

Abstract
Genetic structure within marine species may be driven by local adaptation to their environment, or alternatively by historical processes, such as geographic isolation. The gulfs and seas bordering the Arabian Peninsula offer an ideal setting to examine connectivity patterns in coral reef fishes with respect to environmental gradients and vicariance. The Red Sea is characterized by a unique marine fauna, historical periods of desiccation and isolation, as well as environmental gradients in salinity, temperature, and primary productivity that vary both by latitude and by season. The adjacent Arabian Sea is characterized by a sharper environmental gradient, ranging from extensive coral cover and warm temperatures in the southwest, to sparse coral cover, cooler temperatures, and seasonal upwelling in the northeast. Reef fish, however, are not confined to these seas, with some Red Sea fishes extending varying distances into the northern Arabian Sea, while their pelagic larvae are presumably capable of much greater dispersal. These species must therefore cope with a diversity of conditions that invoke the possibility of steep clines in natural selection. Here, we test for genetic structure in two widespread reef fish species (a butterflyfish and surgeonfish) and eight range‐restricted butterflyfishes across the Red Sea and Arabian Sea using genome‐wide single nucleotide polymorphisms. We performed multiple matrix regression with randomization analyses on genetic distances for all species, as well as reconstructed scenarios for population subdivision in the species with signatures of isolation. We found that (a) widespread species displayed more genetic subdivision than regional endemics and (b) this genetic structure was not correlated with contemporary environmental parameters but instead may reflect historical events. We propose that the endemic species may be adapted to a diversity of local conditions, but the widespread species are instead subject to ecological filtering where different combinations of genotypes persist under divergent ecological regimes.
Keywords: butterflyfishes, coral reefs, ddRAD, Indo‐West Pacific, single nucleotide polymorphism, vicariance
We test for genetic structure in two widespread species (a butterflyfish and surgeonfish) and eight range‐restricted butterflyfishes across the Red Sea and Arabian Sea using genome‐wide single nucleotide polymorphisms, multiple matrix regression with randomization analyses, and demographic reconstruction. We found that (a) widespread species displayed more genetic subdivision than regional endemics and (b) this genetic structure was not correlated with contemporary environmental parameters but instead may reflect historical events.
1. INTRODUCTION
In the marine environment, coral reef fishes are a model group for understanding processes of speciation because they are well‐characterized and represent the most diverse vertebrate communities on the planet (Nelson, 2006). Reef fishes have broad geographic ranges (typically much greater than terrestrial species; Jones, Caley, & Munday, 2002), a nearly ubiquitous pelagic larval stage with relatively few barriers to dispersal, and occupy a variety of habitats. Understanding how such traits influence the evolution and distributions of reef fishes has long motivated researchers (Bowen et al., 2013; Cowman & Bellwood, 2013). Our understanding of reef fish evolution and biogeography has benefitted greatly from the rapid development of molecular analyses, and new genomic approaches have illuminated the processes driving genetic differentiation in natural populations at a variety of temporal and spatial scales (e.g., Gaither et al., 2015).
Genome‐wide single nucleotide polymorphisms (SNPs), generated by restriction site‐associated DNA sequencing (RAD‐seq), have proven valuable for studying divergence driven by natural selection in model fishes adapted to freshwater (cichlids, Wagner et al., 2013) and euryhaline environments (threespine stickleback, Hohenlohe et al., 2010), and a number of nonmodel reef fishes (Beltrán, Schizas, Appeldoorn, & Prada, 2017; Bernal, Gaither, Simison, & Rocha, 2017; DiBattista, Travers, et al., 2017; Gould & Dunlap, 2017; Harrison et al., 2017; Picq, McMillan, & Puebla, 2016; Puebla, Bermingham, & McMillan, 2014; Stockwell et al., 2016). Using RAD‐seq technology, Gaither et al. (2015) demonstrated that the strongest signals of selection in a widespread surgeonfish (Acanthurus olivaceus) were associated with divergent environmental conditions in a peripheral population. Similarly, Saenz‐Agudelo et al. (2015) showed that loci under selection in a range‐restricted clownfish (Amphiprion bicinctus) were geographically structured by environmental gradients across the Red Sea, as well as into the Gulf of Aden and Arabian Sea. Testing the generality of these patterns as precursors to speciation requires evaluating co‐distributed taxa that inhabit contrasting environmental or ecological regimes.
The reefs surrounding the Arabian Peninsula present an excellent arena for testing the genomic consequences of environmental transitions. In contrast to the reef systems of the central Indo‐West Pacific, these peripheral reefs occupy one of the most geologically and oceanographically volatile regions in tropical oceans (DiBattista, Choat, et al., 2016; DiBattista, Gaither, et al., 2017; DiBattista, Roberts, et al., 2016; DiBattista et al., 2015; Simpson, Harrison, Claereboudt, & Planes, 2014; Xu, Ruch, & Jónsson, 2015), and are defined by three prominent features: (a) a sharp increase in nutrient availability in the southern Red Sea, (b) the narrow, shallow Strait of Bab Al Mandab that constitutes the only connection between the Red Sea and Indian Ocean, and (c) seasonal upwelling associated with the northern Indian Ocean monsoon. First, the eutrophic region south of ~17°N in the Red Sea may limit larval dispersal of marine fauna, a hypothesis supported by the disjunctive distribution of some reef fish species (Roberts, Shepherd, & Ormond, 1992), as well as genetic differentiation between populations of reef organisms (Nanninga, Saenz‐Agudelo, Manica, & Berumen, 2014; Giles, Saenz‐Agudelo, Hussey, Ravasi, & Berumen, 2015; Saenz‐Agudelo et al., 2015; Reimer et al., 2017; but see Robitzch, Banguera‐Hinestroza, Sawall, Al‐Sofyani, & Voolstra, 2015). Second, water exchange through the Strait of Bab Al Mandab was repeatedly restricted during Pleistocene glacial cycles when sea level lowered as much as 140 m (Braithwaite, 1987; Rohling et al., 1998). Third, the Indian Ocean monsoon causes profound seasonal changes in ocean temperature, salinity, and productivity (Smeed, 2004; Sofianos, Johns, & Murray, 2002). At the western extreme of the Arabian Sea, the Gulf of Aden and waters of Djibouti have a high and relatively stable temperature regime with extensive limestone reefs and high coral cover (Wilkinson, 2008). At the eastern extreme, the southern coastline of Oman is subject to a “pseudo‐high‐latitude effect,” where seasonally cool sea surface temperatures and monsoonal upwelling events result in rocky reefs with sparse coral cover but dense algal cover (Barber et al., 1995; Savidge, Lennon, & Matthews, 1990; Sheppard, Price, & Roberts, 1992). These substantial changes in reef habitat occur over less than 2,000 km, well within the capacity for larval dispersal and gene flow of most reef fishes (Keith, Herbert, Norton, Hawkins, & Newton, 2011; Keith, Woolsey, Madin, Byrne, & Baird, 2015; Lessios & Robertson, 2006).
The habitat discontinuities, local environmental fluctuations, and vicariance of the coastal seas of the Arabian Peninsula provide an opportunity to investigate the relative importance of dispersal, selection, and historical processes in defining intraspecific or even interspecific genetic architecture. Evidence of selection and local adaptation can be detected with correlations between environmental variables and allele frequencies (Coyne & Orr, 2004; Schluter, 2000). For example, reef fish species distributed across the southern Red Sea and through the Gulf of Aden (including Djibouti) show abrupt changes in demographic features, including life span, over relatively small spatial scales and moderate environmental variation (Taylor, Lindfield, & Choat, 2015; Taylor, Trip, & Choat, 2018). To the east of the Gulf of Aden, the environmentally turbulent Oman upwelling coast is likely to have even greater effects on reef fish demography and assemblage composition (Burt et al., 2011; Priest et al., 2016). Historical processes associated with Pleistocene glacial cycles may have also influenced fish demography and faunal composition (DiBattista, Roberts, et al., 2016). If the contemporary environmental conditions influence genetic architecture, we would then expect strong correlations between those conditions and SNP frequency. Alternatively, if historical conditions were of greater importance, we would predict a weak correlation or no correlation between the two variables.
Reef fishes in the Red Sea, Gulf of Aden, Arabian Sea, and Sea of Oman also provide an opportunity to test for local adaptation across strong environmental gradients in both endemic (i.e., range‐restricted) and widespread reef fishes. We expect that range‐restricted species, which complete their life cycle among these reefs and adjacent oceans, are adapted to local conditions and inherent environmental fluctuations. In contrast, widespread species routinely maintain gene flow with other parts of the Indian Ocean (DiBattista et al., 2013), and larvae arriving from outside the region may be less adapted to these conditions. Although gene flow can prevent local adaptation in widespread species (Lenormand, 2002; Slatkin, 1987), strong differences in ecological conditions between locations can also reduce effective gene flow (Orsini, Vanoverbeke, Swillen, Mergeay, & Meester, 2013). If local adaptation was greater in endemic species compared to widespread species, we would expect the endemics to have less genetic structure than widespread species across a similar geographic range.
Here we use genome‐wide SNPs to investigate genetic structure in reef fishes of the Arabian Peninsula. We adopt a multi‐taxon approach comprising eight regional endemics and two widespread reef fishes, all with larvae capable of long‐distance dispersal. We aim to test (a) for associations between SNPs and contemporary environmental gradients in each species, (b) whether widespread species have greater genetic structure than endemic species across the same geographic region, and (c) determine the influence of vicariant processes on genetic structure by reconstructing scenarios for population subdivision in the species that display signatures of isolation.
2. MATERIALS AND METHODS
2.1. Sample collection and study
We collected tissue samples (fin clip or gill filaments) from individual fish using pole spears at sites between the Gulf of Aqaba in the northern Red Sea (N 28.404°, E 34.738°) and Muscat in the Sea of Oman at the north‐western boundary of the Arabian Sea (N 23.525°, E 58.740°; Figure 1 and Table 1). Nine species are from the butterflyfish family Chaetodontidae and are characterized by a range of geographic distributions (Table 1). We also included a widespread surgeonfish [Ctenochaetus striatus (Quoy & Gaimard, 1825)] from the family Acanthuridae. Some of the species were rare or absent at sampling sites, particularly for the regional endemics, which resulted in missing species and data for some locations. While we accept that our sample sizes are modest (Table 1), the number of collections per site and geographic breadth of sampling are far greater than any RAD‐seq population genomics study of reef fishes to date. Tissues were preserved in a saturated salt‐DMSO solution or 95% ethanol and subsequently stored at −20°C.
Figure 1.
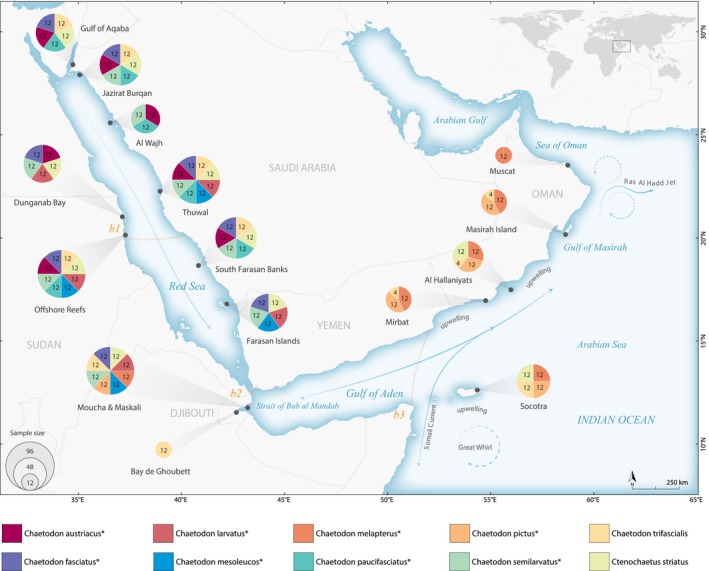
Map indicating collection sites for reef fishes sampled in the Red Sea and Arabian Sea including eight regional endemics (indicated by asterisks) and two widespread species. Colored circles indicate the proportion of samples per species at a site as indicated by the key; circle size is scaled by sample size. Major oceanographic currents and features are represented by arrows. Three putative barriers to larval dispersal are outlined by opaque orange solid lines: b1, 17°N in the Red Sea; b2, Strait of Bab Al Mandab between the Red Sea and Gulf of Aden; and b3, monsoonal upwelling system in the Arabian Sea
Table 1.
STACKS results and genetic diversity metrics for range‐restricted endemics and widespread reef fish sampled in the Red Sea to Arabian Sea (also see Figure 1)
| Species | Sample a (N) | Number of populations (Geographic range of sampling) | Species b distribution | Number of reads used | Number of polymorphic loci passing filter | H O | H E | F IS |
|---|---|---|---|---|---|---|---|---|
| Chaetodon austriacus (exquisite butterflyfish) | 78 | 7 (Gulf of Aqaba to South Farasan Banks, Saudi Arabia) | Northern to central Red Sea | 87,565,224 | 10,711 | 0.0021 (0.2270) | 0.0023 (0.2423) | 0.0007 (0.0786) |
| Chaetodon fasciatus (Red Sea racoon butterflyfish) | 89 | 8 (Gulf of Aqaba to Djibouti) | Northern Red Sea to Gulf of Aden | 77,353,808 | 2,650 | 0.0018 (0.2507) | 0.0018 (0.2553) | 0.0004 (0.0525) |
| Chaetodon larvatus (hooded butterflyfish) | 54 | 5 (Thuwal to Moucha & Maskali, Djibouti) | Southern Red Sea to Gulf of Aden | 77,207,403 | 12,393 | 0.0015 (0.2456) | 0.0016 (0.2577) | 0.0005 (0.0735) |
| Chaetodon melapterus (Arabian butterflyfish) | 69 | 6 (Djibouti to Muscat, Oman) | Southern Red Sea to Arabian Gulf | 88,380,960 | 4,384 | 0.0019 (0.2260) | 0.0021 (0.2415) | 0.0007 (0.0849) |
| Chaetodon mesoleucos (white‐face butterflyfish) | 40 | 4 (Thuwal to Djibouti) | Southern Red Sea to Gulf of Aden | 49,020,448 | 11,151 | 0.0013 (0.2687) | 0.0014 (0.2764) | 0.0003 (0.0659) |
| Chaetodon paucifasciatus (Eritrean butterflyfish) | 71 | 6 (Gulf of Aqaba to South Farasan Banks, Saudi Arabia) | Northern to central Red Sea | 89,837,377 | 13,539 | 0.0023 (0.2038) | 0.0025 (0.2239) | 0.0011 (0.0994) |
| Chaetodon pictus (horseshoe butterflyfish) | 57 | 5 (Djibouti to Masirah Island, Oman) | Southern Red Sea to Arabian Gulf | 40,198,908 | 4,131 | 0.0022 (0.2253) | 0.0024 (0.2486) | 0.0010 (0.1088) |
| Chaetodon semilarvatus (bluecheek butterflyfish) | 93 | 8 (Jazirat Burqan to Djibouti) | Northern Red Sea to Gulf of Aden | 80,036,042 | 2,053 | 0.0012 (0.2753) | 0.0012 (0.2764) | 0.0002 (0.0426) |
| Regional endemic average | 73,700,021 | 7,627 | 0.0018 (0.2403) | 0.0019 (0.2529) | 0.0006 (0.0758) | |||
| Chaetodon trifascialis (chevron butterflyfish) | 102 | 9 (Gulf of Aqaba to Masirah Island, Oman) | Indo‐Pacific | 98,948,301 | 1,271 | 0.0010 (0.2489) | 0.0010 (0.2494) | 0.0002 (0.0420) |
| Ctenochaetus striatus (striated surgeonfish) | 101 | 10 (Gulf of Aqaba to Al Hallaniyats, Oman) | Indo‐Pacific | 110,087,471 | 1,508 | 0.0019 (0.1863) | 0.0021 (0.2072) | 0.0010 (0.0945) |
| Widespread average | 104,517,886 | 1,390 | 0.0015 (0.2176) | 0.0016 (0.2283) | 0.0006 (0.1365) |
Numbers outside and inside parentheses for genetic diversity metrics are based on all single nucleotide polymorphism (SNP) loci versus only variable SNP loci, respectively.
Abbreviations: H E, expected heterozygosity; H O, observed heterozygosity; SNP, single nucleotide polymorphism.
In most cases, 12 individuals were sampled per population prior to quality filtering, except for C. trifascialis, where N = 4 were sampled from Mirbat, Al Hallaniyats, and Masirah Island.
Species distribution is based on a regional database curated over 30 years by R. Myers (see Appendix S2 from DiBattista, Roberts, et al., 2016) but was modified to reflect where species are functionally present versus rare records as waifs.
2.2. Ethics statement
This research was undertaken in accordance with the policies and procedures of the King Abdullah University of Science and Technology (KAUST). Permits for sampling in Saudi Arabian waters were obtained from the Saudi Arabian coastguard. No specific permissions were required, as the study did not involve endangered or protected species. We were unable to obtain ethics approval or a waiver because no ethics board or committee for working with animals existed within KAUST at the time of collection.
2.3. RAD sequencing
DNA was extracted with NucleoSpin Tissue kits (Macherey‐Nagel Düren). RAD‐seq libraries were prepared following Peterson, Weber, Kay, Fisher, and Hoekstra (2012) using 500 ng of DNA per specimen. Library preparation and Illumina sequencing are detailed in DiBattista, Saenz‐Agudelo, et al. (2017).
Sequences were de‐multiplexed and filtered for quality using the process_radtags pipeline in STACKS vers. 1.44 (Catchen, Amores, Hohenlohe, Cresko, & Postlethwait, 2011). Raw reads were trimmed from 101 bp to a common length of 81 bp in FASTQ format. Individual reads with Phred scores ≤ 20 (in a 5 bp sliding window) or with ambiguous barcodes were discarded. All loci were assembled separately individuals using the denovo_map pipeline in STACKS. Although an annotated butterflyfish genome is available (Chaetodon austriacus; DiBattista, Saenz‐Agudelo, et al., 2017), the other species considered in this study are too divergent to enable the recovery of sufficient numbers of SNPs for this comparison, and so we rely on de novo assembly for all species in this case.
For the main analyses presented here, we used a parameter combination tested and optimized as part of DiBattista, Saenz‐Agudelo, et al. (2017) in these same reef fish species: minimum read depth to create a stack (‐m) = 3; number of mismatches allowed between stacks prior to merging (‐M) = 4; maximum number of mismatches when aligning secondary reads to primary stacks (‐N) = 2; maximum number of mismatches allowed between loci when creating a catalog (‐n) = 2. We performed additional data filtering using the “population” component of STACKS retaining only those loci that met the following criteria: (a) minor allele frequency > 0.05, (b) present in at least n−1 population (populations: ‐p), and (c) genotyped in at least 80% of individuals per population (populations: ‐r). We used the “write_single_snp” option and produced a.vcf file with the resulting loci to better conform to the assumption of independent loci. The resulting.vcf file was reformatted to other program input files using PGDSPIDER vers. 2.0.5.1 (Lischer & Excoffier, 2012). We repeated this process to produce a data set that was comprised of SNPs shared between two of the most closely related species (C. austriacus and C. melapterus) in order to benchmark patterns of intra‐ versus interspecific genetic variation. Pairwise FST values were estimated in STACKS with the “populations” module using the “‐‐fstats” flag option.
2.4. Genetic structure analyses
We determined the magnitude of population structure for each of the 10 species by addressing the following two questions: (a) Was there evidence for restricted gene flow based on F ST estimates and clustering analyses? and (b) if so, did this restriction conform to one of the following three models: isolation by barrier (IBB; i.e., vicariance), isolation by distance (IBD), or isolation by environment (IBE; i.e., model testing)? IBE refers to scenarios where strong differences in environmental conditions between locations reduce effective gene flow.
Genetic diversity metrics (number of alleles, observed and expected heterozygosity) were estimated using STACKS. Bayesian clustering analyses were performed using STRUCTURE vers. 2.3.4 (Pritchard, Stephens, & Donnelly, 2000) without population priors. We used the admixture model with correlated allele frequencies (Falush, Stephens, & Pritchard, 2003). A burn‐in of 200,000 MCMC iterations was used, followed by 300,000 iterations for each run. K was set from 1 to the maximum number of sampling sites per species (range: 4–10), and 5 replicate analyses were run for each value of K. The number of clusters was inferred by comparing the ln P[D] among different K using the ad hoc statistic ΔK (Evanno, Regnaut, & Goudet, 2005; also see Table S1 and Appendix S1).
To complement the results from STRUCTURE and graphically summarize the genetic variation among samples within each species, we conducted a principal component analysis (PCA) of the genotype covariance matrix to summarize the genotypic variation across samples. We did this using the “glPca” function of the R package ADEGENET (Jombart & Ahmed, 2011). We also included an analysis, as outlined above, for a combined data set of the closely related C. austriacus and C. melapterus. Previous work has suggested that the divergence between these two species is recent (Waldrop et al., 2016), and together they are distributed across the range of sampling sites for the widespread species included in this study.
2.5. Determinants of genetic differentiation in the Red Sea to Arabian Sea
For species that showed evidence of population genetic structure, we proceeded to evaluate which three models (IBB, IBD, or IBE) best explained pairwise F ST as outlined in Saenz‐Agudelo et al. (2015). Under IBD and IBE, we predict that the degree of population differentiation (measured as pairwise F ST) increases with increasing geographic or environmental distance. As explained in Wang (2013), these models are not mutually exclusive, and so if environmental distance and geographic distance matrices are not correlated, it is possible to compare the relative contribution of each variable via multiple matrix regression with randomisation (MMRR). Under IBB, we predict that genetic variation changes in a discrete manner. Again, IBB is not mutually exclusive from IBD and IBE, but all this information can be visualized via MMRR (e.g., Saenz‐Agudelo et al., 2015).
We retrieved gridded environmental information from the Bio‐Oracle database (Tyberghein et al., 2012) to characterize environmental differences among our sampling locations in the Red Sea to Arabian Sea. Specifically, we used aggregated data (predominantly between 2002 and 2009) that described the mean value and variability of environmental variables in a 5‐arc min. grid (~9 km). These variables were related to climate (sea surface temperature, cloud cover, photosynthetically available radiation), chemistry (salinity, pH, dissolved oxygen), nutrients (silicate, nitrate, phosphate, calcite), and productivity (chlorophyll A, diffuse attenuation). To reduce the dimensionality of the data, we performed a PCA based on the correlation of all environmental variables measured at our 14 sampling locations (Figure 2). All variables were log‐transformed and subsequently normalized to have a mean of zero and unit variance.
Figure 2.
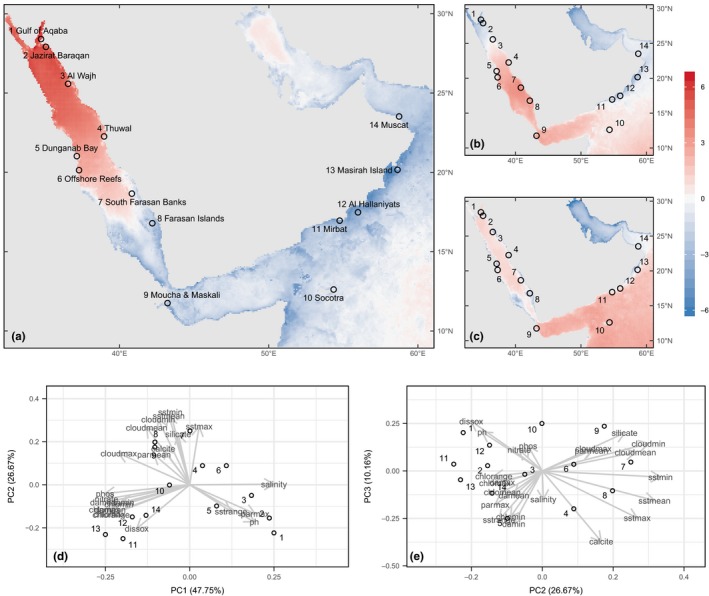
Heat map of environmental data in the Red Sea to Arabian Gulf represented by principal component analysis (PCA) outputs (a) PC1, (b) PC2, and (c) PC3. (d,e) Biplot of the sites and the loading of the environmental drivers underlying the PCA. Collection sites are indicated by numbers
For each species, we calculated the pairwise F ST and the environmental pairwise distance (the distance in PCA space; env) between locations. Because geographic distance between locations might also influence the genetic differentiation within a species, we included pairwise geographic distance (geo) between samples in the analyses. This distance corresponded to the length of the shortest, direct within water path between two given locations. Geographic distances were estimated using the least‐cost distance function (“costDistance”) in the R package gdistance (van Etten, 2017).
Additionally, we explored the influence of three putative barriers to gene flow: (a) at 17°N in the Red Sea, which has been suggested as a potential boundary between ecoregions (see Giles et al., 2015; Nanninga et al., 2014; Roberts et al., 2016), (b) the Strait of Bab Al Mandab, and (c) the monsoonal upwelling system in the Arabian Sea (see Nanninga et al., 2014; Saenz‐Agudelo et al., 2015). All barriers were considered independently and in combination, modeled as a factor with 0 to 3 levels (reflecting the number of barriers between a pair of sites). Sites within the same level were on the same side of the barrier and sites within different levels were on different sides of any given barrier. Seven barrier variables in total were thus included.
We built 67 linear models comprising all biologically plausible combinations of variables (geo, env, and seven barrier variables). Quantitative variables were scaled to a mean of 0 and a variance of 1. For each model, the sample size corrected Akaike information criterion (AICc) was computed as AICc = AIC + 2K (K + 1)/(n−K−1), where AIC = −2log‐likelihood + 2K (K = number of parameters in model; n = number of observations). Models were then ranked according to increasing AICc (Anderson, 2008). For each species, the best model was then chosen, and statistical significance of each parameter was estimated via MMRR (Wang, 2013).
2.6. Testing for the presence of outlier loci
We ran OutFLANK to test for SNP loci showing departures from neutral expectations, which, in some cases, can indicate whether genetic divergence is linked to adaptive processes (Whitlock & Lotterhos, 2015). Indeed, if different species have homologous loci that depart from neutral expectations, this might suggest that a common adaptive process is driving divergence across species. We ran this analysis for both species that displayed genetic structure (Ct. striatus and C. trifascialis), as well as for the C. austriacus and C. melapterus combined data set. OutFLANK (as implemented in R) was used to infer the distribution of F ST values and identify putative loci under selection (Whitlock & Lotterhos, 2015). To estimate the null distribution of F ST values, we trimmed 5% of the loci from the lower and upper ends of the F ST range and included only those loci with heterozygosity higher than 0.1. We used a false discovery rate threshold of 0.05 to calculate q‐values when testing for the neutrality of each SNP locus.
2.7. Reconstructing scenarios for genetic differentiation in the Red Sea to Arabian Sea
We used a modified version of the diffusion approximation method implemented in ∂a∂I (Gutenkunst, Hernandez, Williamson, & Bustamante, 2009) to explore the joint site‐frequency spectrum (JSFS) of population pairs for species with population partitions identified in the genetic structure analyses. We used the approach outlined by Tine et al. (2014) that includes modifications regarding annealing optimization prior to the Broyden–Fletcher–Goldfarb–Shanno step, which has been shown to improve global parameter convergence. Other modifications include the incorporation of varying migration rates across the genome as described by Tine et al. (2014) and Rougemont et al. (2017). This analysis was performed only for Ct. striatus, C. trifascialis, as well as the C. austriacus and C. melapterus combined data set. For these analyses, we grouped samples from the Red Sea and samples from the Indian Ocean together in order to increase the number of individuals used for JSFS estimation. Since PCA analyses of Ct. striatus suggested genetic differentiation between Socotra and Oman samples, we additionally ran all models independently using only Socotra samples or only Oman samples as distinct representatives of the Indian Ocean. All scripts and model descriptions are available from (https://github.com/QuentinRougemont/DemographicInference).
Here, we limited our analyses to seven models of divergence including strict isolation (SI), isolation with migration (IM), ancient migration (AM), and secondary contact (SC). For each of IM, AM, and SC, we explored two options: (a) homogenous migration and (b) heterogeneous migration along the genome (2M; as described in Rougemont et al., 2017). To maximize convergence of parameter estimates, each model was run 20 times; the run providing the lowest AIC score was kept and used to compare against other models as well as to estimate model parameters.
3. RESULTS
A total of 798,635,942 reads of 101 bp were obtained for 678 sampled individuals from the 10 study species (Table 1); 132 individuals were discarded due to a low number of raw reads recovered (≤250,000) or because the number of missing loci exceeded 50%. The average number of usable reads per sample ranged from 705,244 (for Chaetodon pictus) to 1,429,766 (for Chaetodon larvatus). Overall, 1,271 to 13,539 polymorphic SNP loci met the quality filtering criteria for each species. Summary statistics including observed heterozygosity, expected heterozygosity, and F IS are presented in Table 1.
3.1. Genetic structure analyses
STRUCTURE analyses indicated mean probabilities as being highest at K = 1 or ambiguous for all the range‐restricted species, but K = 2 (Ct. striatus) and K = 3 (C. trifascialis) for the two widespread species (Table S1). Moreover, PCA was consistent with a scenario of panmixia for all range‐restricted species but not the two widespread species (Figure 3; Appendix S1). Both widespread species demonstrated a putative barrier to dispersal between the Red Sea and the Arabian Sea, with the western end of the Gulf of Aden at Djibouti acting as a transition zone. The data set that included the two closely related species, C. austriacus and C. melapterus, also indicated that the most likely K was 2 (Table S1); two distinct genetic clusters were apparent in the PCA with a similar transition zone of admixture (Figure 4). For reference, STRUCTURE plots for all species at all possible K and all PCA plots are provided in Appendix S1
Figure 3.
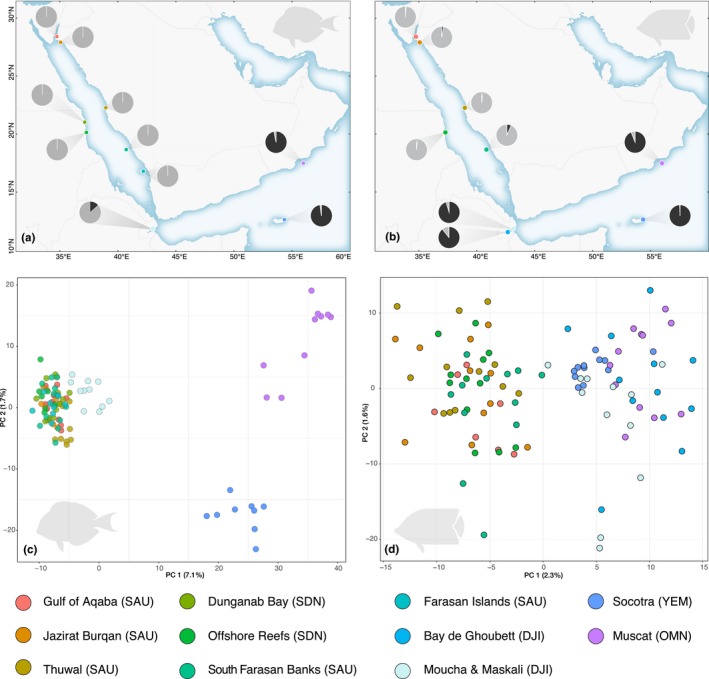
(a,b) Summary of the single nucleotide polymorphism (SNP) admixture estimates from STRUCTURE at each sampling site. The shading in each pie indicate the mean level of admixture per sampling site for K = 2. (c,d) Principal component analysis (PCA) scatter plots for RAD‐seq data. Only data sets from the two widespread species Ctenochaetus striatus (a,c) and Chaetodon trifascialis (b,d) are presented here. For the PCA plots, circles represent individual genotypes and axes show the first two components and the percentage of variance explained in brackets. Three letter abbreviations in parentheses represent the country of sampling
Figure 4.
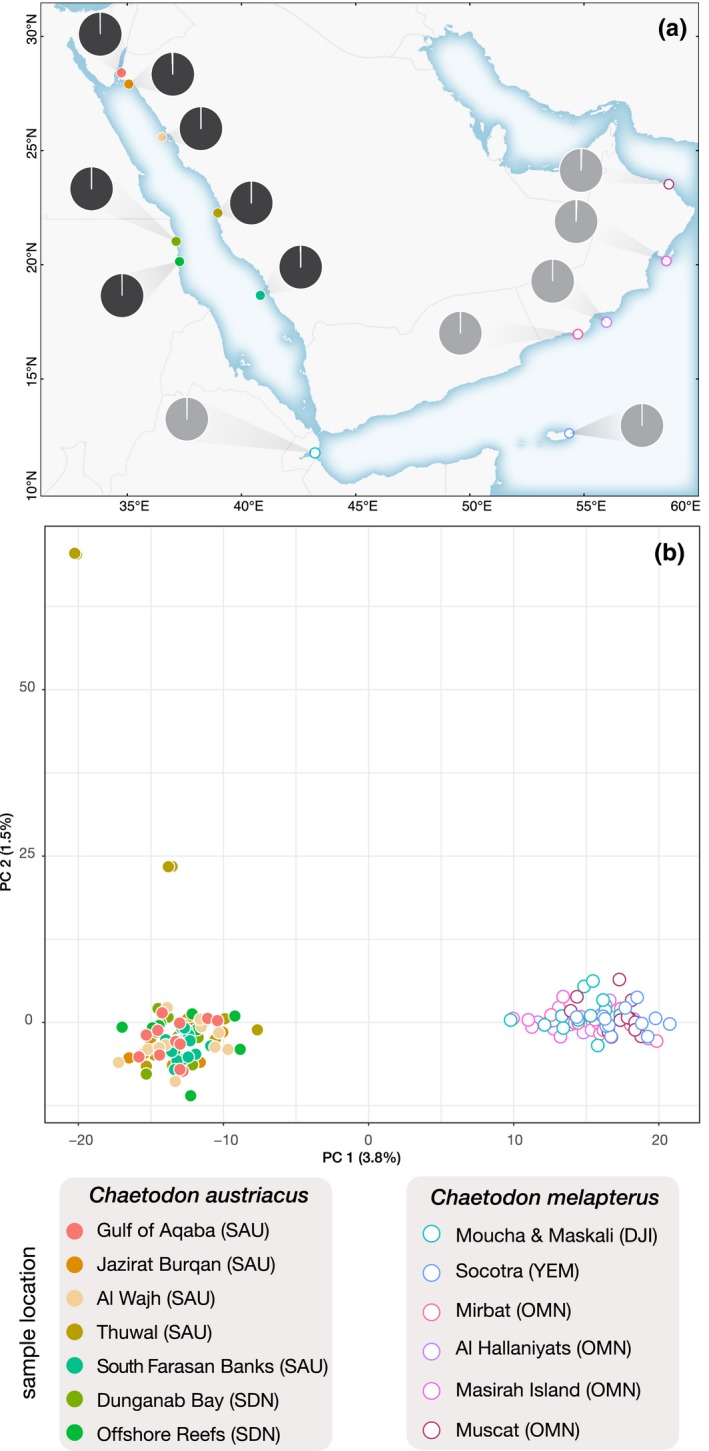
(a) Summary of the single nucleotide polymorphism (SNP) admixture estimates from STRUCTURE at each sampling site. The shading in each pie indicate the mean level of admixture per sampling site for K = 2. (b) Principal component analysis (PCA) scatter plots for RAD‐seq data. Only the data set that was comprised of SNPs shared between two closely related species (Chaetodon austriacus and Chaetodon melapterus) is presented here. For the PCA plots, circles represent individual genotypes and axes show the first two components and the percentage of variance explained in brackets. Three letter abbreviations in parentheses represent the country of sampling
3.2. Determinants of genetic differentiation in the Red Sea to Arabian Sea
The first three principal dimensions explained 85% percent of the environmental variability and were used in subsequent analyses (Figure 2). The first component, which explained 48% of the variance, was positively correlated with salinity and nutrient (phosphate, nitrate, chlorophyll) variables. The second component, which explained 27% of the variance, was positively correlated with variables related to sea surface temperature. The third component, which explained 10% of the variance, was positively correlated with dissolved oxygen and pH, and negatively correlated with calcite.
For C. trifascialis, a widespread butterflyfish species with apparent genetic structure, comparisons of the 67 models that tested different combinations of the effects of IBB, IBD, and IBE indicated that the four models that best explained genetic differentiation based on pairwise F ST (for values see Appendix S2) were those that included the Strait of Bab Al Mandab (b2) as a barrier to gene flow, as well as both geographic and environmental distances (Table S2). Based on these results, the model with the highest probability did not include interactions between variables (model probability, p = .240), although metrics of statistical confidence are lowered by the large number of models included for comparison. This best model suggested that: (a) on average, pairwise F ST values were lower among sites on the same side of Bab Al Mandab compared to pairwise F ST values among sites across Bab Al Mandab (same side of b2: 0.025 ± 0.0011 SE, p = .010; different side of b2: 0.029 ± 0.0015 SE, p = .010), 2) pairwise F ST values were positively but marginally correlated with geographic distance (slope: 1.541 × 10–9 ± 7.932 × 10–10 SE, p = .060) and this slope was the same for comparisons on the same side or across Bab Al Mandab (Figure 5).
Figure 5.
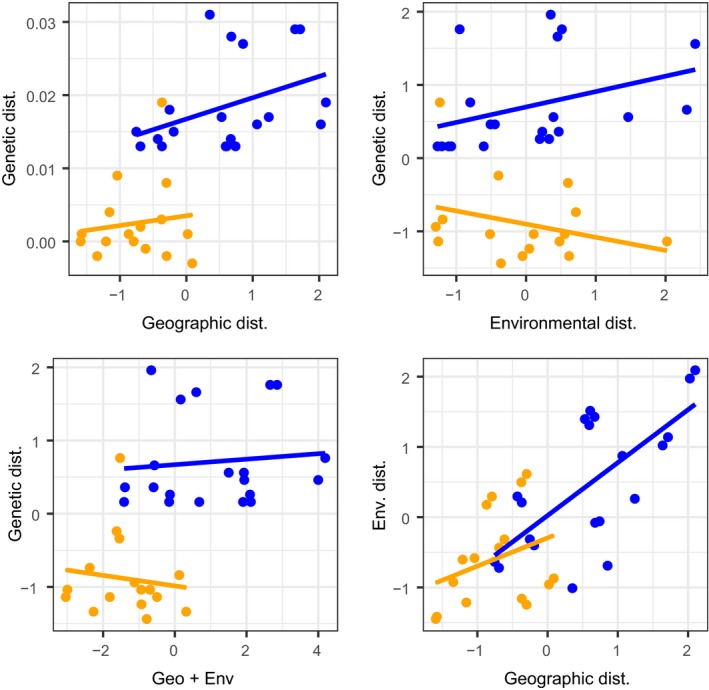
Correlation between pairwise genetic distance (F ST), geographical distance, and environmental distance for Chaetodon trifascialis around the Arabian Peninsula. The top two panels show correlations between genetic and geographic (left) and environmental (right) distances. The bottom two panels show correlations between genetic and combined geographic and environmental distances (left), and the correlation between geographic and environmental distance (right). Blue dots and regression lines correspond to pairwise comparisons among sites on different sides of the Strait of Bab Al Mandab barrier (b2), orange dots and regression lines correspond to pairwise comparisons among sites on the same side of b2
For Ct. striatus, the other species demonstrating genetic structure, the four models that best explained genetic differentiation based on pairwise F ST were the ones that included the monsoonal upwelling system in the Arabian Sea (b3) as a barrier to gene flow, as well as geographic distance (best two models) or environmental distance (fourth model; Table S3). The best model included an interaction between b3 and geographic distance indicating a difference in slopes between comparisons within and between different sides of the barrier (model p = .268). The best model suggested that: (a) on average, pairwise F ST values were lower among sites on the same side of b3 compared to pairwise F ST values among sites across b3 (same side of b3: 0.032 ± 0.0014 SE, p < .001; different side of b3: 0.093 ± 0.0028 SE, p < .001) and (b) pairwise F ST values were positively but marginally correlated with geographic distance, but only for comparisons across b3 (same side of b3: 0.00008 ± 0.0018 SE, p = .965; different side of b3: 0.0048 ± 0.0053 SE, p = .077; Figure 6). The second‐best model included the same variables but not the interaction term, suggesting the same IBD slope both within and between different sides of b3. That said, this model was 1.5 times less favored than the best model (model p = .179). We chose not to run the C. austriacus and C. melapterus combined data set here because this analysis was entirely focused on within‐species differentiation and not between species differentiation.
Figure 6.
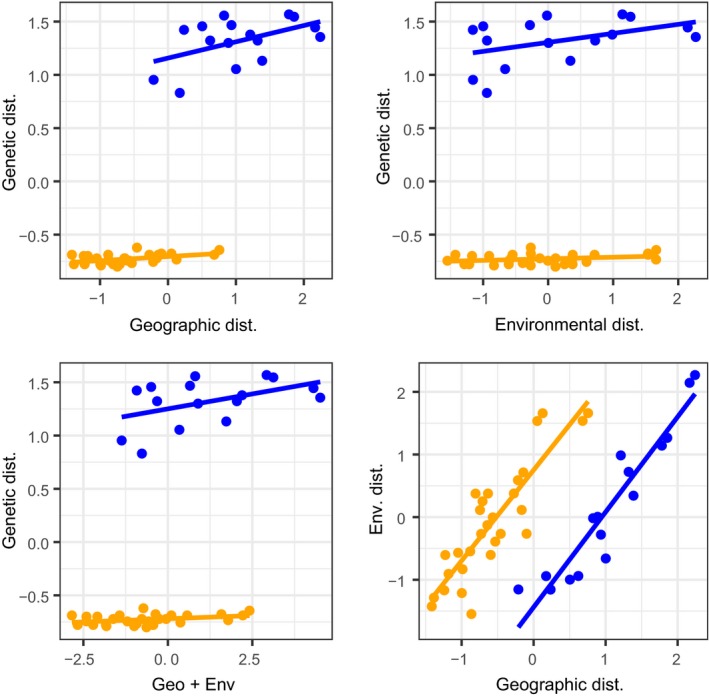
Correlation between pairwise genetic distance (F ST), geographical distance, and environmental distance for Ctenochaetus striatus around the Arabian Peninsula. The top two panels show correlations between genetic and geographic (left) and environmental (right) distances. The bottom two panels show correlations between genetic and combined geographic and environmental distances (left), and the correlation between geographic and environmental distance (right). Blue dots and regression lines correspond to pairwise comparisons among sites on different sides of the monsoonal upwelling system barrier in the Arabian Sea (b3), orange dots and regression lines correspond to pairwise comparisons among sites on the same side of b3
3.3. Testing for the presence of outlier loci
For Ct. striatus, OutFLANK indicated the presence of 73 outlier loci. For C. trifascialis, OutFLANK did not detect a single locus under selection. The results from the C. austriacus and C. melapterus combined data set indicated the presence of 43 outlier loci. A comparison of the consensus sequences of all outlier loci found in the Ct. striatus data set versus those from the C. austriacus and C. melapterus combined data set revealed no shared loci between them.
3.4. Reconstructing scenarios for genetic differentiation in the Red Sea to Arabian Sea
To examine the role of vicariant processes on genetic differentiation, we analyzed the Ct. striatus and C. trifascialis data sets. We included the C. austriacus and C. melapterus combined data set here because reconstructing the demographic history within species can share insights about the processes driving genetic divergence between species. These two butterflyfish are recently diverged sister species whose range spans the study region. For all three data sets, the secondary contact (SC) models were consistently better supported by the data (Table 2). For the Ct. striatus data set as well as the combined C. austriacus and C. melapterus data set, this model included heterogeneous migration along the genome (SC2M), whereas for the C. trifascialis data set, the SC model had the lowest AIC. Similar results were found for the Ct. striatus model that considered Red Sea and Oman samples (where SC2M was the best model), whereas the isolation with migration model (IM2M) was the best model when Socotra samples instead of Oman samples were compared to the Red Sea. We note, however, that the second‐best model for the Socotra to Red Sea comparisons was SC (Table S4). Given that mutation rates are not known for these loci, the values estimated from the JSFS must be interpreted with caution. For all three data sets, effective population size was higher in the Red Sea versus the Indian Ocean, migration rates were higher from the Indian Ocean toward the Red Sea, and the ratio of secondary contact (T sc) to divergence time (T s) indicated short periods of introgression for Ct. striatus and C. trifascialis (3.3% and 4.6% of the total divergence time, respectively), as well as for C. austriacus and C. melapterus (5.4%). Overall, these results indicate that the evolutionary processes that influence divergence between endemic species (C. austriacus and C. melapterus) might be similar to those processes influencing divergence within widespread species (i.e., Ct. striatus and C. trifascialis). The details for each of the six best models tested are shown in Table 2 (also see Figure 7, and Figures S1–S3, and Table S4).
Table 2.
Comparison of seven alternative demographic models obtained from ∂a∂I for Ctenochaetus striatus, Chaetodon trifascialis, as well as Chaetodon austriacus and Chaetodon melapterus data sets using a folded joint frequency spectrum (JSFS)
| Model | AIC | log lik | theta | N Red Sea | N Indian Ocean | m12 | m21 | me12 | me21 | T s | T sc or T am | P |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ctenochaetus striatus | ||||||||||||
| AM a | 994.581 | −491.291 | 34.118 | 14.860 | 0.239 | 0.000 | 6.138 | 9.828 | 0.000 | |||
| AM2M | 934.547 | −458.274 | 34.000 | 13.794 | 0.439 | 0.000 | 6.033 | 0.011 | 0.778 | 9.961 | 0.000 | 0.819 |
| IM | 992.578 | −491.289 | 33.636 | 15.076 | 0.242 | 0.000 | 6.042 | 9.984 | ||||
| IM2M | 932.132 | −458.066 | 34.319 | 13.757 | 0.382 | 0.000 | 7.006 | 0.014 | 0.837 | 9.862 | 0.821 | |
| SC | 920.964 | −454.482 | 36.853 | 9.083 | 0.098 | 1.119 | 17.564 | 9.972 | 0.074 | |||
| SC2M | 874.118 | −428.059 | 38.390 | 8.087 | 0.218 | 0.917 | 9.516 | 0.000 | 1.199 | 4.215 | 0.143 | 0.901 |
| SI | 1,147.322 | −570.661 | 298.115 | 0.030 | 0.010 | 0.002 | ||||||
| Chaetodon trifascialis | ||||||||||||
| AM | 724.579 | −356.289 | 357.163 | 17.442 | 0.010 | 0.000 | 2.991 | 0.000 | 0.000 | |||
| AM2M | 730.536 | −356.268 | 357.235 | 16.036 | 0.025 | 47.853 | 0.037 | 0.000 | 0.000 | 0.001 | 0.000 | 0.785 |
| IM | 722.575 | −356.287 | 357.162 | 18.122 | 0.010 | 0.000 | 0.000 | 0.000 | ||||
| IM2M | 725.819 | −354.910 | 361.850 | 0.249 | 5.479 | 0.000 | 68.565 | 0.000 | 0.000 | 0.013 | 0.891 | |
| SC | 709.632 | −348.816 | 86.016 | 2.015 | 1.384 | 3.309 | 19.594 | 9.815 | 0.453 | |||
| SC2M | 724.290 | −353.145 | 72.397 | 3.653 | 1.856 | 44.695 | 0.000 | 2.744 | 9.635 | 8.784 | 0.584 | 0.192 |
| SI | 718.579 | −356.290 | 357.180 | 10.464 | 0.010 | 0.000 | ||||||
| C. austriacus and C. melapterus | ||||||||||||
| AM | 897.663 | −442.832 | 50.301 | 1.331 | 0.293 | 0.000 | 19.766 | 9.799 | 0.000 | |||
| AM2M | 837.919 | −409.959 | 105.878 | 5.753 | 0.638 | 2.673 | 14.813 | 0.000 | 0.398 | 4.440 | 0.000 | 0.948 |
| IM | 893.045 | −441.523 | 53.745 | 12.669 | 0.294 | 0.606 | 19.656 | 9.344 | ||||
| IM2M | 822.972 | −403.486 | 54.638 | 12.863 | 0.191 | 1.282 | 44.642 | 0.000 | 1.862 | 9.474 | 0.948 | |
| SC | 859.133 | −423.567 | 106.055 | 2.118 | 0.413 | 1.478 | 14.776 | 9.877 | 0.260 | |||
| SC2M | 814.993 | −398.497 | 72.889 | 6.564 | 0.884 | 0.000 | 0.958 | 1.444 | 9.975 | 6.132 | 0.332 | 0.053 |
| SI | 1,002.267 | −498.134 | 418.976 | 0.029 | 0.010 | 0.001 | ||||||
Results of the best run for each model are provided. AIC: Akaike information criterion; log lik: maximum likelihood; theta: 4 Nrefµ; N Red Sea and N Indian Ocean: effective population sizes of each population, respectively; m12 and m21: migration rates from the Red Sea to the Indian Ocean and vice versa, respectively; me12 and me21: effective migration rates in the most differentiated regions of the genome (i.e., genomic islands) from the Red Sea to the Indian Ocean and vice versa, respectively; T s: time of split of the ancestral population into two daughter populations; T sc: duration of secondary contact episodes (only in SC and SC2M models); T am: duration of ancestral migration episodes (only in AM and AM2M models); P: proportion of the genome exchanged under neutrality. The model with the lowest AIC is indicated in bold.
Model abbreviations: SI, strict isolation; IM, isolation with migration; AM, ancient migration; SC, secondary contact. For each of IM, AM, and SC, we explored two options: (1) homogenous migration and (2) heterogeneous migration along the genome (2M).
Figure 7.
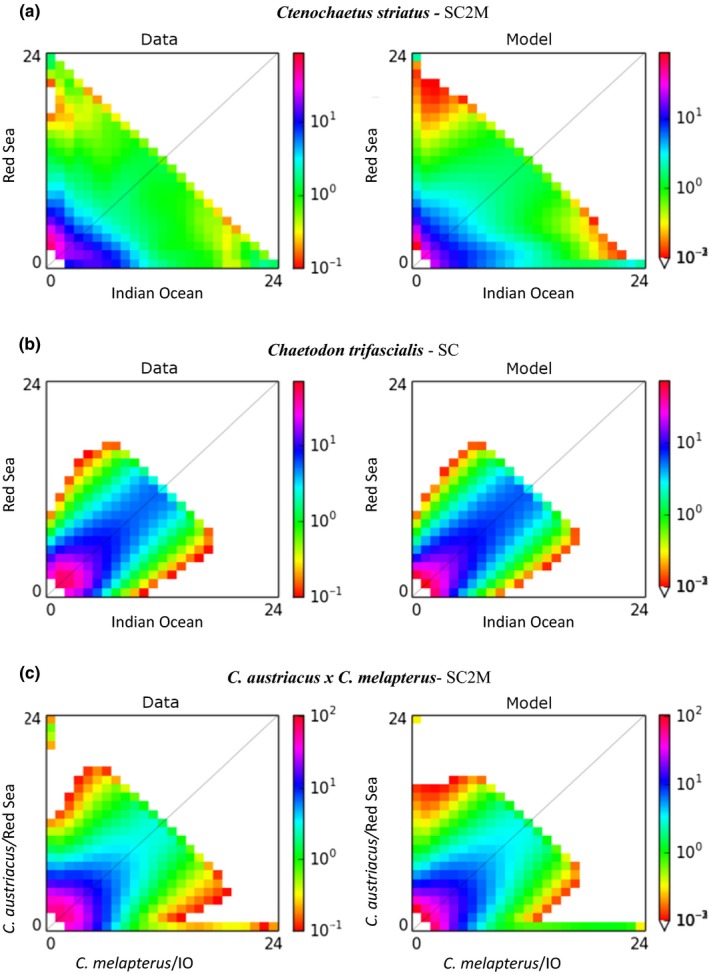
Results of the diffusion approximation models for the (a) Ctenochaetus striatus, (b) Chaetodon trifascialis, and (c) Chaetodon austriacus and Chaetodon melapterus data sets. For each data set, the observed “data” and the best fitting “model” are displayed. Shading indicates probability matrix as indicated by the embedded legend. Plots of all alternative models tested, for each data set, are provided as Figures S1–S3
4. DISCUSSION
The study region from the northern Red Sea to the northern coast of Oman is characterized by a change from high temperature and extensive coral cover to low temperatures and poor coral development typical of marginal, high‐latitude reefs (Barber et al., 1995; Savidge et al., 1990; Sheppard et al., 1992; Wilkinson, 2008). Across this steep environmental gradient, we found genetic structure in the two widespread species but not the eight regional endemics. Moreover, the genetic structure that we identified in these widespread species was not linked to contemporary environmental conditions but instead mirrored the patterns of separation between two closely related butterflyfish species. Based on this finding and complimentary analyses that reconstructed scenarios of isolation, migration, and secondary contact, the apparent genetic structure appears to be associated with historical processes. Although isolation by adaptation has received both theoretical and empirical support in the terrestrial realm (Orsini et al., 2013), our findings did not support this scenario within this marine environment.
4.1. Lack of association between SNPs and contemporary environmental gradients
There was no evidence that the genetic structure found in two study species (Ct. striatus and C. trifascialis) was linked to contemporary environmental gradients. Interestingly, in both widespread species, the best selected models included the presence of a geographic barrier, which explained most of the variation in pairwise genetic distance. None of the best selected models included environmental distance, and in the lower ranked models that did include it, the slope of the relationship between environmental distance and genetic distance was not significant. This null finding is surprising because two lines of evidence support differential selective regimes across the Red Sea to Arabian Sea. Firstly, age‐growth surveys reveal interpopulation differences in life‐history traits for several coastal fish species (Priest et al., 2016; Robertson, Ackerman, Choat, Posada, & Pitt, 2005; Taylor et al., 2018), including Ct. striatus (J. H. Choat, unpublished data). Secondly, there is a considerable difference in the distribution and abundance of Ct. striatus and C. trifascialis across the study region (Roberts et al., 2016; J. H. McIlwain, unpublished data). The most significant changes in life history and abundance occur across the upwelling region in Oman, with major shifts in temperature, productivity, and water clarity that have persisted since the late Miocene (Zhuang, Pagani, & Zhang, 2017).
It is important to note that we did identify the presence of outlier loci for one (Ct. striatus) but not the other (C. trifascialis) widespread species. The presence of outlier loci along with a lack of IBE correlation for Ct. striatus suggests that our simple linear IBE model does not capture the complexity of the processes driving genetic structure in this species. The absence of outlier loci and IBE for C. trifascialis instead suggests that demographic rather than selective processes may be driving genetic structure here. Taken together, the observed patterns of genetic structure in these two widespread species indicate that this region is a complex evolutionary arena where both environmental and historical processes may play important roles.
While the influence of the Arabian Sea upwelling zone was not detected in population genetic partitions, this oceanographic feature nonetheless has a profound biogeographic impact. Six of the eight endemic species considered in this study do not occur east of the upwelling region. This concordant range edge for endemic species indicates that these primarily Red Sea inhabitants cannot tolerate upwelling conditions or simply cannot disperse beyond this potential barrier. Thus, the major changes in contemporary environmental conditions may be important for defining the range edge of endemics and genetic structure in widespread species.
4.2. Genetic structure of endemic and widespread species
Contemporary environmental gradients within the small range of the endemic species, including barriers b1 and b2 (Figure 1), may not be strong enough to induce genetic structuring in reef fishes. In contrast, both widespread species exhibited genetic differentiation in this study. These widespread species are distributed across the Indo‐West Pacific from the Red Sea to the Hawaiian Archipelago, and Ct. striatus had the strongest population genetic partitions. It is likely that these species, like many Indo‐West Pacific reef fishes, arose in the central Indo‐West Pacific and expanded across a vast expanse of ocean to colonize peripheral coral reefs (Briggs, 2005; also see Lawton, Messmer, Pratchett, & Bay, 2011). In contrast, the range‐restricted species persist and evolve under local conditions. Indeed, range‐restricted C. melapterus and C. pictus (mean range size: 0.21 × 106 km2) are thought to be sister species to the wide‐ranging C. trifasciatus (different from our study species C. trifascialis) and C. vagabundus (mean range size: 52.18 × 106 km2), respectively, with their speciation driven by peripheral budding in range edge locations (Bowen et al., 2016; Budd & Pandolfi, 2010). Local adaptation would also explain why endemic reef fishes, including the species studied here, tend to achieve much higher abundances than their widespread congeners (Hobbs, Jones, & Munday, 2011; Kane, Kosaki, & Wagner, 2014).
None of the 10 species surveyed displayed genetic structure within the Red Sea. These results contradict previous studies that described the presence of a genetic discontinuity south of 17°N in the Red Sea for an anemonefish (Amphiprion bicinctus; Nanninga et al., 2014; Saenz‐Agudelo et al., 2015) and a sponge (Stylissa carteri; Giles et al., 2015). One possible explanation for this discrepancy is that both A. bicinctus and S. carteri are brooders, and therefore have a short pelagic larval duration (PLD). In contrast, all 10 species surveyed in our study are broadcast spawners and so their larvae have relatively long PLDs (Chaetodontidae: PLD ~ 23–56 days; Acanthuridae: PLD ~ 31–91 days; Brothers & Thresher, 1985; Doherty, Planes, & Mather, 1995; Fowler, 1989; Wilson & McCormick, 1999). The link between genetic structure and limited dispersal (Bay, Crozier, & Caley, 2006) means that the 10 study species have dispersal abilities that preclude population genetic structure across the Red Sea region.
In contrast, across a similar geographic distance, many endemic fishes (including butterflyfish and surgeonfish) in the Hawaiian Archipelago demonstrated genetic structure (Toonen et al., 2011). Although the Hawaiian archipelago is also a peripheral region rich in endemic species, it differs in several ways to the Red Sea region. Firstly, it is composed of a series of volcanic islands, compared to the continuous continental coastline of the Red Sea region; the expanses of deep water that separate these islands may promote genetic structure in reef fish populations. Secondly, due to the geomorphology of the Hawaiian Islands, historical events (e.g., sea level change) are less likely to create barriers to gene flow compared to the Red Sea region (e.g., Strait of Bab Al Mandab). Thus, congeneric species occupying different peripheral regions can exhibit contrasting patterns of genetic structure due to differences in geomorphology and historical effects.
4.3. Scenarios for population subdivision
One of the most intriguing results in this study was that demographic modeling of both widespread species suggested secondary contact was the most likely scenario of divergence. This model was also favored between the two closely related species, C. austriacus and C. melapterus, whose combined distributional pattern mirrors the genetic break observed for C. trifascialis. The Red Sea and Gulf of Aden have a long history of intermittent isolation at the Strait of Bab Al Mandab, which may partially explain the support for the secondary contact model, at least for C. trifascialis. Interestingly, the best model for C. trifascialis did not include asymmetric rates of genomic differentiation, which points to neutral rather than selective process shaping differentiation; this is also consistent with our outlier analysis. In contrast, the best models for Ct. striatus as well as the combined C. austriacus and C. melapterus data set indicate heterogeneous genomic divergence, supporting a more complex scenario that includes natural selection, but again it is consistent with our outlier analysis. Thus, traits associated with intrinsic dispersal capacity appear to be of minor importance in determining the geographic distributions of the study species. Other factors, including the geographic configuration of coastal and reef systems, as well as the prevailing oceanographic structure, are likely of primary importance in this context.
5. CONCLUSION
Overall, this study found that genetic structure was present in widespread species and absent in endemic species. This genetic structure based on a suite of SNP markers was associated with historical processes and not contemporary environmental conditions or larval dispersal abilities. This novel insight highlights the value of genomic approaches for studying divergence and speciation in nonmodel organisms. This is particularly useful in the marine environment, where research efforts and developments have traditionally lagged behind that of terrestrial and freshwater systems. The marine environment contains some of the most diverse systems in the world (e.g., coral reef ecosystems) and genomic approaches can fast‐track our understanding of the origins and maintenance of this diversity.
CONFLICT OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Joseph D. DiBattista: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal); Project administration (equal); Resources (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Pablo Saenz‐Agudelo: Data curation (equal); Formal analysis (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Marek J. Piatek: Data curation (equal); Formal analysis (equal); Software (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). E. Fernando Cagua: Formal analysis (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Brian W. Bowen: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). John Howard Choat: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Luiz A. Rocha: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Michelle R. Gaither: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Jean‐Paul A. Hobbs: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Tane H. Sinclair‐Taylor: Investigation (equal); Resources (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Jennifer H. McIlwain: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Mark A. Priest: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Camrin D. Braun: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Nigel E. Hussey: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Steven T. Kessel: Investigation (equal); Resources (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Michael L. Berumen: Conceptualization (equal); Funding acquisition (equal); Investigation (equal); Project administration (equal); Resources (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal).
Supporting information
Figs S1‐S3
Table S1
Table S2
Table S3
Table S4
Appendix S1
Appendix S2
ACKNOWLEDGMENTS
This research was supported by the KAUST Office of Competitive Research Funds (OCRF) under Award No. CRG‐1‐2012‐BER‐002 and baseline research funds to M.L.B., a National Geographic Society Grant 9024‐11 to J.D.D., a National Science Foundation grant OCE‐1558852 to B.W.B., California Academy of Sciences funding to L.A.R, and Australian Research Council funding to J.‐P.A.H. (DE200101286). For support in Socotra, we kindly thank the Ministry of Water and Environment of Yemen, staff at the Environment Protection Authority (EPA) Socotra, and especially Salah Saeed Ahmed, Fouad Naseeb and Thabet Abdullah Khamis, as well as Ahmed Issa Ali Affrar from Socotra Specialist Tour for handling general logistics. For logistic support elsewhere, we thank Eric Mason at Dream Divers in Saudi Arabia; the Red Sea State Government and The Red Sea University in Sudan, as well as Equipe Cousteau support for the Sudan Shark and Ray Conservation and Management Program including C. Scarpellini and M. Younis; Nicolas Prévot at Dolphin Divers and the crew of the M/V Deli in Djibouti; the KAUST Coastal and Marine Resources Core Lab and Amr Gusti; and the Ministry of Agriculture and Fisheries in Oman including Abdul Karim. For specimen collections, we thank Tilman Alpermann, Giacomo Bernardi, Richard Coleman, Gerrit Nanninga, and members of the Reef Ecology Lab at KAUST. For assistance with bench work at KAUST, we thank Craig Michell. We also acknowledge important contributions from the KAUST Bioscience Core Laboratory with Sivakumar Neelamegam and Hicham Mansour for their assistance with Illumina sequencing. Thanks to Robert Toonen and Stephen Karl for discussions and critiques that improved this manuscript.
DiBattista JD, Saenz‐Agudelo P, Piatek MJ, et al. Population genomic response to geographic gradients by widespread and endemic fishes of the Arabian Peninsula. Ecol Evol. 2020;10:4314–4330. 10.1002/ece3.6199
Contributor Information
Joseph D. DiBattista, Email: josephdibattista@gmail.com.
Tane H. Sinclair‐Taylor, Email: t.sinclairtaylor@aims.gov.au.
DATA AVAILABILITY STATEMENT
Raw SNP data and all appendices are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.rn8pk0p68.
REFERENCES
- Anderson, D. (2008). Model based inference in the life sciences: A primer on evidence. New York, NY: Springer Science + Business Media. [Google Scholar]
- Bay, L. K. , Crozier, R. H. , & Caley, M. J. (2006). The relationship between population genetic structure and pelagic larval duration in coral reef fishes on the Great Barrier Reef. Marine Biology, 149, 1247–1256. [Google Scholar]
- Beltrán, D. M. , Schizas, N. V. , Appeldoorn, R. S. , & Prada, C. (2017). Effective dispersal of Caribbean reef fish is smaller than current spacing among marine protected areas. Scientific Reports, 7, 4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal, M. A. , Gaither, M. R. , Simison, W. B. , & Rocha, L. A. (2017). Introgression and selection shaped the evolutionary history of sympatric sister‐species of coral reef fishes (genus: Haemulon). Molecular Ecology, 26, 639–652. [DOI] [PubMed] [Google Scholar]
- Bowen, B. W. , Gaither, M. R. , DiBattista, J. D. , Iacchei, M. , Andrews, K. R. , Grant, W. S. , … Briggs, J. C. (2016). Comparative phylogeography of the ocean planet. Proceedings of the National Academy of Sciences of the United States of America, 113, 7962–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen, B. W. , Rocha, L. A. , Toonen, R. J. , Karl, S. A. , Craig, M. T. , DiBattista, J. D. , … Bird, C. E. (2013). Origins of tropical marine biodiversity. Trends in Ecology and Evolution, 28, 359–366. [DOI] [PubMed] [Google Scholar]
- Braithwaite, C. J. R. (1987). Geology and paleogeography of the Red Sea regionIn Sea R., Edwards A. J., & Head S. M. (Eds.), Key environments. Red Sea (pp. 22–44). Oxford, UK: Pergamon Press. [Google Scholar]
- Briggs, J. C. (2005). The marine East Indies: Diversity and speciation. Journal of Biogeography, 32, 1517–1522. [Google Scholar]
- Brothers, E. B. , & Thresher, R. E. (1985). Pelagic duration, dispersal and the distribution of Indo‐Pacific coral reef fishes In Reaka M. L. (Ed.), The ecology of coral reefs (vol. 3). Washington, DC: US Department of Commerce. [Google Scholar]
- Budd, A. F. , & Pandolfi, J. M. (2010). Evolutionary novelty is concentrated at the edge of coral species distributions. Science, 328, 1558–1561. [DOI] [PubMed] [Google Scholar]
- Burt, J. A. , Feary, D. A. , Bauman, A. G. , Usseglio, P. , Cavalcante, G. H. , & Sale, P. F. (2011). Biogeographic patterns of reef fish community structure in the northeastern Arabian Peninsula. ICES Journal of Marine Science, 68, 1875–1883. [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. , Cresko, W. , & Postlethwait, J. H. (2011). Stacks: Building and genotyping loci de novo from short‐read sequences. G3: Genes, Genomes, Genetics, 1, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman, P. F. , & Bellwood, D. R. (2013). The historical biogeography of reef fishes: Global patterns of origination and dispersal. Journal of Biogeography, 40, 209–224. [Google Scholar]
- Coyne, J. A. , & Orr, H. A. (2004). Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- DiBattista, J. D. , Berumen, M. L. , Gaither, M. R. , Rocha, L. A. , Eble, J. A. , Choat, J. H. , … Bowen, B. W. (2013). After continents divide: Comparative phylogeography of reef fishes from the Red Sea and Indian Ocean. Journal of Biogeography, 40, 1170–1181. [Google Scholar]
- DiBattista, J. D. , Choat, J. H. , Gaither, M. R. , Hobbs, J. P. , Lozano‐Cortés, D. F. , Myers, R. F. , … Berumen, M. L. (2016). On the origin of endemic species in the Red Sea. Journal of Biogeography, 43, 13–30. [Google Scholar]
- DiBattista, J. D. , Gaither, M. R. , Hobbs, J.‐P. , Saenz‐Agudelo, P. , Piatek, M. J. , Bowen, B. W. , … Berumen, M. L. (2017). Comparative phylogeography of reef fishes from the Gulf of Aden to the Arabian Sea reveals two cryptic lineages. Coral Reefs, 36, 625–638. [Google Scholar]
- DiBattista, J. D. , Roberts, M. , Bouwmeester, J. , Bowen, B. W. , Coker, D. F. , Lozano‐Cortés, D. F. , … Berumen, M. L. (2016). A review of contemporary patterns of endemism for shallow water reef fauna in the Red Sea. Journal of Biogeography, 43, 423–439. [Google Scholar]
- DiBattista, J. D. , Rocha, L. A. , Hobbs, J. P. , He, S. , Priest, M. A. , Sinclair‐Taylor, T. H. , … Berumen, M. L. (2015). When biogeographical provinces collide: Hybridization of reef fishes at the crossroads of three marine biogeographical provinces in the Arabian Sea. Journal of Biogeography, 42, 1601–1614. [Google Scholar]
- DiBattista, J. D. , Saenz‐Agudelo, P. , Piatek, M. J. , Wang, X. , Aranda, M. , & Berumen, M. L. (2017). Using a butterflyfish genome as a general tool for RAD‐Seq studies in specialized reef fish. Molecular Ecology Resources, 17(6), 1330–1341. 10.1111/1755-0998.12662 [DOI] [PubMed] [Google Scholar]
- DiBattista, J. D. , Travers, M. J. , Moore, G. I. , Evans, R. D. , Newman, S. J. , Feng, M. , … Berry, O. (2017). Seascape genomics reveals fine‐scale patterns of dispersal for a reef fish along the ecologically divergent coast of Northwestern Australia. Molecular Ecology, 26, 6206–6223. [DOI] [PubMed] [Google Scholar]
- Doherty, P. J. , Planes, S. , & Mather, P. (1995). Gene flow and larval duration in seven species of fish from the Great Barrier Reef. Ecology, 76, 2373–2391. [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, A. J. (1989). Description, interpretation and use of the microstructure of otoliths from juvenile butterflyfishes (family Chaetodontidae). Marine Biology, 102, 167–182. [Google Scholar]
- Gaither, M. R. , Bernal, M. A. , Coleman, R. R. , Bowen, B. W. , Jones, S. A. , Simison, W. B. , & Rocha, L. A. (2015). Genomic signatures of geographic isolation and natural selection in coral reef fishes. Molecular Ecology, 24, 1543–1557. [DOI] [PubMed] [Google Scholar]
- Giles, E. C. , Saenz‐Agudelo, P. , Hussey, N. E. , Ravasi, T. , & Berumen, M. L. (2015). Exploring seascape genetics and kinship in the reef sponge Stylissa carteri in the Red Sea. Ecology and Evolution, 5, 2487–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould, A. L. , & Dunlap, P. V. (2017). Genomic analysis of a cardinalfish with larval homing potential reveals genetic admixture in the Okinawa Islands. Molecular Ecology, 26, 3870–3882. [DOI] [PubMed] [Google Scholar]
- Gutenkunst, R. N. , Hernandez, R. D. , Williamson, S. H. , & Bustamante, C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5, e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, H. B. , Berumen, M. L. , Saenz‐Agudelo, P. , Salas, E. , Williamson, D. H. , & Jones, G. P. (2017). Widespread hybridization and bidirectional introgression in sympatric species of coral reef fish. Molecular Ecology, 26, 5692–5704. [DOI] [PubMed] [Google Scholar]
- Hobbs, J.‐P.‐A. , Jones, G. P. , & Munday, P. L. (2011). Extinction risk in endemic marine fishes. Conservation Biology, 25, 1053–1055. [DOI] [PubMed] [Google Scholar]
- Hohenlohe, P. A. , Bassham, S. , Etter, P. D. , Stiffler, N. , Johnson, E. , & Cresko, W. A. (2010). Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genetics, 6, e1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , & Ahmed, I. (2011). adegenet 1.3‐1: New tools for the analysis of genome‐wide SNP data. Bioinformatics, 27, 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G. P. , Caley, M. J. , & Munday, P. L. (2002). Rarity in coral reef fish communities In Sale P. F. (Ed.), Coral reef fishes: Dynamics and diversity in a complex ecosystem (pp. 81–101). San Diego, CA: Academic Press. [Google Scholar]
- Kane, C. , Kosaki, R. K. , & Wagner, D. (2014). High levels of mesophotic reef fish endemism in the Northwestern Hawaiian Islands. Bulletin of Marine Science, 90, 693–703. [Google Scholar]
- Keith, S. A. , Herbert, R. J. , Norton, P. A. , Hawkins, S. J. , & Newton, A. C. (2011). Individualistic species limitations of climate‐induced range expansions generated by meso‐scale dispersal barriers. Diversity and Distributions, 17, 275–286. [Google Scholar]
- Keith, S. A. , Woolsey, P. S. , Madin, J. S. , Byrne, M. , & Baird, A. (2015). Differential establishment potential of species predicts a shift in coral assemblage structure across a biogeographic barrier. Ecography, 38, 1225–1234. [Google Scholar]
- Lawton, R. J. , Messmer, V. , Pratchett, M. S. , & Bay, L. K. (2011). High gene flow across large geographic scales reduces extinction risk for a highly specialised coral feeding butterflyfish. Molecular Ecology, 20, 3584–3598. [DOI] [PubMed] [Google Scholar]
- Lenormand, T. (2002). Gene flow and the limits to natural selection. Trends in Ecology and Evolution, 17, 183–189. [Google Scholar]
- Lessios, H. A. , & Robertson, D. R. (2006). Crossing the impassable: genetic connections in 20 reef fishes across the eastern Pacific barrier. Proceedings of the Royal Society B: Biological Sciences, 273, 2201–2208.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lischer, H. E. L. , & Excoffier, L. (2012). PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics, 28, 298–299. [DOI] [PubMed] [Google Scholar]
- Nanninga, G. B. , Saenz‐Agudelo, P. , Manica, A. , & Berumen, M. L. (2014). Environmental gradients predict the genetic population structure of a coral reef fish in the Red Sea. Molecular Ecology, 23, 591–602. [DOI] [PubMed] [Google Scholar]
- Nelson, J. S. (2006). Fishes of the world. Hoboken, NJ: John Wiley & Sons Inc. [Google Scholar]
- Orsini, L. , Vanoverbeke, J. , Swillen, I. , Mergeay, J. , & Meester, L. (2013). Drivers of population genetic differentiation in the wild: Isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Molecular Ecology, 22, 5983–5999. [DOI] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE, 7, e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picq, S. , McMillan, W. O. , & Puebla, O. (2016). Population genomics of local adaptation versus speciation in coral reef fishes (Hypoplectrus spp, Serranidae). Ecology and Evolution, 6, 2109–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest, M. A. , DiBattista, J. D. , McIlwain, J. L. , Taylor, B. M. , Hussey, N. E. , & Berumen, M. L. (2016). A bridge too far: Dispersal barriers and cryptic speciation in an Arabian Peninsula grouper (Cephalopholis hemistiktos). Journal of Biogeography, 43, 820–832. [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puebla, O. , Bermingham, E. , & McMillan, W. O. (2014). Genomic atolls of differentiation in coral reef fishes (Hypoplectrus spp., Serranidae). Molecular Ecology, 23, 5291–5303. [DOI] [PubMed] [Google Scholar]
- Reimer, J. D. , Herrera, M. , Gatins, R. , Roberts, M. B. , Parkinson, J. E. , & Berumen, M. L. (2017). Latitudinal variation in the symbiotic dinoflagellate Symbiodinium of the common reef zoantharian Palythoa tuberculosa on the Saudi Arabian coast of the Red Sea. Journal of Biogeography, 44, 661–673. [Google Scholar]
- Roberts, M. B. , Jones, G. P. , McCormick, M. I. , Munday, P. L. , Neale, S. , Thorrold, S. , … Berumen, M. L. (2016). Homogeneity of coral reef communities across 8 degrees of latitude in the Saudi Arabian Red Sea. Marine Pollution Bulletin, 105, 558–565. [DOI] [PubMed] [Google Scholar]
- Roberts, C. M. , Shepherd, A. R. D. , & Ormond, R. F. (1992). Large‐scale variation in assemblage structure of Red Sea butterflyfishes and angelfishes. Journal of Biogeography, 19, 239–250. [Google Scholar]
- Robertson, D. R. , Ackerman, J. L. , Choat, J. H. , Posada, J. M. , & Pitt, J. (2005). Ocean surgeonfish Acanthurus bahianus I. The geography of demography. Marine Ecology Progress Series, 295, 229–244. [Google Scholar]
- Robitzch, V. , Banguera‐Hinestroza, E. , Sawall, Y. , Al‐Sofyani, A. , & Voolstra, C. R. (2015). Absence of genetic differentiation in the coral Pocillopora verrucosa along environmental gradients of the Saudi Arabian Red Sea. Frontiers in Marine Science, 11, 2–5. [Google Scholar]
- Rohling, E. J. , Fenton, M. , Jorissen, F. J. , Bertrand, P. , Gnassen, G. , & Caulet, J. P. (1998). Magnitudes of sea‐level low stands of the past 500,000 years. Nature, 394, 162–165. [Google Scholar]
- Rougemont, Q. , Gagnaire, P.‐A. , Perrier, C. , Genthon, C. , Besnard, A. L. , Launey, S. , & Evanno, G. (2017). Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and nonparasitic lamprey ecotypes. Molecular Ecology, 26, 142–162. [DOI] [PubMed] [Google Scholar]
- Saenz‐Agudelo, P. , DiBattista, J. D. , Piatek, M. , Gaither, M. , Harrison, H. , Nanninga, G. , & Berumen, M. L. (2015). Seascape genetics along environmental gradients in the Arabian Peninsula: Insights from ddRAD sequencing of anemonefishes. Molecular Ecology, 24, 6241–6255. [DOI] [PubMed] [Google Scholar]
- Savidge, G. , Lennon, J. , & Matthews, A. J. (1990). A shore‐based survey of upwelling along the coast of Dhofar region, southern Oman. Continental Shelf Research, 10, 259–275. [Google Scholar]
- Schluter, D. (2000). The ecology of adaptive radiation. New York, NY: Oxford University Press. [Google Scholar]
- Sheppard, C. R. C. , Price, A. R. G. , & Roberts, C. J. (1992). Marine ecology of the Arabian Area. Patterns and processes in extreme tropical environments. London, UK: Academic Press. [Google Scholar]
- Simpson, S. D. , Harrison, H. B. , Claereboudt, M. R. , & Planes, S. (2014). Long‐distance dispersal via ocean currents connects Omani clownfish populations throughout entire species range. PLoS ONE, 9, e107610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M. (1987). Gene flow and the geographic structure of natural populations. Science, 236, 787–792. [DOI] [PubMed] [Google Scholar]
- Smeed, D. A. (2004). Exchange through the Bab el Mandab. Deep Sea Research Part II: Topical Studies in Oceanography, 51, 455–474. [Google Scholar]
- Sofianos, S. S. , Johns, W. E. , & Murray, S. P. (2002). Heat and freshwater budgets in the Red Sea from direct observations at Bab el Mandeb. Deep Sea Research Part II: Topical Studies in Oceanography, 49, 1323–1340. [Google Scholar]
- Stockwell, B. L. , Larson, W. A. , Waples, R. K. , Abesamis, R. A. , Seeb, L. W. , & Carpenter, K. E. (2016). The application of genomics to inform conservation of a functionally important reef fish (Scarus niger) in the Philippines. Conservation Genetics, 17, 239–249. [Google Scholar]
- Taylor, B. M. , Lindfield, S. J. , & Choat, J. H. (2015). Hierarchical and scale‐dependent effects of fishing pressure and environment on the structure and size distribution of parrotfish assemblages. Ecography, 38, 520–530. [Google Scholar]
- Taylor, B. M. , Trip, E. D. L. , & Choat, J. H. (2018). Dynamic demography: Investigations of life‐history variation in the parrotfishes. Chapter 4 In Hoey A., & Bonaldo R. (Eds.), The ecology of parrotfishes. Boca Raton, FL: CRC Press. [Google Scholar]
- Tine, M. , Kuhl, H. , Gagnaire, P.‐A. , Louro, B. , Desmarais, E. , Martins, R. S. T. , … Reinhardt, R. (2014). European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature Communications, 5, 5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen, R. J. , Andrews, K. R. , Baums, I. B. , Bird, C. E. , Concepcion, C. T. , Daly‐Engel, T. S. , … Bowen, B. W. (2011). Defining boundaries for applying ecosystem‐based management: A multispecies case study of marine connectivity across the Hawaiian Archipelago. Journal of Marine Biology, 2011, Article ID 460173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyberghein, L. , Verbruggen, H. , Pauly, K. , Troupin, C. , Mineur, F. , & De Clerck, O. (2012). Bio‐ORACLE: A global environmental dataset for marine species distribution modeling. Global Ecology and Biogeography, 21, 272–281. [Google Scholar]
- van Etten, J. (2017). R Package gdistance: Distances and routes on geographical grids. Journal of Statistical Software, 76, 1–21. [Google Scholar]
- Wagner, C. E. , Keller, I. , Wittwer, S. , Selz, O. M. , Mwaiko, S. , Greuter, L. , … Seehausen, O. (2013). Genome‐wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Molecular Ecology, 22, 787–798. [DOI] [PubMed] [Google Scholar]
- Wang, I. J. (2013). Examining the full effects of landscape heterogeneity on spatial genetic variation: A multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution, 67, 3403–3411. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C. , & Lotterhos, K. E. (2015). Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of F(ST). The American Naturalist, 186(Suppl. 1), S24–S36. [DOI] [PubMed] [Google Scholar]
- Wilkinson, C. (2008). Status of coral reefs of the world: 2008. Townsville, Australia: Global Coral Reef Monitoring Network and Reef and Rainforest Research Centre. [Google Scholar]
- Wilson, D. T. , & McCormick, M. I. (1999). Microstructure of settlement‐marks in the otoliths of tropical reef fishes. Marine Biology, 134, 29–41. [Google Scholar]
- Xu, W. , Ruch, J. , & Jónsson, S. (2015). Birth of two volcanic islands in the southern Red Sea. Nature Communications, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, G. , Pagani, M. , & Zhang, Y. G. (2017). Monsoonal upwelling in the western Arabian Sea since the middle Miocene. Geology, 45, 655–658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figs S1‐S3
Table S1
Table S2
Table S3
Table S4
Appendix S1
Appendix S2
Data Availability Statement
Raw SNP data and all appendices are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.rn8pk0p68.