

Abstract
Phosphorus plays a vital role in diverse biological processes including intracellular signaling, membrane integrity, and skeletal biomineralization; therefore, the regulation of phosphorus homeostasis is essential to the well-being of the organism. Cells and whole organisms respond to changes in inorganic phosphorus (Pi) concentrations in their environment by adjusting Pi uptake and altering biochemical processes in cells (local effects) and distant organs (endocrine effects). Unicellular organisms, such as bacteria and yeast, express specific Pi-binding proteins on the plasma membrane that respond to changes in ambient Pi availability and transduce intracellular signals that regulate the expression of genes involved in cellular Pi uptake. Multicellular organisms, including humans, respond at a cellular level to adapt to changes in extracellular Pi concentrations and also have endocrine pathways which integrate signals from various organs (e.g., intestine, kidneys, parathyroid glands, bone) to regulate serum Pi concentrations and whole body phosphorus balance. In mammals, alterations in the concentrations of extracellular Pi modulate type III sodium-phosphate cotransporter activity on the plasma membrane, and trigger changes in cellular function. In addition, elevated extracellular Pi induces activation of fibroblast growth factor receptor, Raf/mitogen-activated protein kinase/ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) and Akt pathways, which modulate gene expression in various mammalian cell types. Excessive Pi exposure, especially in patients with chronic kidney disease, leads to endothelial dysfunction, accelerated vascular calcification, and impaired insulin secretion.
Keywords: phosphorus, signaling, phosphate, chronic kidney disease
Introduction
Phosphorus plays a crucial role in skeletal development, mineral metabolism, and diverse cellular signaling. Phosphorus is an essential component of phospholipids in plasma membranes and organelle membranes, nucleotides that provide cellular energy and serve as fundamental units of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), and phosphorylated intermediates in many intracellular signaling pathways1. In addition, phosphorus is necessary for several enzymatic processes including glycolysis, renal ammoniagenesis, and mitochondrial oxidative phosphorylation, resulting in cellular production of adenosine triphosphate (ATP)2. Moreover, phosphorus regulates oxygen-carrying capacity by its role in determining the generation of red blood cell 2,3-diphosphoglycerate (2,3-DPG) which shifts the hemoglobin-oxygen dissociation curve to the right, thus allowing better oxygenation of tissues.
Phosphorus is present in almost all body fluids. In human serum, phosphorus exists in the form of inorganic phosphorus, lipid phosphorus, and phosphoric ester phosphorus. Total serum phosphorus concentrations range from 8.9 to 14.9 mg/dL (2.87 to 4.81 mmol/L), inorganic phosphorus (Pi) concentrations from 2.56 to 4.16 mg/dL (0.83 to 1.34 mmol/L), phosphoric ester phosphorus concentrations from 2.5 to 4.5 mg/dL (0.81 to 1.45 mmol/L), and lipid phosphorus concentrations from 6.9 to 9.7 mg/dL (2.23 to 3.13 mmol/L). Serum Pi concentrations are usually measured clinically, referred to as the serum phosphorus or phosphate, and the normal range varies with age, with the highest concentration being in infants. Approximately 85% of phosphorus in the body is present in bones, 14% exists in cells of soft tissues, and only 1% is present in extracellular fluids, where it serves an important role as a buffer to maintain total body pH. In the serum, approximately 10% of the Pi content is bound to proteins, 5% is complexed with calcium, magnesium, or sodium, and 85% is free Pi (HPO42- and H2PO4−)3. The ratio of intracellular and extracellular Pi concentration is approximately 100 to 14. At the normal blood pH of 7.40, the pKa for Pi is 6.8 and the ratio of HPO42- to H2PO4− is 4:1.
Dysregulation of the phosphorus homeostasis critically affects cellular metabolism, musculoskeletal and cardiovascular functions. A variety of mechanisms have evolved to increase the efficiency of phosphorus retention in states of phosphorus deficiency, and conversely, to decrease phosphorus retention in states of phosphorus excess in order to maintain phosphorus balance at the cellular and whole body levels. Obviously, an important primary step in the regulation of phosphorus homeostasis is the ability of cells to detect changes in extracellular Pi concentrations. This would imply the existence of a “sensor” within or on the surface of cells that is capable of detecting changes in the extracellular concentration of Pi. Changes in extracellular Pi concentration could affect cellular function directly or alter the overall balance of phosphorus in the body by influencing sites of phosphorus retention or excretion by various hormones. Serum Pi concentrations are regulated within a narrow range via the activity of various hormones such as parathyroid hormone (PTH), fibroblast growth factor 23 (FGF23), and 1,25-dihydroxyvitamin D (1,25(OH)2D). In this review, we will discuss and provide existing evidences regarding the Pi sensing mechanisms, regulation of phosphorus homeostasis, and role of extracellular Pi as a signaling molecule focusing on mammals.
Cellular Sensing and Endocrine Sensing of Phosphate
A relevant question regarding the regulation of phosphorus homeostasis is how the individual cell or the whole organism senses changes in ambient Pi concentrations and adjusts metabolic processes to accommodate such changes. For the sake of simplicity, Pi sensing can be broadly classified into two systems: cellular sensing and endocrine sensing. Cellular Pi sensing mainly functions to maintain appropriate levels of Pi in the intracellular compartment to support cellular metabolism, whereas endocrine Pi sensing primarily maintains extracellular Pi concentrations and phosphorus balance at a whole-body level. Whether the “cellular” and the “endocrine” sensors are identical and use the same or different signal transduction pathway is largely unknown.
In unicellular organisms, such as bacteria and yeast, transcellular Pi uptake is controlled at the plasma membrane, where changes in extracellular Pi concentration stimulate signal transduction pathways that regulate the expression of phosphate transporters, and enzymes involved in cellular Pi uptake5,6. Emerging evidence suggests that similar but more complex events are at work to mediate the cellular effects of Pi in multicellular organisms including humans. Moreover, the responsiveness of mammalian cells to fluctuations of extracellular Pi concentrations suggests that they have Pi sensor proteins on their plasma membranes which sense ambient Pi concentrations, with or without requiring intracellular uptake of Pi for regulating intracellular signal transduction in individual cells7. Whether cellular Pi sensing requires the cellular uptake of Pi is dependent upon the cell type being examined. For example, intracellular Pi uptake via PiT-1 in vascular smooth muscle cells is necessary for initiation of vascular calcification, whereas Pi binding on PiT-1/PiT-2 heterodimers in plasma membrane is sufficient to stimulate the production of matrix Gla protein and osteopontin in MC3T3-E1 osteoblast-like cells8.
In multicellular organisms, concentrations of intracellular Pi are influenced by pH, hormones (e.g., adrenaline, insulin, insulin-like growth factor 1)9,10, and subcellular compartmentalization; Pi levels in these compartments may be regulated by separate phosphate transporters in mitochondria11, lysosomes12, and the endoplasmic reticulum13. For example, a fall in the partial pressure of carbon dioxide in the blood during acute respiratory alkalosis results in a reduction in serum Pi concentrations. The change in the partial pressure of carbon dioxide in blood is associated with a reduction in intracellular carbon dioxide concentrations because carbon dioxide readily diffuses across plasma membranes. The resulting increase in intracellular pH stimulates phosphofructokinase activity which in turn stimulates glycolysis14. Production of glycolytic intermediates and ATP is enhanced, thus consuming cytoplasmic Pi, which in turn induces a shift of Pi from serum into cells with a subsequent elevation of intracellular Pi concentrations. A rise in plasma adrenaline or insulin concentrations also causes shifts of Pi from the extracellular to intracellular compartment, thereby increasing intracellular Pi concentration.
Endocrine Pi signaling, which develop in multicellular organisms including humans, should be viewed as the interorgan signaling that integrates the signals from multiple sites such as the intestine, kidneys, parathyroid glands, and bone, to maintain phosphorus homeostasis via hormonal regulation. In adult, the serum Pi concentration is maintained within the narrow range of 2.56 to 4.16 mg/dL and is the result of intestinal absorption, influx into and efflux from bone, renal tubular reabsorption, and modest intestinal secretion15. The serum Pi concentration fluctuates slightly with age, dietary phosphorus intake, acid-base status, and diurnal variation. Phosphorus homeostasis in humans is tightly regulated by three key hormones, including PTH, FGF23, and 1,25(OH)2D, all of which interact with each other through feedback loops16 (Fig. 1).
Fig. 1.
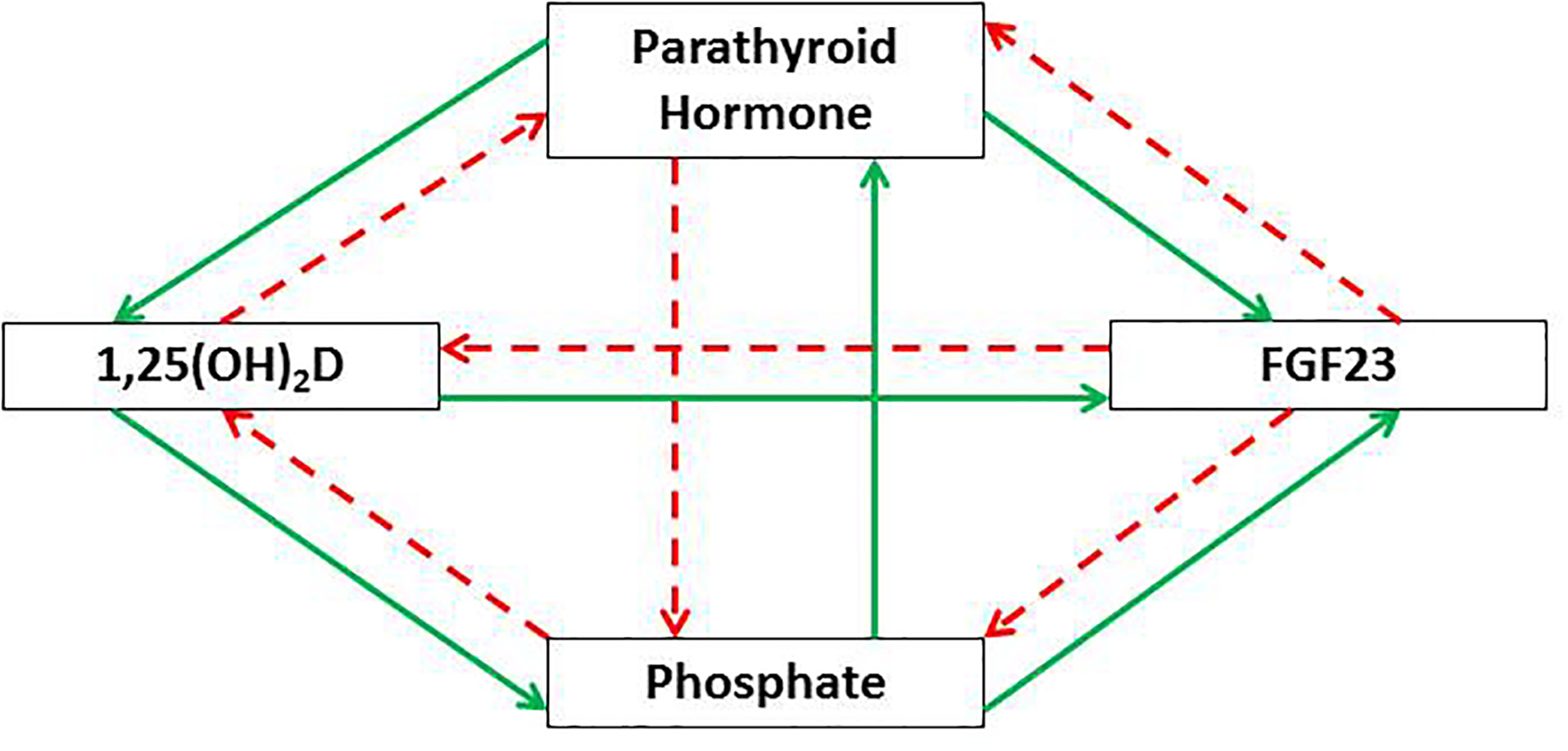
Regulation of phosphorus homeostasis by the PTH-FGF23-vitamin D axis. PTH and FGF23 act on the proximal tubule to reduce renal tubular Pi reabsorption. PTH also stimulates the renal production of 1,25(OH)2D which subsequently promotes Pi absorption in the intestine and kidney as well as inhibits PTH production via negative feedback. In contrast, FGF23 and Pi suppress the renal production of 1,25(OH)2D. In the parathyroid gland, Pi stimulates secretion of PTH, whereas this secretion is inhibited by FGF23. The production of FGF23 in the bone is stimulated by PTH, 1,25(OH)2D, and Pi. (Dashed red arrows indicate inhibition, and solid green arrows indicate stimulation).
Cellular Phosphate Sensing in Bacteria and Yeast
Bacterial cells and the unicellular yeast, Saccharomyces cerevisiae, are able to respond to changes in Pi concentrations in the environment and appropriately increase the retention or rejection of Pi. In Escherichia coli, extracellular Pi concentration is sensed by ATP-binding cassette (ABC)-type phosphate specific transporter (Pst) and a protein called PhoU in the periplasmic membrane17. Under phosphorus-depleted condition, the Pst which functions as a Pi sensor facilitates the phosphorylation of histidine residues on PhoR present in the periplasmic membrane; phosphorylated PhoR, in turn, phosphorylates aspartate residues on the cytosolic protein, PhoB. Phosphorylated PhoB acts as a transcription factor by binding to specific DNA sequences known as “PHO boxes” to regulate the transcription of target genes in the Pho regulon. Conversely, under phosphorus-saturated condition, PhoU inhibits PhoR activity and dephosphorylated PhoR no longer phosphorylates PhoB; unphosphorylated PhoB is incapable of binding PHO boxes and activating the transcription (Fig. 2). Many of the genes in the Pho regulon encode a number of protein products, such as alkaline phosphatase and phosphate transporters, that facilitate bacterial adaptation to changes in ambient Pi concentrations17.
Fig. 2.
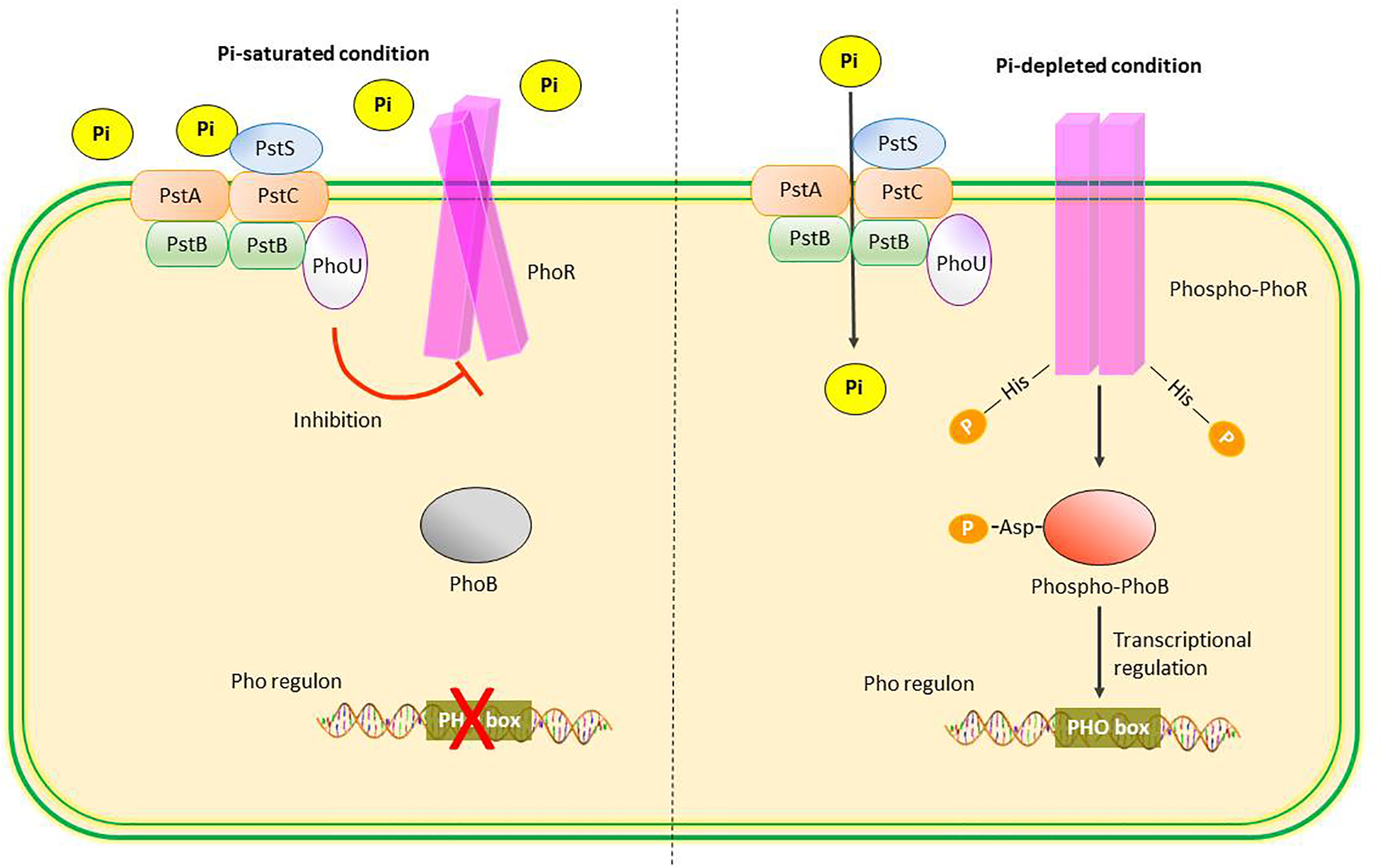
Phosphate sensing in Escherichia coli. The Pi specific transporter (Pst) in periplasmic membrane consists of 4 subunits: PstS, PstA, PstB, and PstC, and functions as a Pi sensor. Under conditions of Pi saturation, PhoU inhibits PhoR activity, leading to increased levels of unphosphorylated PhoB and inactivation of Pho regulon genes. Under conditions of Pi depletion, PhoR is activated by autophosphorylation which in turn phosphorylates PhoB. The phosphorylated PhoB acts as a transcription factor binding to the PHO box of Pho regulon and regulating the transcription of target genes involved in Pi conservation.
In Saccharomyces cerevisiae, when environmental phosphorus is limited, the intracellular synthesis of diphosphoinositol pentakisphosphate (IP7) is increased; IP7, in turn, activates the cyclin-dependent kinase (CDK) inhibitor, Pho81. Pho81 inactivates the Pho80-Pho85 cyclin-CDK complex. As a result the transcription factor, Pho4, is dephosphorylated and activated, leading to the induction of PHO genes, one of which encodes Pho84 protein that functions as a high-affinity phosphate transporter in the plasma membrane and scavenges Pi from the environment5. When environmental phosphorus is no longer limiting, Pho84 is rapidly internalized from the plasma membrane and the transcription factor, Pho4, is phosphorylated and exported from the nucleus to the cytoplasm, thereby turning off the expression of the PHO genes5 (Fig. 3). It is noteworthy that, in bacteria and yeast, the cellular Pi sensing for adaptation to phosphorus deprivation is stimulated by default, and binding of a saturated amount of Pi to plasma membrane transceptors elicits the intracellular signal to switch off the system.
Fig. 3.
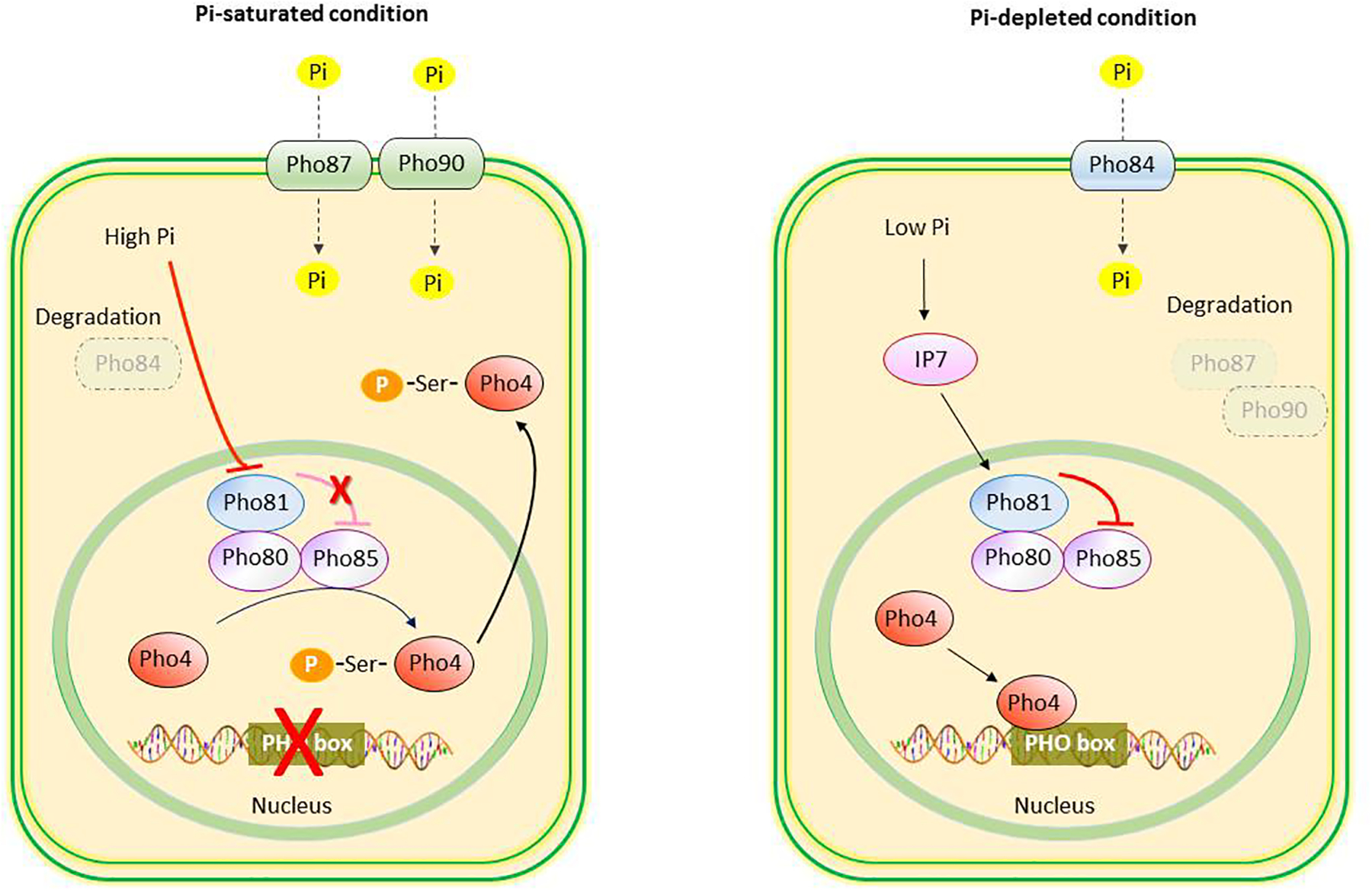
Phosphate sensing in Saccharomyces cerevisiae. Under conditions of Pi saturation, Pho84 is degraded, and Pho87 and Pho90 transporters facilitate the transcellular transport of Pi. Increased concentrations of Pi leads to inhibition of Pho81 activity, which renders Pho80-Pho85 kinase complex to phosphorylate transcription factor Pho4. The phosphorylated Pho4 is exported from the nucleus to the cytoplasm, and the expression of PHO regulon is turned off. Under conditions of Pi depletion, increased concentrations of diphosphoinositol pentakisphosphate (IP7) activate Pho81 which prevents the phosphorylation of Pho4 by the Pho80-Pho85. The unphosphorylated Pho4 binds to the PHO box of Pho regulon and activates the transcription of genes including high-affinity Pho84 Pi transporters and secreted acid phosphatase.
Phosphate Transceptors for Phosphate Sensing in Mammals
In multicellular organisms, phosphorus levels should be sensed at a whole-body level to maintain the homeostasis of extracellular phosphorus, as well as at a cellular level to regulate the effects of phosphorus on cellular function. Considering that bacteria and yeast use some specific types of phosphate transporters as Pi sensors, it is reasonable to postulate that mammalian cells also may use phosphate transporters on the plasma membrane to sense and adapt to phosphorus availability. However, mammals may have less risk of phosphorus deficiency because they can seek out phosphorus-containing foods and conserve phosphorus in the skeleton.
In mammals, two families of sodium-dependent phosphate cotransporters, type II (SLC34) and type III (SLC20), are responsible for the inward transport of extracellular Pi6,18 (Table 1). The type II sodium-dependent phosphate cotransporters are the primary phosphate transporters in the kidney (NaPi-2a and NaPi-2c) and the intestine (NaPi-2b). The type III sodium-dependent phosphate cotransporters, which include PiT-1 and PiT-2, are ubiquitously expressed across a variety of cell types and are generally considered to be involved in supplying cells with Pi to facilitate the needs of individual cell functions. Among the known mammalian phosphate transporters, PiT-1 is a potential candidate for a phosphate transceptor because many studies in cultured mammalian cells suggested the involvement of PiT-1 in the signal transduction and gene regulation triggered by altered extracellular Pi concentrations19–21. A recent study in MC3T3-E1 cells demonstrated that PiT-1 and PiT-2 form high-abundance homodimers and low-abundance heterodimers; heterodimerization is facilitated by increased extracellular Pi concentrations8. Deletion of either transporter blunted phosphate-dependent ERK1/2-mediated phosphorylation and changes in gene expression of the biomineralization inhibitory factors, matrix Gla protein and osteopontin. Interestingly, ERK1/2-mediated phosphorylation could be rescued by overexpressing phosphate-transport deficient PiT mutants. We discuss the role of PiT-1 and PiT-2 and the requirement for phosphate transport in osteoblasts in a later section.
Table 1.
Characteristics of phosphate transporters
| SLC34 Family (Type II Na/Pi cotransporter) | SLC20 Family (Type III Na/Pi cotransporter) | ||||
|---|---|---|---|---|---|
| NaPi-2a | NaPi-2b | NaPi-2c | PiT-1 | PiT-2 | |
| Human gene | SLC34A1 | SLC34A2 | SLC34A3 | SLC20A1 | SLC20A2 |
| Gene locus | Chromosome 5 | Chromosome 4 | Chromosome 2 | Chromosome 2 | Chromosome 8 |
| Predominant transported species of Pi | HPO42- | HPO42- | HPO42- | H2PO4- | H2PO4- |
| Na+:Pi stoichiometry | 3:1 | 3:1 | 2:1 | 2:1 | 2:1 |
| Ion transport property | Electrogenic | Electrogenic | Electroneutral | Electrogenic | Electrogenic |
| Major organ expression | Kidney | Intestine, lung | Kidney | Ubiquitous expression | Kidney, neuron |
| Genetic disorders associated with inactivating mutation |
|
Pulmonary alveolar microlithiasis | Hereditary hypophosphatemic rickets with hypercalciuria | Unknown | Familial idiopathic basal ganglia calcification (Fahr’s disease) |
Abbreviations: NaPi, sodium-phosphate cotransporter; PiT, phosphate transporter
Interestingly, there is a convincing evidence showing that PiT-1 can modulate cell proliferation in human cell lines22. Using RNA interference in HeLa and HepG2 cells, it has been shown that PiT-1 depletion markedly reduces cell proliferation, delays cell cycle, and impairs mitosis and cytokinesis. In vivo, injection of HeLa cells lacking PiT-1 in nude mice results in reduced tumor growth. In PiT-1 non-transporting mutants, the modulation of cell proliferation through PiT-1 expression is still maintained and cannot be compensated by PiT-2 overexpression. PiT-1 is likely to function as a critical sensor protein that regulates cell proliferation in certain cell types, independent from its previously known phosphate transport activity22. However, the precise mechanism by which PiT-1 modulates cell proliferation require further investigation.
Intestinal Phosphate Absorption
Intestinal Pi transport occurs via two pathways: a passive paracellular pathway and an active transcellular transport pathway, with the majority of Pi absorption by passive paracellular transport. The intestine has high paracellular Pi permeability that favors H2PO4− influx over HPO42- influx and allows a higher Pi absorption in the proximal small intestine, where lower luminal pH and higher luminal Pi concentrations drive paracellular Pi absorption23. Passive transport mechanisms generally depend on electrochemical Pi gradients across an intestinal epithelial layer with paracellular movement occurring through tight junction complexes that are formed by the interaction of adhesive proteins of adjacent enterocytes. Two major components of tight junction proteins, occludins and claudins express along the gastrointestinal tract and may have an important role in controlling specific ion transport. These intestinal tight junction proteins are regulated by signal transduction pathways, actively interact with cytoskeleton proteins, and provide selective paracellular permeability24,25. However, specific tight junction proteins for paracellular Pi transport have so far been not identified.
The transcellular mechanism of intestinal Pi transport depends on active transport predominantly through NaPi-2b transporters26. In addition, PiT-1 and, to a lesser extent, PiT-2 transporters also contribute to intestinal Pi transport27–29. NaPi-2b is expressed on the apical membrane of intestinal epithelial cells, and aids in the coupled transport of sodium and Pi from the intestinal lumen into the intestinal epithelial cells, especially when dietary phosphorus is low. However, the precise mechanism of Pi transport across the basolateral membrane of intestinal epithelial cells remains unknown. NaPi-2b expression is upregulated by the administration of 1,25(OH)2D and dietary phosphorus restriction30,31. PTH does not increase NaPi-2b expression in the intestine directly, but activates it indirectly by its stimulatory effect on renal synthesis of 1,25(OH)2D, which enhances NaPi-2b expression32. An earlier study has demonstrated that increases in the expression of the NaPi-2b occur when vitamin D receptor knockout mice are fed a low phosphorus diet31. This would imply that the intestine is capable of responding to low dietary phosphorus levels by upregulating NaPi-2b expression independent of 1,25(OH)2D.
Renal Phosphate Handling
In the steady state of neutral phosphorus balance, the amount of phosphorus absorbed in the intestine (approximately 0.8–1.5 g per 24 h) is equivalent to the amount excreted in the urine, reflecting that phosphorus homeostasis and serum Pi levels are maintained primarily through the renal Pi handling. Approximately 90% of serum Pi (free and complexed forms) is freely filtered by the glomerulus and subsequently 80% of filtered Pi is reabsorbed, occurring mainly in the proximal tubule. Renal tubular Pi reabsorption or secretion do not occur in the distal nephron. The extent of renal tubular Pi reabsorption depends on dietary intake of phosphorus and a variety of other factors, such as acid-base status and hormones. For the average diet, fractional excretion of Pi in adults is in the range of 10–30% of the filtered load.
NaPi-2a and NaPi-2c transporters, are expressed at the apical membrane of proximal tubule, and are collectively responsible for the reabsorption of approximately 80% of filtered Pi. Studies using NaPi-2a–/– mice have shown that NaPi-2a and NaPi-2b account for 70% and 30% of renal Pi reabsorption, respectively33,34. NaPi-2a expressed at the highest level in the apical membrane of S1 segments of the proximal tubule and level of expression gradually decrease toward the S3 segment, whereas NaPi-2c is mainly detected in S1 and S2 segments and is absent in S3 segments of proximal tubule35. Changes in the urinary excretion of Pi are almost invariably parallel changes in the apical membrane expression of NaPi-2a and NaPi-2c in the proximal tubules36.
Even in the absence of NaPi-2a and NaPi-2c expression, there is some residual renal tubular Pi reabsorption37. Thus, other renal phosphate transporters are able to maintain some Pi reabsorption in the kidney. PiT-2 transporter has been localized in the apical membrane of proximal tubule, and its expression is regulated by dietary phosphorus38. In states of normal phosphorus intake, PiT-2 expression is largely restricted to the S1 segment, and chronic dietary phosphorus restriction induces apical membrane expression of PiT-2 in all segments of the proximal tubule38. PiT-2 adapts to dietary phosphorus composition more slowly than the NaPi-2a, and the involvement of PiT-2 in phosphorus uptake can vary with luminal pH39.
Until recently, the precise mechanism of basolateral Pi efflux has been elusive; recent data indicate that a retroviral receptor, xenotropic and polytropic retrovirus receptor 1 (XPR1), potentially could function as a basolateral phosphate transporter in the kidney of humans and mice40–42 (Fig. 4). Mice with conditional inactivation of XPR1 gene in the renal tubule exhibit generalized proximal tubular dysfunction (Fanconi syndrome) with decreased renal Pi reabsorption and hypophosphatemic rickets, suggesting a crucial role of XPR1 in the renal tubular Pi reclamation41.
Fig. 4.

Cellular mechanism of Pi reabsorption in the proximal tubule. The majority of filtered Pi is reabsorbed by the electrogenic NaPi-IIa transporter in the apical membrane and depend on the basolateral Na+/K+-ATPase activity to maintain the Na+ gradient that drives the secondary active transport process. Electroneutral NaPi-IIc and electrogenic PiT-2 transporter also contribute to the apical Pi influx. Basolateral Pi efflux possibly occur by facilitated diffusion through XPR1.
Abbreviation: XPR1, xenotropic and polytropic retrovirus receptor 1
Factors Regulating Renal Phosphate Excretion
Renal Pi handling is tightly regulated by many factors including dietary phosphorus intake, acid-base homeostasis, potassium status, and a variety of hormones. The renal excretion of Pi is increased by high dietary intake of phosphorus, during metabolic acidosis, hypokalemia, by PTH, FGF23, Klotho (by both FGF23-dependent and FGF23-independent signaling), calcitonin, dopamine, estrogen, and glucocorticoids18. In contrast, the renal tubular reabsorption of Pi is stimulated by low dietary intake of phosphorus, during metabolic alkalosis, by 1,25(OH)2D, growth hormone, insulin, insulin-like growth factor 1, epidermal growth factor, and thyroid hormone16,43–47. The primary mechanism responsible for adjusting renal tubular Pi reabsorption is the expression of NaPi-2a and, to a lesser extent, NaPi-2c transporters in the apical membrane of proximal tubule.
Short-term Adaptation to Changes in Dietary Phosphorus
There are several lines of evidence demonstrating the presence of intestine-kidney axis that rapidly responds to changes in intestinal luminal ion concentrations by increasing the renal excretion of those particular ions rapidly after oral administration. For example, it is known that natriuretic responses following oral sodium chloride feeding are more marked than the responses seen following the intravenous infusion of sodium chloride, suggesting the presence of both sensing and effector mechanisms in the intestine for sodium. Guanylin and uroguanylin have been identified as intestinal natriuretic factors produced in the intestine that contribute to the enhanced natriuresis elicited by high salt loads in the postprandial state48. Gastrointestinal sensing has also been identified in the case of potassium49. Finally, the extracellular calcium-sensing receptor is expressed abundantly throughout gastrointestinal tract and responds to dietary L-amino acids by regulation of gut hormone secretion, intestinal epithelial transport, and satiety50.
According to the signaling mechanism of rapid enteric-renal response for various ions, a mechanism similar to that mentioned above may exist for phosphorus as well. It is important to conceptualize the regulation of Pi transport in terms of rapid, short-term changes that occur within a short period (minutes to hours) after the ingestion of a phosphorus-containing meal and those that occur over the long term (over a period of days) in association with chronic changes in the dietary intake of phosphorus. It is recognized that the short-term changes are presumably mediated by processes that are distinct from those required for more long-term adaptations to alterations in dietary phosphorus. Previous human study has shown that early and rapid increases in urinary Pi excretion occur within 1 hour following a high-phosphorus diet, without significant changes in plasma concentrations of PTH, FGF23, or 1,25(OH)2D suggesting the presence of an intestinal Pi sensor51. Additionally, the adaptation of renal tubular Pi reabsorption to acute variations in dietary phosphorus levels does not involve alterations in messenger RNA (mRNA) levels of NaPi-2a and NaPi-2b in the proximal tubule52.
Consistently, Berndt et al36 shown that intact or parathyroidectomized rats administered Pi into the duodenum rapidly altered renal Pi reabsorption. The short-term changes in the urinary fractional excretion of Pi were examined. It was shown that in intact rats, the administration of Pi into the duodenum was associated with a rapid increase in the urinary fractional excretion of Pi within 10 minutes. At the early time points, there were trivial changes in serum Pi concentrations. Conversely, rats administered sodium chloride did not show a significant change in the urinary fractional excretion of Pi. Plasma concentrations of PTH, FGF23 and secreted frizzled-related protein 4 (sFRP4) did not significantly change despite rises in the urinary fractional excretion of Pi following the intraduodenal Pi loading. These observations augmented by showing that parathyroidectomy did not alter the response to the intraduodenal Pi loading. An important caveat is that plasma concentrations of matrix extracellular phosphoglycoprotein (MEPE), FGF7, and insulin were not measured and it is not known whether concentrations of these phosphaturic substances increased following the intraduodenal Pi loading. The investigators also demonstrated that the increase in the urinary fractional excretion of Pi following the intraduodenal Pi loading was maintained in rats receiving bilateral renal denervation. Moreover, homogenates of the duodenal mucosa infused into rats increased the urinary fractional excretion of Pi demonstrating the presence of one or more intestinal factors that signal to the kidney to rapidly increase the urinary Pi excretion. This mechanism would allow for a rapid crosstalk between intestine and kidney and provide a feedforward mechanism preventing a potentially detrimental hyperphosphatemia after ingestion of a phosphorus-containing meal. However, the exact molecular identity of the intestinal Pi factor is presently unknown.
In contrast to results in rodents, a recent study in healthy human where the response to acute intragastric and intravenous Pi loading was compared found no strong evidence for intestinal-specific phosphaturic control mechanisms53. In this study, the earliest phosphaturia was observed only after plasma Pi and PTH concentrations had risen following both intragastric and intravenous Pi administration, and there was nearly identical phosphaturic response between two methods, both quantitatively and temporally. The early rise of plasma PTH was partly due to hyperphosphatemia in the absence of significant hypocalcemia and later due to the expected hypocalcemia in response to Pi loading. Interestingly, the plasma FGF23 level was increased after the onset of phosphaturia, followed by a decrease in plasma 1,25(OH)2D levels. These findings suggest that PTH may be an important direct secretagogue for FGF23 and a late decrease in plasma 1,25(OH)2D levels is due to an inhibitory effect of FGF23 and Pi on renal 1-alpha-hydroxylase activity. Moreover, a recent study in rats demonstrated that either intravenous or intragastric Pi loading resulted in the onset of phosphaturia which is paralleled by a rise in plasma PTH and precedes changes in plasma FGF23 and 1,25(OH)2D levels, suggesting a predominant role of PTH for short-term adaptations to alterations in dietary phosphorus intake by inducing early phosphaturia54. Interestingly, no phosphaturia could be elicited by intragastric Pi loading in parathyroidectomized rats, supporting the PTH-requiring mechanism of phosphaturia. To prove the existence of a PTH-independent, intestine-to-kidney signaling to regulate phosphorus homeostasis after an acute Pi loading, identification of the intestinal phosphatonin will be definitely required.
Long-term Adaptation to Changes in Dietary Phosphorus
Long-term adaptations to alterations in dietary phosphorus intake are apparently associated with distinct mechanisms. Persistent changes in serum Pi concentration as a result of chronic alterations in dietary phosphorus intake primarily modulate the gene expression of NaPi-2a and NaPi-2b in the proximal tubule. In mice responding to increases and decreases in dietary phosphorus intake, a DNA sequence responsible for phosphate response has been reported in the NaPi-2a gene. The phosphate-response element of the NaPi-2a gene consists of the motif related to the E-box, 5’-CACGTG-3’. The authors identified the mouse transcription factor muE3 (TFE3) bound to the phosphate-response element and confirmed the upregulation of the renal TFE3 expression in mice fed a low-phosphorus diet55. These results suggested that low dietary intake of phosphorus increases the renal TFE3 expression and activates the transcription of NaPi-2a gene in the kidney. Nevertheless, it has not been clarified yet whether the direct trigger for the induction of renal TFE3 expression is a decrease in serum Pi or unknown molecules transducing a signal from the intestine to the kidney. Moreover, a number of studies have demonstrated that the transcriptional regulation of NaPi-2a gene by TFE3 appears to be less physiologically important than the hormonal regulation of the abundance of NaPi-2a on the apical membrane, which is mediated by microtubule-dependent translocation of presynthesized NaPi-2a protein into the apical membrane of proximal tubule56–58.
The adaptive changes of the kidney to chronic variations in dietary phosphorus intake are accompanied by modifications in the plasma concentration of several hormones including PTH, FGF23, and 1,25(OH)2D. Animals fed a low phosphorus diet have decreased serum Pi concentrations that are associated with a reciprocal increase in serum calcium concentrations. The increase in serum calcium concentrations inhibits PTH release, which in turn, reduces the renal excretion of Pi. Pi also directly stimulates PTH release; hence, hypophosphatemia potentially reduces PTH secretion. Most studies demonstrated that high phosphorus diet increased and a low phosphorus diet decreased serum FGF23 levels independent of serum Pi levels in human within a period of days after changing dietary phosphorus intake59–61, suggesting the relative importance of FGF23 for long-term adaptations to alterations in dietary phosphorus intake. Additionally, a low phosphorus diet and reductions in serum Pi concentration are associated with increased 1,25(OH)2D synthesis as a result of stimulation of 1-alpha-hydroxylase activity and reduction of 24-hydroxylase activity in the kidney62. The increases in 1-alpha-hydroxylase activity in animals fed a low phosphorus diet are independent of PTH effect because they occur in parathyroidectomized animals63,64. Moreover, a low phosphorus diet elicits an increase in serum 1,25(OH)2D concentration, renal 1-alpha-hydroxylase activity and mRNA abundance in NaPi-2 knockout mice, demonstrating that the renal expression of NaPi-2a and NaPi-2c is not required for the regulation of 1-alpha-hydroxylase by Pi65.
Signal Transduction Pathways Induced by Extracellular Phosphate
The current evidences demonstrate that extracellular Pi itself modulates the functions of various cells via several specific signaling pathways. Increased extracellular Pi concentration activates the Raf/Mitogen-activated protein kinase/ERK kinase (MEK)/Extracellular signal-regulated kinase (ERK) pathway and directly regulates gene expression in various mammalian cell lines, including ATDC5 chondrogenic cells, MC3T3-E1 osteoblastic-like cells, and human embryonic kidney 293 (HEK293) cells66. Elevated extracellular Pi triggers signal transduction via PiT-1 and FGFR in the plasma membrane and influences intracellular FGF23 signaling in HEK293 cells. Increasing the extracellular Pi concentration results in the phosphorylation of FGF receptor substrate 2α (FRS2α), the plasma membrane-associated adapter protein, and subsequently activates the Raf/MEK/ERK pathway, suggesting that the signaling evoked by elevated extracellular Pi partially shares the same downstream cascade as FGF23 signaling66.
Various transcription factors have been shown to mediate the cellular effects of increased extracellular Pi concentration. In mammalian cell lines, activator protein 1 (AP1), nuclear factor erythroid 2-related factor 2 (Nrf2), and early growth response 1 (Erg1), function as the transcription factors that are upregulated by increased extracellular Pi concentration67–69. The activation of Raf/MEK/ERK pathway induced by increased extracellular Pi, which involves FGFR and PiT-1, causes the translocation of these transcription factors from the cytoplasm to the nucleus and subsequently regulates the expression of phosphate-responsive genes (Fig. 5). Similar responses to increased extracellular Pi levels were also observed in human cell lines70.
Fig. 5.

Possible signal transduction pathway triggered by extracellular Pi in mammalian cells. Type III Na/Pi cotransporter, especially PiT-1, plays a major role in the sensing of the cells to the altered extracellular Pi concentration. Increased extracellular Pi binds to type III Na/Pi cotransporter on the plasma membrane, then induces activation of FGFR, Raf/MEK/ERK pathway, and Akt pathway, which subsequently regulates gene transcription through binding of several transcription factors (e.g., AP1, Nrf2, Egr1) to the phosphate response element in the nucleus. The signaling molecule(s) that link between type III Na/Pi cotransporter and FGFR are currently unknown.
Abbreviations: AP1, activator protein 1; Egr1, early growth response 1; ERK, extracellular signal-regulated kinase; FGFR, fibroblast growth factor receptor; FRS2α, fibroblast growth factor receptor substrate 2α; MEK, mitogen-activated protein kinase/ERK kinase; Nrf2, nuclear factor erythroid 2-related factor 2
Recent data suggest that both PiT-1 and PiT-2 transporters are important in modulating extracellular phosphate mediated cellular events in osteoblasts8. Deletion of either transporter blunted phosphate-dependent ERK1/2-mediated phosphorylation and subsequent changes in gene expression of the biomineralization inhibitory factors matrix Gla protein and osteopontin. Interestingly, ERK1/2-mediated phosphorylation could be rescued by overexpressing phosphate-transport deficient PiT mutants. Extracellular phosphate concentrations regulate the homodimerization and heterodimerization of the PiT-1 and PiT-2 transporters on the osteoblast plasma membrane. These findings suggest that phosphate binding rather than phosphate uptake is important in mediating phosphate signaling through the PiT proteins in osteoblasts. It is interesting to note that the concentrations of phosphate used to induce changes in cellular events in osteoblasts were exceptionally high (10 mM). Whether such changes occur in vivo in osteoblasts is to be determined.
Several other signaling pathways are also regulated by extracellular Pi. In hyperphosphatemic alpha-Klotho-KO mice, high levels of extracellular Pi enhance the phosphorylation of Akt (protein kinase B) and thereby activating the downstream cascade mammalian target of rapamycin complex 1 (mTORC1) pathway by suppressing plasma membrane-bound phosphatase and tensin homolog (PTEN) levels, this process is PiT-1-dependent. In brown adipose tissue, the Pi-induced activation of Akt/mTORC1 signaling pathway results in the increased oxidative stress and premature aging71. It should be noted that the signal transduction pathways including the Raf/MEK/ERK pathway and the Akt/mTORC1 pathway are not only regulated by extracellular Pi concentration but also by a vast repertoire of cytokines and growth factors, depending upon the cell type. Moreover, it needs to be clarified whether cellular and endocrine sensing of Pi in humans involve the same or different signal transduction pathways.
Physiological and Pathological Effects Induced by Phosphate Signaling
Effects of Extracellular Phosphate on PTH secretion
The primary role of the parathyroid glands is to function as a “calciostat”; hence, PTH secretion by the parathyroid glands is mainly regulated by extracellular calcium on a transcriptional and post-transcriptional level. Similarly, 1,25(OH)2D inhibits PTH gene expression via vitamin D responsive elements in the PTH promoter. Moreover, in vitro incubation of human parathyroid glands with escalating concentrations of Pi while keeping ionized calcium constant stimulated PTH secretion, suggesting that the parathyroid gland is capable of sensing changes in extracellular Pi concentration in the absence of changes in serum calcium and 1,25(OH)2D72,73.
Chronic elevation of serum Pi stimulates PTH secretion by indirect and direct mechanisms. Persistent hyperphosphatemia stimulates PTH secretion presumably by lowering extracellular calcium concentration, while hypophosphatemia suppresses PTH secretion indirectly by up-regulation of 1,25(OH)2D synthesis. Increased 1,25(OH)2D levels also stimulate FGF23 synthesis, which acts at the parathyroid glands to suppress PTH mRNA transcription and PTH secretion in an alpha-klotho-dependent mechanism.
In addition to the indirect effects, extracellular Pi directly acts on the parathyroid glands to stimulate PTH secretion through the phospholipase A2-arachidonic acid signaling pathway. High extracellular Pi concentration stimulates the secretion of PTH secretion by diminishing the production of phospholipase A2-derived arachidonic acid in parathyroid cells74. Moreover, high-phosphorus diet stimulates expression of transforming growth factor-alpha (TGF-alpha) in the uremic rat parathyroid gland and constitutes an autocrine signal that enhances parathyroid cell proliferation75. Conversely, low-phosphorus diet induces p21 gene transcription and mediate the antiproliferative effects of dietary phosphorus restriction on uremia-induced parathyroid cell growth.
PiT-1 may play a role in the responsiveness of rat parathyroid glands to extracellular Pi, however, it remains unproven whether entry of Pi into the parathyroid cells is required for the regulation of PTH synthesis and parathyroid cell growth76. In rats fed a low-phosphorus diet, the abundance of parathyroid PiT-1 mRNA is higher than when on a high-phosphorus diet. Interestingly, a recent study has revealed binding sites for Pi on the extracellular domain of human CaSR, and their results suggest that the binding of Pi reinforces the inactive conformation of CaSR77. The Pi binding to CaSR might have substantial influence on the sensing of calcium and Pi levels as well as on PTH secretion.
Pi regulates PTH gene expression posttranscriptionally by increasing the stability of PTH mRNA in rats. Using Northwestern blot analysis, RNA electrophoretic mobility shift and affinity cross-linking studies of parathyroid extracts, cytosolic trans-acting factors were identified that bind to a cis-acting instability element in the PTH mRNA 3’-untranslated region (UTR)78. There is a balanced interaction of PTH mRNA with the stabilizing proteins, including AU-rich binding factor 1 (AUF1) and Upstream of N-ras (Unr), and the destabilizing protein, called K-homology splicing regulator protein (KSRP)79,80. In the setting of hyperphosphatemia or hypocalcemia the peptidyl-prolyl cis/trans isomerase (Pin1) is inactive, resulting in KSRP phosphorylation and hence its inactivation81. This presumably allows AUF1 and Unr to bind the PTH mRNA 3’-UTR with a greater affinity, leading to enhanced PTH mRNA stability. Conversely, hypophosphatemia and hypercalcemia lead to destabilization of the PTH mRNA, which then is targeted for degradation by the RNA exosome complexes. However, the mechanistic links between the regulation of PTH mRNA stability by Pi in human and the Pin1/KSRP pathway remain to be elucidated.
Effects of Extracellular Phosphate on FGF23 secretion
FGF23 is a bone-derived hormone, which primarily secreted by osteocytes and osteoblasts, and has the major role in the regulation of phosphate and vitamin D metabolism82. FGF23 downregulates NaPi-2a and NaPi-2c expression in the apical membrane of proximal tubule, which resembles the action of PTH, and results in increased renal excretion of Pi. In addition, FGF23 inhibits renal 1,25(OH)2D synthesis by 1-alpha-hydroxylase (CYP27B1) and stimulates renal 1,25(OH)2D catabolism by 24-hydroxylase (CYP24A1), thereby reducing serum levels of 1,25(OH)2D. N-acetylgalactosaminyltransferase 3 (GALNT3) is the glycosyltransferase enzyme that is responsible for O-glycosylation of FGF23 in the Golgi apparatus, preventing its proteolytic cleavage into inactive fragments and therefore allowing the secretion of intact FGF23.
FGF23 synthesis is stimulated by high dietary or serum Pi levels and by PTH, 1,25(OH)2D, and calcium83. Using in vivo and ex vivo murine models, the Pi-induced stimulation of FGF23 secretion was blunted in PiT-2 KO mice and isolated bones, highlighting PiT-2 as a putative Pi sensor in the bone cells20. Moreover, a recent study unraveled the mechanism of Pi-induced FGF23 secretion by demonstrating that high extracellular Pi activated the unliganded FGF receptor and ERK pathway, in association with enhanced expression of GALNT3 mRNA in osteoblastic UMR 106 cells84. Thus, O-glycosylation of FGF23 by the GALNT3 gene product prevented the proteolytic cleavage, leading to an increased in the serum level of FGF23 in mice fed a high-phosphorus diet.
Effects of Extracellular Phosphate on Renal 1-Alpha-Hydroxylase Activity
Cholecalciferol is produced from its precursors 7-dehydrocholesterol in the epidermis, 25-hydroxylated in the liver and subsequently converted to the active vitamin D metabolite, 1,25(OH)2D, in proximal tubular epithelial cells by the 1-alpha-hydroxylase85. PTH and hypocalcemia induce renal expression of CYP27B1, the gene encoding 1-alpha-hydroxylase86. In contrast, FGF23, 1,25(OH)2D, and hypercalcemia reduce renal expression of CYP27B1. However, whether Pi directly regulates renal CYP27B1 expression remains controversial. In primary mouse kidney cells, low phosphorus conditions directly increase the activity of 1-alpha-hydroxylase87,88, but direct effects of Pi on renal CYP27B1 expression have not been replicated by other studies89,90. Moreover, the precise upstream sensing mechanism and how Pi regulates renal CYP27B1 expression in human remain to be further studied.
Effects of Extracellular Phosphate on Pancreatic Insulin Secretion
In pancreatic beta cells, cellular uptake of Pi via PiT-1 and PiT-2 transporters prompted cytosolic alkalinization, which in turn facilitated Pi transport into the mitochondrial matrix91. Pi transport into mitochondria is essential for the generation of ATP, a signaling factor in energy-requiring insulin secretion. However, excessive extracellular Pi increased the mitochondrial membrane potential, induced the mitochondrial hyperpolarization, and stimulated ROS generation. In addition, high Pi also promoted the opening of the mitochondrial permeability transition pore in the inner mitochondrial membrane, leading to loss of mitochondrial antioxidants and increase of cytosolic calcium concentrations, which can activate calcium-dependent caspases and consequently induce apoptosis of pancreatic beta cells92. Therefore, the reduced insulin secretion and diminished pancreatic beta cell viability are commonly observed under hyperphosphatemic states, and these contribute to the development of glycemic dysregulation in patients with CKD93,94.
Effects of Extracellular Phosphate on Vascular Endothelial Dysfunction
Acute or sustained exposure to Pi causes impaired endothelium-dependent vasodilatation95,96. The mechanism underlying this detrimental effect of Pi on vascular endothelial function is apparently due to the disruption of nitric oxide signaling. Exposing vascular endothelial cells to a Pi load increases the generation of reactive oxygen species, which depend on transcellular Pi influx via PiT-1 and PiT-2 transporters, and decreased nitric oxide production via inhibitory phosphorylation of endothelial nitric oxide synthase97–100. Therefore, the deleterious effects of hyperphosphatemia, independent of other factors in the uremic milieu, may be partly responsible for enhanced oxidative stress, systemic endothelial dysfunction, and the increased cardiovascular risk in patients with chronic kidney disease (CKD)96.
Effects of Extracellular Phosphate on Vascular Calcification
Vascular calcification has been consistently shown to be a significant contributing factor to cardiovascular disease which is the leading cause of mortality in patients with CKD101–103. Patients with CKD exhibit accelerated calcification of the tunica intima, tunica media, heart valves, and myocardium as well as the rare but devastating condition of calcific uremic arteriolopathy (calciphylaxis). The initiation and progression of vascular calcification in patients with CKD is caused by a combination of various pathological factors104,105. Disturbances of mineral homeostasis, most notably hyperphosphatemia, are considered primary determinants of vascular calcification in CKD106,107. Even in patients with preserved renal function, high serum Pi levels are associated with more severe coronary artery calcification108.
Among patients with CKD, there are two types of vascular calcification with different pathogenesis: intimal calcification and medial calcification109,110. However, these may also coexist and share overlaps in their risk factors and signaling pathway, especially vascular inflammation111. Intimal calcification is secondary to established atherosclerosis which is highly prevalent in CKD patients. Although the pathogenesis of atherosclerosis appears to be the same in CKD as in non-CKD patients, it usually takes a more aggressive course in the CKD population, partly due to the effect of hyperphosphatemia on atherosclerotic plaque progression112. Intimal calcification in conjunction with atherosclerotic plaque formation lead to vascular obstruction, impaired organ perfusion, and eventually plaque rupture and ischemia113,114. Medial calcification develops from the excessive calcium-phosphate deposition in the medial layer of the arteries, in an active process similar to bone ossification. Medial calcification mainly decreases vascular distensibility leading to increased arterial stiffness and an increased pulse pressure, thereby increases cardiac afterload115,116. This significantly contributes to the risk of left ventricular hypertrophy, heart failure, and myocardial infarction117. Additionally, the arterial stiffness caused by medial calcification directly contributes to the arterial wall shear stress, atherosclerosis, and progression of intimal calcification118.
In human vascular smooth muscle cells (VSMCs), intracellular Pi uptake occurs primarily through PiT-1, which function as both Pi sensors and transporters104. In addition, the activation of Toll-like receptor 4 in VSMCs may be involve in Pi sensing mechanism119. PiT-1 expression is increased by the activation of intracellular mineralocorticoid receptor (MR) in VSMCs120,121. Recent study has shown that spironolactone, a MR antagonist, dose‑dependently attenuated vascular calcification by downregulation of PiT‑1 expression in VSMCs122. In contrast to PiT-1, PiT-2 in vascular smooth muscle cells might have a protective role against phosphate-induced vascular calcification123. The mechanism whereby PiT-2 protects against vascular calcification is currently not known.
Cellular uptake of Pi and a subsequent rise in intracellular Pi levels is required to initiate phenotypic transformation of contractile VSMCs into osteoblast-like cells, a phenomenon called “osteogenic transdifferentiation”. The transdifferentiated VSMCs express a variety of osteogenic and chondrogenic transcription factors including core-binding factor subunit alpha-1 (CBFA1), also known as Runt-related transcription factor 2 (RUNX2), Msh homeobox 2 (MSX2), and osterix124–127. Subsequently, the downregulation of smooth muscle-specific proteins, such as alpha-smooth muscle actin and smooth muscle protein 22-alpha, is also observed128,129. The osteochondrogenic transcription factors further induce the expression of osteogenic- and chondrogenic-specific proteins in VSMCs such as osteocalcin, type I collagen, and alkaline phosphatase. Eventually, the transdifferentiated VSMCs promote the vascular calcification by producing local procalcifying environment and nidus sites for accelerated precipitation of calcium and Pi as well as growth of calcium phosphate crystals in the arterial wall. In summary, PiT-1 is necessary for Pi-induced VSMC osteogenic transdifferentiation and initiation of vascular calcification in patients with CKD.
Conclusions
Phosphorus is a crucial element for all living organisms. Unicellular organisms such as bacteria and yeast sense to changes in ambient Pi concentration via a protein complex in plasma membrane and a specialized transcription factor. In contrast to a simple mechanism of Pi sensing in unicellular organisms, the multicellular organisms including humans develop more complex systems of Pi sensing (cellular sensing and endocrine sensing) in order to maintain serum Pi levels within an appropriate range, which is essential for numerous cellular functions, normal cell growth, and bone biomineralization. Cellular Pi sensing primarily aims to regulate the effects of extracellular Pi on cell metabolism, whereas endocrine Pi sensing in the intestine, parathyroid gland, kidney, and bone is vital for maintenance of phosphorus homeostasis at a whole-body level. In mammals, a body of evidence support the type III sodium-phosphate cotransporter (PiT-1 or PiT-2, or both, depending on the specific cell type) as a cell-surface Pi sensor triggered by changes in extracellular Pi concentration. Additionally, the Pi-induced activation of Raf/MEK/ERK pathway and Akt pathway has been consistently shown to modulate gene expression in various mammalian cells. However, whether cellular and endocrine sensing of Pi in humans share the same Pi sensor or signal transduction pathway remains to be established.
References
- 1.Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signal. 2011;4:re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angelova PR, Baev AY, Berezhnov AV, Abramov AY. Role of inorganic polyphosphate in mammalian cells: from signal transduction and mitochondrial metabolism to cell death. Biochem Soc Trans. 2016;44:40–5. [DOI] [PubMed] [Google Scholar]
- 3.Amanzadeh J, Reilly RF. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract. 2006;2:136–48. [DOI] [PubMed] [Google Scholar]
- 4.Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int. 2013;17:479–82. [DOI] [PubMed] [Google Scholar]
- 5.Mouillon J-M, Persson BL. New aspects on phosphate sensing and signalling in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:171–6. [DOI] [PubMed] [Google Scholar]
- 6.Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol. 2007;293:F643–654. [DOI] [PubMed] [Google Scholar]
- 7.Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a Signaling Molecule and Its Sensing Mechanism. Physiol Rev. 2018;98:2317–48. [DOI] [PubMed] [Google Scholar]
- 8.Bon N, Couasnay G, Bourgine A, Sourice S, Beck-Cormier S, Guicheux J, et al. Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J Biol Chem. 2018;293:2102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polgreen KE, Kemp GJ, Leighton B, Radda GK. Modulation of Pi transport in skeletal muscle by insulin and IGF-1. Biochim Biophys Acta. 1994;1223:279–84. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Stimulation of sodium-dependent inorganic phosphate transport by activation of Gi/o-protein-coupled receptors by epinephrine in MC3T3-E1 osteoblast-like cells. Bone. 2001;28:589–94. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira GC, Pedersen PL. Phosphate transport in mitochondria: past accomplishments, present problems, and future challenges. J Bioenerg Biomembr. 1993;25:483–92. [DOI] [PubMed] [Google Scholar]
- 12.Pisoni RL. Characterization of a phosphate transport system in human fibroblast lysosomes. J Biol Chem. 1991;266:979–85. [PubMed] [Google Scholar]
- 13.Burchell A Endoplasmic reticulum phosphate transport. Kidney Int. 1996;49:953–8. [DOI] [PubMed] [Google Scholar]
- 14.Brautbar N, Leibovici H, Massry SG. On the mechanism of hypophosphatemia during acute hyperventilation: evidence for increased muscle glycolysis. Miner Electrolyte Metab. 1983;9:45–50. [PubMed] [Google Scholar]
- 15.Peters J, Binswanger U. Calcium and inorganic phosphate secretion of rat ileum in vitro. Influence of uremia and 1,25 (OH)2D3 inhibition. Res Exp Med (Berl). 1988;188:139–49. [DOI] [PubMed] [Google Scholar]
- 16.Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamarche MG, Wanner BL, Crépin S, Harel J. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev. 2008;32:461–73. [DOI] [PubMed] [Google Scholar]
- 18.Levi M, Gratton E, Forster IC, Hernando N, Wagner CA, Biber J, et al. Mechanisms of phosphate transport. Nat Rev Nephrol. 2019;15:482–500. [DOI] [PubMed] [Google Scholar]
- 19.Kimata M, Michigami T, Tachikawa K, Okada T, Koshimizu T, Yamazaki M, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone. 2010;47:938–47. [DOI] [PubMed] [Google Scholar]
- 20.Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab. 2018;11:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Yang H-Y, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–12. [DOI] [PubMed] [Google Scholar]
- 22.Beck L, Leroy C, Salaün C, Margall-Ducos G, Desdouets C, Friedlander G. Identification of a novel function of PiT1 critical for cell proliferation and independent of its phosphate transport activity. J Biol Chem. 2009;284:31363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knöpfel T, Himmerkus N, Günzel D, Bleich M, Hernando N, Wagner CA. Paracellular transport of phosphate along the intestine. Am J Physiol. 2019; [DOI] [PubMed] [Google Scholar]
- 24.Amasheh S, Fromm M, Günzel D. Claudins of intestine and nephron - a correlation of molecular tight junction structure and barrier function. Acta Physiol. 2011;201:133–40. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. 2017;1397:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2010;299:F285–296. [DOI] [PubMed] [Google Scholar]
- 27.Bai L, Collins JF, Ghishan FK. Cloning and characterization of a type III Na-dependent phosphate cotransporter from mouse intestine. Am J Physiol Cell Physiol. 2000;279:C1135–1143. [DOI] [PubMed] [Google Scholar]
- 28.Olah Z, Lehel C, Anderson WB, Eiden MV, Wilson CA. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J Biol Chem. 1994;269:25426–31. [PubMed] [Google Scholar]
- 29.Christakos S, Lieben L, Masuyama R, Carmeliet G. Vitamin D endocrine system and the intestine. BoneKEy Rep. 2014;3:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Bai L, Collins JF, Ghishan FK. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). Am J Physiol Cell Physiol. 2002;282:C487–493. [DOI] [PubMed] [Google Scholar]
- 31.Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, et al. Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol. 2004;287:F39–47. [DOI] [PubMed] [Google Scholar]
- 32.Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2010;299:F285–296. [DOI] [PubMed] [Google Scholar]
- 33.Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K. Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol. 2003;285:F1271–1278. [DOI] [PubMed] [Google Scholar]
- 34.Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95:5372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trepiccione F, Capasso G. SGK3: a novel regulator of renal phosphate transport? Kidney Int. 2011;80:13–5. [DOI] [PubMed] [Google Scholar]
- 36.Berndt T, Thomas LF, Craig TA, Sommer S, Li X, Bergstralh EJ, et al. Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A. 2007;104:11085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segawa H, Onitsuka A, Kuwahata M, Hanabusa E, Furutani J, Kaneko I, et al. Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol. 2009;20:104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, et al. The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol. 2009;296:F691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villa-Bellosta R, Sorribas V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch. 2010;459:499–508. [DOI] [PubMed] [Google Scholar]
- 40.Giovannini D, Touhami J, Charnet P, Sitbon M, Battini J-L. Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 2013;3:1866–73. [DOI] [PubMed] [Google Scholar]
- 41.Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, et al. Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron. J Am Soc Nephrol. 2017;28:1073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JRM, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet. 2015;47:579–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farrow EG, White KE. Recent advances in renal phosphate handling. Nat Rev Nephrol. 2010;6:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69:495–503. [DOI] [PubMed] [Google Scholar]
- 45.Breusegem SY, Takahashi H, Giral-Arnal H, Wang X, Jiang T, Verlander JW, et al. Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Renal Physiol. 2009;297:F350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murer H, Hernando N, Forster I, Biber J. Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol. 2003;65:531–42. [DOI] [PubMed] [Google Scholar]
- 47.Levi M, Gratton E, Forster IC, Hernando N, Wagner CA, Biber J, et al. Mechanisms of phosphate transport. Nat Rev Nephrol. 2019; [DOI] [PubMed] [Google Scholar]
- 48.Forte LR. A novel role for uroguanylin in the regulation of sodium balance. J Clin Invest. 2003;112:1138–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee FN, Oh G, McDonough AA, Youn JH. Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol. 2007;293:F541–547. [DOI] [PubMed] [Google Scholar]
- 50.Conigrave AD, Brown EM. Taste receptors in the gastrointestinal tract. II. L-amino acid sensing by calcium-sensing receptors: implications for GI physiology. Am J Physiol Gastrointest Liver Physiol. 2006;291:G753–761. [DOI] [PubMed] [Google Scholar]
- 51.Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006;70:2141–7. [DOI] [PubMed] [Google Scholar]
- 52.Ritthaler T, Traebert M, Lötscher M, Biber J, Murer H, Kaissling B. Effects of phosphate intake on distribution of type II Na/Pi cotransporter mRNA in rat kidney. Kidney Int. 1999;55:976–83. [DOI] [PubMed] [Google Scholar]
- 53.Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R. The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol. 2014;25:2730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas L, Bettoni C, Knöpfel T, Hernando N, Biber J, Wagner CA. Acute Adaption to Oral or Intravenous Phosphate Requires Parathyroid Hormone. J Am Soc Nephrol. 2017;28:903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kido S, Miyamoto K, Mizobuchi H, Taketani Y, Ohkido I, Ogawa N, et al. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. 1999;274:28256–63. [DOI] [PubMed] [Google Scholar]
- 56.Beck L, Tenenhouse HS, Meyer RA, Meyer MH, Biber J, Murer H. Renal expression of Na+-phosphate cotransporter mRNA and protein: effect of the Gy mutation and low phosphate diet. Pflugers Arch. 1996;431:936–41. [DOI] [PubMed] [Google Scholar]
- 57.Hoag HM, Martel J, Gauthier C, Tenenhouse HS. Effects of Npt2 gene ablation and low-phosphate diet on renal Na(+)/phosphate cotransport and cotransporter gene expression. J Clin Invest. 1999;104:679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kilav R, Silver J, Biber J, Murer H, Naveh-Many T. Coordinate regulation of rat renal parathyroid hormone receptor mRNA and Na-Pi cotransporter mRNA and protein. Am J Physiol. 1995;268:F1017–1022. [DOI] [PubMed] [Google Scholar]
- 59.Ito N, Fukumoto S, Takeuchi Y, Takeda S, Suzuki H, Yamashita T, et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab. 2007;25:419–22. [DOI] [PubMed] [Google Scholar]
- 60.Ferrari SL, Bonjour J-P, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–24. [DOI] [PubMed] [Google Scholar]
- 61.Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–9. [DOI] [PubMed] [Google Scholar]
- 62.Zhang S, Gillihan R, He N, Fields T, Liu S, Green T, et al. Dietary phosphate restriction suppresses phosphaturia but does not prevent FGF23 elevation in a mouse model of chronic kidney disease. Kidney Int. 2013;84:713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haussler M, Hughes M, Baylink D, Littledike ET, Cork D, Pitt M. Influence of phosphate depletion on the biosynthesis and circulating level of 1alpha,25-dihydroxyvitamin D. Adv Exp Med Biol. 1977;81:233–50. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka Y, Deluca HF. The control of 25-hydroxyvitamin D metabolism by inorganic phosphorus. Arch Biochem Biophys. 1973;154:566–74. [DOI] [PubMed] [Google Scholar]
- 65.Tenenhouse HS, Martel J, Gauthier C, Zhang MY, Portale AA. Renal expression of the sodium/phosphate cotransporter gene, Npt2, is not required for regulation of renal 1 alpha-hydroxylase by phosphate. Endocrinology. 2001;142:1124–9. [DOI] [PubMed] [Google Scholar]
- 66.Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem. 2010;111:1210–21. [DOI] [PubMed] [Google Scholar]
- 67.Conrads KA, Yi M, Simpson KA, Lucas DA, Camalier CE, Yu L-R, et al. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol Cell Proteomics. 2005;4:1284–96. [DOI] [PubMed] [Google Scholar]
- 68.Julien M, Magne D, Masson M, Rolli-Derkinderen M, Chassande O, Cario-Toumaniantz C, et al. Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology. 2007;148:530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, et al. Extracellular Phosphate Induces the Expression of Dentin Matrix Protein 1 Through the FGF Receptor in Osteoblasts. J Cell Biochem. 2017;118:1151–63. [DOI] [PubMed] [Google Scholar]
- 70.Camalier CE, Yi M, Yu L-R, Hood BL, Conrads KA, Lee YJ, et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol. 2013;228:1536–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawai M, Kinoshita S, Ozono K, Michigami T. Inorganic Phosphate Activates the AKT/mTORC1 Pathway and Shortens the Life Span of an α‑Klotho-Deficient Model. J Am Soc Nephrol. 2016;27:2810–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almaden Y, Canalejo A, Hernandez A, Ballesteros E, Garcia-Navarro S, Torres A, et al. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res. 1996;11:970–6. [DOI] [PubMed] [Google Scholar]
- 73.Almaden Y, Hernandez A, Torregrosa V, Canalejo A, Sabate L, Fernandez Cruz L, et al. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. J Am Soc Nephrol. 1998;9:1845–52. [DOI] [PubMed] [Google Scholar]
- 74.Almadén Y, Canalejo A, Ballesteros E, Añón G, Rodríguez M. Effect of high extracellular phosphate concentration on arachidonic acid production by parathyroid tissue in vitro. J Am Soc Nephrol. 2000;11:1712–8. [DOI] [PubMed] [Google Scholar]
- 75.Dusso AS, Pavlopoulos T, Naumovich L, Lu Y, Finch J, Brown AJ, et al. p21(WAF1) and transforming growth factor-alpha mediate dietary phosphate regulation of parathyroid cell growth. Kidney Int. 2001;59:855–65. [DOI] [PubMed] [Google Scholar]
- 76.Tatsumi S, Segawa H, Morita K, Haga H, Kouda T, Yamamoto H, et al. Molecular cloning and hormonal regulation of PiT-1, a sodium-dependent phosphate cotransporter from rat parathyroid glands. Endocrinology. 1998;139:1692–9. [DOI] [PubMed] [Google Scholar]
- 77.Geng Y, Mosyak L, Kurinov I, Zuo H, Sturchler E, Cheng TC, et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moallem E, Kilav R, Silver J, Naveh-Many T. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–9. [DOI] [PubMed] [Google Scholar]
- 79.Nechama M, Ben-Dov IZ, Briata P, Gherzi R, Naveh-Many T. The mRNA decay promoting factor K-homology splicing regulator protein post-transcriptionally determines parathyroid hormone mRNA levels. FASEB J. 2008;22:3458–68. [DOI] [PubMed] [Google Scholar]
- 80.Dinur M, Kilav R, Sela-Brown A, Jacquemin-Sablon H, Naveh-Many T. In vitro evidence that upstream of N-ras participates in the regulation of parathyroid hormone messenger ribonucleic acid stability. Mol Endocrinol Baltim Md. 2006;20:1652–60. [DOI] [PubMed] [Google Scholar]
- 81.Nechama M, Uchida T, Mor Yosef-Levi I, Silver J, Naveh-Many T. The peptidyl-prolyl isomerase Pin1 determines parathyroid hormone mRNA levels and stability in rat models of secondary hyperparathyroidism. J Clin Invest. 2009;119:3102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perwad F, Azam N, Zhang MYH, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358–64. [DOI] [PubMed] [Google Scholar]
- 84.Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci U S A. 2019;116:11418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol Rev. 2016;96:365–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21:319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Condamine L, Menaa C, Vrtovsnik F, Vztovsnik F, Friedlander G, Garabédian M. Local action of phosphate depletion and insulin-like growth factor 1 on in vitro production of 1,25-dihydroxyvitamin D by cultured mammalian kidney cells. J Clin Invest. 1994;94:1673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fukase M, Birge SJ, Rifas L, Avioli LV, Chase LR. Regulation of 25 hydroxyvitamin D3 1-hydroxylase in serum-free monolayer culture of mouse kidney. Endocrinology. 1982;110:1073–5. [DOI] [PubMed] [Google Scholar]
- 89.Gray RW. Control of plasma 1,25-(OH)2-vitamin D concentrations by calcium and phosphorus in the rat: effects of hypophysectomy. Calcif Tissue Int. 1981;33:485–8. [DOI] [PubMed] [Google Scholar]
- 90.Halloran BP, Spencer EM. Dietary phosphorus and 1,25-dihydroxyvitamin D metabolism: influence of insulin-like growth factor I. Endocrinology. 1988;123:1225–9. [DOI] [PubMed] [Google Scholar]
- 91.Nguyen TT, Quan X, Xu S, Das R, Cha S-K, Kong ID, et al. Intracellular alkalinization by phosphate uptake via type III sodium-phosphate cotransporter participates in high-phosphate-induced mitochondrial oxidative stress and defective insulin secretion. FASEB J. 2016;30:3979–88. [DOI] [PubMed] [Google Scholar]
- 92.Nguyen TT, Quan X, Hwang K-H, Xu S, Das R, Choi S-K, et al. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am J Physiol Endocrinol Metab. 2015;308:E933–941. [DOI] [PubMed] [Google Scholar]
- 93.Kovesdy CP, Sharma K, Kalantar-Zadeh K. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis. 2008;52:766–77. [DOI] [PubMed] [Google Scholar]
- 94.Williams ME, Garg R. Glycemic management in ESRD and earlier stages of CKD. Am J Kidney Dis. 2014;63:S22–38. [DOI] [PubMed] [Google Scholar]
- 95.Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M, et al. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol. 2009;20:1504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stevens KK, Denby L, Patel RK, Mark PB, Kettlewell S, Smith GL, et al. Deleterious effects of phosphate on vascular and endothelial function via disruption to the nitric oxide pathway. Nephrol Dial Transplant. 2017;32:1617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peng A, Wu T, Zeng C, Rakheja D, Zhu J, Ye T, et al. Adverse effects of simulated hyper- and hypophosphatemia on endothelial cell function and viability. PloS One. 2011;6:e23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thambyrajah J, Landray MJ, McGlynn FJ, Jones HJ, Wheeler DC, Townend JN. Abnormalities of endothelial function in patients with predialysis renal failure. Heart. 2000;83:205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van TV, Watari E, Taketani Y, Kitamura T, Shiota A, Tanaka T, et al. Dietary phosphate restriction ameliorates endothelial dysfunction in adenine-induced kidney disease rats. J Clin Biochem Nutr. 2012;51:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Annuk M, Soveri I, Zilmer M, Lind L, Hulthe J, Fellström B. Endothelial function, CRP and oxidative stress in chronic kidney disease. J Nephrol. 2005;18:721–6. [PubMed] [Google Scholar]
- 101.Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–83. [DOI] [PubMed] [Google Scholar]
- 102.Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol. 2009;20:1453–64. [DOI] [PubMed] [Google Scholar]
- 103.London GM. Cardiovascular calcifications in uremic patients: clinical impact on cardiovascular function. J Am Soc Nephrol. 2003;14:S305–309. [DOI] [PubMed] [Google Scholar]
- 104.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schlieper G, Schurgers L, Brandenburg V, Reutelingsperger C, Floege J. Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transplant. 2016;31:31–9. [DOI] [PubMed] [Google Scholar]
- 106.Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307:F891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Giachelli CM. Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol. 2003;14:S300–304. [DOI] [PubMed] [Google Scholar]
- 108.Cancela AL, Santos RD, Titan SM, Goldenstein PT, Rochitte CE, Lemos PA, et al. Phosphorus is associated with coronary artery disease in patients with preserved renal function. PloS One. 2012;7:e36883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vervloet M, Cozzolino M. Vascular calcification in chronic kidney disease: different bricks in the wall? Kidney Int. 2017;91:808–17. [DOI] [PubMed] [Google Scholar]
- 110.Jablonski KL, Chonchol M. Vascular calcification in end-stage renal disease. Hemodial Int Int Symp Home Hemodial. 2013;17 Suppl 1:S17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Benz K, Hilgers K-F, Daniel C, Amann K. Vascular Calcification in Chronic Kidney Disease: The Role of Inflammation. Int J Nephrol. 2018;2018:4310379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Holden RM, Hétu M-F, Li TY, Ward E, Couture LE, Herr JE, et al. The Heart and Kidney: Abnormal Phosphate Homeostasis Is Associated With Atherosclerosis. J Endocr Soc. 2019;3:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernelot Moens SJ, Verweij SL, van der Valk FM, van Capelleveen JC, Kroon J, Versloot M, et al. Arterial and Cellular Inflammation in Patients with CKD. J Am Soc Nephrol JASN. 2017;28:1278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Voelkl J, Cejka D, Alesutan I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr Opin Nephrol Hypertens. 2019;28:289–96. [DOI] [PubMed] [Google Scholar]
- 115.Cho I-J, Chang H-J, Park H-B, Heo R, Shin S, Shim CY, et al. Aortic calcification is associated with arterial stiffening, left ventricular hypertrophy, and diastolic dysfunction in elderly male patients with hypertension. J Hypertens. 2015;33:1633–41. [DOI] [PubMed] [Google Scholar]
- 116.Morita S, Asou T, Kuboyama I, Harasawa Y, Sunagawa K, Yasui H. Inelastic vascular prosthesis for proximal aorta increases pulsatile arterial load and causes left ventricular hypertrophy in dogs. J Thorac Cardiovasc Surg. 2002;124:768–74. [DOI] [PubMed] [Google Scholar]
- 117.Moody WE, Edwards NC, Chue CD, Ferro CJ, Townend JN. Arterial disease in chronic kidney disease. Heart. 2013;99:365–72. [DOI] [PubMed] [Google Scholar]
- 118.Huveneers S, Daemen MJAP, Hordijk PL. Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ Res. 2015;116:895–908. [DOI] [PubMed] [Google Scholar]
- 119.Zhang D, Bi X, Liu Y, Huang Y, Xiong J, Xu X, et al. High Phosphate-Induced Calcification of Vascular Smooth Muscle Cells is Associated with the TLR4/NF-κb Signaling Pathway. Kidney Blood Press Res. 2017;42:1205–15. [DOI] [PubMed] [Google Scholar]
- 120.Alesutan I, Voelkl J, Feger M, Kratschmar DV, Castor T, Mia S, et al. Involvement Of Vascular Aldosterone Synthase In Phosphate-Induced Osteogenic Transformation Of Vascular Smooth Muscle Cells. Sci Rep. 2017;7:2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lang F, Ritz E, Voelkl J, Alesutan I. Vascular calcification--is aldosterone a culprit? Nephrol Dial Transplant. 2013;28:1080–4. [DOI] [PubMed] [Google Scholar]
- 122.Wang P, Quan Z, Luo D, Chen W, Peng D. Spironolactone dose‑dependently alleviates the calcification of aortic rings cultured in hyperphosphatemic medium with or without hyperglycemia by suppressing phenotypic transition of VSMCs through downregulation of Pit‑1. Mol Med Rep. 2019;19:3622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018;94:716–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen NX, Moe SM. Pathophysiology of Vascular Calcification. Curr Osteoporos Rep. 2015;13:372–80. [DOI] [PubMed] [Google Scholar]
- 125.Speer MY, Li X, Hiremath PG, Giachelli CM. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J Cell Biochem. 2010;110:935–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nishio Y, Dong Y, Paris M, O’Keefe RJ, Schwarz EM, Drissi H. Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene. 2006;372:62–70. [DOI] [PubMed] [Google Scholar]
- 127.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–54. [DOI] [PubMed] [Google Scholar]
- 129.Speer MY, Yang H-Y, Brabb T, Leaf E, Look A, Lin W-L, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]