

Abstract
Background:
The guidelines on idiopathic pulmonary fibrosis (IPF) diagnosis established the crucial role of multidisciplinary discussion (MDD) in the diagnosis of interstitial lung diseases (ILD). However, real-life evaluation of MDD remains scarce. Our aim was to study the impact of a well-structured MDD on etiological assessment, diagnosis, and management of ILD.
Methods:
We collected and analysed all relevant data on patients concerning diagnosis and treatment before and after MDD during the year 2017.
Results:
One hundred fifty patients were included in the analysis. MDD had a significant impact on management: 42% of diagnoses were revised and the number of unclassifiable ILD was significantly reduced. Lung biopsy was performed in 26 patients (12 cryobiopsies and 14 surgical biopsies). The most prevalent diagnoses were connective-tissue disease associated ILD (32%), idiopathic pulmonary fibrosis (23%), hypersensitivity pneumonitis (13%) and granulomatous ILD (7%). MDD led to a change or initiation of treatment in 55% of cases. Nine patients were evaluated for transplantation, 23 patients were screened for academic or sponsored clinical trials and an 8-fold increase in rehabilitation inclusion was observed.
Conclusion:
Our results confirm the benefits of MDD on ILD management and diagnosis. MDD also facilitates access to non-pharmacological therapies and clinical trials.
Key words: interstitial lung diseases, multidisciplinary management
Introduction
The diagnostic and management of interstitial lung diseases (ILD) are complex, as this group of disorders encompasses a wide heterogeneity of diseases, presenting with different causes, requiring personalized management and leading to variable outcomes. In 2001, The American Thoracic Society/European Respiratory Society (ATS/ERS) already highlighted the need for a multidisciplinary and dynamic process in diagnosing idiopathic interstitial pneumonias (IIP) (1). Few years later, the ATS/ERS guidelines recommended multidisciplinary discussion (MDD) among experts to diagnose idiopathic pulmonary fibrosis (IPF) (1). This recommendation was reconducted in the last 2018 guidelines (2, 3). The emergence of anti-fibrotic drugs and the potential danger of misused immunosuppressive therapy (4) makes discrimination between IPF and non IPF-ILD critically important in clinical practice (4, 5).
Some studies have tackled the issue of the role of MDD in ILD diagnosis. Flaherty et al have shown that, in idiopathic interstitial pneumonia’s (IIP), level of diagnostic agreement between observers and diagnostic confidence improves as more data are shared during a multidisciplinary discussion, especially for the non IPF-ILD (6). Disagreement in term of diagnosis was at the highest level in non-academic centres with no access to MDD meetings, reflecting the need for referring ILD in expert centres and for promoting the use of these MDD meetings (7). Walsh et al have demonstrated that MDD increases frequency and confidence of IPF diagnosis. They have also shown that inter-MDD agreement was good, especially in IPF. Regarding the subgroup of IPF diagnosed without requirement of a biopsy (typical clinical context and typical HRCT pattern), the level of inter-observers and inter-MDD diagnostic agreement was high and the difference between levels of inter-individual and inter MDD agreement was low (8). This is probably explained by the existence of validated guidelines that are easy to apply for clinicians with experience in ILD. Another observation that emerged from these studies was that diagnosis of chronic hypersensitivity pneumonitis and disease with a non-specific interstitial pneumonia (NSIP) HRCT pattern were still challenging despite the input of MDD. Therefore, evaluating MDD performance in real-world setting is valuable.
In the light of recent evidence, a well-structured MDD was set up in our department. The aim of the present study was to assess the impact of these MDD in our daily clinical practice. We hypothesized that MDD would significantly impact (1) ILD diagnosis and (2) ILD management. The purpose of this study was not only to observe the effects on diagnoses, but also on diagnostic processes, choices of treatment and recommendations for non-pharmacological treatment.
Methods
Study design
This is a single-centre retrospective study. All information of ILD patients discussed in MDD between January 1st and December 31st2017 were included in a database and eligible for the study. For each case, relevant clinical and demographic characteristics were collected. We reported also data about pre- and post-referral investigations, diagnosis and treatment. For every patient, we had a “pre-MDD” diagnosis (i.e. the suspected diagnosis, based on the form filled by the clinician) and a “post-MDD” diagnosis, corresponding to the conclusion.
Recommendations on rehabilitation program, transplantation valuation and academic or sponsored clinical trials were analysed. Data collection was performed between January 1st and July 1st 2018.
We included patients only once even if the case was presented again during the year. We excluded patients for which no structured form had been completed and validated after the MDD.
We applied the STROBE criteria for observational studies (http://www.equator-network.org/wp-content/uploads/2015/10/STROBE_checklist_v4_combined.pdf).
Multidisciplinary discussion meetings
MDD are organized every other week and last about 90 minutes. The panel comprises two pulmonologists with experience in ILD, at least one chest radiologist, 1 rheumatologist, 1 surgeon and 1 histopathologist. A study coordinator involved in ILD-related clinical trials also attends the meeting. Other specialties (general internal medicine, nephrologists) occasionally refer patients for an ILD workup. For each patient, information about medical history, symptoms and signs, toxic exposure (smoking status, drugs, environmental and occupational), functional respiratory test, bronchoalveolar lavage findings and autoantibody profile are collected in a structured computer form by the referral specialist (supplementary figure 1). The clinical context is exposed briefly and then images from high resolution computed tomography (HRCT) are presented by the radiologist who defined a typical CT pattern whenever possible. In case a biopsy was performed, selected images are presented by the pathologist. For each case presented, the structured form is completed with a definitive CT pattern, histopathology pattern when available, final diagnosis and recommendations for further management and follow up. Finally, a pulmonologist specialized in ILD validates this form and inserts it in the patient’s medical file. Some cases are discussed twice or more: Patients who underwent a lung biopsy and cases requiring treatment response assessment or a significant change in management.
Figure S1.

Structured form to fill prior to MDD for every patient. Items in blue are mandatory for form validation. Some items are filled with text (i.e. past medical history); others are simply ticked (i.e. smoking status). Biological values are automatically filled from our informatics system. Lung function values can be added in the appropriate box.
Analysis and statistics
For every patient included, we compared suspected diagnosis at referral to final diagnosis established by the MDD. Chi-square test was used to estimate the impact of MDD impact on the number of unclassifiable ILD after MDD.
Ethics
The present study was approved by our local ethics committee (study PNEU-ILD-02, approval number 2018/15MAR/116).
Results
Study population
One hundred fifty-three patients were discussed in MDD. A mean of 9 (range 7-16) patients are discussed at every meeting. We excluded three cases for who the structured form had not been filled properly and validated, meaning that one hundred fifty patients were included in the analysis (table 1). Sex ratio was 78/72 (M/F). The mean age was 63.1 years (SD=15.3). One third of the subjects were former or current smokers of at least 20 pack-years. Most patients were referred by pulmonologists (n= 58, 38 %) and rheumatologists (n=46, 30%) working in our hospital. Others (19%) were addressed by pulmonologists from primary and secondary centres. The others were referred by various department of our centre (internal medicine 10; intensive care 3; oncology 1; haematology 1; nephrology 1; geriatric service 1). At the time of analysis, follow-up ranged from 7 months to 19 months.
Table 1.
Demographic characteristics of the cohort
| Subjects | |
| Sexfemale/male | 78/72 |
| Age mean±SD (years) | 63.1±15.3 |
| Smoking status | |
| Ex/currentsmokers | 50 |
| Non smokers | 100 |
| Environmental exposure | |
| Occupational/Environmental | 37 |
| Drugs | 8 |
| Comorbidities/pre-existing diseases | |
| Chronicrespiratorydisease | 42 |
| Rheumatologicdisease | 61 |
| Ethnic group | |
| Europeans | 115 |
| Africans | 26 |
| Asians | 7 |
| Americans | 2 |
Diagnostic assessment
The most prevalent diagnosis was connective-tissue disease associated ILD (CTD-ILD) with 48 cases reported (32%). Other frequent diagnoses were idiopathic pulmonary fibrosis (IPF, n=35, 23%), chronic hypersensitivity pneumonitis (HP, n=20, 13%) and granulomatosis (n=11, 7%). Despite MDD, 15 cases (10%) remained unclassifiable ILD (figure 1).
Fig. 1.
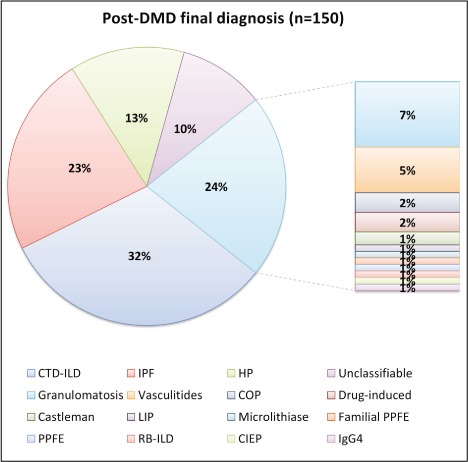
Proportion of MDD final diagnoses
Ninety-five cases of ILD (63%) were from known causes: CTD-ILD, HP, granulomatosis and ILD from rarer causes (drugs n=3; hemopathy and lymphoproliferative disease n=8). While 55 cases (37%) remained idiopathic: unclassifiable ILD, IPF, cryptogenic organizing pneumonia (n=3) and pleuroparenchymal fibroelastosis (n=2).
Sarcoidosis was the most common granulomatosis (n=9). Two cases of eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg-Strauss syndrome) and one case of granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis) were also reported.
Nine cases of familial interstitial lung diseases were detected (6 % of study cohort), including 6 IPF, 2 cases of pleuroparenchymal fibroelastosis and 1 case of chronic hypersensitivity pneumonitis. Following genetic testing, we identified a telomerase mutation in 5 cases of familial IPF (TERC n=2, TERT mutations n=2, RTEL1 mutation n=1). In 3 cases, gene sequencing was done but no known mutations were found, and in 1 case genetic testing is still ongoing.
Impact of MDD on diagnoses
Reviewing cases in MDD led to a change between suspected diagnosis (pre-MDD) and final diagnosis (post-MDD) in 63 of cases (42%) (figure 2 and figure S2). We observed a 5-fold increase in diagnosis of IPF after MDD: From 7 suspected IPF (5%) to 35 cases of confirmed IPF (23%). These changes were in most cases due to face-to-face discussion between radiologists and clinicians (n=15). In nine cases a biopsy was required by the multidisciplinary team and led to the diagnosis of IPF. In 3 cases, a pre-existing biopsy was examined by our expert pathologist and the pattern was UIP, leading to MDD diagnosis of IPF.
Fig. 2.
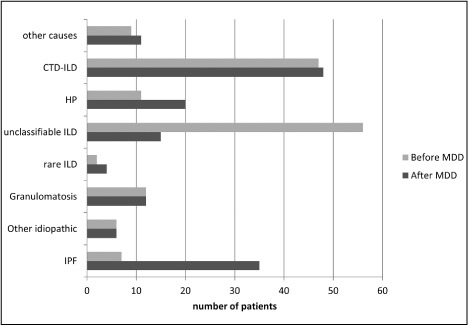
Comparison between suspected diagnosis at referral (light grey bars) and final MDD diagnosis (dark grey bars)
Figure S2.
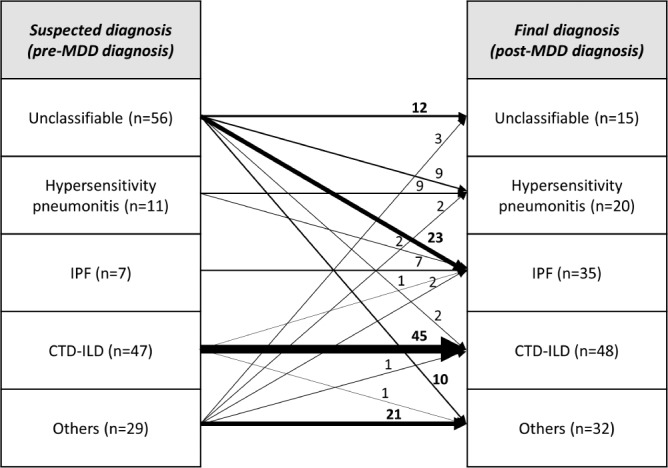
Schematic view of changes in diagnoses following MDD. The width of the arrows is proportional to the number of patients. As expected, most changes in final diagnosis occurred in the “unclassifiable fibrosis” subgroup
MDD led to a significant increase of the number of HP diagnoses (from 11 to 20 cases). Four cases required histological confirmation because of atypical presentation. The other sixteen cases were diagnosed by combination of symptoms, clinical examination, proved sensitivity towards an antigen, broncho-alveolar lavage (BAL) composition and CT pattern. MDD emphasized the need to search for an incriminated antigen by dosing serum precipitins and searching antigen at home. Despite this, we only identified a relevant antigen exposure in 9 cases of HP. At referral, 11 cases of HP were proposed, 2 became IPF after performing a biopsy. From the 11 cases newly diagnosed as HP, the majority were referred as ILD of undetermined aetiology.
Following MDD, the amount of unclassifiable ILD was significantly reduced (from 56 to 15, p<0.0001). The majority of unclassifiable ILD (n=23) at referral were diagnosed as IPF by the multidisciplinary team: 13 patients met the ATS-ERS criteria for probable or definite UIP pattern at the CT-scan, seven patients required histological confirmation and underwent lung biopsy. Finally, we confirmed a histological UIP pattern in three patients that had been biopsied elsewhere. (figure 3). As previously said, a part of unclassifiable ILDs were finally diagnosed as HP without requirement of a biopsy. MDD recommended performing biopsy in 19 cases of unclassifiable at referral but only 13 subjects underwent a biopsy. Among the 15 cases of unclassifiable after MDD, three patients remained unclassifiable despite a lung biopsy (2 cryobiopsies and 1 surgical biopsy). In three cases, the patient declined the procedure. Two patients had formal contraindication for either surgical biopsy or transbronchial-cryobiopsy. For five patients, biopsies were not proposed because of spontaneous clinical recovering, old age or very mild disease.
Fig. 3.
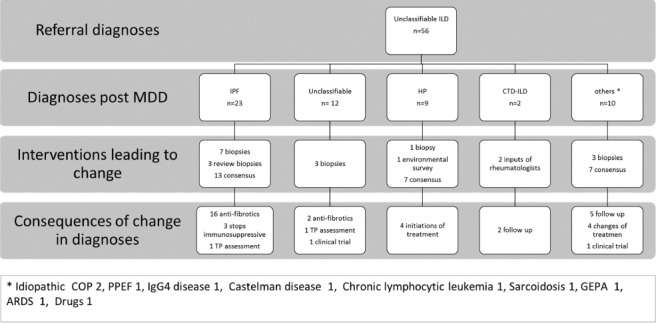
Flowchart describing patients addressed for unclassifiable fibrosis, including interventions leading to a change in diagnosis and the consequences on treatment
Impact of MDD on ILD management
In total, lung biopsy was proposed in 37 cases (25%) and effectively performed in 26 patients (12 cryobiopsies and 14 surgical biopsies). Input of histopathology allowed to change diagnosis in 18 cases (69% of biopsied patient) and to change treatment in 14 cases (54% of biopsied patients). The number of lung biopsies increased significantly between 2016 and 2017 with implementation of the structured MDD (from 4 to 14 surgical biopsies and from 6 to 12 cryobiopsies).
MDD led to a change or initiation of treatment in 81 cases (54%). Anti-fibrotic were prescribed for IPF but also for unclassifiable ILD with working diagnosis of IPF (total of 32 new antifibrotic treatments initiated). 6 patients were included in sponsored clinical trial, providing them an access to treatment. MDD strictly recommended to stop corticosteroids and immunosuppressive therapies in case of IPF or unclassifiable ILD in 6 cases. In contrast, in HP group MDD recommended to start corticosteroid in 9 cases and steroid sparing agent in 2 cases (1 mycophenolate mofetil and 1 azathioprine). Similarly, MDD recommended starting steroid sparing agent in 7 cases of sarcoidosis.
In the CTD-ILD subgroup, there was an increased recommendation for initiation of corticosteroid-sparing immunosuppressive therapies: In 14 cases, intravenous cyclophosphamide pulse therapy was advocate because of clinical, radiological and/or functional decline. Advices were given about oral medication after the pulse therapy. Thirteen patients received azathioprine or mycophenolate mofetil after recommendation of MDD. Half of patients included in rehab program following the MDD were CTD-ILD patients (n=16). Four CTD-ILD patients were assessed for lung transplantation due to an end-stage respiratory disease (figure 4).
Fig. 4.
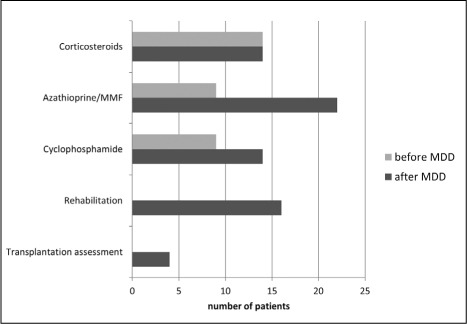
Changes in pharmacological and non-pharmacological treatment in CT-ILD. Light grey bars represent treatment before MDD, dark grey bars correspond to treatment after MDD
Identifying definite diagnoses for 44 cases of unclassifiable ILD at referral led to change or initiation of treatment in 28 cases. Despite the absence of definite diagnosis, treatments with pirfenidone in clinical trials were proposed for three cases.
MDD strongly supported inclusion on rehabilitation program for 29 patients. Sixteen patients were really included in the outpatient pulmonary rehabilitation program of our centre, an 8-fold increase compared with inclusion in 2016. MDD led to an increasing recommendation for early transplantation evaluation (9 patients in total, including 4 CTD-ILD, 3 IPF and 2 HP). Finally, 23 patients were screened for academic or sponsored clinical trials.
Discussion
In this study, we assessed the effect of a well-structured MDD meeting on ILD management. Our local results confirm that MDD has a significant impact on final diagnosis (42% modification), pharmacological treatment as well as non-pharmacological therapies (54% change in therapy). The most prevalent diagnoses were CTD-ILD, IPF and HP. Our epidemiological data are consistent with those presented in three recent publications from the Greater Paris region (9) (Seine-Saint-Denis), United States (10) and Leuven (11). However, small differences are worth noticing: We collected a higher percentage of IPF (23%) than in Seine Saint Denis (11%) and approximately the same percentage as in USA (20%). One explication given by authors for the relative low frequency of IPF in Seine Saint-Denis is that their population is especially young and not representative of the general population. Our higher percentage of IPF is also explained by local regulations that condition access to antifibrotic drugs to MDD discussion. We found a higher proportion of HP (13%) than in Seine-Saint-Denis (3%) but less than in USA (20%). Comparatively, we have reported a larger proportion of CTD-ILD (32%) than in Leuven (7%), Seine-Saint-Denis (17%) and in USA (20%). This is explained by the facts that (1) our hospital is a tertiary referral centre for systemic sclerosis and systemic lupus erythematosus, and (2) MDD is systematically attended by at least one rheumatologist, as recommended by the recent Fleishner’s Society position paper (3). The impact of MDD on CTD-ILD management in our study underlines the benefits of a proper collaboration between pulmonology and rheumatology departments: On one side, input of rheumatologists allows detecting unrecognized connective tissue disease in case of ILD associated with atypical autoimmune serological findings or clinical signs that can be difficult to integrate by respiratory physicians. On the other side, input of pulmonologists enables to standardize monitoring of functional test, to detect cases requiring treatment and to optimize the non-pharmacological management (rehabilitation, transplantation assessment, oxygen therapy). Finally, the presence of a rheumatologist is required by national regulatory rules (Belgian rules for the reimbursement of antifibrotic drugs, www.inami.fgov.be). Of note, several studies have placed great value on this close collaboration: Jo et al have shown in their survey that when a rheumatologist is attending the meeting, he always or frequently contributes to discussion (12). Walsh et al have highlighted in a case cohort-study the importance of rheumatologists input to distinguish IIP from CT-ILD. They suggested that rheumatological consultation might be part of the diagnostic process in selected patients, whereas one study from Castelino et al. advocated systematic rheumatological assessment for all ILD-patients (8, 13). We reported a relatively low proportion of granulomatous diseases (i.e. sarcoidosis). This low rate is explained by the facts of those diseases are not systematically discussed in MDD because of time limitations and relatively simple diagnosis of lung sarcoidosis compared to other forms of ILD.
Analysis of our data confirms that MDD meetings have a real impact on several aspect of daily clinical management of ILD. First, as shown in previous studies, MDD increases degree of diagnostic certainty and leads to more definite diagnosis in challenging cases by advising complementary investigations. In this study, diagnoses were changed in 42% of cases thanks to MDD. Similarly to what was described in previous study from Ryerson et al, 10% of our cases remain unclassifiable ILD after MDD (14), mostly because there was contraindication or patient refusal for lung biopsies. In these cases, a working diagnosis was proposed and treatment, non-pharmacological management and follow-up were recommended.
We observed almost a 2-fold increases in HP diagnosis. This highlights the importance of occupational and environmental interrogation, BAL findings and searching for serum precipitins. HP diagnosis requires integration of clinical history, environmental and occupational exposure assessment, biological and broncho-alveolar lavage findings and radiological features. The clinical and radiological presentation varies over time and can mimic other ILD. MDD emphasized the need to scrutinize for a culprit antigen by interviewing patients, dosing serum precipitins and searching antigen at home. In cases of occult exposure, atypical presentation and pejorative evolution despite adequate treatment MDD argued for confirming the diagnosis by histopathologic findings. In line with the results of previous studies,we reported an increase in IPF diagnoses after MDD (7, 15, 16). Lung biopsies were recommended by MDD when clinical context and radiologic pattern were discordant. The number of lung biopsies increased with implementation of regular and standardized MDD, reflecting the obsession of the MDD to approach more accurately the diagnosis and adapt management.
Secondly, changing diagnosis led to changes in treatment in 54% cases. With the increase of IPF diagnoses we observed a significant increase in anti-fibrotic therapies prescription and recommendation against immunosuppressive therapies and corticosteroids was made. In contrast, combination of corticosteroids and immunosuppressive therapies were shown to increase risk of death and hospitalization (4). MDD helps to optimize immunosuppressive therapy in CTD-ILD.
Finally, MDD brought non-pharmacological measures that improved global management of ILD: Based on two controlled trials (17, 18), ATS/ERS guidelines for IPF management promoted inclusion in pulmonary rehabilitation program. These two studies have shown improvement in walked distance and symptoms or quality of life (18-20). Despite this, recommendations for pulmonary rehabilitation remain weak in guidelines (21). Over the last few years, many studies were published and have strengthened the conviction that ILD patients benefits from exercise training (17, 22). After implementation of structured MDD, recommendations for pulmonary rehabilitation attendance and effective participation increases. Efforts still need to be made to propose more systematically rehabilitation programs and to convince patients to participate.
Clinical trials are crucial in ILD to improve therapies and outcomes in this area where our therapeutic action remains limited in certain cases. MDD allowed screening more patients for inclusion in clinical trial.
This study comprises several limitations: It is a retrospective study, so we could not compare MDD and absence of MDD face-to-face. In line with our inclusion criteria (files discussed between January and December 2017), we lack long-term follow up data that may provide hints regarding morbidity and mortality outcomes. Furthermore, our study was not designed for longitudinal evaluation of patients. Finally, the implementation of a MDD per se is likely to have improved ILD management locally.
In conclusion, we report our experience on one-year use of a well-structured MDD. Our results emphasize the multiple benefits related to MDD in ILD management. Furthermore, MDD management fosters collaboration between different departments of the hospital, favouring integrated medicine and holistic care of patients.
References
- 1.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 3.Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2017 doi: 10.1016/S2213-2600(17)30433-2. [DOI] [PubMed] [Google Scholar]
- 4.Idiopathic Pulmonary Fibrosis Clinical Research N. Raghu G, Anstrom KJ, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–77. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KR, King TE, Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170(8):904–10. doi: 10.1164/rccm.200402-147OC. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KR, Andrei AC, King TE, Jr, et al. Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? Am J Respir Crit Care Med. 2007;175(10):1054–60. doi: 10.1164/rccm.200606-833OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4(7):557–65. doi: 10.1016/S2213-2600(16)30033-9. [DOI] [PubMed] [Google Scholar]
- 9.Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017;50(2) doi: 10.1183/13993003.02419-2016. [DOI] [PubMed] [Google Scholar]
- 10.Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811–23. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 11.De Sadeleer LJ, Meert C, Yserbyt J, et al. Diagnostic Ability of a Dynamic Multidisciplinary Discussion in Interstitial Lung Diseases: A Retrospective Observational Study of 938 Cases. Chest. 2018;153(6):1416–23. doi: 10.1016/j.chest.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 12.Jo HE, Corte TJ, Moodley Y, et al. Evaluating the interstitial lung disease multidisciplinary meeting: a survey of expert centres. BMC Pulm Med. 2016;16:22. doi: 10.1186/s12890-016-0179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castelino FV, Goldberg H, Dellaripa PF. The impact of rheumatological evaluation in the management of patients with interstitial lung disease. Rheumatology (Oxford) 2011;50(3):489–93. doi: 10.1093/rheumatology/keq233. [DOI] [PubMed] [Google Scholar]
- 14.Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. 2013;42(3):750–7. doi: 10.1183/09031936.00131912. [DOI] [PubMed] [Google Scholar]
- 15.Jo HE, Glaspole IN, Levin KC, et al. Clinical impact of the interstitial lung disease multidisciplinary service. Respirology. 2016;21(8):1438–44. doi: 10.1111/resp.12850. [DOI] [PubMed] [Google Scholar]
- 16.Chaudhuri N, Spencer L, Greaves M, et al. A Review of the Multidisciplinary Diagnosis of Interstitial Lung Diseases: A Retrospective Analysis in a Single UK Specialist Centre. J Clin Med. 2016;5(8) doi: 10.3390/jcm5080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryerson CJ, Cayou C, Topp F, et al. Pulmonary rehabilitation improves long-term outcomes in interstitial lung disease: a prospective cohort study. Respir Med. 2014;108(1):203–10. doi: 10.1016/j.rmed.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Dowman LM, McDonald CF, Hill CJ, et al. The evidence of benefits of exercise training in interstitial lung disease: a randomised controlled trial. Thorax. 2017 doi: 10.1136/thoraxjnl-2016-208638. [DOI] [PubMed] [Google Scholar]
- 19.Nishiyama O, Kondoh Y, Kimura T, et al. Effects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology. 2008;13(3):394–9. doi: 10.1111/j.1440-1843.2007.01205.x. [DOI] [PubMed] [Google Scholar]
- 20.Holland AE, Hill CJ, Conron M, et al. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax. 2008;63(6):549–54. doi: 10.1136/thx.2007.088070. [DOI] [PubMed] [Google Scholar]
- 21.Spruit MA. Pulmonary rehabilitation. Eur Respir Rev. 2014;23(131):55–63. doi: 10.1183/09059180.00008013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huppmann P, Sczepanski B, Boensch M, et al. Effects of inpatient pulmonary rehabilitation in patients with interstitial lung disease. Eur Respir J. 2013;42(2):444–53. doi: 10.1183/09031936.00081512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.