

SUMMARY:
A fundamental feature of cellular plasma membranes (PM) is asymmetric lipid distribution between the bilayer leaflets. However, neither the detailed, comprehensive compositions of individual PM leaflets, nor how these contribute to structural membrane asymmetries have been defined. We report the distinct lipidomes and biophysical properties of both monolayers in living mammalian PMs. Phospholipid unsaturation is dramatically asymmetric, with the cytoplasmic leaflet being ~2-fold more unsaturated than the exoplasmic. Atomistic simulations and spectroscopy of leaflet-selective fluorescent probes reveal that the outer PM leaflet is more packed and less diffusive than the inner leaflet, with this biophysical asymmetry maintained in the endocytic system. The structural asymmetry of the PM is reflected in asymmetric structures of protein transmembrane domains (TMD). These structural asymmetries are conserved throughout Eukaryota, suggesting fundamental cellular design principles.
INTRODUCTION
The asymmetric distribution of lipids between the two leaflets of the plasma membrane (PM) bilayer is a prevalent and fundamental feature of living cells1. In mammals, compositional asymmetry is most widely studied for phosphatidylserine (PS), a negatively charged lipid found almost exclusively on the cytoplasmic (inner) leaflet of the PM. Other phospholipid headgroups are also asymmetrically distributed, with sphingolipids enriched in the exoplasmic leaflet and charged and amino-phospholipids more abundant on the cytoplasmic2,3. These distributions were originally established in red blood cells (RBCs)3 and platelets4 and later confirmed in PMs of various nucleated cells1,5. Asymmetric distributions were also proposed between the lumenal and cytoplasmic leaflets of organellar membranes6.
Because of their reliance on chromatographic methods, previous measurements of lipid asymmetries in biomembranes have been limited to the few major lipid types defined by their polar headgroups. However, advances in lipidomics reveal a vast diversity of lipid species in mammalian membranes, comprising hundreds of lipid species with distinct headgroups, acyl chains, and backbone linkages. How these individual lipid species are distributed across the two PM leaflets remains poorly understood. Similarly, one of the most important and challenging questions in membrane biology concerns the biophysical consequences of membrane asymmetry. While the connections between membrane composition and physical properties have been extensively addressed in symmetric bilayers, little is known about how these insights translate to compositionally asymmetric membranes. Recently, robust protocols for producing asymmetric synthetic membranes have been developed7–9 and used to characterize physical coupling between leaflets10,11, effects of lipid asymmetry on protein conformation7,12, and the influence of proteins on lipid distribution between leaflets13. However, the extent to which membrane properties are coupled across the bilayer in living cells remains unknown.
Disparities in biophysical properties between the two leaflets of cellular PMs have long been proposed14, but remain ambiguous due to experimental limitations and inconsistencies. Studies have often relied on RBCs (due to their lack of internal membranes) and external quenchers or headgroup-specific chemistries to probe individual leaflets. However, these methods have yielded contradictory conclusions, with some studies inferring more fluid inner PM leaflets15–18 and others more fluid outer leaflets19–21. Ultimately, the presence and nature of biophysical asymmetry in mammalian membranes remains unresolved because of limitations inherent to methodologies relying on biological lipid-based probes and fluorescence quenchers (see legend of Supplementary Fig 7). Finally, previous studies have focused largely on the PM, thus the biophysical asymmetry of intracellular organelles has been rarely studied and remains unknown.
Here, we investigated the lipidomes and biophysical properties of both leaflets in intact mammalian PMs. Using enzymatic digestion, the asymmetric distribution of ~400 lipid species is defined for human RBC PMs and compiled into a detailed model for the compositions of PM leaflets. While the observed headgroup distributions are largely consistent with previous reports, a striking asymmetry is observed for phospholipid acyl chains, with the cytoplasmic leaflet composed largely of highly unsaturated lipids. These observations suggested asymmetric properties in the two leaflets of the PM, an inference that was confirmed by atomistic simulations of complex lipid bilayers. These predictions were then directly examined using a novel approach that relies on selective staining of PM leaflets by an environment-sensitive fluorescent reporter. Obvious differences in lipid packing are observed between the outer and inner leaflets of live cell PMs, wherein a tightly packed exoplasmic leaflet apposes a more loosely packed cytoplasmic leaflet. These biophysical asymmetries persist in the membranes of intracellular endosomes. Finally, the asymmetric organization of mammalian PMs is reflected in asymmetric structures of protein TMDs, a property that appears to be conserved throughout Eukaryota.
RESULTS
Detailed lipidomic asymmetry of a mammalian PM
The asymmetric distribution of phospholipid headgroups in eukaryote PMs was described by classical studies on mammalian erythrocytes and platelets3,4 and confirmed in various eukaryotic cells1,5. However, the differences in phospholipid backbones and acyl chains between exo- and cytoplasmic leaflets of the PM have not been investigated. Combining phospholipase digestion with mass spectrometric lipidomics, we report the detailed, comprehensive lipid asymmetry of the RBC membrane. To this end, RBCs were treated with either phospholipases or sphingomyelinase (SMase) to specifically digest only the lipid species on the exoplasmic leaflet of the PM. These treatments were followed by quantitative mass spectrometry of ~400 unique phospholipid species. The species remaining after enzymatic digestion are presumed to be on the inner leaflet (protected from the enzyme), whereas the enzymatically digested lipids are inferred to comprise the outer leaflet (Fig 1A and Supplementary Figure 1). Combining several independent enzyme treatments reveals the comprehensive lipidomes of both leaflets. The measured asymmetric lipid headgroup distributions (Fig 1A) were consistent with previous estimates3. PLA2 treatment of intact cells minimally affected PE or PS, consistent with the presumed near-absolute inner leaflet confinement of these aminophospholipids3,4. PI, PA, and PEp were similarly unaffected, suggesting their inner leaflet residence. SM was largely degraded (~90%) by treatment of intact cells with SMase, confirming its concentration in the outer leaflet, while ~60% of the PC was on the outer leaflet.
Fig 1 -. Lipidomic asymmetry of erythrocyte PMs.
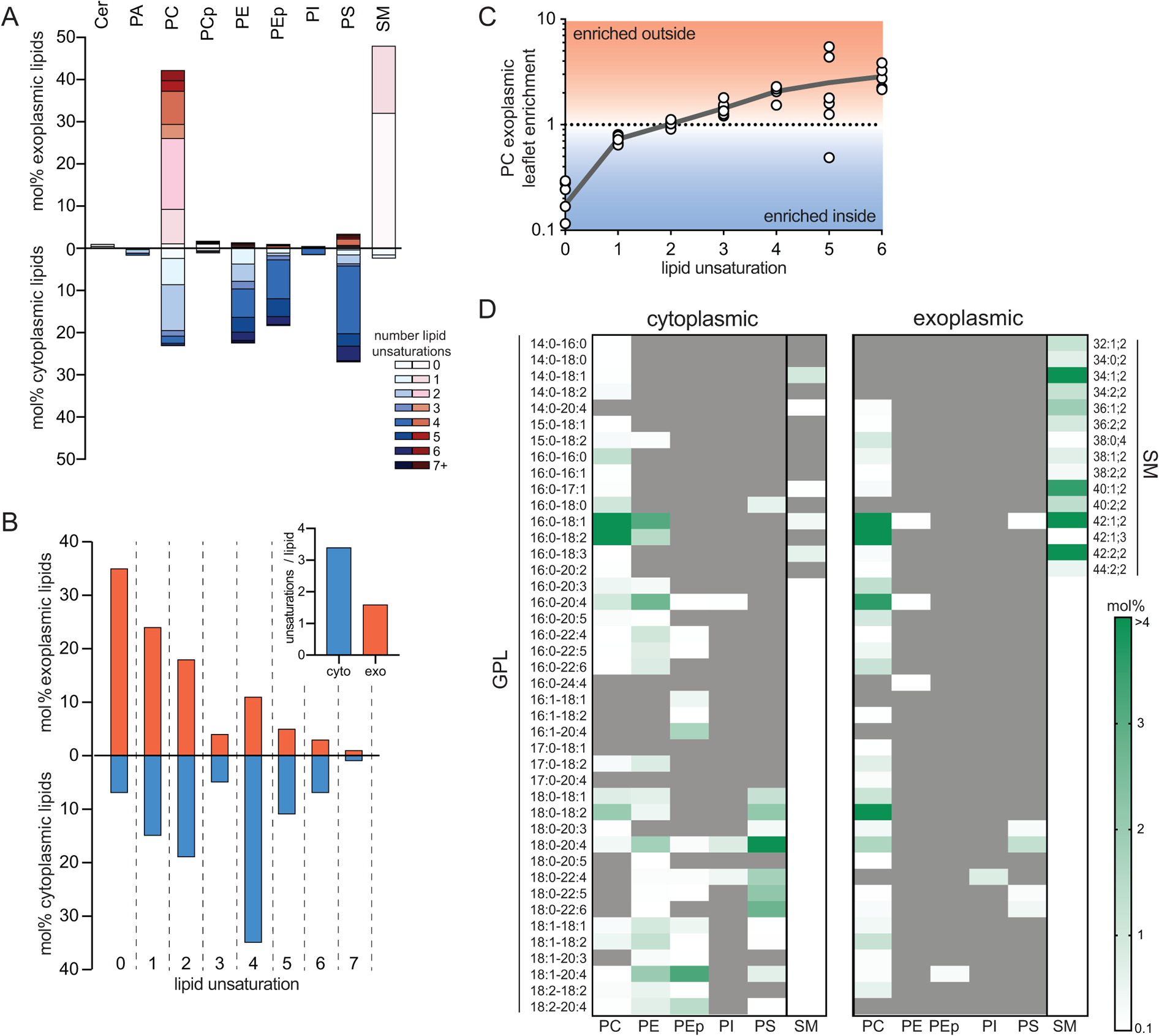
(A) Phospholipid compositions of exo- (red) and cytoplasmic (blue) PM leaflets as defined by enzymatic digestion and mass spectrometry. The exoplasmic leaflet is almost exclusively composed of PC and SM; the inner leaflet is approximately equimolar between PC, PE, PS, and PEp (plasmalogen). (B) Leaflet asymmetry of acyl chain unsaturation. The plurality of phospholipids in the outer leaflet are fully saturated, whereas the majority of the cytoplasmic leaflet is polyunsaturated. (inset) Abundance-weighted average unsaturation is ~2-fold greater for inner leaflet phospholipids. (C) Asymmetry of acyl chain saturation for PC species. Fully saturated acyl chains are highly enriched in the inner leaflet PC, and there is a general correlation between outer leaflet enrichment of PC species and the extent of unsaturation. Data in D are mean ± SD of 7 independent samples. Pearson correlation calculated to be r=0.997 for unsaturation compared to exoplasmic leaflet enrichment. (D) Lipidomic bar codes of the inner and outer PM leaflets. Shown in white-green scale are all lipid species (GPL = glycerophospholipid, SM = sphingomyelin) comprising <0.1 mol% of lipids with mol% encoded in green intensity (darkest = most abundant). Gray are species below the 0.1 mol% threshold (including not detected).
The quantitative accuracy of these measurements is supported by several key controls: (1) complete digestion of target lipids in lysed cells (Supplementary Fig 1); (2) lack of hemoglobin leakage in enzyme-treated cells (Supplementary Fig 2); and (3) decrease in target lipids (e.g. SM) that quantitatively matches stoichiometric appearance of reaction products (e.g. Cer; Supplementary Fig 1). The third point is crucial, as target and product lipids are quantified via different standards and often different detection modes (e.g. positive versus negative ion).
Analysis of lipid hydrophobic chains revealed a striking difference in acyl chain unsaturation between leaflets, with ~35% of exoplasmic leaflet phospholipids being saturated (not including the sphingoid backbone double bond, which is a trans double bond near the headgroup and does not disrupt lipid packing) and the majority having less than 2 unsaturations per lipid (Fig 1B). In contrast, the cytoplasmic leaflet was highly enriched in lipids with polyunsaturated acyl chains, with the majority of lipids containing 4 or more unsaturations. Overall, the average outer leaflet lipid bears 1.6 unsaturations in comparison to 3.4 on the inner leaflet (Fig 1B, inset). Most of the lipid headgroup classes were confined to one of the PM leaflets, precluding any within-class asymmetry. The only exception present in both leaflets was PC, which showed a surprisingly asymmetric distribution: fully saturated PC species (e.g. dipalmitoyl PC) were almost exclusively present on the cytosolic leaflet, whereas exoplasmic PC showed a preference for polyunsaturated species (Supplementary Fig 4). More generally, there was a clear correlation between the unsaturation of PC species and their relative enrichment in the outer leaflet (Fig 1C). We speculate that this unexpected asymmetry of PC species may arise from fatty acid remodeling, with the outer leaflet reflecting de novo synthesized species while inner leaflets are remodeled by cytoplasmic machineries, consistent with fatty acyl selectivity in these two pathways22.
The complete phospholipid compositions (‘lipidomic barcode’) of the two leaflets are shown in Fig 1D and summarized in Fig 2A&C, highlighting several notable features: (1) the PM lipidome is dominated by lipids with one saturated and one unsaturated acyl chain, with the exception of saturated SMs comprising ~35% of outer leaflet PLs; (2) polyunsaturated lipids comprise two-thirds of inner leaflet PLs; (3) ~33% of outer leaflet PLs are SMs bearing very-long chain (24:0 and 24:1) fatty acids; (4) lipids containing two unsaturated acyl chains are rare, comprising ~10% of the inner leaflet and essentially not present on the outer leaflet; (5) ether linked lipids (i.e. plasmalogens) comprise a sizeable fraction (~20%) of the inner leaflet; (6) headgroups are highly selective for lipid acyl chains, with polyunsaturated fatty acids concentrated in PE and PS and wholly excluded from SM.
Fig 2 – Atomistic simulation of biophysical asymmetry of erythrocytes PM.
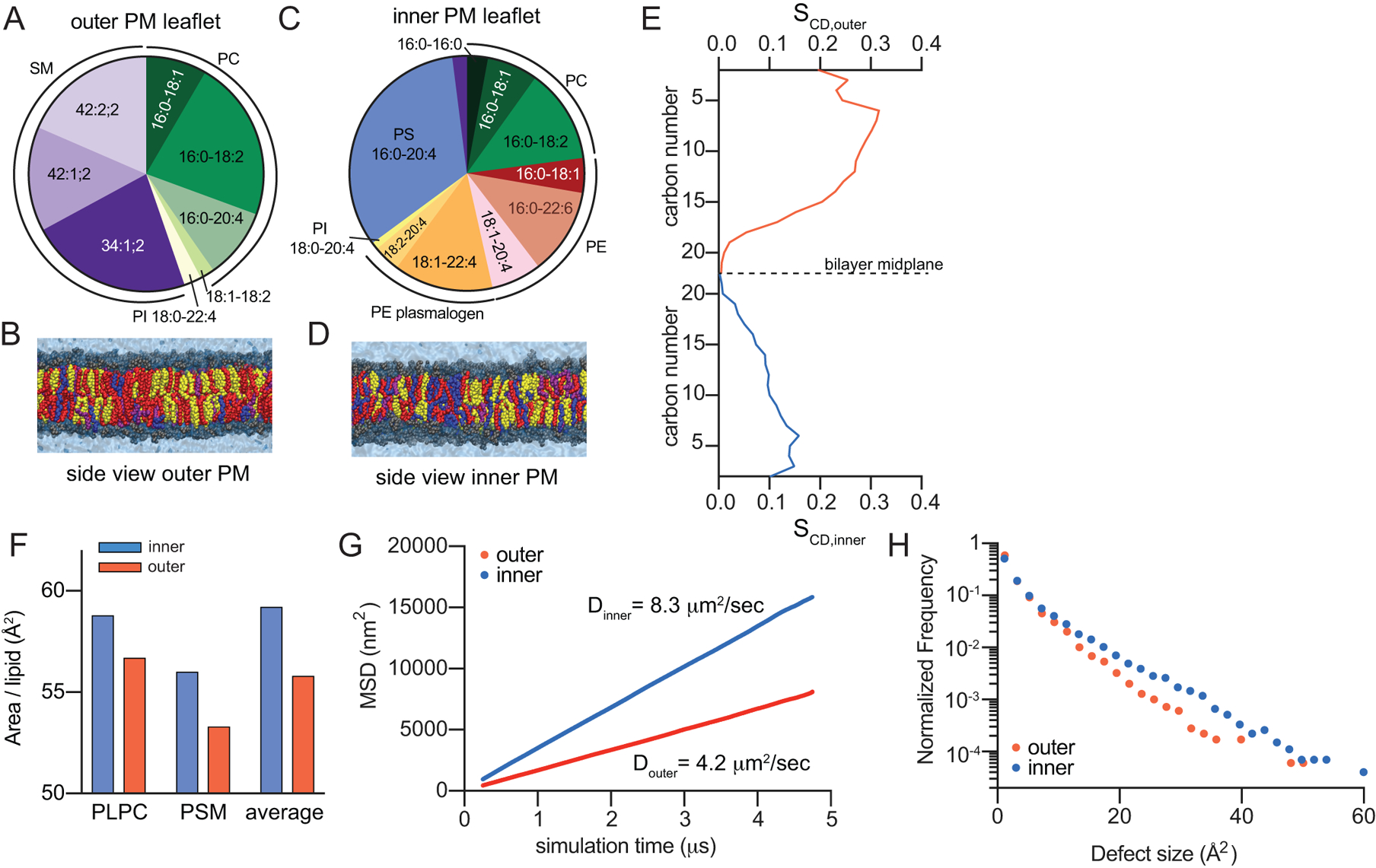
(A&C) Compiled compositions of outer (exoplasmic; A) and inner (cytoplasmic; C) PM leaflets from lipidomics. Below are final snapshots of outer-leaflet (B) and inner-PM leaflet (D) mimetic simulations (yellow = cholesterol, red = saturated acyl chains, purple = mono- and di-unsaturated, blue = polyunsaturated). (E) Concentration-weighted average order parameters for lipids in the outer versus inner leaflet simulation suggest a more ordered outer leaflet. (F) Slope of average MSD over time reveals ~2-fold slower diffusivity of the simulated outer leaflet. (G) Histogram of hydrophobic defects reveals more abundant large defects in the simulated inner leaflet. (H) Area/phospholipid for PLPC and PSM (the two lipids shared between both simulations) and the abundance-weighted average phospholipid.
Simulations reveal differences in leaflet properties
The striking differences in phospholipid composition between PM leaflets suggested the possibility of biophysical asymmetries across the PM. Differences in membrane properties associated with lipid headgroup charge and size have been extensively documented23–25, but lipid acyl chains are also major determinants of membrane structure and organization. Namely, membranes rich in saturated lipids are tightly packed, rigid, and ordered, whereas high unsaturation levels yield relatively fluid, loosely packed membranes, suggesting that the outer leaflet of mammalian PMs may be more tightly packed and ordered than the inner leaflet. This possibility was evaluated by atomistic molecular dynamics simulations. To specifically isolate the biophysical effects of distinct leaflet compositions from transbilayer asymmetry per se, we compared symmetric bilayers composed of inner- or outer-mimetic lipid complements (Table I). The full lipidomic complexity of RBCs was distilled to a set of representative lipids for each leaflet that recapitulates the headgroup and acyl chain profiles of the compete leaflet lipidomes (Fig 2A–D, Table I; for details, see Supplementary Table II). Cholesterol was included in both systems at 40 mol%, since this was the overall abundance in RBCs and cholesterol distributes approximately equally between leaflets in previous PM-mimetic simulations26 (cholesterol asymmetry is addressed in Discussion).
Table I –
Distilled inner and outer leaflet lipidomes of the RBC PM.
| Outer (exoplasmic) leaflet | Inner (cytoplasmic) leaflet | ||
|---|---|---|---|
| representative lipid | mol% of outer leaflet | representative lipid | mol% of inner leaflet |
| PC 16:0–18:2 | 22.2 | PS 16:0–20:4 | 33.1 |
| SM 34:1;2 | 22.4 | PE O- 18:1–22:4 | 13.7 |
| SM 42:2;2 | 18.5 | PC 16:0–18:2 | 13.0 |
| SM 42:1;2 | 14.4 | PE 16:0–22:6 | 11.9 |
| PC 16:0–20:4 | 9.7 | PE 18:1–20:4 | 6.9 |
| PC 18:0–18:1 | 8.4 | PC 16:0–18:1 | 7.0 |
| PC 18:1–18:2 | 2.0 | PE 16:0–18:1 | 4.8 |
| PS 18:0–20:4 | 1.5 | PE O- 18:2–20:4 | 3.7 |
| PI 18:0–22:4 | 0.9 | PC 16:0–16:0 | 2.9 |
| SM 34:1;2 | 2.0 | ||
| PI 18:0–20:4 | 1.0 | ||
To quantitatively compare the two systems, we evaluated bulk parameters that correspond to lipid order, diffusivity, and membrane packing. Lipid order was assessed by calculating the concentration-weighted average acyl chain order parameters (() along both hydrophobic chains for all lipids in the systems (e.g. Supplementary Fig 5). The outer leaflet-mimetic was roughly twice as ordered as the inner leaflet (Fig 2E), consistent with inner leaflet enrichment of relatively disordered unsaturated acyl chains. These differences were also reflected in the order parameters of identical lipids in the two mixtures, with 1-palmitoyl-2-linoleoyl PC (16:0/18:2 PC) and palmitoyl-SM being more ordered in the outer leaflet mixture (Supplementary Fig 5). This order disparity corresponded to different molecular areas for lipids comprising the inner versus outer leaflet simulations. The average area per phospholipid (APL) was greater in the inner leaflet versus outer leaflet (59 versus 55 Å2), as was the APL for individual lipid species common to both simulations (Fig 2H). Finally, striking differences in membrane fluidity were evidenced by the diffusion coefficient (Dleaflet) (calculated from concentration-weighted mean square displacements (MSDs) of all lipids) being ~2-fold greater in the inner compared to the outer leaflet (Fig 2F). Finally, differences in lipid packing at the headgroup level were revealed by measuring hydrophobic packing defects, i.e. regions of the membrane where hydrophobic acyl chains are transiently exposed to water27. The defect distribution is clearly shifted in the inner leaflet, indicating more abundant and larger packing defects, reflective of reduced headgroup packing (Fig 2G).
PM leaflets have distinct biophysical properties
The atomistic simulations predict that inner leaflet lipids would have different physical properties from outer leaflet, consistent with extensive experimental observations of symmetric membranes. However, the biophysical consequences of lipid asymmetry in either model or biological membranes remain poorly understood because methodologies for constructing asymmetric model membranes have only recently become widely accessible7–9,11,12. We probed the biophysical asymmetry of live cell PMs using a fluorescent reporter of membrane packing (Di-4-ANEPPDHQ, Di4). The photophysical characteristics (e.g. fluorescence lifetime and emission spectra) of this dye are dependent on lipid packing28–30, making it a robust reporter of membrane properties (Supplementary Fig 6). The key feature making it suitable for leaflet-selective measurements is the presence of two charged moieties (Supplementary Fig 7A) that prevent passive flip-flop across the bilayer28. Thus, the exoplasmic leaflet of living cells can be selectively stained by adding the dye directly to the extracellular solution, whereas the inner leaflet is selectively stained by microinjecting the dye into the cytoplasm (schematized in Fig 3A). These features were confirmed in extensive control experiments described in the Supplement (Supplementary Figs 6–11).
Figure 3 – Biophysical asymmetry of the PM.
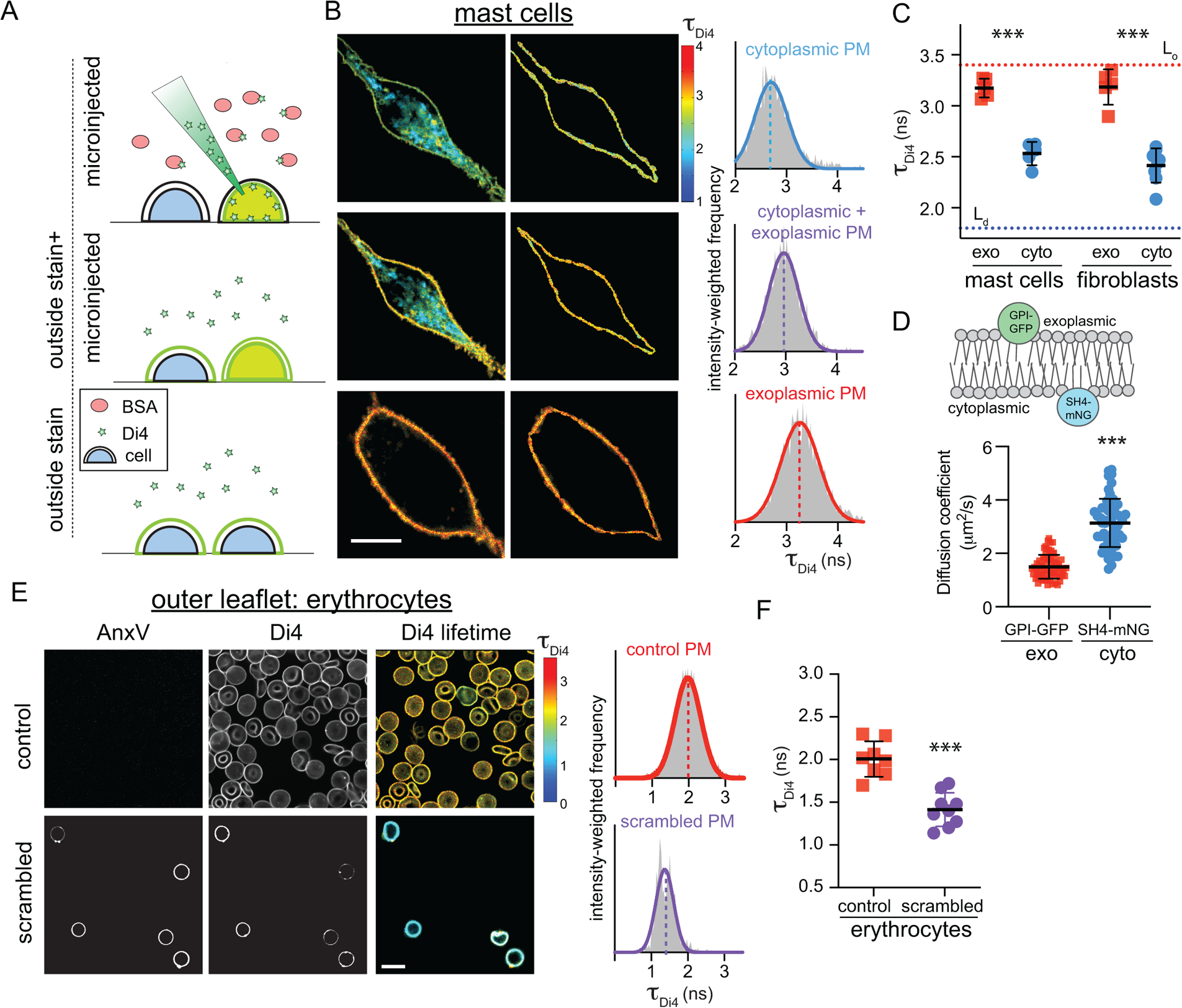
(A) Microinjection of Di4 in presence of BSA stains only the cytoplasmic PM leaflet (microinjected). Subsequent addition of Di4 to the outside stains both membrane leaflets (outside stain + microinjected). Staining from the outside labels only the outer PM monolayer (outside stain). Exemplary FLIM images of (B) RBL mast cells showing (left) whole cells, (middle) PM masks, and (right) intensity-weighted histograms of the PM mask from n ≥ 3 independent experiments. (C) Average Di4 lifetime in exoplasmic (red) versus cytoplasmic (blue) PM leaflets. Dotted lines represent Di4 lifetime in Lo and Ld phases in GUVs. Corroborating measurements of Di4 emission wavelength shifts are shown in Supplementary Fig 11. (D) Diffusion coefficients measured by FCS of exoplasmically anchored GPI-GFP (red) versus cytoplasmically anchored SH4-mNG (blue) (E) AnxV staining for exoplasmic PS (left), Di4 intensity and lifetime (middle), and lifetime histogram (right), in control versus PMA-scrambled erythrocytes stained from the outside with Di4. PMA-induced scrambling induces PS exposure (AnxV binding) and reduces the packing of the outer leaflet. (F) Average Di4 lifetime in untreated (red) versus scrambled (purple) PM outer leaflets in erythrocytes. Data points in C and F represent averages of individual experiments, with 5–10 cells/experiment. Mean ± SD shown; ***p<0.001 for Sidak’s multiple comparison test. Points in E represent individual cells, with the mean ± SD shown; ***p<0.001 for unpaired two-tailed t-test. All data are representative of >3 independent experiments.
Having confirmed Di4 as a leaflet-selective indicator of membrane physical properties, we probed their asymmetry in the PMs of two cultured mammalian cell types (RBL and 3T3). Note that while the lipidomic asymmetry was established in RBCs, we switched to nucleated cell types for the biophysical measurements because RBCs are not amenable to microinjection. In both cell types, microinjecting Di4 broadly stained intracellular membranes, with the PM identifiable by the general morphology of the cell and the high Di4 lifetime in the PM (left column of Fig 3B and Supplementary Fig 10), revealing that the cytoplasmic leaflet of the PM is more tightly packed than those of intracellular membranes. An intensity-weighted histogram (right column) of a PM mask (middle column) revealed an average lifetime (τDi4) of ~2.5 ns in microinjected cells (Fig 3B, top row). When the same cells were then stained externally with Di4, the lifetime of the internal membranes was unaffected (Supplementary Fig 8B), whereas the Di4 lifetime in the PM increased significantly (Fig 3B, middle row), suggesting that the exoplasmic PM leaflet is more tightly packed than the cytoplasmic leaflet. This inference was confirmed by comparison to cells stained only from the outside, where Di4 signal was confined to the outer PM leaflet. In outside-only stained cells (Fig 3B, bottom row), Di4 lifetime was ~3.2 ns, significantly greater than the PM in microinjected cells, revealing a striking asymmetry in membrane packing between the inner and outer leaflet. Similar values and trends were observed for both cell types (Fig 3C and Supplementary Fig 10). To relate τDi4 values to membrane properties, the cellular measurements were compared with Lo and Ld phases of synthetic phase-separated GUVs (Supplementary Fig 6). The outer leaflets of cellular PMs were slightly less packed than the synthetic Lo phase, whereas the inner leaflets were approximately intermediate between the Lo and Ld phases, in excellent agreement with recent reports18. All FLIM results were corroborated by measuring Di4 emission wavelength shift, calculated as Generalized Polarization (GP, see Materials and Methods), in the same experimental setups (Supplementary Fig 11). To test the computational prediction that inner leaflet components diffuse faster than outer (Fig 2F), Fluorescence Correlation Spectroscopy (FCS) was used to measure the diffusion of fluorescent proteins anchored either to the outer (GPI-GFP) or inner leaflet (SH4-mNG) by saturated fatty acids (Fig 3D). Outer leaflet-anchored GPI-GFP diffused ~2-fold slower than inner leaflet SH4-mNG, quantitatively consistency with the computational result.
These measurements strongly suggest biophysical asymmetry in mammalian PMs, with the outer leaflet being less fluid and more tightly packed. We confirmed that Di4 photophysical properties are not affected by charged lipids, pH, ionic composition of the media, or transmembrane potential (Supplementary Fig 9). Further, to rule out artifacts associated with microinjection or the presence of internal membranes in nucleated cells, we investigated biophysical asymmetry in human erythrocyte PMs. These cells are too small for efficient microinjection, thus only the outer leaflet was probed by external staining. To assess biophysical asymmetry, asymmetric PMs in untreated RBCs were compared with those whose asymmetry was pharmacologically abrogated by treatment with phorbol myristate acetate (PMA), which induces efficient PM scrambling31 detected by the exposure of PS on the exoplasmic leaflet via the PS-binding probe Annexin V (AnxV; Fig 3E, left column). Outer leaflet packing was significantly reduced by lipid scrambling, suggesting that the inner leaflet of the RBC PM is more loosely packed than the outer (Fig 3E–F). NR12S, an independent leaflet-selective order-sensitive probe, showed similar trends, ruling out probe-specific effects (Supplementary Fig 12).
Lipid packing asymmetry is preserved upon endocytosis
Having observed differential packing between the two PM leaflets, we investigated whether biophysical asymmetries persist in endocytic membranes, which are composed in part of material arriving from the PM but are also remodeled for sorting, processing, and degradation32. To probe membrane asymmetry in the endocytic system, these compartments were marked by pulse-chase with fluorescent dextran. Dextran is taken up by passive pinocytosis and accumulates in endocytic compartments, which appear expanded by the increased load of non-degradable material (Fig 4A). PM outer leaflets were labelled by external Di4 (as in Fig 3), and the cells were incubated for up to 1 h (without Di4 or dextran) to allow dye endocytosis and staining of endo-lysosome membrane lumenal leaflets (Fig 4A). The contours of the dextran staining were used to evaluate Di4 properties in endolysosomal regions (Fig 4A, mask). Strikingly, Di4 lifetime in endocytic organelles was only slightly reduced compared to the PM outer leaflet (Fig 4). In contrast, the cytoplasmic leaflets of dextran-positive endosomes (probed by Di4 microinjection) had much lower membrane packing (i.e. τDi4) than any of the lumenally stained endosomes. With longer incubation times, there was a slight reduction of τDi4 in the lumenally labelled endosomes, due to either membrane remodeling during endocytic maturation (Fig 4B) or minor probe flipping between leaflets after long incubations (Supplementary Fig 7). These observations were consistent between cell types (Fig 4 and Supplementary Fig 13) and imply that biophysical membrane asymmetry is maintained in the endosomal system.
Figure 4 – Asymmetry of membrane packing through the endocytic pathway.
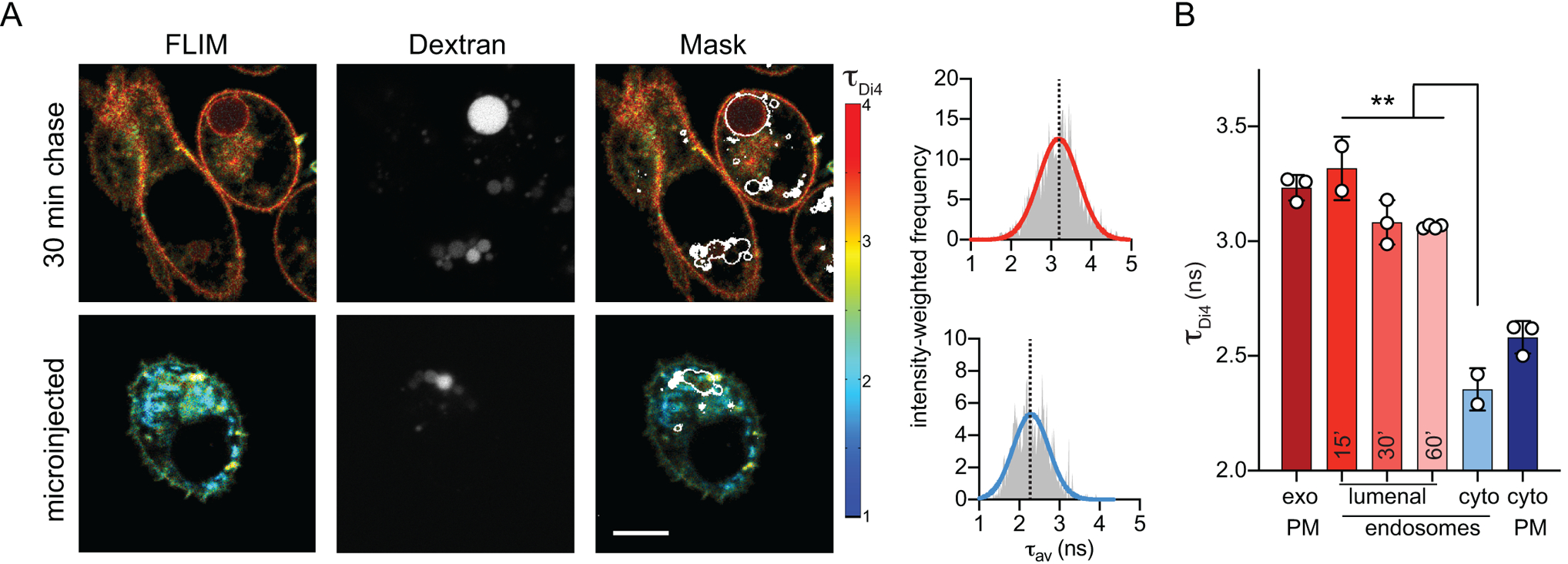
(A) Exemplary FLIM images of Di4 lifetime and dextran fluorescence in RBL cells following 30 min incubation to “chase” stains into endosomes. The accumulation of dextran (middle) is used to create an endosomal membrane mask (right images) to derive intensity-weighted histograms of τDi4 (right). (B) The high lifetime (i.e. lipid packing) of the exoplasmic PM leaflet is maintained in the lumenal leaflets of endosomes, even up to 60 min after endocytosis. Images and quantifications of 3T3 cells are shown in Supplementary Fig. 12. Mean ± SD of >10 individual cells; ***p<0.001 for unpaired t-test. Representative of at least 3 independent experiments.
Asymmetry of TMDs conforms to lipid packing asymmetry
We recently reported that structural features of single-pass transmembrane protein domains (TMDs) determine protein partitioning between coexisting lipid phases in isolated PMs33. In particular, the lipid-accessible surface area of TMDs dictates partitioning to raft versus non-raft phases, with relatively thin TMDs (i.e. rich in Ala/Gly) more efficiently partitioning into tightly-packed raft-like domains, whereas larger TMDs (rich in Leu/Phe) are excluded from these regions. Our observations of lipid packing asymmetry in live cell membranes prompt the hypothesis that proteins may have co-evolved to conform to these asymmetries. Namely, TMDs with relatively thin exoplasmic regions would minimally perturb the tight packing of the exoplasmic leaflet, whereas the TMD regions interfacing with the cytoplasmic leaflet could be larger due to their solvation by relatively loosely packed lipids. To evaluate this possibility, we calculated the ratio of exoplasmic to cytoplasmic TMD surface area for all annotated human single-pass PM proteins. This analysis revealed a clear asymmetry of TMD structures, with a distinct bias towards TMDs with relatively thin exoplasmic portions (Fig 5A), consistent with a previous report of an exoplasmic bias in the abundance of amino acids with smaller side chains14.
Fig 5 – Structural asymmetry in PM protein TMDs is related to subcellular localization.
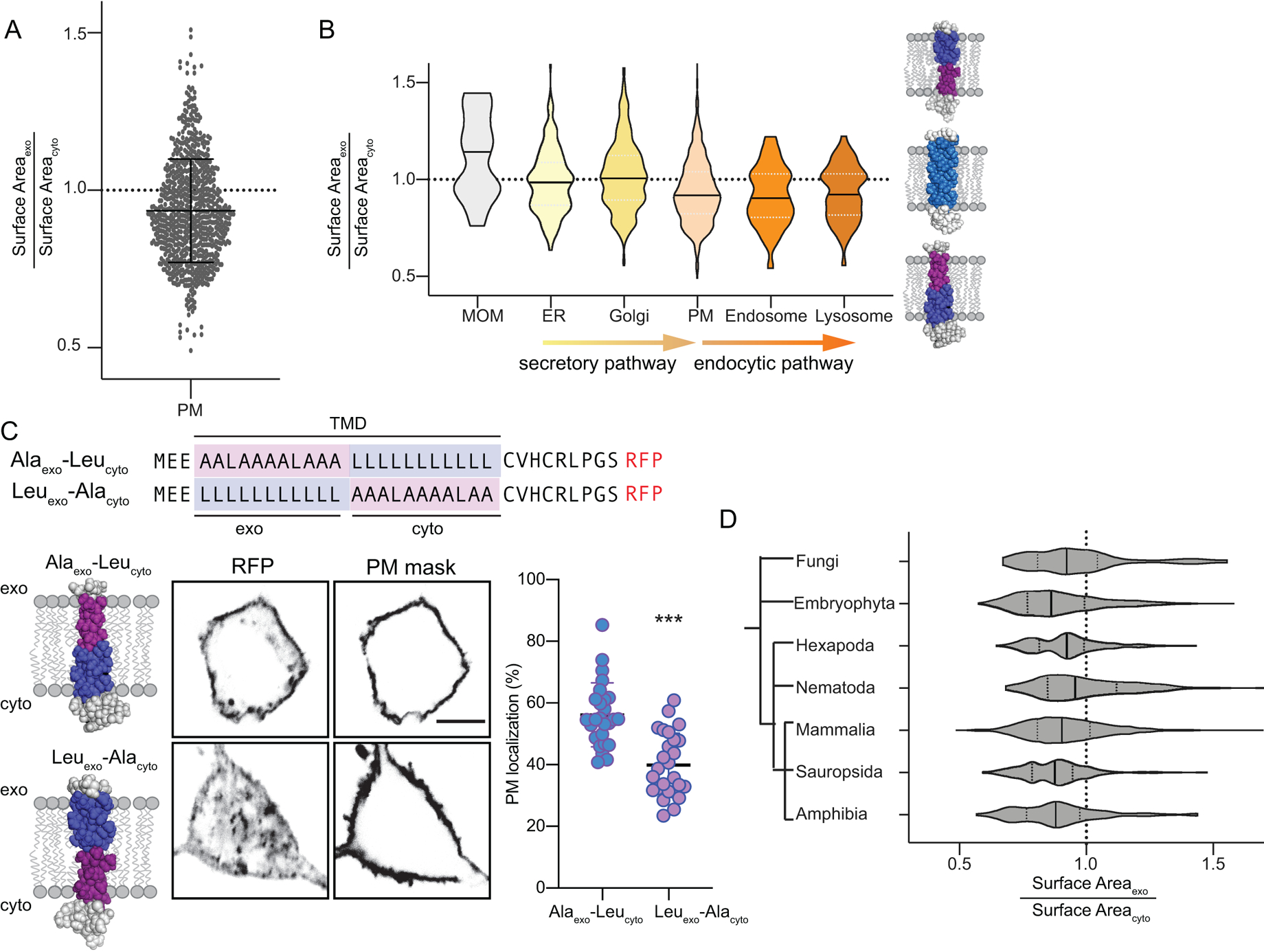
(A) Asymmetry of single-pass transmembrane domain surface area between exoplasmic and cytoplasmic halves of human PM proteins. Shown are all annotated PM-resident, single-pass proteins in the human proteome (mean ± SD overlaid). (B) Violin plots demonstrating the distributions of relative exoplasmic / cytoplasmic surface areas for single-pass TMDs in various organelles. PM, endosomal, and lysosomal proteins are asymmetric with smaller exoplasmic halves, whereas ER and Golgi proteins are on average symmetric (MOM = mitochondrial outer membrane). Median and quartiles are shown. (C) Subcellular localization of model TMDs that match (Alaexo-Leucyto) or counter (Leuexo-Alacyto) the biophysical asymmetry of the PM. The matching TMD localizes efficiently at the PM, whereas the countering TMD is largely intracellular. Mean ± SD of individual cells expressing the TMD construct; ***p<0.001 unpaired two-tailed t-test. (D) Bias towards asymmetric TMDs is observed throughout eukaryote PMs. The phylogenetic dendrogram is intended to show only general evolutionary relationships. Median and quartiles are shown.
The structural asymmetry of protein TMDs was not isolated to PMs but was also evident for proteins localized in endosomes and lysosomes (Fig 5B), consistent with our observations of leaflet asymmetry in endosomes (Fig4A–B). In contrast, TMDs of the endoplasmic reticulum (ER) and Golgi apparatus were on average symmetric (Fig 5B), suggesting that membrane biophysical asymmetry arises late in the secretory pathway. Notably, proteins of the outer mitochondrial membrane (OMM) have a clear bias towards the opposite asymmetry, with relatively small cytoplasmic regions. These observations are fully consistent with our measurements of relatively tightly packed exoplasmic leaflets in the PM and endocytic organelles, suggesting that subcellular protein localization is determined to some extent by matching the physical properties of protein TMDs with their solvating lipids. To support this inference, we measured the subcellular distribution of model asymmetric TMDs designed to either conform to the measured biophysical asymmetry of the PM (i.e. thinner outer half; Alaexo-Leucyto) or oppose it (thick outer half; Leuexo-Alacyto) (Fig 5C). These TMDs are identical in all other “bulk” physical features (TMD length, surface area, hydrophobicity, etc). The majority (~60%) of the asymmetric TMD that conforms to the packing asymmetry of the PM (thin outer half) localized to the PM, with a minor fraction in punctate endosome-like structures (Supplementary Fig 15). In contrast, the TMD with the thicker outer half was more confined to internal membranes (Fig 5C), identified as late endosomes via Rab7 staining (Supplementary Fig 15).
Finally, the structural asymmetry of mammalian PM TMDs is also evident in non-mammalian eukaryotes. Across the eukaryotic domain of life, TMDs of single-pass PM proteins are strikingly asymmetric, with relatively thin exoplasmic regions and relatively thick cytoplasmic ones (Fig 5D). Thus, the compositional and biophysical plasma membrane asymmetry we describe in cultured mammalian cell lines may be a fundamental and conserved design principle in Eukaryota.
DISCUSSION
Combining enzymatic digestion with quantitative mass spectrometry revealed the detailed lipidomes of the inner and outer leaflets of RBC PMs. Consistent with expectations2,3, SMs were highly enriched on the outer leaflet, whereas most glycerophospholipids were exclusive to the cytoplasmic leaflet, excepting PCs, which were distributed approximately equally between the two leaflets. Such significant lipid asymmetries are entropically unfavorable but can be maintained because of the intrinsically low rate of lipid flip-flop across the bilayer (typically estimated to be on the order of hours)34. To establish and regulate asymmetry, cells rely on the coordinated action of numerous enzymes and channels. Enzymes called flippases and floppases move lipids between leaflets in an ATP-dependent fashion, while scramblases are channels that allow lipids to flow down concentration gradients34.
Importantly, the reported compositions are not fully comprehensive. Complex glycolipids are not detected in the shotgun platform, nor are phosphorylated inositols. The most abundant glycolipid in human RBCs is globoside (Gb4), estimated to be <5% of all lipids35; PIP2 is the most abundant phosphoinositide, estimated at <1%36. Similarly, while the total concentration of cholesterol is quantified (~40 mol%), its distribution between leaflets is not accessible by our methodology and remains controversial37, with some groups reporting inner leaflet enrichment of cholesterol38 and others the opposite39. Our measurements do not directly inform on this debate, though the higher packing of the outer leaflet may argue for higher cholesterol content therein. However, the packing of the inner leaflet is such that extremely low cholesterol concentrations are also unlikely. In the absence of consensus, and because cholesterol is believed to flip rapidly between leaflets40, the molecular dynamics simulations (Fig 2) assumed no a priori cholesterol preference for either leaflet.
The most notable disparity between the acyl chains of lipids in the two RBC PM leaflets is ~2-fold more double bonds per lipid in the inner versus outer leaflet (Fig 1B). This difference is likely responsible for the robust biophysical PM asymmetry (Fig 3, Supplementary Fig 8&10–11). These live cell results are consistent with model membrane observations that lipid order can be decoupled between the two leaflets of asymmetric membranes10. The biophysical asymmetry appears to persist after internalization of the PM into the early endosomal pathway, suggesting that lipid asymmetry is also present in some intracellular compartments, consistent with other reports41.
Comparing the lifetime of Di4 in the PM leaflets of live cells to synthetic model membranes suggests that the outer PM leaflet has biophysical properties similar to a Lo phase, whereas the inner PM leaflet is intermediate between the ordered and disordered phases (Fig 3C). This comparison prompts examination of long-standing questions about the physical organization of the mammalian PM and its partitioning into ordered domains termed lipid rafts42. Asymmetric model membrane experiments have shown that ordered domains in phase-separating leaflets can induce domains in apposed leaflets that otherwise would not form Lo phases43,44. Long-chain SM species are implicated as potential mediators of such transbilayer domain coupling43. The asymmetric RBC lipidome reveals an outer leaflet rich in high-melting lipids (i.e. saturated SM) and cholesterol with a non-trivial abundance of low melting lipids (i.e. unsaturated glycerophospholipids). Such a membrane monolayer is poised to form coexisting ordered and disordered domains45. The inner leaflet contains largely low-melting unsaturated lipids that are not amenable to ordered domain formation, though the high abundance of long-chain SM species in the outer leaflet may promote domain coupling between leaflets. The ultimate organization of the membrane is then a combination of these membrane-intrinsic effects and extrinsic inputs like protein scaffolds46 and cytoskeletal dynamics47.
The lipid packing asymmetry of the mammalian PM is reflected in the structure of its resident TMDs. We have recently shown that tightly packed membrane phases tend to select TMDs based on their lipid-accessible surface area, with larger, bulky TMDs being excluded in favor of thin TMDs33. Consistent with these principles, mammalian PM proteins have asymmetric surface areas, with the TMD region in the exoplasmic leaflet being relatively thin. Similar tendencies have been previously reported in bioinformatic studies of both mammalian and yeast PM proteins14, with the authors anticipating their mechanistic origins as being the biophysical asymmetry reported here. The TMD asymmetry of PM proteins is also evident in proteins of the late secretory and endocytic systems, consistent with our findings of asymmetric biophysical properties in endocytic organelles (Fig 4). In contrast, other organelles have relatively symmetric TMDs, serving as an important control and also suggesting that these membranes may not be asymmetric. The physical characteristics of protein TMDs have been previously implicated in subcellular protein sorting14,32,33, with longer and thinner TMDs preferentially trafficking to the thicker, more tightly packed PM. A similar ‘matching’ between transbilayer membrane packing profiles and TMD structures appears to also affect PM localization (Fig 5C), as model TMDs conforming to the asymmetry of the PM are trafficked efficiently to the PM, whereas those with the opposite asymmetry are retained in intracellular membranes. Mechanistically, it is possible that sub-domains of intracellular sorting organelles (e.g. trans-Golgi and endosomes) select proteins with specific properties for transport to the PM. Alternatively, PM proteins that do not conform may be preferentially endocytosed.
The observation that the structural asymmetry of PM TMDs is ubiquitous in eukaryotic organisms prompts the conclusion that biophysical PM asymmetry is a conserved and fundamental principle of eukaryotic membranes. Since the generation and maintenance of lipid disparities in the two leaflets is likely energetically costly, their ubiquity raises a key question: what adaptive advantage is gained from PM asymmetry? There are several hypothetical explanations: from the standpoint of material properties, coupling distinct leaflets may combine desirable features into a single material. For example, a tightly packed outer leaflet may serve as an effective permeability barrier, while the more fluid inner leaflet allows for rapid signal transmission. It may be interesting to develop methods to individually tune leaflet compositions48 and physical properties to determine whether certain cellular functions (e.g. signal transduction) are more dependent on one leaflet versus the other. A non-exclusive alternative is that PM asymmetry is used for energy storage, in analogy with pumped-storage hydroelectricity. Cells may store the potential energy generated by pumping lipids against their concentration gradients to be released upon regulated scrambling of the bilayer. Finally, membrane asymmetries could be used as organellar identifiers for selective sorting of lipids and proteins between organelles. Finally, it has been shown that membrane asymmetry can affect the properties of membrane domains9,11,43 and possibly the partitioning of proteins between ordered and disordered lipid environments49,50, making the magnitude and mechanisms of physical coupling between disparate leaflets in asymmetric membranes an essential open question42. Ultimately, deciphering the purpose of membrane asymmetry is central to understanding the functions of cell membranes.
ONLINE METHODS
General experimental design
Lipidomics:
Freshly isolated, intact human erythrocytes were treated phospholipase A2 or sphingomyelinase to specifically digest only the lipid species present on the exoplasmic leaflet of the PM. Comparison of enzyme-treated cells to untreated controls revealed the extent of digestion (i.e. abundance on the exoplasmic leaflet) of each of the ~400 unique phospholipid species detected in our measurements. Detailed asymmetry was measured for nearly all major phospholipid species in RBCs, including all PC, PE, PI, PS, SM, and the ether form of PE (plasmalogen; PE-O). The only exception was the PC plasmalogen (PC-O), which were not susceptible to any of our enzyme treatments. These lipids comprise less >1% of the total lipidomes and were assumed to distribute like other PC lipids. For all enzyme treatments, hemolysis was monitored to ensure that RBCs remained intact (Supplementary Fig. 3). Treatment of sonically disrupted cells ensured that the enzyme conditions were sufficient to completely degrade all available target lipids, with the increase in degradation products quantitatively validating the enzymatic digestions (Supplementary Fig. 1).
Spontaneous phospholipid flipping is negligible on our experimental time scales. PLA2 treatment of intact cells did not degrade PE or PS to any appreciable extent, consistent with the near-absolute inner leaflet confinement of these aminophospholipids noted previously. This result further confirmed that minimal flipping of inner leaflet lipids occurred during our procedure. The observed headgroup distributions are in excellent quantitative correspondence with previous reports3.
The lipidome of the inner leaflet was inferred from the lipid species remaining after digestion of intact cells. Conversely, the outer leaflet was calculated by subtracting the abundance of each lipid species remaining after digestion from the untreated controls. These data were highly consistent between multiple healthy adult donors (N=3) and repeated samples from the same donor (Supplementary Figs. 1–2). These measurements produced the raw lipidomes of the inner and outer leaflets given in Supplementary Data. These were then collated by sorting all species above a threshold of 0.5 mol% into self-similar groups defined by headgroup, acyl chains, and compositions (Table I and Supplementary Table II). The details for these groupings are in the legend to Supplementary Table II. These collated lipidomes largely recapitulate the headgroup and acyl chain profiles of the complete leaflet lipidomes.
Leaflet selective biophysical properties:
The biophysical asymmetry of live cell PMs was probed using a fluorescent reporter of membrane packing (Di-4-ANEPPDHQ, Di4). To calibrate the sensitivity of Di4 to membrane packing, we constructed synthetic Giant Unilamellar Vesicles (GUVs) composed of DOPC, DPPC, and cholesterol. Such vesicles separate into microscopic liquid-ordered (Lo) and liquid-disordered (Ld) domains that have been widely employed as synthetic analogues of membrane structures in mammalian cells . Di4 can be used to probe physical differences between phases because its emission spectrum is dependent on membrane packing, red-shifting in a more disordered environment, as typically described by the Generalized Polarization (GP)30. We observed a clear difference in GP between coexisting phases in our GUVs (Supplementary Fig. 6). The difference between Lo and Ld phases was much better resolved by measuring Di4 emission lifetime using Fluorescence Lifetime Imaging Microscopy (FLIM), as previously noted29. Supplementary Fig. 6B shows that average lifetime histograms in the two phases are distinct and non-overlapping, as opposed to those of GP in the same vesicle.
Having confirmed that Di4 sensitively reports membrane physical properties, selectivity for staining individual PM leaflets in live cells was verified. Cells were first stained externally by adding dye directly into the medium. The charged groups on Di4 should prevent flipping across the bilayer midplane, thus only staining the outer leaflets of the PM. This supposition was supported by BSA back-extraction experiments, wherein BSA complexes and removes any accessible dye from the exoplasmic leaflet of cellular membranes51. Application of BSA to externally stained cells completely eliminated Di4 fluorescence specifically at the PM, leaving only minimal residual signal from internal membranes (Supplementary Fig. 7B). This effect was observable even after relatively long incubation times: after 30 min of incubation, some dye was efficiently internalized into punctate endocytic structures (Supplementary Fig. 7B). Treatment with BSA did not noticeably affect the intensity of these structures, but did eliminate all observable signal from the PM, suggesting that Di4 does not flip across the bilayer in live cells on our experimental time-scales. These effects were completely independent of cell type.
To selectively stain the inner leaflet of the PM, cells were microinjected with Di4, together with a fluorescent dextran to ensure membrane integrity of the injected cells (Supplementary Fig. 7C). Due to leakage of dye from the micropipette, cells in the vicinity of the injected cell were also stained, though presumably only on the outer leaflet of the PM, as no internal membrane staining was observed (Supplementary Fig. 7C), suggesting the dye was unable to passively diffuse through the PM. Inclusion of BSA into the extracellular medium captures the leaked dye from the micropipette, preventing staining of nearby cells (Supplementary Fig. 7D&8A); however, the microinjected cells were still efficiently labeled on cytoplasmic membranes, including clear staining of their PMs (see Fig 3B–C, Supplementary Figs. 7D & 8A). This PM staining is exclusive to the cytosolic leaflet, as extracellular BSA did not eliminate this PM signal, in contrast to externally added Di4 (microinjections in Fig 3B–C, Supplementary Fig. 7D, and 8A were conducted in presence of BSA). Leaflet-specific localization of Di4 after microinjection versus external addition is also supported by their different lifetimes in the PM, as discussed in the results. Also generalized polarization (GP) of Di4, a complementary reporter of membrane packing, showed the same trends as emission lifetime, confirming the robustness of the result (Supplementary Fig. 10).
Finally, we confirmed that Di4 photophysical properties are not affected by charged lipids or transmembrane potential (Supplementary Fig. 9). For completeness, we directly quantified the spontaneous Di4 flipping rate in synthetic membranes and observed minimal flip-flop on the experimentally relevant time scales (Supplementary Fig. 7F), consistent with the structural features of this probe.
Materials:
Synthetic lipids were obtained from Avanti. Di-4-ANEPPDHQ (Di4), BSA (fraction V), Phenol red-free MEM, AnxV-Pacific Blue, and dextran-cascade blue (3000 Da) were purchased from Thermo Fisher. NR12S was kindly provided by A. Klymchenko and Mikhail Bogdanov. All other chemicals were purchased from Sigma. Rat basophilic cells (RBL) and NIH 3T3 fibroblasts were purchased from ATCC. Glass micropipettes were from Sutter Instruments and microloaders from Eppendorf. For analysis of red blood cells, 6 channel IBIDI™ slides were used.
Detailed methods: Lipidomics
Lipidomics summary:
300 uL of packed, freshly isolated, intact human erythrocytes from healthy donors with informed consent were treated with 10 I.U. phospholipase A2 (PLA2; from Apis mellifera) or 0.5 I.U. sphingomyelinase (SMase; from Bacillus cereus) in 5 mL isotonic saline solution (50mM Tris HCl, 0.25 mM CaCl2, 0.25mM MgCl2, 150mM NaCl, pH 7.4) for 30 minutes at 30°C to specifically digest only the lipid species present on the exoplasmic leaflet of the PM. After treatment for the indicated time, the cells were fast frozen in liquid nitrogen, and their detailed lipid compositions were analyzed by shotgun electron spray ionization and tandem MS-MS (ESI-MS/MS). All lipidomics were performed at Lipotype GmbH (Dresden, Germany) as described previously52,53 and detailed below. Lipidomes were prepared from at least 3 independent human donors for all experiments using the following procedures.
Nomenclature:
The following lipid names and abbreviations are used: ceramide (Cer), cholesterol (Chol), sphingomyelin (SM), diacylglycerol (DAG), lactosyl ceramide (DiHexCer), glucosyl/galactosyl ceramide (HexCer), sterol ester (SE), and triacylglycerol (TAG), as well as phosphatidic acid (PA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and phosphatidylinositol (PI), phosphatidylserine (PS), and their respective lysospecies (lysoPA, lysoPC, lysoPE, lysoPI, and lysoPS) and ether derivatives (PC O-, PE O-, LPC O-, and LPE O-). Lipid species were annotated according to their molecular composition as follows: [lipid class]-[sum of carbon atoms in the FAs]:[sum of double bonds in the FAs];[sum of hydroxyl groups in the long chain base and the FA moiety] (e.g., SM-32:2;1). Where available, the individual FA composition according to the same rule is given in brackets (e.g., 18:1;0–24:2;0).
Lipid standards:
Synthetic lipid standards were purchased from Sigma-Aldrich (Chol D6), Larodan (Solna, Sweden) Fine Chemicals (DAG and TAG), and Avanti Polar Lipids (all others).
Lipid extraction for mass spectrometry lipidomics:
Lipids were extracted using a two-step chloroform/methanol procedure . Samples were spiked with internal lipid standard mixture containing: cardiolipin 16:1/15:0/15:0/15:0 (CL), ceramide 18:1;2/17:0 (Cer), diacylglycerol 17:0/17:0 (DAG), hexosylceramide 18:1;2/12:0 (HexCer), lyso-phosphatidate 17:0 (LPA), lyso-phosphatidylcholine 12:0 (LPC), lyso-phosphatidylethanolamine 17:1 (LPE), lyso-phosphatidylglycerol 17:1 (LPG), lyso-phosphatidylinositol 17:1 (LPI), lyso-phosphatidylserine 17:1 (LPS), phosphatidate 17:0/17:0 (PA), phosphatidylcholine 17:0/17:0 (PC), phosphatidylethanolamine 17:0/17:0 (PE), phosphatidylglycerol 17:0/17:0 (PG), phosphatidylinositol 16:0/16:0 (PI), phosphatidylserine 17:0/17:0 (PS), cholesterol ester 20:0 (CE), sphingomyelin 18:1;2/12:0;0 (SM), triacylglycerol 17:0/17:0/17:0 (TAG) and cholesterol D6 (Chol). After extraction, the organic phase was transferred to an infusion plate and dried in a speed vacuum concentrator. 1st step dry extract was re-suspended in 7.5 mM ammonium acetate in chloroform/methanol/propanol (1:2:4, V:V:V) and 2nd step dry extract in 33% ethanol solution of methylamine in chloroform/methanol (0.003:5:1; V:V:V). All liquid handling steps were performed using Hamilton Robotics STARlet robotic platform with the Anti Droplet Control feature for organic solvents pipetting.
MS data acquisition:
Samples were analyzed by direct infusion on a QExactive mass spectrometer (Thermo Scientific) equipped with a TriVersa NanoMate ion source (Advion Biosciences). Samples were analyzed in both positive and negative ion modes with a resolution of Rm/z=200=280000 for MS and Rm/z=200=17500 for MS/MS experiments, in a single acquisition. MS/MS was triggered by an inclusion list encompassing corresponding MS mass ranges scanned in 1 Da increments53. Both MS and MS/MS data were combined to monitor CE, DAG and TAG ions as ammonium adducts; PC, PC O-, as acetate adducts; and CL, PA, PE, PE O-, PG, PI and PS as deprotonated anions. MS only was used to monitor LPA, LPE, LPE O-, LPI and LPS as deprotonated anions; Cer, HexCer, SM, LPC and LPC O- as acetate adducts and cholesterol as ammonium adduct of an acetylated derivative .
Lipid identification and quantification:
Data were analyzed with in-house developed lipid identification software based on LipidXplorer . Data post-processing and normalization were performed using an in-house developed data management system. Only lipid identifications with a signal-to-noise ratio >5, and a signal intensity 5-fold higher than in corresponding blank samples were considered for further data analysis.
Lipidomics data processing:
The lipidomic analysis yields a list of >600 individual lipid species and their picomolar abundances. These were processed by first transforming into mol% of all lipids detected. Next, the contaminating TAG and sterol esters were removed from the analysis, and the remaining data was analyzed as mol% of membrane lipids. From here, the datasets were broken down further into class composition. In some cases, the distribution and structural characteristics (e.g. number of carbons or unsaturations in the acyl chains) of the individual species were analyzed. Each class was then compared separately for each individual biological replicate (i.e. the enzymatically treated samples were directly compared to the untreated parallel sample for each individual experiment to control for variance across RBC isolation or human donor).
Hemolysis measurements:
For all enzyme treatments, hemolysis was monitored to ensure that the RBCs remained intact, and only conditions that yielded no hemolysis were considered for the asymmetry studies (Supplementary Fig. 3). Hemolysis was measured by measuring the absorbance of the supernatant of the enzyme-treated cells at 540 nm on a Tecan plate reader. As an additional test, RBCs were incubated with FITC-dextran (MW=3KDa) during treatment with SMase and imaged on a Nikon A1R laser scanning confocal microscope immediately after.
Detailed methods: Simulations
Simulations summary:
Two simulations of symmetric bilayers (i.e. identical compositions in each leaflet) were performed, one with the composition of the inner leaflet and one of the outer leaflet (Supplementary Table I). For each system, a small patch of membrane 1/9 the area of the final system was built and equilibrated. This small membrane patch was duplicated in a 3×3 array to generate the final system, which was equilibrated further before transferring to Anton2 for production simulation. Initial configurations for the small systems were generated with the CHARMMGUI membrane builder54, including at least 45 water molecules per lipid and sufficient ions to neutralize the system and set the overall salt concentration to 150 mM, with KCl for the inner leaflet simulation and NaCl for the outer leaflet. The lipids were modeled with the Charmm36 force field55. All equilibration steps were performed on local resources with NAMD v2.12 . Each small system was minimized by 4,000 steps of conjugate gradient, then heated to 310 K by rescaling velocities to increase the temperature by 3 K every 4 timesteps, followed by another 50,000 timesteps of equilibration at 310 K, rescaling velocities every 100 timesteps. During heating and equilibration the system was coupled semi-isotropically to a Nosé-Hoover/Langevin piston with a target pressure of 1.013 bar, a damping timescale of 100 fsec, and period of 200 fsec, and a temperature of 310 K. Long range electrostatics were computed by particle mesh Ewald on a 1.0 Å grid with 4th order interpolation and a tolerance of 10−6. Long range Lennard-Jones interactions were shifted to zero from 10–12 Å, with both force and potential continuous across the cutoff. The small system was then tiled in a 3×3 array using the periodic unit cell to generate a final system approximately 18 nm × 18 nm, with the final numbers of lipids shown in Supplementary Table I. These final systems were then equilibrated for an additional 50,000 timesteps by velocity rescaling at 310 K, followed by 20 nsec of Langevin dynamics at 310 K with a damping constant of 0.5 psec−1 using a 2.0 fsec timestep, hydrogens constrained by SHAKE with a 10−5 tolerance.
The systems were then transferred to Anton2 for production with v.1.37.3c7. The NAMD restart files were converted to dms format using the NAMDtoDMS python script available from the Anton wiki page. Force field information was added with viparr v1.9.1. The systems were integrated with the multigrator with a 2.5 fsec timestep using Martyna-Tobias-Klein for semi-isotropic pressure control at 1.013 bar (no long range dispersion correction) and Nosé-Hoover for temperature control at 310 K. Lennard-Jones interactions were cutoff at 9.0 Å. Electrostatics were computed with the k-GSE algorithm56. with long-range interactions updated every 3 timesteps. Parameters including cutoffs were chosen by the Anton2 guessers. Each system was run for 5 μsec, with the observables averaged over the last 4 μsec of the simulation.
Simulation Analysis:
Order parameters.
Hydrocarbon chain order was evaluated by computing the 2H NMR order parameter , where θ is the angle between the carbon-hydrogen bond vector and the membrane normal and the average is taken over both time and lipids of the same type. A single SCD value is computed for every position along both chains for each lipid type, then weighted by the population of each lipid in the mixture to generate the position-specific weighted average SCD curves shown in Fig 2C. The analysis was performed using a modified tcl script kindly provided by Jeff Klauda.
Area per chain.
The area associated with each hydrocarbon chain was computed by a Voronoi tessellation of each leaflet, associating a unique area to the center of mass of each chain, then averaging over both chains of the same type and simulation time. The Voronoi analysis was performed with code generously shared by Alex Sodt.
Mean squared displacement.
Lipid mobilities were estimated by computing the mean squared displacement (MSD) of each lipid type according to MSD(t) = 〈(r(t +t0) − r(t0))2〉, where r(t) is the two-dimensional position of a single lipid in the membrane plane at time t and the average is taken over both time and lipids of the same type. The MSDs so obtained were then fit to obtain an effective two-dimensional diffusion coefficient. Note that these diffusion coefficients are not expected to be in quantitative agreement with experimental measurements due to finite-size effects in the simulations,57 but that they can be compared across similarly sized simulations as is done here.
Hydrocarbon chain packing defects:
The transient exposure of hydrocarbon chains to the aqueous environment was evaluated as described58. For each simulation snapshot, the solvent exposure of the hydrocarbon chains was determined using the solvent accessible surface area (SASA) VMD plugin with a probe radius of 3.0 Å. The points representing the SASA of the hydrocarbon chains are then projected onto the membrane midplane separately for each leaflet, generating localized clusters of points representing packing defects. Points are then partitioned into clusters by a breadth first search, connecting all pairs of points that are less than 2.5 Å apart. The area of each defect is then estimated on a grid with a spacing of 0.4 Å by summing the number of occupied pixels for each defect. This procedure is repeated for each simulation snapshot, to generate the histogram in Fig 2E.
Detailed methods: Di4 staining and imaging
Cell culture:
Rat basophilic leukemia (RBL) cells were maintained in medium containing 60% MEM, 30% RPMI, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. NIH 3T3 cells were maintained in 10% FBS in DMEM with 100 U/ml penicillin, and 100 μg/ml streptomycin.
Plasmid construction:
All TMD constructs were based on the trLAT backbone previously described32,33. The amino acid sequences of WT trLAT is NH2-MEEAILVPCVLGLLLLPILAMLMALCVHCHRLPGS followed by a short linker (GSGS) and monomeric RFP (mRFP). TMD mutants were generated by synthesizing the gene of interest (Genscript) and subsequent cloning of the mutant sequence into the trLAT construct. Mutants were confirmed by sequencing. The TMD sequences used here were:
AexoLcyto: MEEAALAAAALAAALLLLLLLLLLLCVHCRLPGSGS
LexoAcyto: MEELLLLLLLLLLLAAALAAAALAACVHCRLPGSGS.
Transfection and immunofluorescence:
RBLs were transfected by nucleofection (Amaxa) using the protocols provided with the reagents. After 4–6 h of transfection, cells were washed with PBS and then incubated with serum-free medium overnight. To synchronize the cells, 1 h to fixation, the cells were given full-serum medium. Cells were then fixed with 4% formaldehyde for 20 minutes at room temperature. For immunofluorescence labeling of various organelles, the following primary antibodies were used: anti-Rab7 rabbit monoclonal antibody (late endosome; Cell Signaling Technology), anti-Rab5 rabbit monoclonal antibody (late endosome; Cell Signaling Technology), anti-Giantin rabbit polyclonal antibody (Golgi; Abcam), and anti-LAMP1 rabbit polyclonal antibody (lysosome; Abcam). Fluorescent (Atto 488) secondaries were purchased from Invitrogen.
PM localization:
PM localization was quantified by an automated imaging protocol32. Briefly, transfected RBL cells were surface-labeled by a membrane-impermeable biotinylation reagent (Biotin-NHSLC) at 1 mg/ml in PBS for 20 min at 4 °C. Cells were fixed with 4% paraformaldehyde, and the PM were fluorescently labeled by streptavidin-Alexa488 (10 μg/ml for 20 min). Cells were imaged on a Nikon A1R confocal microscope using a ×60 Apochromat oil immersion objective. PM localization was quantified by a custom image processing protocol (MATLAB) in which the fluorescence intensity of the protein construct that was co-localized with the plasma membrane stain was divided by the total fluorescence intensity in the whole cell.
Leaflet-selective staining of plasma membrane leaflets:
Outer leaflet staining of cultured cells was achieved by incubating cells at 4°C for 8 min with Di4 at 1 μg/ml in staining buffer (10 mM HEPES, pH=7.4, 150 mM NaCl, 2 mM CaCl2). Cells were washed twice in the same buffer at ambient temperature before imaging in phenol red-free MEM. For dextran chase assays, cells were stained in the same manner.
To specifically stain the inner leaflet, Di4 was microinjected directly into the cytoplasm. Briefly, cells were kept in MEM media (no phenol red) containing 1.5% BSA to capture any dye leakage from the microinjection needle, which prevents staining the outer leaflet of surrounding cells (compare Supplementary Fig. 7C–D and 8) and also extracts any external PM dye (Supplementary Fig. 7). The microinjection solution was prepared by evaporating 4 μg of Di4 (dissolved in EtOH) under nitrogen flow, re-dissolving in 8 μl microinjection buffer containing 10 mM potassium phosphate (mono and dibasic to adjust at pH = 7) and 100 mM KCl. We then centrifuge at 10,000 xg for 10 min to remove undissolved material. Microinjection needles were pulled from glass micropipettes (1 mm outer, 0.56 mm inner diameter) and loaded by micro-loaders (2 μl). Microinjection was performed using a Transjector 5246 (Eppendorf) installed on a light microscope to visualize the cells. After microinjection, cells were bathed in MEM medium (no phenol red) without BSA and imaged as below. Some microinjected cells were further stained from the outside after imaging (outside/inside staining) by adding Di4 to the bathing medium. To exclude apoptosis or membrane disruption by microinjection, cells were stained with Annexin V (AnxV-pacific blue) after FLIM and/or GP imaging; all AnxV-positive cells were excluded from the analysis.
To remove outer leaflet Di4 from labeled cells, cells were incubated 3 times for 10 min with 1.5% BSA in MEM media at 4°C (to prevent endo- or exocytosis). To test whether BSA itself affects membrane properties, RBLs were incubated 3x for 10 min with 1.5% BSA at 4°C, washed with PBS, subsequently stained with 1 μg/mL Di4 at 4°C for 8 min, and imaged via FLIM (as described below).
Isolation and treatment of red blood cells for imaging:
RBCs were freshly isolated from human donors and diluted at 5e6 cells/ml in Ringer’s solution. Cells were then stained with Di4 (1 ug/ml) at 4°C for 8 min or NR12S (100 nM) for 10 min at at 37°C, centrifuged at 2000g for 5 min, resuspended in Ringer’s solution. IBIDI™ slides were used for FLIM and GP imaging. To induce membrane scrambling, the RBCs were treated with 10 μM PMA for 10 min under constant mixing at 300 rpm31. RBCs were stained with AnxV-pacific blue, only after imaging Di4 imaging, to verify scrambling and PS exposure while not interfering with Di4 spectra/lifetime (e.g. via FRET).
Confocal spectral imaging and lifetime imaging microscopy (FLIM):
Confocal and lifetime microscopy were performed on a Nikon A1 laser scanning microscope with an integrated Picoquant time-correlated single photon counting (TC-SPC) system (Berlin, Germany). Generalized polarization (GP)-imaging was done as previously described52. Briefly, emission was collected at I1=595 and I2=700 nm and GP was calculated as GP=(I1-G*I2)/(I1+G*I2) where G, the instrumental response factor, was determined according to protocol30.
For FLIM, Di4 emission was collected at >560 nm and the instrument response function (IRF) was determined with a saturated erythrosine B and KI solution at pH=10 according to the manufacturer’s (Picoquant) protocol. Di4 images were acquired using 20 MHz pulse frequency. The photon count rate was kept under 10% of the pulse rate by adjusting a manual shutter, and enough frames were acquired to obtain at least 104 photons cumulative signal intensity. The fluorescence decay curves were fitted to a bi-exponential re-convolution function adjusted to the IRF and the average lifetime was calculated and represented in the FLIM images as τDi4.
Dextran chase experiment:
RBL and 3T3 cells were incubated with dextran-cascade blue (0.5 mg/ml) for 2 h at 37°C. Cells were then washed twice with HEPES buffer (10 mM HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7) and stained on the inner or outer plasma membrane leaflet by Di4 (see selective staining). After staining, cells were incubated for 15, 30, or 60 min (chase) at 37°C in MEM media, allowing dextran to accumulate in endosomes and Di4 to access endosomal membranes.
Preparation of GUV:
GUV of DOPC/DPPC/Chol were prepared by electroformation as described previously59. Briefly, 1 μl of the lipid solution (1:2 methanol/chloroform at 2.5 mg/ml) was applied to Pt electrodes of an electroformation chamber. The solution was dried in vacuum for 3 h, then rehydrated in a 0.4 M sucrose. Electroformation was performed at 500Hz and 2.5 V for 2 h at 50°C. GUVs containing charged lipids (DOPC/DOPS, 4:1) were also prepared by hydration swelling to avoid potential issues with electroformation60. Briefly, glass slides were incubated with 1% agarose ultra-low gelling temperature (Sigma-Aldrich) for 2 min at ambient temperature. Slides were then heated at 40°C. After drying, 30 μl of lipid solution (4 mg/ml in CHCl3/MeOH 9:1) was spread over the film and evaporated. The slide was kept in vacuum overnight and rehydrated with 0.4 M sucrose solution the next day at ambient temperature for 2h. All GUVs were later stained by adding Di4 at 250 nM final concentration and incubated for 5 min. 10 μl of stained GUVs were rediluted into 200 μl of 0.4 M glucose solution (containing 0.5% agarose to prevent moving of GUV during FLIM) and imaged. To show efficient integration of DOPS into GUVs, we stained GUVs with Anx-V-640 after formation and visualized the fluorescent GUV in a fluorescent microscope (data not shown). To test the lifetime of Di4 under various buffer conditions, the GUVs were diluted into the following solutions (all brought to 400mOsm/L with glucose) prior to imaging: intracellular buffer (20 mM HEPES, 110 mM KCl, 10 mM NaCl, 2 mM MgCl2, 5 mM KH2PO4, pH 7.3), extracellular buffer (20 mM HEPES, 5 mM KCl, 140 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM KH2PO4, pH 7.3), 20 mM HEPES pH 5.2, and 20 mM HEPES pH 7.0.
Determination of Di4 flipping rate:
One milliliter of a chloroform/methanol (2:1) solution containing 10 mM DOPC +/− Di4 (molar ratio 1:200) was evaporated in a rotary evaporator under vacuum for 1 h and stored in a vacuum dessicator overnight. Multilamellar vesicles (MLV) were made according to the freeze-thawing method by hydrating the lipid film with buffer containing 10 mM Tris, 400 mM NaCl, pH 7.4. The MLV were then extruded 15 times through a 200 nm filter to obtain LUVs with a uniform size distribution. Size distribution was verified by dynamic light scattering and phospholipid content was determined by the inorganic phosphate assay. To determine the flipping speed, LUV were diluted to 100 μM phospholipids and we measured the magnitude of Di4 quenching (λex= 480, λem=650 nm) by potassium iodide (KI; 800 mM) over time at 20˚C. To that end, LUVs were post-labeled with Di4 by incubating pre-made LUVs with 500 nM Di4 for 8 min incubation at 4°C. KI was then added at different time points after labeling and the resulting fluorescence drop was measured in a fluorimeter (Clariostar, BMG Labtech, Ortenberg, Germany). LUVs prepared with Di4 on both leaflets (i.e. Di4 included during vesicle production) were used as a control to estimate the amount of quenching when equal abundance of Di4 was present on both leaflets. When these vesicles were sonicated in the presence of KI, we observed similar quenching as for outside-labeled vesicles.
Quenching efficiency was calculated as:
where Fquenched/Funuenched is the ratio of fluorescence intensity of Di4 in presence and absence of KI, respectively. The percentage of Di4 on the outer leaflet at various timepoints was calculated as:
where QEDi4out is the quenching efficiency of Di4 in LUVs where Di4 was only added to the outer leaflet and QEDi4, both is the quenching efficiency of LUVs in which Di4 was present at both leaflet. The factor 2 comes from the fact that only half of the Di4 molecules are on the outer leaflet in that configuration. Three independent experiments were done with triplicate measurements for quenched and unquenched LUVs.
Fluorescent Correlation Spectroscopy:
We measured the diffusion coefficient of GPI-GFP and SH4-mNeonGreen on CHO cells using fluorescence correlation spectroscopy. CHO cells were seeded on glass slides (#1.5) two days before the measurement. One day before the measurement, cells were transfected using Lipofectamine 3000 using the protocol supplied by the manufacturer. Both proteins were localized almost exclusively on the PM (not shown). FCS on transfected cells was carried out at room temperature using Zeiss LSM 880 microscope, 40X water immersion objective (numerical aperture 1.2). The laser spot was focused on the basal membrane of the cells by finding the focal plane of maximum fluorescence intensity. Then, 3–5 curves were obtained for each spot (five seconds each). The obtained curves were fit using the freely available FoCuS-point software using 2D and triplet model.
Bioinformatics:
We analyzed all transmembrane domains from single-pass transmembrane proteins annotated in the Uniprot database as previously described33. Lipid accessible surface area (ASA) of TMDs was calculated from structural / statistical predictions based on amino acid sequence, as described previously33. The exoplasmic/luminal and cytoplasmic portions of the TMD were defined as first and second half of the transmembrane domain according to the annotated orientation of the protein in the plasma membrane (i.e. N-terminal or C-terminal outside). For TMDs with an uneven number of amino acids, the ASA of the amino acid in the center of the membrane was divided by 2 and half of its ASA was attributed to each half of the exo/cytoplasmic side.
Supplementary Material
ACKNOWLEDGEMENTS
All fluorescence microscopy was performed at the Center for Advanced Microscopy, Department of Integrative Biology & Pharmacology at McGovern Medical School, UTHealth. We thank Neal Waxham for his generous sharing of the microinjection system. We gratefully acknowledge Kai Simons, Theodore Steck, Yvonne Lange, and Gerald Feigenson for their critical feedback on this manuscript. Funding for this work was provided by the NIH/National Institute of General Medical Sciences (GM114282, GM124072, GM120351, GM134949), the Volkswagen Foundation (grant 93091), and the Human Frontiers Science Program (RGP0059/2019). ES is funded by Newton-Katip Ҫelebi Institutional Links grant (352333122). Anton2 computer time was provided by the National Resource for Biomedical Supercomputing (NRBSC), the Pittsburgh Supercomputing Center (PSC), and the Biomedical Technology Research Center for Multiscale Modeling of Biological Systems through grant P41GM103712-S1 from the National Institutes of Health. All authors have no competing interests.
Footnotes
COMPETING FINANCIAL INTERESTS
None of the authors have competing interests.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.
References
- 1.Devaux PF Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30, 1163–1173 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Op den Kamp JA Lipid asymmetry in membranes. Annu Rev Biochem 48, 47–71, doi: 10.1146/annurev.bi.48.070179.000403 (1979). [DOI] [PubMed] [Google Scholar]
- 3.Verkleij AJ et al. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta 323, 178–193 (1973). [DOI] [PubMed] [Google Scholar]
- 4.Schick PK, Kurica KB & Chacko GK Location of phosphatidylethanolamine and phosphatidylserine in the human platelet plasma membrane. The Journal of clinical investigation 57, 1221–1226, doi: 10.1172/JCI108390 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandra A & Pagano RE Phospholipid asymmetry in LM cell plasma membrane derivatives: polar head group and acyl chain distributions. Biochemistry 17, 332–338 (1978). [DOI] [PubMed] [Google Scholar]
- 6.Bollen IC & Higgins JA Phospholipid asymmetry in rough- and smooth-endoplasmic-reticulum membranes of untreated and phenobarbital-treated rat liver. Biochem J 189, 475–480 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Q & London E The influence of natural lipid asymmetry upon the conformation of a membrane-inserted protein (perfringolysin O). J Biol Chem 289, 5467–5478, doi: 10.1074/jbc.M113.533943 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doktorova M et al. Preparation of asymmetric phospholipid vesicles for use as cell membrane models. Nat Protoc 13, 2086–2101, doi: 10.1038/s41596-018-0033-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enoki TA & Feigenson GW Asymmetric bilayers by hemifusion: method and leaflet behaviors. Biophys J 117, 1037–1050, doi: 10.1016/j.bpj.2019.07.054 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiantia S & London E Acyl chain length and saturation modulate interleaflet coupling in asymmetric bilayers: effects on dynamics and structural order. Biophys J 103, 2311–2319, doi: 10.1016/j.bpj.2012.10.033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heberle FA et al. Subnanometer Structure of an Asymmetric Model Membrane: Interleaflet Coupling Influences Domain Properties. Langmuir 32, 5195–5200, doi: 10.1021/acs.langmuir.5b04562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng HT, Megha & London E Preparation and properties of asymmetric vesicles that mimic cell membranes: effect upon lipid raft formation and transmembrane helix orientation. J Biol Chem 284, 6079–6092, doi: 10.1074/jbc.M806077200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doktorova M et al. Gramicidin Increases Lipid Flip-Flop in Symmetric and Asymmetric Lipid Vesicles. Biophys J, doi: 10.1016/j.bpj.2019.01.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharpe HJ, Stevens TJ & Munro S A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169, doi: 10.1016/j.cell.2010.05.037 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrot G et al. Asymmetric lateral mobility of phospholipids in the human erythrocyte membrane. Proc Natl Acad Sci U S A 83, 6863–6867 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.el Hage Chahine JM, Cribier S & Devaux PF Phospholipid transmembrane domains and lateral diffusion in fibroblasts. Proc Natl Acad Sci U S A 90, 447–451 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka KI & Ohnishi S Heterogeneity in the fluidity of intact erythrocyte membrane and its homogenization upon hemolysis. Biochim Biophys Acta 426, 218–231 (1976). [DOI] [PubMed] [Google Scholar]
- 18.Gupta A, Korte T, Herrmann A & Wohland T Plasma membrane asymmetry of lipid organization: fluorescence lifetime microscopy and correlation spectroscopy analysis. J Lipid Res 61, 252–266, doi: 10.1194/jlr.D119000364 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schroeder F Differences in fluidity between bilayer halves of tumour cell plasma membranes. Nature 276, 528–530 (1978). [DOI] [PubMed] [Google Scholar]
- 20.Schachter D, Abbott RE, Cogan U & Flamm M Lipid fluidity of the individual hemileaflets of human erythrocyte membranes. Annals of the New York Academy of Sciences 414, 19–28 (1983). [DOI] [PubMed] [Google Scholar]
- 21.Rimon G, Meyerstein N & Henis YI Lateral mobility of phospholipids in the external and internal leaflets of normal and hereditary spherocytic human erythrocytes. Biochim Biophys Acta 775, 283–290 (1984). [DOI] [PubMed] [Google Scholar]
- 22.Harayama T et al. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metab 20, 295–305, doi: 10.1016/j.cmet.2014.05.019 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Yeung T et al. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213, doi: 10.1126/science.1152066 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Levental I, Janmey PA & Cebers A Electrostatic contribution to the surface pressure of charged monolayers containing polyphosphoinositides. Biophys J 95, 1199–1205, doi: 10.1529/biophysj.107.126615 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sodt AJ, Venable RM, Lyman E & Pastor RW Nonadditive Compositional Curvature Energetics of Lipid Bilayers. Phys Rev Lett 117, 138104, doi: 10.1103/PhysRevLett.117.138104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingolfsson HI et al. Lipid organization of the plasma membrane. J Am Chem Soc 136, 14554–14559, doi: 10.1021/ja507832e (2014). [DOI] [PubMed] [Google Scholar]
- 27.Cui H, Lyman E & Voth GA Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys J 100, 1271–1279, doi: 10.1016/j.bpj.2011.01.036 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin L et al. Characterization and application of a new optical probe for membrane lipid domains. Biophys J 90, 2563–2575, doi: 10.1529/biophysj.105.072884 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owen DM et al. Fluorescence lifetime imaging provides enhanced contrast when imaging the phase-sensitive dye di-4-ANEPPDHQ in model membranes and live cells. Biophys J 90, L80–82, doi: 10.1529/biophysj.106.084673 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sezgin E, Sadowski T & Simons K Measuring lipid packing of model and cellular membranes with environment sensitive probes. Langmuir 30, 8160–8166, doi: 10.1021/la501226v (2014). [DOI] [PubMed] [Google Scholar]
- 31.Wesseling MC et al. Novel Insights in the Regulation of Phosphatidylserine Exposure in Human Red Blood Cells. Cell Physiol Biochem 39, 1941–1954, doi: 10.1159/000447891 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Diaz-Rohrer BB, Levental KR, Simons K & Levental I Membrane raft association is a determinant of plasma membrane localization. Proc Natl Acad Sci U S A 111, 8500–8505, doi: 10.1073/pnas.1404582111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorent JH et al. Structural determinants and functional consequences of protein affinity for membrane rafts. Nature communications 8, 1219, doi: 10.1038/s41467-017-01328-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi T & Menon AK Transbilayer lipid asymmetry. Curr Biol 28, R386–R391, doi: 10.1016/j.cub.2018.01.007 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Yamakawa T & Nagai Y Glycolipids at the cell surface and their biological functions. Trends Biochem Sci 3, 128–131 (1978). [Google Scholar]
- 36.Levental I, Cebers A & Janmey PA Combined electrostatics and hydrogen bonding determine intermolecular interactions between polyphosphoinositides. J Am Chem Soc 130, 9025–9030, doi:doi: 10.1021/ja800948c (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steck TL & Lange Y Transverse distribution of plasma membrane bilayer cholesterol: Picking sides. Traffic 19, 750–760, doi: 10.1111/tra.12586 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Courtney KC et al. C24 Sphingolipids Govern the Transbilayer Asymmetry of Cholesterol and Lateral Organization of Model and Live-Cell Plasma Membranes. Cell Rep 24, 1037–1049, doi: 10.1016/j.celrep.2018.06.104 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Liu SL et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat Chem Biol 13, 268–274, doi: 10.1038/nchembio.2268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steck TL & Lange Y How slow is the transbilayer diffusion (flip-flop) of cholesterol? Biophys J 102, 945–946; author reply 947–949, doi: 10.1016/j.bpj.2011.10.059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iaea DB & Maxfield FR Membrane order in the plasma membrane and endocytic recycling compartment. PloS one 12, e0188041, doi: 10.1371/journal.pone.0188041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levental I, Levental KR & Heberle FA Lipid rafts: controversies resolved, mysteries remain. Trends in cell biology in press (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Q & London E Ordered raft domains induced by outer leaflet sphingomyelin in cholesterol-rich asymmetric vesicles. Biophys J 108, 2212–2222, doi: 10.1016/j.bpj.2015.03.056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiessling V, Crane JM & Tamm LK Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys J 91, 3313–3326, doi: 10.1529/biophysj.106.091421 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veatch SL & Keller SL Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett 94, 148101 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Kusumi A et al. Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu. Rev. Cell Dev.Biol 28, 215–250 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Honigmann A et al. A lipid bound actin meshwork organizes liquid phase separation in model membranes. eLife 3, e01671, doi: 10.7554/eLife.01671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levental KR et al. Homeostatic remodeling of mammalian membranes in response to dietary lipids is essential for cellular fitness. Nat Comm in press, doi: 10.1101/342873 (2020). [DOI] [Google Scholar]
- 49.Hussain NF, Siegel AP, Ge Y, Jordan R & Naumann CA Bilayer asymmetry influences integrin sequestering in raft-mimicking lipid mixtures. Biophys J 104, 2212–2221, doi: 10.1016/j.bpj.2013.04.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frewein M, Kollmitzer B, Heftberger P & Pabst G Lateral pressure-mediated protein partitioning into liquid-ordered/liquid-disordered domains. Soft Matter 12, 3189–3195, doi: 10.1039/c6sm00042h (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McIntyre JC & Sleight RG Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30, 11819–11827 (1991). [DOI] [PubMed] [Google Scholar]
- 52.Levental KR et al. omega-3 polyunsaturated fatty acids direct differentiation of the membrane phenotype in mesenchymal stem cells to potentiate osteogenesis. Science advances 3, eaao1193, doi: 10.1126/sciadv.aao1193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Surma MA et al. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. European journal of lipid science and technology : EJLST 117, 1540–1549, doi: 10.1002/ejlt.201500145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu EL et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. Journal of Computational Chemistry 35, 1997–2004, doi: 10.1002/jcc.23702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venable Richard M. et al. CHARMM All-Atom Additive Force Field for Sphingomyelin: Elucidation of Hydrogen Bonding and of Positive Curvature. Biophysical Journal 107, 134–145, doi: 10.1016/j.bpj.2014.05.034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shan Y, Klepeis JL, Eastwood MP, Dror RO & Shaw DE Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. The Journal of Chemical Physics 122, 054101, doi:doi: 10.1063/1.1839571 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Camley BA, Lerner MG, Pastor RW & Brown FLH Strong influence of periodic boundary conditions on lateral diffusion in lipid bilayer membranes. The Journal of Chemical Physics 143, 243113, doi:doi: 10.1063/1.4932980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui H, Lyman E & Voth GA Mechanism of Membrane Curvature Sensing by Amphipathic Helix Containing Proteins. Biophysical journal 100, 1271–1279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Q, Wang X, Ma S, Zhang Y & Han X Electroformation of giant unilamellar vesicles in saline solution. Colloids and surfaces. B, Biointerfaces 147, 368–375, doi: 10.1016/j.colsurfb.2016.08.018 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Steinkuhler J, De Tillieux P, Knorr RL, Lipowsky R & Dimova R Charged giant unilamellar vesicles prepared by electroformation exhibit nanotubes and transbilayer lipid asymmetry. Scientific reports 8, 11838, doi: 10.1038/s41598-018-30286-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.