

Abstract
Background:
The association between interstitial lung disease (ILD) and myeloperoxidase (MPO)-antineutrophil cytoplasmic antibodies (ANCAs) has been described, but pathologic characteristics are not well characterized.
Objectives:
We assessed the radiologic and pathologic characteristics of ILD in MPO-ANCA–positive patients and the association between ILD and vasculitis, particularly microscopic polyangiitis (MPA).
Methods:
We retrospectively searched electronic health records to identify MPO-ANCA–positive patients with ILD who underwent surgical lung biopsy at our institution from January 1997 through August 2017. Demographic, clinical, imaging, and pathologic characteristics were analyzed.
Results:
We identified 18 MPO-ANCA–positive patients with ILD. The median (range) age was 58 (43-75) years, and the cohort included 10 men (56%), 10 former smokers (56%), and 11 patients (61%) had clinical evidence of systemic vasculitis (MPA) at the time of diagnosis of ILD. On high-resolution computed tomography, the most common radiologic pattern was “inconsistent with usual interstitial pneumonia” (UIP) (n=14 [78%]); the other 4 patients (22%) fulfilled the radiologic criteria for the UIP pattern. Honeycombing was seen in 15 patients (83%). Ten patients (56%) had the UIP pattern on biopsy: 4 of these patients had additional inflammatory changes that were not typical of UIP (as seen in patients with idiopathic pulmonary fibrosis), and the other 6 patients had other inflammatory patterns or findings. The presence or absence of MPA did not correlate with pathologic findings.
Conclusions:
MPO-ANCA–positive patients with ILD do not show the typical UIP pattern as seen in patients with idiopathic pulmonary fibrosis on surgical lung biopsy.
Key words: ILD, interstitial lung disease, microscopic polyangiitis, myeloperoxidase antibodies, UIP, usual interstitial pneumonia
Abbreviations
- ANCA, antineutrophil cytoplasmic antibody
- DLCO, diffusing capacity of lung for carbon monoxide
- HR, hazard ratio
- HRCT, high-resolution computed tomography
- ILD, interstitial lung disease
- IPF, idiopathic pulmonary fibrosis
- MPA, microscopic polyangiitis
- MPO, myeloperoxidase
- NSIP, nonspecific interstitial pneumonia
- OP, organizing pneumonia
- UIP, usual interstitial pneumonia
Introduction
Microscopic polyangiitis (MPA) is a necrotizing, systemic small-vessel vasculitis that often involves the lungs, skin, kidneys, and peripheral nervous system and is associated with myeloperoxidase (MPO)-antineutrophil cytoplasmic antibodies (ANCAs) (1). MPO-ANCAs are thought to be pathogenic because they promote the development of capillaritis (2). The association between interstitial lung disease (ILD) and MPO-ANCA was first reported in a case series in the 1990s (3). Other studies subsequently reported similar associations (4-11). ILD is observed in 2 settings: in patients with systemic vasculitis, usually MPA; and in patients with only positive MPO-ANCA serologic findings, some of whom later have MPA. It is unclear whether ILD observed in the presence of only MPO-ANCA differs from idiopathic ILD and ILD with overt MPA. Although the association between radiologic evidence of ILD and MPO-ANCA has been described, pathologic descriptions are limited.
The objective of this study was to describe the radiologic and pathologic characteristics of ILD associated with positive MPO-ANCA serologic findings. We also aimed to identify differences related to the presence of systemic vasculitis, particularly MPA.
Methods
Patient Selection
We used Advanced Cohort Explorer (Mayo Clinic) to retrospectively search for the electronic health records of all MPO-ANCA–positive adult patients with ILD who underwent surgical lung biopsy at Mayo Clinic, Rochester, Minnesota, from January 1, 1997, through August 31, 2017. Our search was conducted iteratively by using the following terms: first, “microscopic polyangiitis,” “MPO,” “MPO vasculitis,” and “p-ANCA vasculitis”; second, “pulmonary fibrosis” and “interstitial lung disease”; and third, “surgical lung biopsy,” “lung biopsy,” and “video-assisted thoracoscopic surgery.” For all identified patients, we performed a complete review of the health records to confirm the diagnoses and lung biopsy procedures. To avoid the limitations of transbronchial biopsy results, we focused on only surgical lung biopsies. We excluded patients with transbronchial lung biopsy and patients positive for perinuclear ANCA activity determined by only indirect immunofluorescence (ie, without MPO reactivity).
The Mayo Clinic Institutional Review Board approved this study (No. 17-002430). Patients who did not authorize the use of their electronic health records were excluded.
Clinical and Laboratory Data
Data extracted from the electronic health records included age, sex, race/ethnicity, smoking status, symptoms, laboratory results, MPO-ANCA status, urinalysis results, serum creatinine level, presence or absence of inflammatory markers, pulmonary function test results, treatment, outcome, and follow-up duration.
Diagnostic Criteria for MPA
MPA, when present, was diagnosed according to the definitions for vasculitides adopted by the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides (1).
Radiologic Analysis
A thoracic radiologist (C.W.C.) reviewed all thoracic high-resolution computed tomography (HRCT) scans. The patterns were described according to the diagnostic criteria of the European Respiratory Society/American Thoracic Society for idiopathic interstitial pneumonias (12, 13). The extent and location of fibrosis were also determined. Follow-up HRCT scans, when available, were also reviewed.
Pathologic Analysis
A thoracic pathologist (E.E.Y. and/or T.V.C.) reviewed all available surgical lung biopsy slides and reports. Pathologic changes were described according to the appropriate classifications of the European Respiratory Society/American Thoracic Society for idiopathic interstitial pneumonias (12,13).
Statistical Analysis
Categorical variables were compared between patients with MPA and patients without MPA with the Fisher exact test. The Wilcoxon rank sum test was used to compare continuous variables between groups. P<.05 was considered statistically significant. Univariate Cox proportional hazards regression analysis was used to evaluate associations between variables and death. Kaplan-Meier analysis was used to estimate survival. JMP software (SAS Institute Inc) was used to perform the statistical analysis.
Results
We identified and evaluated 18 MPO-ANCA–positive patients. The median (range) age was 58 (43-75) years, and 10 men (56%) were included (Table 1). Most patients in our cohort were white (89%), and 10 patients (56%) had a history of smoking. The median (range) duration of respiratory symptoms before the diagnosis of ILD was 1 (0.16-30) year. Eleven (61%) patients had clinical evidence of active systemic vasculitis (MPA) at the time of diagnosis of ILD.
Table 1.
Demographic and Clinical Characteristics
| Characteristic | Valuea (N=18) |
| Age, y | 58 (43-75) |
| Men | 10 (56) |
| White race/ethnicity | 16 (89) |
| History of smoking | 10 (56) |
| Microscopic polyangiitis | 11 (61) |
| Respiratory symptomsb | 17 (94) |
| Dyspnea | 16 (94) |
| Cough | 10 (59) |
| Chest pain | 5 (29) |
| Results of pulmonary function tests | |
| Percentage predicted FVCc | 64 (32-88) |
| Percentage predicted DLCOd | 54 (37-88) |
| RVSP, mm Hgd | 36 (21-69) |
Abbreviations: DLCO, diffusing capacity of lung for carbon monoxide; FVC, forced vital capacity; RVSP, right ventricular systolic pressure.
a Values are shown as No. (%) or median (range); b Some patients had more than 1 symptom; c Determined for 11 patients at the time of diagnosis of pulmonary fibrosis; d Determined for 13 patients at the time of diagnosis of pulmonary fibrosis.
Fourteen patients (78%) underwent pulmonary function tests at the time of diagnosis of ILD. Six patients had a restrictive pattern evidenced by reduction in total lung capacity and reduction in diffusing capacity of lung for carbon monoxide (DLCO); 5, isolated reduction in DLCO; 1, isolated reduction in total lung capacity (DLCO not measured); and 2, normal pulmonary function test results. The median (range) percentage predicted forced vital capacity was 64% (32%-88%), and the median (range) percentage predicted DLCO was 54% (37%-88%). Eleven patients underwent echocardiography at the time of diagnosis of ILD, and the median (range) right ventricular systolic pressure was 36 (21-69) mm Hg. Three patients met the echocardiographic criteria for pulmonary hypertension (right ventricular systolic pressure ≥50 mm Hg) (14).
The HRCT findings are summarized in Table 2. The most common pattern was “inconsistent with usual interstitial pneumonia” (UIP), which was noted in 14 patients (78%) (unclassified or atypical, n=9; nonspecific interstitial pneumonia [NSIP], n=4; organizing pneumonia [OP], n=1). Only 4 patients (22%) met the radiologic criteria for the UIP pattern, but most patients had honeycombing (n=15 [83%]). The presence of ground-glass opacities (n=11 [61%]) and mosaic attenuation (n=3 [17%]) were the most common reasons for classifying findings on HRCT images as inconsistent with a UIP pattern (Figure 1). Presence of the UIP pattern on HRCT was not associated with the presence or absence of clinical vasculitis (MPA) at the time of diagnosis. Among the patients who had MPA at the time of diagnosis of ILD, 2 (18%) had the UIP pattern on HRCT and 9 (82%) had a pattern inconsistent with UIP. However, among patients who did not have active MPA at the time of diagnosis of ILD, 2 (29%) had the UIP pattern on HRCT and 5 (71%) had a pattern inconsis-tent with UIP (P=.61). Fibrosis was predominantly distributed in the lower lobe (n=10 [56%]), or a diffuse pattern was detected (n=8 [44%]); upper-lobe predominance was not detected in any patients. Regarding the axial distribution of fibrosis, peripheral changes were most commonly seen (n=13 [72%]).
Table 2.
Findings on High-Resolution Computed Tomography
| Characteristic | No. of Patients (%) (N=18) |
| Pattern of interstitial abnormalities | |
| UIP | 4 (22) |
| Inconsistent with UIP | 14 (78) |
| Unclassified or atypical | 9 (64) |
| NSIP | 4 (29) |
| OP | 1 (7) |
| Honeycombing | 15 (83) |
| Cause of inconsistency with the UIP patterna | |
| Ground-glass opacities | 11 (61) |
| Mosaic attenuation | 3 (17) |
| Diffuse distribution | 2 (11) |
| Midlung distribution | 2 (11) |
| Apical thickening | 1 (6) |
| Consolidation | 1 (6) |
| Subpleural sparing | 1 (6) |
| Distribution of fibrosis | |
| Craniocaudal | |
| Lower | 10 (56) |
| Diffuse | 8 (44) |
| Axial | |
| Peripheral | 13 (72) |
| Diffuse | 3 (16) |
| Central | 2 (11) |
Abbreviations: NSIP, nonspecific interstitial pneumonia; OP, organizing pneumonia; UIP, usual interstitial pneumonia.
a Some patients had more than 1 finding.
Fig. 1.
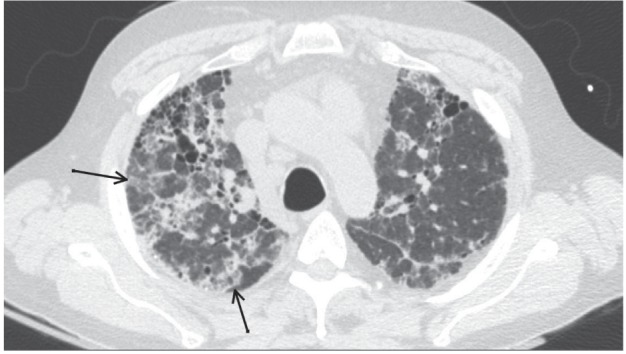
Pattern Inconsistent With Usual Interstitial Pneumonia. Axial high-resolution computed tomographic image of the upper pulmonary lobes of a 55-year-old man with microscopic polyangiitis. This complex interstitial pattern is inconsistent with usual interstitial pneumonia according to the 2011 criteria of the European Respiratory Society/American Thoracic Society. Ground-glass opacities (black arrows), diffuse architectural distortion, scattered consolidation, and honeycombing are shown
The surgical lung biopsy results are summarized (Table 3). The UIP pattern was predominant in 10 patients (56%). In 4 of 10 patients with the UIP pattern, additional inflammatory changes were identified that are not typical of the UIP pattern in patients with idiopathic pulmonary fibrosis (IPF), including bronchiolitis, lymphoid hyperplasia, desquamative interstitial pneumonia, and OP. The NSIP pattern was predominant in 5 patients, and bronchiolitis was predominant in 1 patient. NSIP and the early UIP pattern could not be confidently distinguished in 1 patient. Only 1 of 18 biopsies showed active granulomatosis with polyangiitis (fibroinflammatory changes with granulomas) as the cause of the radiologic findings. The presence of a UIP pattern on biopsy was not associated with the presence or absence of clinical vasculitis (MPA) at the time of diagnosis. Among patients who had MPA at the time of diagnosis of ILD, 3 (27%) had the UIP pattern on biopsy, 1 (9%) had the UIP pattern with additional features, and 7 (64%) had a pattern inconsistent with UIP. However, among patients who did not have MPA at the time of diagnosis of ILD, 3 (43%) had the UIP pattern on biopsy, 3 (43%) had the UIP pattern with additional features, and 1 (14%) had a pattern inconsistent with UIP (P=.07).
Table 3.
Pathologic pulmonary Findings of 18 patients
| Patient No. | Histologic Findings |
| 1 | UIP pattern with areas showing features of DIP |
| 2 | Follicular bronchiolitis and cellular bronchiolitis with foci of OP and foci resembling NSIP |
| 3 | UIP pattern with a slight increase in lymphoid aggregates and focal OP |
| 4 | NSIP with follicular hyperplasia and DIP features |
| 5 | UIP pattern |
| 6 | NSIP with OP and follicular bronchiolitis |
| 7 | Fibroinflammatory changes with granulomas consistent with GPA |
| 8 | UIP pattern |
| 9 | UIP pattern with some associated chronic bronchiolitis, chronic pleuritis, and focal OP |
| 10 | UIP pattern |
| 11 | Unable to distinguish fibrosing NSIP and the early UIP pattern |
| 12 | NSIP with chronic bronchiolitis, OP, and early, focal, acute bronchopneumonia |
| 13 | Cellular NSIP with mild fibrosis and OP |
| 14 | Cellular and fibrosing NSIP |
| 15 | UIP pattern |
| 16 | UIP pattern |
| 17 | UIP pattern |
| 18 | UIP pattern with lymphocytic infiltrates and follicular lymphoid hyperplasia |
Abbreviations: DIP, desquamative interstitial pneumonia; GPA, granulomatosis with polyangiitis; NSIP, nonspecific interstitial pneumonia; OP, organizing pneumonia; UIP, usual interstitial pneumonia
Follow-Up After Initial Evaluation
The median (range) follow-up period was 4.3 (0.6-13.4) years. During this period, active vasculitis developed in 3 of 7 patients without vasculitis (MPA) at the time of diagnosis of ILD at 7.2, 9.6, and 20.4 months after the initial diagnosis of ILD. Fifteen patients (83%) underwent follow-up HRCT, with a median (range) interval of 1.78 (0.24-11.39) years (HRCT was not performed at fixed intervals). Eleven patients had progression of interstitial findings (including 8 patients with moderate progression); 3, stable findings; and 1, improved findings.
Pulmonary function tests were repeated in 11 patients, with a median (range) interval of 2.37 (0.6-12.0) years (pulmonary function tests were not performed at fixed intervals). The median (range) change in percentage predicted forced vital capacity was −1% (−29% to 27%), and the median (range) change in percentage predicted DLCO was −10% (−26% to 25%).
Nine patients (50%) died during follow-up, and the most common cause of death was respiratory failure (n=6). One patient died of active vasculitis, and the causes of death could not be determined for 2 patients. The median survival of our cohort was 5.5 years (Figure 2). According to the univariate analysis, the predictors of death were DLCO at diagnosis (HR [hazard ratio], 0.89; 95% CI, 0.78-0.97; P=.007), DLCO at follow-up (HR, 0.82; 95% CI, 0.53-0.96; P=.008), and forced vital capacity at follow-up (HR, 0.95; 95% CI, 0.90-0.99; P=.02). All of these predictors are related to impaired lung function.
Fig. 2.
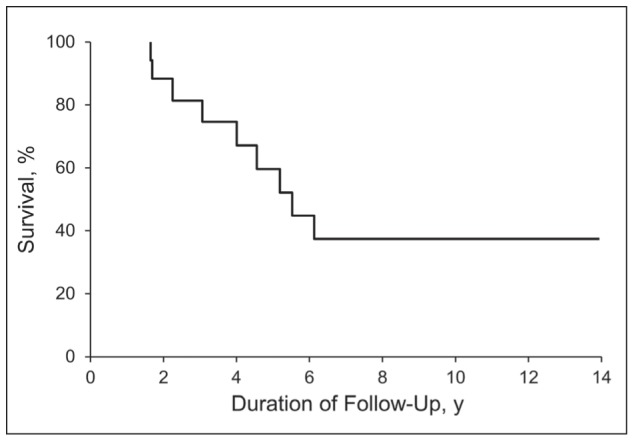
Kaplan-Meier Estimate of Survival
Discussion
In this retrospective study, we assessed the relationship between radiologic and pathologic characteristics of MPO-ANCA–positive patients with ILD who underwent surgical lung biopsy. The most common radiologic pattern in this cohort was inconsistent with the UIP pattern (as seen in patients with IPF), although more than half of patients had a predominant UIP pattern on biopsy. NSIP and bronchiolitis were also predominant pathologic findings in some patients. Of the 10 patients with a predominant UIP pattern, 4 had inflammatory changes that were not typical of the UIP pattern observed in patients with IPF. Radiologic and pathologic findings appeared to be independent of active clinical vasculitis at the time of diagnosis of ILD. Only 1 of 18 patients had pathologic characteristics of vasculitis (fibroinflammatory changes with granulomas) reported as the cause of the radiologic changes.
UIP has been described as a predominant pattern of fibrosis on HRCT in MPO-ANCA–positive patients (15, 16). However, only 4 of our patients (22%) had the typical UIP pattern on HRCT. Nevertheless, most patients in our cohort (15 patients [83%]) had honeycombing indicative of a clinically significant fibrosing process. In addition to honeycombing, some findings, particularly ground-glass opacities, made the radiographic diagnosis of UIP less likely. This discrepancy most likely reflects the bias associated with selecting patients for surgical lung biopsy who did not meet the radiographic criteria for UIP. Similar findings have been described by Foulon et al (10), who reported some degree of ground-glass opacities, honeycombing, reticular opacities, and traction bronchiectasis in all 17 patients in their study.
In our study, radiologic signs of fibrosis were predominantly distributed in the lower lobe. A similar finding is described in another study, in which 10 of 14 patients had lower-zone–predominant lung fibrosis (11). Similar lower-zone–predominant and peripheral-zone–predominant patterns of pulmonary fibrosis were described in another study of 19 patients (4). This is an important observation because the presence of lower-zone pulmonary fibrosis may distinguish ILD associated with MPO-ANCA and MPA from other types of ILD with upper-lobe predominance, such as chronic hypersensitivity pneumonitis.
Although the UIP pattern was the most common, predominant pathologic pattern in our patients, additional inflammatory changes that are not typical of the UIP pattern in patients with IPF were seen in 4 patients, including bronchiolitis, OP, lymphoid aggregates, and lymphoid hyperplasia. Five of our patients had a predominant NSIP pattern. In 1 patient, NSIP and early UIP could not be confidently distinguished, and bronchiolitis was the main finding in another patient. Thus, in 17 of 18 patients, the predominant histologic finding was the UIP pattern or NSIP, although patients with the UIP pattern showed additional inflammatory changes usually not seen in patients with IPF. Only 1 patient (Patient No. 7) showed vasculitis on surgical lung biopsy as the cause of the radiologic changes.
Prior studies of pathologic findings in MPO-ANCA–positive patients are rare. Homma et al (17) described biopsy and postmortem specimens obtained from 15 patients. Honeycombing was seen in 12 of these patients, but most of them also had additional findings, including OP (n=9 [60%]), pleuritis (n=7 [47%]), lymphoid hyperplasia (n=6 [40%]), or vasculitis (n=5 [33%]). Foulon et al (10) reported 3 lung biopsies that showed only the UIP pattern. Ando et al (18) reported the UIP pattern in 6 patients who underwent surgical lung biopsy, but 2 patients had prominent lymphoid proliferation. Although relatively few patients were reported in these studies, the findings do not appear to be appreciably different from ours, with the provision that NSIP has not been described as a predominant pattern.
The median survival of our patients was 5.5 years. This is comparable to a median survival of 72 months for patients with MPA and pulmonary fibrosis reported by Tzelepis et al (8) and the mean survival time of 4.2 (range, 0.0-27.8) years reported by Arulkumaran et al (11). In the latter study, the authors reviewed 99 cases described in the literature and reported a median (range) survival of 5.3 (0.08-13.7) years. We identified 3 factors associated with death, all indicators of severity of pulmonary fibrosis. Kagiyama et al (5) identified older age, proteinase 3–ANCA positivity, and poor pulmonary function test results as negative prognostic factors. Over the years, pulmonary fibrosis tends to worsen in MPO-ANCA–positive patients; whether immunosuppressive therapy provides any beneficial effect on the progression of lung fibrosis is unclear. Treatment response could also differ according to the type of fibrosis. Most patients in our cohort died of respiratory failure, and other studies have reported similar findings (9, 10, 16). Thus, the overall prognosis of these patients could be directly related to the pulmonary fibrosis and not the underlying vasculitis. The prognosis of MPO-ANCA–positive patients with pulmonary fibrosis has been described as better than (19), similar to (17, 18) and worse than (5, 17) the prognosis of MPO-ANCA–negative patients with pulmonary fibrosis. This suggests that acute management of vasculitis may influence the reported outcomes of MPO-ANCA–positive patients with ILD and MPA.
Our study raises several important questions about the care of MPO-ANCA–positive patients with ILD. First, how should clinicians manage ILD? This question, which is particularly relevant in the era of antifibrotics, has 2 components: 1) Which patients (if any) should be treated with immunosuppressive therapy; and 2) Which patients (if any) should be considered for antifibrotic therapy? Because most of our patients had radiologic patterns of fibrosis that were not consistent with the UIP pattern, we cannot draw conclusions about patients with the typical UIP pattern. However, Chino et al (19) have shown that MPO-ANCA–positive patients with UIP but without overt MPA have more inflammation on biopsy than MPO-ANCA–negative patients with UIP. Their observation and our findings might suggest that MPO-ANCA–positive patients with ILD need an immunosuppressive treatment trial for ILD. Further standardized, multicenter investigations should evaluate the treatment responses of patients with ILD to immunosuppressive therapy and antifibrotics, particularly the responses of patients with radiographic or pathologic findings of UIP.
The prevalence of MPO-ANCA among patients with ILD is unclear. However, our study confirms the findings of other studies, which indicate that over the ensuing years overt MPA subsequently develops in more than 40% of MPO-ANCA–positive patients without overt MPA at the time of diagnosis of ILD (5, 10). Thus, the second important management question is how to address the risk of vasculitis among MPO-ANCA–positive patients. At the very least, patients should know about the signs and symptoms of active MPA and should be carefully monitored for the manifestations of MPA, particularly glomerulonephritis. Immunosuppressive treatment of ILD with inflammatory characteristics could also prevent the development of overt MPA.
The final management question raised by our findings is whether MPO-ANCA–positive patients with ILD who do not have a typical UIP pattern on HRCT, and therefore are considered for biopsy, should undergo this procedure, or whether they should first receive a treatment trial of immunosuppressive therapy.
Our study has several limitations. First, this is a retrospective study with a small sample size, and data are unavailable for some patients. Although we tried to identify excessive inflammation beyond what is generally seen in patients with the UIP pattern (eg, OP, NSIP, lymphoid aggregates), some patients may have had autoimmune features within the accepted criteria for UIP (eg, Patient No. 3 in Table 3). Second, our study is dependent on the biases of the clinicians who selected patients for surgical lung biopsy. Most patients at our institution with a radiologic pattern of definite UIP do not undergo surgical lung biopsy, and the primary role of this procedure is to identify or exclude other causes of ILD that may be amenable to treatment. Consequently, our study may not be representative of the most common pattern of pulmonary fibrosis in MPO-ANCA–positive patients that can be detected with HRCT. Third, the chronologic evolution of lung fibrosis and active vasculitis was difficult to assess exactly because most patients were first seen elsewhere and the exact sequence of events was difficult to establish. However, because most patients had clinical features of active vasculitis (which develops acutely or subacutely) at the time of the biopsy, and because most patients had radiologic and pathologic evidence of the UIP pattern (a lesion that develops slowly), we speculate that pulmonary fibrosis usually precedes the onset of MPA. This is further supported by the observation (made here and by others) that systemic MPA will subsequently develop in many patients with only MPO-ANCA positivity (5).
In conclusion, in assessing the relationship between pathologic and radiologic findings in MPO-ANCA–positive patients with pulmonary fibrosis, we identified inflammation on surgical lung biopsies. This was mainly seen when the HRCT findings were not consistent with the UIP pattern. Our study shows the variability in the characteristics of ILD among MPO-ANCA–positive patients. Further multicenter investigations of larger cohorts of patients are needed to define outcomes and treatment responses according to the type of ILD.
Role of the Funding Source:
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author Contributions:
M.B.: collected data, analyzed and interpreted data, wrote the manuscript. E.E.Y.: collected pathologic data, analyzed and interpreted data, helped write the manuscript. T.V.C.: collected pathologic data, analyzed and interpreted data, helped write the manuscript. C.W.C.: collected radiologic data, analyzed and interpreted data, helped write the manuscript. J.H.R.: analyzed and interpreted data, helped write the manuscript. U.S.: senior author, research design, collected data, analyzed and interpreted data, statistical analysis, wrote the manuscript.
References
- 1.Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 2.Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nada AK, Torres VE, Ryu JH, Lie JT, Holley KE. Pulmonary fibrosis as an unusual clinical manifestation of a pulmonary-renal vasculitis in elderly patients. Mayo Clin Proc. 1990;65(6):847–856. doi: 10.1016/s0025-6196(12)62575-0. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Wang YX, Jiang CG, et al. A retrospective study of microscopic polyangiitis patients presenting with pulmonary fibrosis in China. BMC Pulm Med. 2014;14:8. doi: 10.1186/1471-2466-14-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagiyama N, Takayanagi N, Kanauchi T, Ishiguro T, Yanagisawa T, Sugita Y. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2015;2(1):e000058. doi: 10.1136/bmjresp-2014-000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ando Y, Okada F, Matsumoto S, Mori H. Thoracic manifestation of myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA)-related disease. CT findings in 51 patients. J Comput Assist Tomogr. 2004;28(5):710–716. doi: 10.1097/01.rct.0000135280.79012.c7. [DOI] [PubMed] [Google Scholar]
- 7.Nozu T, Kondo M, Suzuki K, Tamaoki J, Nagai A. A comparison of the clinical features of ANCA–positive and ANCA–negative idiopathic pulmonary fibrosis patients. Respiration. 2009;77(4):407–415. doi: 10.1159/000183754. [DOI] [PubMed] [Google Scholar]
- 8.Tzelepis GE, Kokosi M, Tzioufas A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J. 2010;36(1):116–121. doi: 10.1183/09031936.00110109. [DOI] [PubMed] [Google Scholar]
- 9.Hervier B, Pagnoux C, Agard C, et al. Pulmonary fibrosis associated with ANCA–positive vasculitides. Retrospective study of 12 cases and review of the literature. Ann Rheum Dis. 2009;68(3):404–407. doi: 10.1136/ard.2008.096131. [DOI] [PubMed] [Google Scholar]
- 10.Foulon G, Delaval P, Valeyre D, et al. ANCA-associated lung fibrosis: analysis of 17 patients. Respir Med. 2008;102(10):1392–1398. doi: 10.1016/j.rmed.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Arulkumaran N, Periselneris N, Gaskin G, et al. Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 2011;50(11):2035–2043. doi: 10.1093/rheumatology/ker236. [DOI] [PubMed] [Google Scholar]
- 12.Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–987. doi: 10.1183/13993003.00150-2015. [DOI] [PubMed] [Google Scholar]
- 13.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadrous HF, Pellikka PA, Krowka MJ, et al. The impact of pulmonary hypertension on survival in patients with idiopathic pulmonary fibrosis. Chest. 2005;128(6 Suppl):616S–617S. doi: 10.1378/chest.128.6_suppl.616S-a. [DOI] [PubMed] [Google Scholar]
- 15.Comarmond C, Crestani B, Tazi A, et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: a series of 49 patients and review of the literature. Medicine (Baltimore) 2014;93(24):340–349. doi: 10.1097/MD.0000000000000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez Casares M, Gonzalez A, Fielli M, Caputo F, Bottinelli Y, Zamboni M. Microscopic polyangiitis associated with pulmonary fibrosis. Clin Rheumatol. 2015;34(7):1273–1277. doi: 10.1007/s10067-014-2676-1. [DOI] [PubMed] [Google Scholar]
- 17.Homma S, Matsushita H, Nakata K. Pulmonary fibrosis in myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitides. Respirology. 2004;9(2):190–196. doi: 10.1111/j.1440-1843.2004.00581.x. [DOI] [PubMed] [Google Scholar]
- 18.Ando M, Miyazaki E, Ishii T, et al. Incidence of myeloperoxidase anti-neutrophil cytoplasmic antibody positivity and microscopic polyangitis in the course of idiopathic pulmonary fibrosis. Respir Med. 2013;107(4):608–615. doi: 10.1016/j.rmed.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Chino H, Hagiwara E, Kitamura H, et al. Myeloperoxidase Anti-Neutrophil Cytoplasmic Antibody-Positive Interstitial Pneumonia Associated with Granulomatosis with Polyangiitis Diagnosed by Surgical Lung Biopsy. Respiration. 2016;92(5):348–355. doi: 10.1159/000449529. [DOI] [PubMed] [Google Scholar]