | 5 (1.8) |
| 1CLCA† | 35–128 | 140–575 | 11 | 129–139 | 1 (1.7) |
| 1CLVA | 1–373 | 386–471 | 12 | 374–385 | 1 (1.4) |
| 1CR5B | 23–101 | 121–207 | 19 | 102–120 | 1 (0.9) |
| 1DLUB | 4–260 | 274–392 | 13 | 261–273 | 1 (1.5) |
| 1E5MA | 6–249 | 260–416 | 10 | 250–259 | 1 (1.1) |
| 1E9IB | 1–132 | 147–430 | 14 | 133–146 | 1 (1.4) |
| 1EBGA | 1–132 | 143–436 | 11 | 133–143 | 1 (1.4) |
| 1EE0A | 20–230 | 244–395 | 13 | 231–243 | 1 (1.6) |
| 1ET6B | 4–93 | 105–209 | 11 | 94–104 | 1 (1.7) |
| 1ET9A | 1–92 | 101–204 | 8 | 93–100 | 1 (1.5) |
| 1ETPB | 1–86 | 102–190 | 15 | 87–101 | 224 (1.1) |
| 1F2EA | 1–74 | 90–201 | 15 | 75–89 | 1 (1.0) |
| 1FDRA | 2–91 | 108–248 | 16 | 92–107 | 1 (1.6) |
| 1FFHA | 2–85 | 99–295 | 13 | 86–98 | 1 (0.8) |
| 1FFUF | 1–173 | 181–287 | 7 | 174–180 | 1 (0.9) |
| 1FIQB | 224–410 | 419–528 | 8 | 411–418 | 1 (1.7) |
| 1GK8C | 7–145 | 155–475 | 9 | 146–154 | 1 (1.7) |
| 1GSQA | 1–71 | 83–202 | 11 | 72–82 | 1 (1.6) |
| 1H1OB | 213–285 | 302–383 | 16 | 286–301 | 2 (0.9) |
| 1I8DB† | 1–86 | 105–206 | 18 | 87–104 | 2189 (0.9) |
| 1IK6A | 1–180 | 201–326 | 20 | 181–200 | 16 (1.0) |
| 1J3NA | 1–240 | 254–408 | 13 | 241–253 | 1 (1.0) |
| 1JLVA | 1–73 | 89–207 | 15 | 74–88 | 1 (1.0) |
| 1KGZA | 12–75 | 87–344 | 11 | 76–86 | 1 (1.2) |
| 1KZLA | 1–86 | 105–202 | 18 | 87–104 | 213 (1.1) |
| 1MB8A | 56–171 | 188–293 | 16 | 172–187 | 1 (3.9) |
| 1N5WF | 1–173 | 183–286 | 9 | 174–182 | 1 (1.7) |
| 1OE7A | 4–79 | 98–207 | 18 | 80–97 | 1 (0.8) |
| 1OI7A | 1–113 | 139–288 | 25 | 114–138 | 1 (9.5) |
| 1P5UA | 9–143 | 155–234 | 11 | 144–154 | 211 (2.1) |
| 1PBJA | 2–56 | 74–121 | 17 | 57–73 | 1 (1.1) |
| 1PDYA | 1–132 | 143–433 | 10 | 133–142 | 1 (1.3) |
| 1PGSA† | 4–145 | 150–314 | 14 | 136–149 | 1 (1.3) |
| 1PMTA | 1–74 | 8–8201 | 14 | 75–88 | 1 (1.7) |
| 1QNNA | 1–79 | 91–191 | 11 | 80–90 | 1 (1.4) |
| 1R5AA | 2–75 | 91–215 | 15 | 76–90 | 1 (1.7) |
| 1R6LA | 1–143 | 160–239 | 16 | 144–159 | 1 (1.4) |
| 1RWZA | 1–109 | 129–244 | 19 | 110–128 | 1 (1.1) |
| 1V8GA | 1–61 | 73–329 | 11 | 62–72 | 1 (1.4) |
a The first 4-letters are PDB ID and the 5th letter is chain ID. † It is also included in Wollacott dataset (Table 1).
Parameter optimization
The score function weights wzrank, wete, wppi, and wdock were optimized using the training dataset. Each weight was assigned an integer value of 1–10. First, the structural model was reranked using a score function with all weights set to 1. As a result, 51 of 62 proteins in the training dataset had at least one structure model whose RMSD was less than 10 Å, which is considered as an acceptable model, within the top 10 poses. From this initial state, we changed each weight by 1 and searched for the weight (optimized weight) that maximized the number of proteins with at least one acceptable model within the top 10 poses.
Evaluation performance
RMSD was used to evaluate the generated structural model, which was defined as follows:
where N is the number of atoms and di is the distance between atoms with the same residue number for the structural model and native structure. Additionally, when calculating the RMSD of the structural model, we only considered Cα atoms between the native structure of the domain with fewer residues and structural model among the two domains of the structural model. We fitted the domain with a large number of residues (receptor) and calculated the RMSD of the domain with a small number of residues (ligand).
Evaluation of the score function was performed using a test dataset. When 10,800 structural models were reranked using a score function for one multidomain protein, the prediction was defined as a ‘success’ if at least one ‘acceptable’ (RMSD <10 Å) model within the top 10 positions was obtained. For the 55 proteins in the test dataset, we determined the score function as the number of successful predictions.
Results and Discussion
Optimized parameters for score function
We optimized the four weights wzrank, wete, wppi, and wdock of the score function using the training set for optimization (62 proteins). In the initial state in which all weights were 1, prediction was successful for 51 of 62 proteins. After optimizing the weights, the optimized weights were wzrank=9, wete=2, wppi=1, and wdock=4. When the training dataset was predicted using the score function with this combination of weights, prediction was successful for 52 proteins. The protein (PDB ID: 1BKB) that was newly successfully predicted by weight optimization, had a structural model with a smaller RMSD reranked higher than with the initial state.
Score function evaluation
After reranking the test dataset (55 proteins) using the optimized weights shown in the previous section, the proposed method obtained ‘acceptable’ predictions (where the top 10 models include at least one model with RMSD <10 Å) for 50 multidomain proteins among the 55 tested. On the other hand, the baseline method (only using Szrank and Sete) in which there was no template and the interaction surface obtained acceptable predictions for 47 proteins among 55 (while the original DINE successfully predicted 46 proteins [16]). Thus the proposed method, PINE, successfully identified 3 more proteins than the baseline method. In addition, PINE obtained 49 ‘good’ predictions (where the top 10 models include at least one model with RMSD <5 Å) among 55 proteins, whereas DINE obtained 39 among 55 [16].
Residue pair
When reranking was performed using only interaction amino acid residue scores, only one of the 55 proteins showed an acceptable model within the top 10 positions. The successfully predicted protein (PDB ID: 1BAG) reranked a structural model with an RMSD of 4.3 Å at rank 9.
Docking score
When the structural model was reranked by docking score only, the prediction was successful for 48 of 55 proteins. The score can predict more proteins than other terms when it was calculated using only one term.
Contribution of score term
To investigate the contribution of each term (Szrank, Sete, Sppi, Sdock) to the score function, the prediction was performed using a score function in which the combination of terms used was changed (Table 3). The weight of the score function was also optimized in the same manner. The test dataset was reranked using this score function, and Figure 3 shows the distribution of acceptable models.
Table 3.
Differences in success rate by combination of scores
| Terms of score function | Rank 10 | Rank 20 | Rank 500 | |||
|---|---|---|---|---|---|---|
| Szrank | 0.82 | 0.85 | 0.95 | |||
| Sete | 0.75 | 0.87 | 0.93 | |||
| Scont | 0.02 | 0.05 | 0.51 | |||
| Sdock | 0.87 | 0.91 | 0.96 | |||
| Szrank | +Sete | 0.85 | 0.85 | 0.95 | ||
| Szrank | +Scont | 0.84 | 0.84 | 0.93 | ||
| Szrank | +Sdock | 0.85 | 0.89 | 0.95 | ||
| Sete | +Scont | 0.09 | 0.13 | 0.67 | ||
| Sete | +Sdock | 0.87 | 0.91 | 0.96 | ||
| Scont | +Sdock | 0.84 | 0.85 | 0.95 | ||
| Szrank | +Sete | +Scont | 0.85 | 0.87 | 0.95 | |
| Szrank | +Sete | +Sdock | 0.91 | 0.91 | 0.98 | |
| Szrank | +Scont | +Sdock | 0.85 | 0.89 | 0.95 | |
| Sete | +Scont | +Sdock | 0.87 | 0.91 | 0.96 | |
| Szrank | +Sete | +Scont | +Sdock | 0.91 | 0.93 | 0.98 |
Figure 3.

Total number of acceptable structures in each score function. The score function using the binding energy score reranks many acceptable structures to higher ranks. Moreover, although the interaction amino acid residue score could predict only one protein in the test dataset, the score function containing this term reranks many acceptable structures to higher ranks. Particularly, comparing the proposed method with the score function excluding interaction amino acid residue score from the proposed method, the prediction accuracy was the same, whereas the number of acceptable structures within the top 500 predicted by the former method was larger than that predicted by the latter method.
Binding energy score
The weight of each score term in the proposed method was optimized using a training dataset. Among these weights, a largest value was assigned to the binding energy score, wzrank=9, which was more than 2-fold higher than the other weights. Additionally, when the score function containing Szrank was compared to that not containing this value, a higher success rate was achieved. Therefore, the binding energy score was a major contributor to this score function. However, 5 of 10 proteins that could not be predicted using only the binding energy score were predicted using the proposed method.
Docking score
Prediction using only docking score was successful for 48 of 55 proteins. This was the highest prediction accuracy among those using a single score term. In addition, the weight of Sdock and wdock had the second highest value (=4) after wzrank. Therefore, docking scores were considered to contribute significantly to the accuracy of the proposed method. Among the 10 proteins that failed to be predicted by the binding energy score alone, 5 proteins were successfully predicted by the proposed method. Because these proteins were reranked to the top by prediction based on the docking score, prediction using the proposed method was successful. In fact, the proposed method showed a higher success rate than the score function using the three terms other than Sdock.
Inter-domain distance score
Prediction accuracy using only the term based on the inter-domain distance Sete was not high (success rate of 75%), and prediction using a single score was difficult. However, the score function with Szrank, Sdock, and Sete using two terms with high prediction accuracy in a single case and inter-domain distance score showed higher prediction accuracy than that using only Szrank and Sdock. This may be because the distance between the domains was limited.
Interaction amino acid residue score
The value of wppi was the smallest among the four weights. In addition, the number of proteins successfully predicted with only this term was one of the 55 proteins, showing the lowest prediction accuracy. As shown in Figure 4, there was variation from the predicted protein structure (PDB ID: 1BAG), which was the only successful prediction using only Sppi. The only model with an RMSD of less than 10 Å was the 9th model, and the models ranked 1st to 3rd were not similar to the native structure. However, the proposed method using the interaction amino acid residue score predicts more acceptable structures at higher ranks (Fig. 3). This indicates that the type of residue pair interacting in the protein complex also affects interactions between domains.
Figure 4.
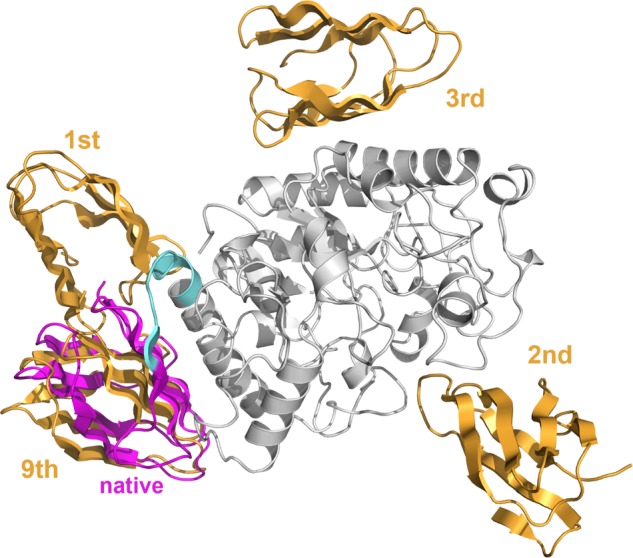
PDB ID: 1BAG’s receptor domain (gray), 1BAG’s native ligand domain (magenta), four ligand domain models (orange), and the linker region (cyan). This is the only protein in which the permissive structure exists within the top 10 in the prediction based on the interaction amino acid residue score alone. However, the top three structures are far from the native position, and their positions are disjointed. The acceptable model predicted to be 9th. Even for other proteins, predictions with only interaction amino acid residue scores may dislocate the position of the structural model to prevent concentration near the native structure.
Score function evaluation
Table 4 shows the proteins for which prediction failed. These five cases, also failed when the prediction was performed based on binding energy. This indicates that prediction using the proposed method fails if the wzrank value is large and prediction accuracy is poor. Additionally, the interaction surface of the native structure of the proteins for which prediction failed were smaller than 1,400 Å2. In these cases, because of the weak interaction, the results of protein docking became worse [30], and thus prediction based on binding energy and the generation accuracy of the structural model by docking showed worse results. Therefore, this prediction failed.
Table 4.
Best rank of acceptable pose for which prediction failed (values in parentheses are RMSD (Å))
| PDB ID | The best rank of acceptable structure | Interface area | ||
|---|---|---|---|---|
| only Szrank | only Sdock | PINE | ||
| 1ETPB | 1,243 (1.2) | 36 (1.1) | 224 (1.1) | 784 Å2 |
| 1I8DB | 1,296 (2.4) | 891 (0.9) | 2,189 (0.9) | 821 Å2 |
| 1IK6A | 106 (3.1) | 14 (1.0) | 16 (1.0) | 1,090 Å2 |
| 1KZLA | 1,728 (2.9) | 12 (1.2) | 213 (1.1) | 979 Å2 |
| 1P5UA | 472 (2.1) | 3,255 (2.1) | 211 (2.1) | 585 Å2 |
Factors affecting prediction accuracy
Figure 5 shows an example of successful prediction in this study. This protein (PDB ID: 1AOR) was also reranked as 1st in the docking score and binding energy score by ZRANK. Of the generated structural models, the centroids of the top 100 ligand models ranked by docking scores were plotted. The results indicated that the position of the ligand domain was correctly predicted. In this case, the structure model ranked 1st according the docking score was also predicted based on the binding energy score and the proposed method. Moreover, the interaction surface was predicted accurately, and the high-rank structure models were gathered near the native structure. Another case visualizes how PINE works successfully. The protein in Figure 6 (PDB ID: 1BKB) suggests that the ligand centroids of the top 100 models by PINE were closer to the native position than the ligand centroids of the top 100 from the initial docking.
Figure 5.
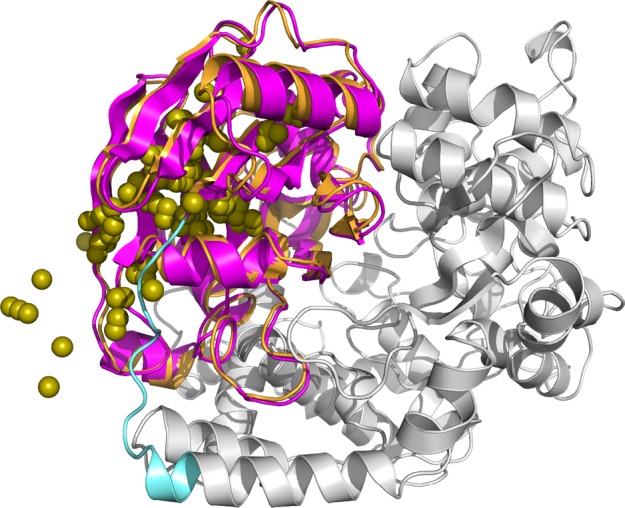
Native protein structure of PDB ID: 1AOR and center of gravity for top 100 ligands in docking score. Gray indicates the receptor domain of 1AOR, magenta indicates the native ligand domain, orange indicates top 1 ligand structure model predicted by PINE, cyan indicates the linker region, and yellow spheres indicate the center of gravity of the top 100 ligand models generated by initial docking.
Figure 6.
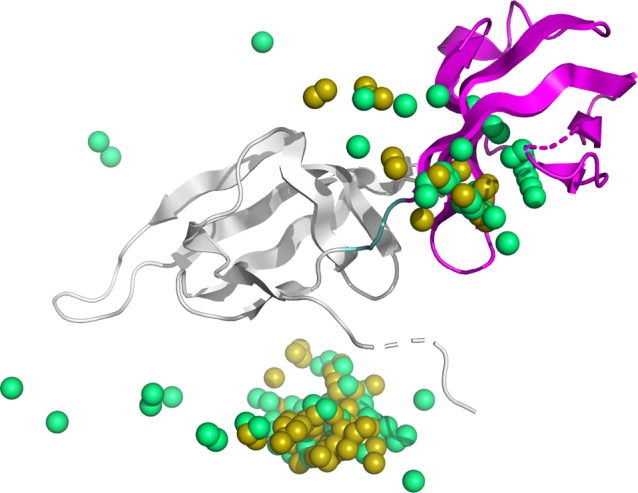
Native protein structure of 1BKB and center of gravity for top 100 ligands in docking score and PINE. Gray indicates the receptor domain of 1P5U, magenta indicates the ligand domain, cyan indicates the linker region, yellow sphere indicates the center of gravity of the ligand in the structural model generated by initial docking, and green sphere indicates the center of gravity of the ligand in the structural model generated by PINE.
However, the structural model of the ligand domain for the failed protein (PDB ID: 1P5U) was concentrated at a position that differed from that of the native structure (Fig. 7). This indicates that the model generation accuracy by MEGADOCK was low for this protein. Moreover, the prediction was poor even when reranking by binding energy was performed; as a result, prediction failed when using the proposed method. As described in the previous section, protein 1P5U may have failed the prediction because of its small interaction surface. The interaction surface area of the successfully predicted protein 1AOR was 3,201 Å2, whereas that of the failed protein 1P5U was 585 Å2. This indicates that 1P5U domains interact on a small interface compared to ordinary PPI interface. The domain-domain interaction form was constrained by its linker, leading to distance limitation, and was different from PPI with freely forming interaction. Hence, domains in a multidomain protein can interact with each other not only in a broad interface but also on a narrow interface. In such a case, accurate prediction using free form docking may be difficult. Figure 7 shows that structural models may be generated on the wrong surface. As observed for other failed proteins, the center of gravity of the top 100 structural models of the docking score was not concentrated at the position of the ligand domain in the native structure. In contrast, in most proteins which were predicted at the 1st rank, the center of gravity was concentrated at the position in the native structure. We expect that it is possible to generate a highly accurate structural model by modifying it so that the interaction surface can be limited when generating the model.
Figure 7.
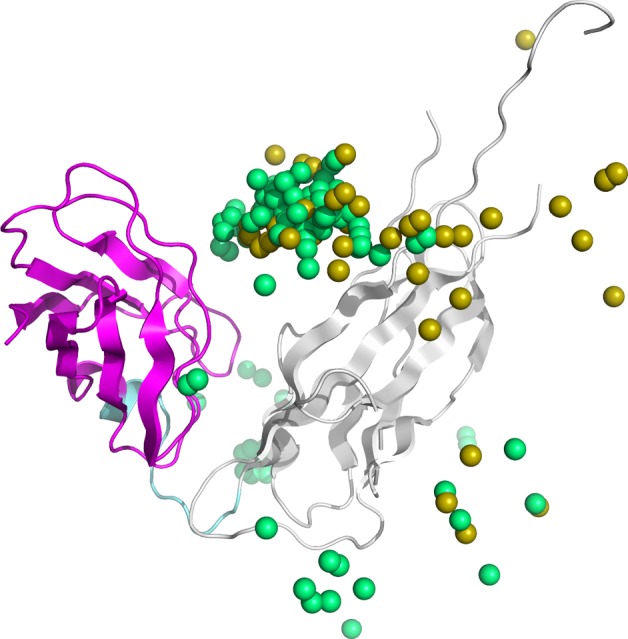
Native protein structure of 1P5U and center of gravity for top 100 ligands in docking score and PINE. Gray indicates the receptor domain of 1P5U, magenta indicates the ligand domain, cyan indicates the linker region, yellow sphere indicates the center of gravity of the ligand in the structural model generated by initial docking, and green sphere indicates the center of gravity of the ligand in the structural model generated by PINE.
Further validation for domain-domain linking
Unlike PPI, the interaction between domains of multidomain proteins is limited in distance by linking. Thus, it is not always the case that the domain binding is coupled with an interface which obtained optimal binding free energy. Therefore, we verified whether the model selection could be improved by changing the domain-domain linking score, which limits the distance between domains. Specifically, the Sete formula was modified as follows to give a penalty depending on the distance between domains.
As a result of this improvement, the protein (PDB ID: 1QCS), as an example, had the number of correct models within the top 100 increased from 12 by original PINE to 22 by modified PINE. However, the S'ete did not improve overall performance. If the linker length is short, scoring with Sete will work, but it may not work well if the linker length is long. As shown in Figures 6 and 7, some models were not properly located within reach of the linker. As a result, it can be considered that it is difficult to score appropriately only by statistical information of linker length and interdomain distance. Development of a more refined score function depending on the secondary structure of the linker and the vector direction at the domain terminal is needed. In addition, since a turn that suddenly changes direction at the terminal of the linker is unlikely to occur intrinsically, it is necessary to consider other methods such as removing a structural model that takes such a difficult conformation.
Conclusion
Most proteins have multiple domains. Because these domains are folded and stable, various methods can be used to predict the whole protein. The rigid-body docking method uses the 3D structure of the domains and does not require a template for predicting the whole structure of a multidomain protein; this method can also predict whole structures from partial structures. In this study, we improved the method for predicting multidomain protein structures with two domains through computation. Existing methods for predicting multidomain protein structures by rigid-body docking using protein domains require template proteins with homology to the query proteins, particularly in the interface area.
In this study, we proposed a method for predicting multidomain protein structure using an interaction amino acid residue score without requiring a template protein structure. This score estimates domain-domain interactions based on protein-protein interactions. The interaction and docking scores calculated by docking analyses were used as the score function rather than the interface prediction of the existing method. As a result, the prediction was possible even when no template protein structure was available; by using the interaction amino acid residue score, more acceptable structures could be reranked to higher values. Using the interaction amino acid residue score alone made reranking into the top 10 difficult, but the score contributed to improving the reranking. This suggests that the prediction of the interaction surface between domains is based on the PPI. Additionally, in cases where the prediction accuracy of docking and energy scoring is poor, the interaction surfaces tend to be small. In such a case, a score that can predict interaction surfaces between domains is important. For the structural prediction of multidomain proteins that are difficult to predict, designing interaction amino acid residue scores corresponding to cases with small interaction surfaces should be further examined. A previous study [16] gave a uniform score for the structural model that fits the linker length condition based on the inter-domain distance. Studies are needed to improve this approach, such as by gradually changing this score or giving a higher score to a structural model closer to the native structure.
Acknowledgments
We would like to thank Dr. Masafumi Shionyu for providing the detailed multidomain protein dataset information and for insightful discussion. This work was partially supported by JSPS KAKENHI (Grant Nos. 17H01814, 18K18149), JST Research Complex Program “Wellbeing Research Campus: Creating new values through technological and social innovation,” MEXT Program for Building Regional Innovation Ecosystems “Program to Industrialize an Innovative Middle Molecule Drug Discovery Flow through Fusion of Computational Drug Design and Chemical Synthesis Technology,” and AMED Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) (Grant No. JP18am0101112). We would like to thank Editage (www.editage.jp) for English language editing.
Conflicts of Interest
SM, MO and YA declare that they have no conflict of interest.
Authors’ Contributions
SM, MO, YA: conceived and designed the experiments; SM: performed the experiments; SM, MO, YA: analyzed the data; SM, MO, YA: wrote the paper. All the authors approved the final version of the manuscript.
References
- [1].Apic G., Gough J. & Teichmann S. A.. Domain Combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mol. Biol. 310, 311‒325 (2011). [DOI] [PubMed] [Google Scholar]
- [2].Ekman D., Björklund A. K., Frey-Skött J. & Elofsson A.. Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions. J. Mol. Biol. 348, 231‒243 (2005). [DOI] [PubMed] [Google Scholar]
- [3].Marchler-Bauer A., Panchenko A. R., Shoemaker B. A., Thiessen P. A., Geer L. Y. & Bryant S. H.. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 30, 281‒283 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vogel C., Bashton M., Kerrison N. D., Chothia C. & Teichmam S. A.. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 14, 208–216 (2004). [DOI] [PubMed] [Google Scholar]
- [5].Dyson H. J. & Wright P. E.. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005). [DOI] [PubMed] [Google Scholar]
- [6].Gruszka D. T., Mendonca C. A., Paci E., Whelan F., Hawkhead J., Potts J. R., et al. . Disorder drives cooperative folding in a multidomain protein. Proc. Natl. Acad. Sci. USA 113, 11841–11846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., et al. . The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wollacott A. M., Zanghellini A., Murphy P. & Baker D.. Prediction of structures of multidomain proteins from structures of the individual domains. Protein Sci. 16, 165–175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hardin C., Pogorelov T. V. & Luthey-Schulten Z.. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 12, 176–181 (2002). [DOI] [PubMed] [Google Scholar]
- [10].Lee, J., Freddolino, P. L. & Zhang, Y. Ab initio protein structure prediction. in From Protein Structure to Function with Bioinformatics (Rigden, J. D. ed.) pp. 3–35 (Springer, Dordrecht, 2017). [Google Scholar]
- [11].Xu D., Jaroszewski L., Li Z. & Godzik A.. AIDA: ab initio domain assembly for automated multidomain protein structure prediction and domain-domain interaction prediction. Bioinformatics 31, 2098–2105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Roy A., Kucukural A. & Zhang Y.. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., et al. . Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cheng T. M. K., Blundell T. L. & Fernandez-Recio J.. Structural assembly of two-domain proteins by rigid-body docking. BMC Bioinformatics 9, 441 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Almsned, F., Gogovi, G., Bracci, N., Kehn-Hall, K., Blaisten-Barojas, E. & Shehu, A. Modeling the tertiary structure of a multi-domain protein: structure prediction of multi-domain proteins. Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, 615–620 (2018). [Google Scholar]
- [16].Hirako S. & Shionyu M.. DINE: a novel score function for modeling multidomain protein structures with domain linker and interface restraints. IPSJ Trans. Bioinformatics 5, 18–26 (2012). [Google Scholar]
- [17].Kundrotas P. J., Zhu Z., Janin J. & Vakser I. A.. Templates are available to model nearly all complexes of structurally characterized proteins. Proc. Natl. Acad. Sci. USA 109, 9438–9441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Murzin A. G., Brenner S. E., Hubbard T. & Chothia C.. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 (1995). [DOI] [PubMed] [Google Scholar]
- [19].Kabsch W. & Sander C.. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- [20].Chen R., Li L. & Weng Z.. ZDOCK: An initial-stage protein-docking algorithm. Proteins 52, 80–87 (2003). [DOI] [PubMed] [Google Scholar]
- [21].Prierce B. & Weng Z.. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins 67, 1078–1086 (2007). [DOI] [PubMed] [Google Scholar]
- [22].Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S. & Karplus M.. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 (1983). [Google Scholar]
- [23].Zhang C., Vasmatzis G., Cornette J. L. & DeLisi C.. Determination of atomic desolvation energies from the structures of crystallized proteins. J. Mol. Biol. 267, 707–726 (1997). [DOI] [PubMed] [Google Scholar]
- [24].Anfinsen C. B. Principles that govern the folding of protein chains. Science 181, 223–230 (1973). [DOI] [PubMed] [Google Scholar]
- [25].Aloy P., Ceulemans H., Stark A. & Russell R. B.. The relationship between sequence and interaction divergence in proteins. J. Mol. Biol. 332, 989–998 (2003). [DOI] [PubMed] [Google Scholar]
- [26].Korkin D., Davis F. P. & Sali A.. Localization of protein-binding sites within families of proteins. Protein Sci. 14, 2350–2360 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ohue M., Shimoda T., Suzuki S., Matsuzaki Y., Ishida T. & Akiyama Y.. MEGADOCK 4.0: An ultra-high-performance protein-protein docking software for heterogeneous supercomputers. Bioinformatics 30, 3281–3283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ohue M., Matsuzaki Y., Uchikoga N., Ishida T. & Akiyama Y.. MEGADOCK: An all-to-all protein-protein interaction prediction system using tertiary structure data. Protein Pept. Lett. 21, 766–778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Word J. M., Lovell S. C., Richardson J. S. & Richardson D. C.. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285, 1735–1747 (1999). [DOI] [PubMed] [Google Scholar]
- [30].Vajda S. Classification of protein complexes based on docking difficulty. Proteins 60, 176–180 (2005). [DOI] [PubMed] [Google Scholar]