

Abstract
Although relatively few patients with pulmonary sarcoidosis develop advanced disease that progresses to respiratory insufficiency despite receiving best practice pharmacologic interventions, lung transplantation may be the only therapeutic option for such patients to both prolong survival and provide improved quality of life. Lung transplant can be successfully performed for patients with end-stage pulmonary sarcoidosis, and post-transplant survival is similar to that for other transplant indications such as idiopathic pulmonary fibrosis. However, appropriate timing of referral, comprehensive assessment of potential candidates for lung transplant, placement of patients on the lung transplant waiting list when within the transplant window as appropriate, choosing the best procedure (bilateral versus single lung transplant), and optimal peri-operative and post-transplant management are key to successful lung transplant outcomes for patients with sarcoidosis.
Key words: lung transplantation, sarcoidosis, interstitial lung disease, pulmonary fibrosis
Introduction
Sarcoidosis is a granulomatous, multi-system disease characterized by a wide variety of clinical presentations and phenotypes (1-4). While sarcoidosis has a tendency to spontaneously remit, its clinical course is highly variable. Although up to 95% of patients with sarcoidosis develop some form of lung disease over the course of their lives, only approximately one-third of patients develop chronic or progressive disease. Advanced lung disease in sarcoidosis can be characterized by extensive fibrosis, vascular remodeling with pulmonary hypertension, cyst formation, airway involvement with loss of patency/stricture or dilatation due to bronchiectasis, or combinations thereof (1-3, 5). However, only approximately 5% of patients diagnosed with sarcoidosis will develop advanced lung disease due to pulmonary fibrosis (5). The majority of these patients, however, eventually succumb to respiratory complications of chronic pulmonary sarcoidosis, although some patients can remain clinically stable for long periods of time (5, 6).
Lung transplantation is a treatment option that can improve quality of life and prolong survival for patients with advanced lung disease refractory to other therapeutic interventions (7). Indeed, end-stage sarcoidosis with severe fibrocystic lung disease and/or the presence of World Health Association Group 5 pulmonary hypertension (PH) remains a difficult-to-treat form of advanced lung disease for which lung transplantation may be the only intervention that can improve survival and quality of life. Although the total number of lung transplants reported to the International Society for Lung Transplantation (ISHLT) for patients with sarcoidosis is relatively low (approximately 2.5% of all transplants performed from 1995 through 2014) (8), actuarial post-transplant survival has been reported to be comparable to that for patients with other forms of pulmonary fibrosis (9-11), and median survival according to recent ISHLT data is 6.1 years following primary transplantation (8). An examination of United Network for Organ Sharing (UNOS) data for patients listed for transplant from 1995 through 2000 showed that waitlisted patients with sarcoidosis had a mortality rate that was similar to the high risk of mortality observed for patients diagnosed with idiopathic pulmonary fibrosis (IPF) (12). Additionally, Shorr et al. (13) reported that African Americans appeared to face a significantly increased risk of death (odds ratio of 2.5) while waitlisted, even when the data were adjusted for potential confounding factors. Determining the right time for referral and transplantation of sarcoidosis patients with advanced lung disease presents a considerable challenge.
Respiratory tract manifestations and complications of chronic sarcoidosis
Many risk factors that are associated with worse outcomes in patients with sarcoidosis have been identified (Table 1). Additionally, advanced pulmonary sarcoidosis has many manifestations and characteristics, and patients can develop a variety of complications (Table 2). Imaging of advanced pulmonary sarcoidosis with high-resolution computed tomography (HRCT) reveals a variety of patterns with extensive adenopathy, parenchymal fibrosis, and/or airway disease (Figure 1). While no specific risk factors for the development of advanced pulmonary sarcoidosis have been identified, a number of factors have been linked to increased risk of developing progressive and/or chronic disease (6), and some of these individuals will go on to develop advanced disease despite non-transplant therapeutic interventions. These risk factors include involvement of multiple organ systems, higher Scadding stage at diagnosis or progressing to higher Scadding radiographic stage, need for systemic therapy, lack of lymphadenopathy, female gender, older age, and black race. Nonetheless, the clinical course of sarcoidosis is highly variable, and many patients can remain stable despite symptomatic and/or persistent disease, even in the absence of chronic pharmacologic therapies.
Table 1.
Risk factors for worse outcomes in sarcoidosis
|
Table 2.
Manifestations and complications of pulmonary sarcoidosis
Parenchymal diseasea
|
Airway involvement
|
Vascular disease & pulmonary hypertension
|
Pulmonary infection
|
| Hemoptysis |
Pleural disease
|
Fig. 1 a-d.
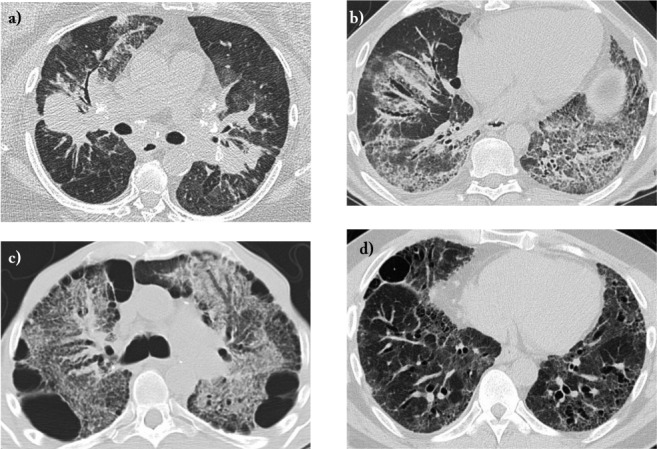
Transverse high-resolution computed tomography images of potential lung transplant candidates with advanced pulmonary sarcoidosis and severe lung function impairment. a) Long-standing sarcoidosis with bulky adenopathy, diffuse granulomatous infiltrates, and mild sarcoidosis-associated pulmonary hypertension (SAPH); b) Scadding Stage 4 sarcoidosis with extensive fibrosis and traction bronchiectasis, initially interpreted as a usual interstitial pneumonia pattern; explanted lung showed diffuse granulomata typical of sarcoidosis plus extensive fibrosis; c) Stage 4 sarcoidosis with extensive diffuse fibrosis, peripheral bleb formation, and bronchiectasis; d) Stage 4 sarcoidosis with extensive bronchiectasis but without evidence of chronic infection.
Fig. 1 e-h.
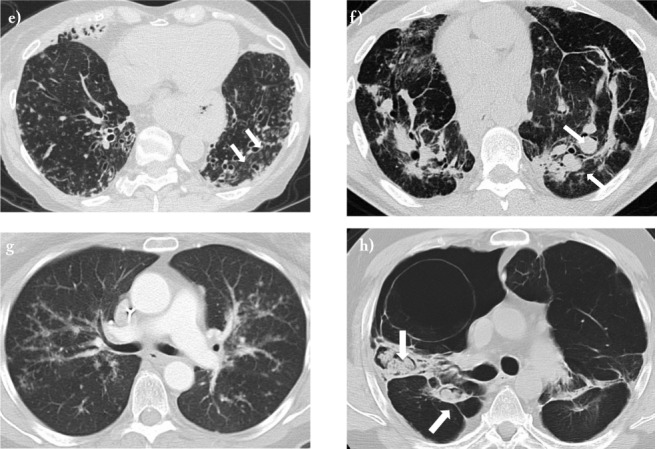
Transverse high-resolution computed tomography images of potential lung transplant candidates with advanced pulmonary sarcoidosis and severe lung function impairment. e) Stage 4 sarcoidosis with extensive bronchiectasis (arrows) and chronic bacterial infection with P. aeruginosa; f) Stage 4 sarcoidosis with parenchymal fibrosis and severe SAPH with diffuse bilateral pulmonary artery aneurysm formation (arrows); g) Stage 3 sarcoidosis with severe SAPH; h) Stage 4 fibrobullous sarcoidosis with multiple aspergillomas (arrows), SAPH, and recurrent hemoptysis.
Proximal airway disease
Sarcoidosis can involve the nasal passages, paranasal sinuses, mouth, larynx, trachea, or bronchi (14). Severe stenosis of the trachea or cartilaginous bronchi may occur, but this is estimated to occur in less than one percent of patients (15). Although patients with extensive airway narrowing may be quite symptomatic, they usually do not have enough functional impairment to qualify them for lung transplant candidacy.
Intrathoracic lymphadenopathy
Intrathoracic lymphadenopathy is observed in approximately 80% of patients during the course of their illness. Hilar adenopathy is bilateral in most cases, although unilateral hilar adenopathy can be seen in up to 5% of patients (16). Lymph node calcification can be seen at the time of diagnosis, and the likelihood of calcification increases later during the course of the disease (17).
Large nodules and alveolar consolidation
Nodules that follow a perilymphatic distribution and predominate in the mid to upper lung zones are seen in approximately 90% of patients (18). Sarcoid nodules can occasionally aggregate and form larger pulmonary nodules (up to 3 cm diameter) or large masses. Large nodules can remain stable for long periods of time, show partial or complete regression, or cavitate. Massive consolidation may occur with coalescent interstitial granulomas compressing alveoli (19).
Pulmonary fibrosis
Approximately 5% of patients have evidence of fibrotic changes on routine chest radiography at presentation (5, 20). Fibrosis tends to predominate in the upper and mid lung regions, and conglomerate masses that surround and encompass vessels and bronchi with associated bronchial distortion is seen in over half of patients with fibrotic pulmonary sarcoidosis (21). Advanced fibrotic sarcoidosis is characterized by the presence of fibrotic cysts, bullae, traction bronchiectasis, and paracicatricial emphysema, and cystic abnormalities are commonly seen in the upper lobes (22). Honeycomb change can be seen, and honeycomb-like cysts are usually found in an upper lobe distribution, but lower lobe honeycomb change that mimics a usual interstitial pneumonia (UIP) pattern that is typical of idiopathic pulmonary fibrosis (IPF) can also be seen (20, 23).
Pulmonary hypertension
The estimated prevalence of PH in sarcoidosis ranges from one to 28% (defined as mean pulmonary arterial pressure [mPAP] ≥25 mm Hg) at rest and as high as 43% if measured during exercise (24-28). Most patients with sarcoidosis-associated PH (SAPH) have radiographic changes of advanced disease (Scadding stages III or IV), although extensive parenchymal abnormalities are not always present (24-26). One case series reported that 60% of patients with SAPH lacked evidence of significant fibrosis on chest radiography (27), and the extent of parenchymal lung involvement may not correlate with the degree of PH as reflected by right heart catheterization measurements. Up to 75% of patients who are listed for lung transplantation meet criteria for SAPH when subjected to right heart catheterization, and its presence is associated with a poor prognosis (26,29). Shorr and colleagues examined a cohort of 363 patients listed for lung transplantation for sarcoidosis and found that 66% had mPAP ≥25 mm Hg and 36% had mPAP≥40 mm Hg (26). Furthermore, nearly 70% of patients with mPAP ≥40 mm Hg needed at least some if not total assistance with functional activities, and patients with severe PH had a nearly 7-fold increase in need for supplemental oxygen.
Bronchiectasis
Sarcoidosis-related bronchiectasis is usually diffuse but can occasionally be localized. Bronchiectasis in patients with Scadding stage 4 disease has been reported to range from 18-40% on high-resolution computed tomographic (HRCT) scanning, and bronchiectasis is present in nearly 100% of patients listed for lung transplantation (21, 30, 31). Diffuse cystic bronchiectasis is perceived as being caused from either traction due to surrounding parenchymal fibrosis or direct airway damage caused by granulomatous inflammation, whereas localized bronchiectasis can be post-obstructive, caused by external compression by enlarged lymph nodes, or due to persistent endobronchial sarcoidosis (32). Suppurative bronchiectasis with recurrent infectious exacerbations can be seen in some patients (30).
Pleural disease
Granulomas can infiltrate both visceral and parietal pleura, and pleural involvement plus lymphatic channel compromise can cause pleural effusions to form. However, pleural effusion is an unusual finding in sarcoidosis and may be caused by comorbidities such as pneumonia or congestive heart failure (33). Chylothorax has also been described in sarcoidosis but is an exceedingly rare complication (34). Finally, spontaneous pneumothorax has been reported and attributed to rupture of subpleural blebs, especially when advanced fibrocystic disease is present (35).
Other complications
Aspergillus species are ubiquitous in the environment and can be commonly found in the both the oral and lung mycobiomes of normal humans (36). Both aspergillomas and other aspergillosis syndromes have been reported in patients with sarcoidosis. Mycetoma formation, which usually occurs in pre-existing cysts that are colonized by fungi (usually Aspergillus spp), occurs in approximately 2-5 percent of patients with sarcoidosis, and life-threatening pulmonary hemorrhage can occur (37, 38). Mycetoma formation does not have a predilection for right or left lung, but they occur most commonly in the upper lobes and can be multiple. No specific consensus recommendations currently exist for management of aspergillomas in patients with sarcoidosis. While anecdotal reports of poor outcomes in lung transplant recipients when pre-transplant mycetomas have been published, successful lung transplantation has been reported with a combination of careful native lung explantation and post-operative antifungal pharmacologic therapy (39).
Acute exacerbations of pulmonary sarcoidosis are not uncommon, but the definition of an acute exacerbation (AE) and information regarding diagnostic criteria and management are sparse. Panselinas and Judson (40) have proposed the combination of (1) worsened pulmonary symptoms in patients with known sarcoidosis that cannot be explained by alternative causes, (2) a ≥10% decline in forced expiratory volume in one second (FEV1) and/or forced vital capacity (FVC), and (3) the presence of symptoms for at least one month as diagnostic criteria for an episode of an AE of pulmonary sarcoidosis. Risk factors for AE include tapering corticosteroid therapy, administration of interferon-alpha, initiation of antiretroviral therapy, and treatment with tumor necrosis factor-alpha (TNF-α) antagonists (40).
Pharmacologic management of pulmonary sarcoidosis
Although pulmonary disease is the most common manifestation of sarcoidosis, not all patients with pulmonary disease will require drug therapy. Major indications for treating pulmonary sarcoidosis include cough, dyspnea, declining lung function, or radiologic evidence of worsening lung disease, and it is estimated that about half of patients in the US with pulmonary disease receive systemic therapy (38). Additionally, systemic therapy may be required for significant involvement of other organ systems even though pulmonary disease appears to be stable. Asymptomatic lung disease accompanied by stable lung function does not require therapy. If indicated, pharmacologic therapies can range from inhaled corticosteroids and/or non-steroidal anti-inflammatory drugs for minimal symptoms with stable lung function to systemic corticosteroids, anti-malarial drugs, cytotoxic drugs, biologic agents, or combinations of such for significantly symptomatic disease and/or progressive decline in lung function (41-44). However, whether the use of systemic corticosteroids or other agents such as TNF-α inhibitors can prevent the development or halt the progression of pulmonary fibrosis remains debatable (45,46).
Patients who report persistent dyspnea despite therapy and have normal left ventricular function have an estimated prevalence of PH that approximates 53% (47), and patients listed for lung transplant have an even higher incidence of PH at approximately 74% (26). Although most forms of PH associated with underlying parenchymal lung disease are classified as WHO group 3 PH, SAPH is categorized as WHO group 5 due to its complex and multifactorial pathogenesis, and there can be substantial dissociation between the magnitude of physiologic measures of restriction as a surrogate marker for parenchymal disease burden and the presence and severity of SAPH. Such discordance is likely due to the multifactorial nature of circulatory impairment in SAPH, which can be due to various combinations of distal capillary bed destruction due to fibrotic parenchymal remodeling combined with areas of hypoxemic vasoconstriction, direct involvement of vessels by granulomatous inflammation, and increased vasoreactivity that may respond to vasodilators such as nitric oxide or prostacyclin, upregulation of vasoactive cytokines such as endothelin-1, or mechanical extrinsic compression of pulmonary vessels by bulky intrathoracic adenopathy (28). Because of the multifactorial nature of SAPH, some patients may show a significant response to interventions such as supplemental oxygen, treatment of obstructive sleep apnea if present, treatment of cardiac dysfunction, identification and treatment of thromboembolic disease, or immunosuppressive therapies targeting active sarcoidosis. The administration of vasoactive agents that show efficacy for WHO Group 1 PH remains controversial, but responses have been reported for pharmacologic therapies that target the endothelin pathway (endothelin receptor antagonists such as bosentan), the nitric oxide pathway (selective phosphodiesterase inhibitors), or prostacyclin pathway inhibitors such as epoprostenol (28). However, such therapies, while having potential benefit for some patients, may also cause harm by worsening ventilation-perfusion mismatching and hypoxemia, and such pharmacologic intervention should only be considered on a case-by-case basis by experienced referral center clinicians (and preferably in the setting of a randomized clinical trial) (28). Additionally, vasoactive drugs for targeted treatment of SAPH should probably be avoided for patients with mPAP values <40 mm Hg.
Evaluation and listing for lung transplantation
Progressive pulmonary fibrosis, SAPH, and recurrent/chronic respiratory infection are leading causes of respiratory failure and mortality in patients with advanced pulmonary sarcoidosis (6, 48). Independent predictors of mortality that were identified in long-term follow-up (≥8 years) after adjustment for various confounders were older age, extensive fibrosis on HRCT scanning, and the presence of PH (49). However, quantitative models that can predict clinical behavior of disease and mortality are lacking (50). Therefore, decisions concerning timing of a referral to a transplant center are generally made via case-by-case assessments of patients with advanced lung disease.
A series of consensus documents created by task forces working under the auspices of the ISHLT have provided guidance for decisions regarding referral and evaluation of patients with various forms of advanced lung disease with the most recent published in 2015 (51). While these recommendations have not necessarily been validated, they are widely followed and provide a very useful roadmap for referral, evaluation, bridging to transplant, and transplantation. All potential candidates must lack absolute contraindications to lung transplantation (Table 3), and relative contraindications must be carefully weighed on a case-by-case basis. The majority of patients with advanced pulmonary disease due to sarcoidosis fall into the category of interstitial lung disease (ILD), for which guidance for timing of referral and timing of placing on the transplant waitlist are provided (Table 4). However, some patients with sarcoidosis may have predominantly vascular involvement but lack extensive pulmonary fibrosis, and ISHLT recommendations provided for pulmonary vascular diseases may be more appropriate for such patients (Table 4). Because waitlist mortality is quite high among patients with pulmonary fibrosis, timely referral for transplant evaluation is essential for patients who have severe disease despite maximal therapy and wish to be considered for lung transplantation.
Table 3.
Contraindications to lung transplantation
Absolute
|
Relative
|
Table 4.
Guidelines for timing of referral for potential lung transplantation
Referral to a transplant center (all patients with ILD)*
|
Referral to a transplant center (all patients with PVD)*
|
Suggested timing of referral to a transplant center for patients with sarcoidosis
|
Evaluation of potential candidates for lung transplant should include (1) an objective determination of disease severity, (2) elucidation of the nature of disease characteristics that are causing the patient’s symptoms, (3) a determination of whether the benefits (prolonged survival, improved quality of life) of undergoing lung transplantation clearly outweigh the risks associated with receiving a lung transplant (Table 5). Many patients will unfortunately not be eligible for lung transplantation due to the presence of an absolute contraindication or combinations of relative contraindications and comorbidities that make the possibility of achieving a successful transplant unlikely (e.g. severe corticosteroid-induced diabetes and obesity). On the other hand, some patients with sarcoidosis can be very symptomatic from their disease yet not have enough physiologic impairment to receive a high enough lung allocation score (LAS) value (for countries that use a LAS system to prioritize transplant candidates) to have a reasonable chance of receiving a donor lung offer if placed on the waitlist.
Table 5.
Evaluation of potential lung transplant candidates with sarcoidosis
Disease-specific considerations for patients with sarcoidosis
|
Evaluation and testing
|
The LAS system (Table 6) was implemented in the US in 2005 with the goals of (1) balancing the urgency of need for transplantation due risk of death without receiving a transplant with the likelihood of an acceptable outcome following transplantation, (2) optimally placing organs according to LAS values combined with matching characteristics of potential recipients (e.g. blood type, thoracic cage dimensions), and (3) reducing the number of candidates on transplant center waitlists who die without the opportunity to undergo a transplant (52,53). The LAS is weighted more by transplant urgency than likelihood of surviving for at least one year post-transplant, but its successful aspects have led to its adoption by a number of countries outside of the US. Because patients with ILD (especially those with IPF) tend to have higher LAS values than candidates with other disease indications, the total number of transplants for ILD (mostly IPF) in the US surpassed that for other indications (e.g. chronic obstructive pulmonary disease, cystic fibrosis, PH due to pulmonary vascular disease) in 2007, and IPF is the leading indication for lung transplantation in the US at present (54). The major indications for lung transplantation for sarcoidosis are advanced fibrotic lung disease, severe pulmonary hypertension, or a combination of both.
Table 6.
|
* Some values are adjusted according to Disease Group (A-D); sarcoidosis is classified as Group A if mPAP is ≤30 mm Hg but switches to Group D if mPAP is >30 mm Hg.
** The LAS calculation incorporates three different measures (waiting list urgency, post-transplant survival, and transplant benefit) to derive a Raw Allocation Score that is then normalized on a continuous scale of 0 to 100.
For additional information see concerning LAS components and calculations see https://optn.transplant.hrsa.gov/media/1200/optn_policies.pdf#nameddest=Policy_10. Organ Procurement and Transplantation Network Policies; Policy 10: Allocation of Lungs. Date accessed, 1/26/18.
A key question when evaluating a patient with sarcoidosis for potential lung transplantation is whether their lung disease has been adequately treated. Our center has had candidates who were using supplemental oxygen and very incapacitated but improved markedly and were able to be weaned off supplemental oxygen and achieve acceptable quality of life when placed on adequate pharmacotherapy. Another key question is whether significant extrapulmonary sarcoidosis is present that may have an impact on post-transplant outcome, and appropriate screening should be performed to detect significant left ventricular dysfunction or sustained ventricular dysrhythmias.
Prior to placement on a transplant waitlist, co-morbidities should be aggressively and optimally managed. This includes angioplasty and/or stent placement for coronary artery disease if needed, anti-resorptive therapies to reduce fracture risk if osteoporosis is present, joining a weight loss program if overweight, and medical treatment of systemic hypertension or diabetes mellitus. Because dyspnea may limit physical activity and promote deconditioning, pulmonary rehabilitation with physical training and breathing exercises should be prescribed, and pulmonary rehabilitation programs can provide educational and psychological support and optimize exercise tolerance and functional status prior to transplantation. Because of the prolonged and variable disease course for patients with sarcoidosis, the decision as to when to proceed with transplantation is challenging, even for experienced clinicians at referral/transplant centers. Guidance for timing the placement of lung transplant candidates on the waitlist has been provided by the ISHLT (Table 7).
Table 7.
Guidelines for timing of waitlist placement for transplant candidates
Timing of placing a patient on the lung transplant waitlist (all patients with ILD)*
|
Timing of placing a patient on the lung transplant waitlist (all patients with PVD)*
|
Suggested timing of waitlist placement for patients with sarcoidosis
|
Surgical considerations
Previous thoracic surgical procedures are generally not a contraindication to performing a lung transplant, but higher risk of hemorrhage, increased need for chest re-exploration, and renal dysfunction can be encountered in patients who have had previous chest surgical procedures, especially if prolonged cardiopulmonary bypass times are required. The decision of whether to perform a single, bilateral, or heart-lung transplant involves consideration of the nature of lung involvement and extent of physiologic impairment, whether significant extrapulmonary disease is an issue, what comorbid conditions are present, and the likelihood of procuring a donor organ that matches a candidate’s thoracic cage dimensions and ABO blood group status. Explanting lungs from patients with advanced sarcoidosis can be extremely challenging due to pleural adhesions and perihilar fibrosis, and substantial intraoperative bleeding is more likely to occur if resection of the native lung(s) proves to be difficult (55).
The presence of one or more mycetomas, especially if abutting the pleura, increases the risk of seeding the pleural spaces during explantation. The risk and degree of pleural bleeding (especially if patients require cardiopulmonary bypass) is likely to be increased, and a prolonged dissection to explant the native lungs may significantly increase donor lung cold ischemic time, thereby increasing the risk of significant reperfusion injury. One case series reported that post-transplant outcomes were significantly worse for patients with mycetomas (56), but aggressive pre-transplant antifungal therapy and prolonged post-transplant prophylaxis may successfully prevent post-transplant Aspergillus infection (57). Additionally, irrigation of the pleural space with an anti-fungal agent (e.g. amphotericin B) when donor lungs are implanted should be considered. Patients with mycetomas, even if apparently unilateral, should only be listed for bilateral lung transplant (BLT).
If suppurative bronchiectasis is present, sputum cultures should be obtained as native lungs are explanted to identify all infecting organisms and their sensitivities to antibiotics. Peri-operative and post-operative antibiotics should be administered according to culture and sensitivity results. Because a bronchiectatic native lung can serve as a reservoir of infection that places a transplanted single lung at risk for post-transplant infection, BLT is the preferred approach for patients with bronchiectasis and chronic suppurative infection.
Bilateral transplant may also be a better choice than single lung transplant (SLT) for patients with significant SAPH, although recipients can do well with SLT despite the presence of PH with mPAP values greater than 40 mm Hg (58). Indeed, a SLT may be a reasonable choice for patients in whom BLT is not required, and listing for SLT may improve chances for a donor organ offer and reduce risk of dying on the waitlist (59). A heart-lung transplant can be considered for patients with significant left ventricular dysfunction or cardiac dysrhythmias.
Post-transplant management, complications, and outcomes
Post-transplant management is multi-faceted and complicated, yet few randomized, prospective controlled trials are available to provide robust evidence for optimal recipient management. Peri-operative care in the ICU requires both ventilator and circulatory support, and early surgical and/or medical complications must be promptly identified and addressed. Protocols should be in place to facilitate prevention of infections (e.g. prophylaxis for cytomegalovirus and Pneumocystis jiroveci) as well as protocols to rapidly identify and treat infectious complications that may develop. Immunosuppressive regimens typically consist of pulse corticosteroid and an induction agent given at the time of surgery, and maintenance immunosuppression with a calcineurin inhibitor (tacrolimus or cyclosporine A), anti-metabolite (usually mycophenolate or azathioprine), and a corticosteroid that is gradually weaned to a low dose.
Transplant recipients are at risk for a multitude of immediate, acute, and subacute/chronic complications following successful transplantation (Table 8) (60,61). Approximately one third of lung transplant recipients will develop grade 3 primary graft dysfunction (PGD) (62), but while a number of markers have been identified that correlate with increased risk of high-grade PGD (63), interventions other than providing supportive care have been relatively ineffective in preventing or treating PGD.
Table 8.
Complications of lung transplantation
|
Pulmonary complications Lung allograft complications
Lung allograft and/or native lung complications
Native lung complications (single lung transplant recipients)
|
Extrapulmonary/systemic complications (may impact lung function)
|
Acute/subacute complications include antibody-mediated rejection, acute cellular rejection, lymphocytic bronchiolitis, and infection. Because up to 90% of recipients have pre-formed anti-HLA antibodies of which approximately one third are donor-specific antibodies (DSAs) (64), effective and carefully monitored immune suppression is essential to establish allograft immune tolerance. Monitoring recipients for evidence of lung function decline as well as monitoring for the appearance of numerous complications and co-morbidities is essential to optimize post-transplant allograft function and recipient quality of life and survival. Many centers subject recipients to surveillance bronchoscopy with BAL and transbronchial biopsies (TBBs) at protocol-determined intervals to detect occult infection and/or evidence of rejection, although other centers perform few if any protocol-driven surveillance bronchoscopies and may only perform such when clinically indicated by deterioration in lung function with the suspicion that infection or allograft rejection may be the cause. Consensus guidelines for using or not using post-transplant surveillance bronchoscopies have not yet become available.
For lung transplant recipients who survive beyond the first year post-transplant, the development of chronic lung allograft dysfunction (CLAD) is the greatest threat to long-term allograft and recipient survival (65, 66). The ISHLT/American Thoracic Society (ATS)/European Respiratory Society (ERS) clinical practice guideline systematically examined available evidence for the prevention and treatment of BOS/CLAD and provided recommendations for the diagnosis and treatment of BOS/CLAD. Identified risk factors included PGD, various forms of alloimmune rejection (acute cellular rejection, antibody-mediated rejection, lymphocytic bronchiolitis), infections (viral, bacterial, fungal), pathologic GER, autoimmunity, and persistent bronchoalveolar lavage (BAL) neutrophilia. Although evidence from randomized controlled trials (RCTs) for preventing and treating BOS/CLAD was found to be of low or very low quality, a number of conditional recommendations were made by consensus among task force members following a comprehensive review of available publications. These include ruling out other causes of delayed, persistent allograft function decline, administering azithromycin, adjustment of immunosuppressive regimens, and the detection/treatment of significant gastrointestinal reflux that may be affecting the lung allograft (65). Evidence for other salvage therapies for CLAD, such as extracorporeal photopheresis or total lymphoid irradiation, are weak at best (65, 67, 68).
A meta-analysis of 13 different reports that included a total of 10,042 lung transplant recipients of which 98 were transplanted for sarcoidosis concluded that sarcoidosis patients had a 50% prevalence of PGD (69). Additionally, the risk of short-term mortality has been reported to be significantly increased for African-American recipients (9). Furthermore, a higher incidence of hemothorax in sarcoidosis recipients was found to be associated with longer need for ventilator support, increased length of stay in intensive care units, and more prolonged length of hospital stay following lung transplant (70). Nonetheless, despite concerns that short-term outcomes and risk of early mortality may be somewhat worse for sarcoidosis lung recipients and especially African-American recipients, long-term post-transplant survival for recipients with sarcoidosis appears to be generally similar to survival rates for patients with other forms of fibrotic ILD. Tamieh et al. (11) examined a cumulative cohort of 695 patients with sarcoidosis (out of a total of 20,896 recipients) transplanted over a 25-year time period and reported that median survival rates for sarcoidosis recipients were not significantly different from that of non-sarcoid recipients. Additionally, the incidence of BOS does not appear to be increased for patients transplanted for sarcoidosis (11, 71).
Recurrence of non-caseating granulomata in transplanted lungs despite intense chronic immune suppression is a frequent observation in sarcoid recipients (72-78). The majority of cases were detected via transbronchial biopsy, many of which were surveillance procedures, but recurrent granulomas may also be significant enough to allow detection via HRCT scanning. Ionescu et al. (75) showed via DNA analysis that recurrence of granulomas in the lung allografts appeared to be of recipient origin. Additionally, granulomas tend to appear within the first 6-12 months post-transplant, are usually detected via surveillance biopsies, and rarely seem to have a significant impact on allograft function, although disease recurrence has been occasionally reported to cause significant allograft dysfunction (78, 79). Currently available data suggest that granuloma recurrence in the transplanted lungs occurs in approximately a third of recipients, but the impact of disease recurrence on survival is minimal.
We have detected subclinical recurrence of allograft granulomas in 5 of 22 recipients with sarcoidosis at our center, and all spontaneously regressed with the passage of time. Interestingly, an additional non-sarcoid recipient (a 37-year-old Caucasian female) with severe constrictive bronchiolitis caused by an inhalation injury (whose explanted lungs showed no evidence of granulomatous inflammation) had asymptomatic granulomas appear on surveillance transbronchial biopsies at one year post-transplant. These persisted until two years post-transplant (present on multiple sequential surveillance bronchoscopies) and then regressed spontaneously over a period of approximately one year without any change in her chronic immunosuppression regimen. Although BAL lymphocyte percentages on differential cell count and CD4/CD8 lymphocyte ratios are generally very low in lung transplant recipients on surveillance biopsies and this recipient’s percent lymphocytes on BAL were 3%, 3%, and 6% at 2, 6, and 24 weeks post-transplant, her BAL lymphocyte percentage had increased to 24% at 52 weeks with a CD4/CD8 ratio of 2.7 (versus 0.6±0.1 for clinically stable non-sarcoid lung recipients [N=20]) along with the appearance of typical well-formed, non-caseating granulomata on transbronchial biopsies. While repeat surveillance bronchoscopies up to 48 months post-transplant showed BAL lymphocyte percentages that ranged up to 49% with CD4/CD8 lymphocyte ratios as high as 5.7 along with persistence of well-formed non-caseating granulomata on transbronchial biopsies, serial HRCT imaging showed no changes and lung function remained completely stable. BAL culture and special stains showed no evidence of infection, and her maintenance immunosuppression and other medications were not altered. At 2.5 years post-transplant, the granulomas had regressed and were no longer detectable, the BAL lymphocytosis resolved, and the BAL lymphocyte CD4/CD8 ratio returned to a low ratio consistent with stable lung transplant recipient status. We suspect that this individual, who was of northern European ethnicity, likely developed a sarcoidosis syndrome with lung-limited infiltration of recipient immune cells into the lung allograft that gradually peaked and then eventually regressed spontaneously.
Key Points
A small number of patients diagnosed with sarcoidosis develop advanced lung disease.
Advanced pulmonary disease phenotypes include extensive pulmonary fibrosis, pulmonary hypertension, and purulent bronchiectasis.
Lung transplantation is an appropriate treatment for sarcoidosis patients with advanced lung disease that progresses to respiratory insufficiency despite other therapies.
Post-transplant survival is generally similar to that of recipients with other transplant indications such as IPF.
Although bilateral lung transplantation is generally a preferred procedure, single lung transplant may be an appropriate procedure for patients without complications of their lung disease such as purulent bronchiectasis, chronic fungal infection, or severe pulmonary hypertension.
Although recurrence of granulomas in transplanted lungs may occur, this rarely has a significant impact on lung allograft function or recipient survival.
Acknowledgment
Supported in part by the George and Julie Mosher Pulmonary Research Fund.
Financial/nonfinancial disclosures:
Within the past 3 years Dr. Meyer has been an investigator in clinical trials sponsored by Boehringer-Ingelheim, Bristol Meyers Squibb, Genentech, National Institutes of Health, Nivalis, Parion, Promedior, Roche, and Vertex. Dr. Meyer does not report any other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this manuscript. No writing assistance was utilized in the production of this manuscript.
References
- 1.Hunninghake GW, Costabel U, Ando M, Baughman R, Cordier JF, du Bois R, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999 Sep;16(2):149–73. [PubMed] [Google Scholar]
- 2.Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med. 1997 Apr 24;336(17):1224–34. doi: 10.1056/NEJM199704243361706. [DOI] [PubMed] [Google Scholar]
- 3.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014 Mar 29;383(9923):1155–67. doi: 10.1016/S0140-6736(13)60680-7. [DOI] [PubMed] [Google Scholar]
- 4.Culver DA, Baughman RP. It’s time to evolve from Scadding: phenotyping sarcoidosis. Eur Respir J. 2018 Jan 25;51(1) doi: 10.1183/13993003.00050-2018. [DOI] [PubMed] [Google Scholar]
- 5.Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H, Jr, Bresnitz EA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001 Nov 15;164(10 Pt 1):1885–9. doi: 10.1164/ajrccm.164.10.2104046. [DOI] [PubMed] [Google Scholar]
- 6.Patel DC, Budev M, Culver DA. Advanced (“end-stage”) pulmonary sarcoidosis. In Pulmonary Sarcoidosis: A Guide for the Practicing Clinician, Respiratory Medicine 17, Ed Judson M. 2014:79–110. [Google Scholar]
- 7.Kotloff RM, Thabut G. Lung transplantation. Am J Respir Crit Care Med. 2011 Jul 15;184(2):159–71. doi: 10.1164/rccm.201101-0134CI. [DOI] [PubMed] [Google Scholar]
- 8.Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. J Heart Lung Transplant. 2015 Oct;34(10):1264–77. doi: 10.1016/j.healun.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Shorr AF, Helman DL, Davies DB, Nathan SD. Sarcoidosis, race, and short-term outcomes following lung transplantation. Chest. 2004 Mar;125(3):990–6. doi: 10.1378/chest.125.3.990. [DOI] [PubMed] [Google Scholar]
- 10.Shah L. Lung transplantation in sarcoidosis. Semin Respir Crit Care Med. 2007;28:134–140. doi: 10.1055/s-2007-970339. [DOI] [PubMed] [Google Scholar]
- 11.Taimeh Z, Hertz MI, Shumway S, Pritzker M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax. 2016 Apr;71(4):378–9. doi: 10.1136/thoraxjnl-2015-207497. [DOI] [PubMed] [Google Scholar]
- 12.Shorr AF, Davies DB, Nathan SD. Outcomes for patients with sarcoidosis awaiting lung transplantation. Chest. 2002 Jul;122(1):233–8. doi: 10.1378/chest.122.1.233. [DOI] [PubMed] [Google Scholar]
- 13.Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest. 2003 Sep;124(3):922–8. [PubMed] [Google Scholar]
- 14.Baughman RP, Lower EE, Tami T. Upper airway. 4: Sarcoidosis of the upper respiratory tract (SURT) Thorax. 2010;65(2):181–6. doi: 10.1136/thx.2008.112896. [DOI] [PubMed] [Google Scholar]
- 15.Chambellan A, Turbie P, Nunes H, Brauner M, Battesti JP, Valeyre D. Endoluminal stenosis of proximal bronchi in sarcoidosis: bronchoscopy, function, and evolution. Chest. 2005 Feb;127(2):472–81. doi: 10.1378/chest.127.2.472. [DOI] [PubMed] [Google Scholar]
- 16.Patil SN, Levin DL. Distribution of thoracic lymphadenopathy in sarcoidosis using computed tomography. J Thorac Imaging. 1999 Apr;14(2):114–7. doi: 10.1097/00005382-199904000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Murdoch J, Müller NL. Pulmonary sarcoidosis: changes on follow-up CT examination. AJR Am J Roentgenol. 1992 Sep;159(3):473–7. doi: 10.2214/ajr.159.3.1503008. [DOI] [PubMed] [Google Scholar]
- 18.McLoud TC, Epler GR, Gaensler EA, Burke GW, Carrington CB. A radiographic classification for sarcoidosis: physiologic correlation. Invest Radiol. 1982 Mar-Apr;17(2):129–38. doi: 10.1097/00004424-198203000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Battesti JP, Saumon G, Valeyre D, et al. Pulmonary sarcoidosis with an alveolar radiographic pattern. Thorax. 1982 Jun;37(6):448–52. doi: 10.1136/thx.37.6.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nunes H, Brillet PY, Valeyre D, Brauner MW, Wells AU. Imaging in sarcoidosis. Semin Respir Crit Care Med. 2007 Feb;28(1):102–20. doi: 10.1055/s-2007-970336. [DOI] [PubMed] [Google Scholar]
- 21.Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am J Roentgenol. 2000 Jun;174(6):1751–7. doi: 10.2214/ajr.174.6.1741751. [DOI] [PubMed] [Google Scholar]
- 22.Primack SL, Hartman TE, Hansell DM, Müller NL. End-stage lung disease: CT findings in 61 patients. Radiology. 1993 Dec;189(3):681–6. doi: 10.1148/radiology.189.3.8234691. [DOI] [PubMed] [Google Scholar]
- 23.Padley SP, Padhani AR, Nicholson A, Hansell DM. Pulmonary sarcoidosis mimicking cryptogenic fibrosing alveolitis on CT. Clin Radiol. 1996 Nov;51(11):807–10. doi: 10.1016/s0009-9260(96)80011-0. [DOI] [PubMed] [Google Scholar]
- 24.Handa T, Nagai S, Miki S, Fushimi Y, Ohta K, Mishima M, Izumi T. Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. 2006 May;129(5):1246–52. doi: 10.1378/chest.129.5.1246. [DOI] [PubMed] [Google Scholar]
- 25.Nunes H, Humbert M, Capron F, Brauner M, Sitbon O, Battesti JP, Simonneau G, Valeyre D. Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax. 2006 Jan;61(1):68–74. doi: 10.1136/thx.2005.042838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shorr AF, Helman DL, Davies DB, Nathan SD. Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics. Eur Respir J. 2005 May;25(5):783–8. [Google Scholar]
- 27.Sulica R, Teirstein AS, Kakarla S, Nemani N, Behnegar A, Padilla ML. Distinctive clinical, radiographic, and functional characteristics of patients with sarcoidosis-related pulmonary hypertension. Chest. 2005 Sep;128(3):1483–9. doi: 10.1378/chest.128.3.1483. [DOI] [PubMed] [Google Scholar]
- 28.Shlobin O, Nathan SD. Sarcoidosis-associated pulmonary hypertension. In Pulmonary Sarcoidosis: A Guide for the Practicing Clinician, Respiratory Medicine 17, Ed Judson M. 2014:111–128. [Google Scholar]
- 29.Baughman RP, Engel PJ, Taylor L, Lower EE. Survival in sarcoidosis-associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. 2010 Nov;138(5):1078–85. doi: 10.1378/chest.09-2002. [DOI] [PubMed] [Google Scholar]
- 30.Lewis MM, Mortelliti MP, Yeager H, Jr, Tsou E. Clinical bronchiectasis complicating pulmonary sarcoidosis: case series of seven patients. Sarcoidosis Vasc Diffuse Lung Dis. 2002 Jun;19(2):154–9. [PubMed] [Google Scholar]
- 31.Xu L, Kligerman S, Burke A. End-stage sarcoid lung disease is distinct from usual interstitial pneumonia. Am J Surg Pathol. 2013 Apr;37(4):593–600. doi: 10.1097/PAS.0b013e3182785a2d. [DOI] [PubMed] [Google Scholar]
- 32.Udwadia ZF, Pilling JR, Jenkins PF, Harrison BD. Bronchoscopic and bronchographic findings in 12 patients with sarcoidosis and severe or progressive airways obstruction. Thorax. 1990 Apr;45(4):272–5. doi: 10.1136/thx.45.4.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soskel NT, Sharma OP. Pleural involvement in sarcoidosis. Curr Opin Pulm Med. 2000 Sep;6(5):455–68. doi: 10.1097/00063198-200009000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Parker JM, Torrington KG, Phillips YY. Sarcoidosis complicated by chylothorax. South Med J. 1994 Aug;87(8):860–2. doi: 10.1097/00007611-199408000-00026. [DOI] [PubMed] [Google Scholar]
- 35.Froudarakis ME, Bouros D, Voloudaki A, et al. Pneumothorax as a first manifestation of sarcoidosis. Chest. 1997 Jul;112(1):278–80. doi: 10.1378/chest.112.1.278. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen LD, Viscogliosi E, Delhaes L. The lung mycobiome: an emerging field of the human respiratory microbiome. Front Microbiol. 2015 Feb 13;6:89. doi: 10.3389/fmicb.2015.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pena TA, Soubani AO, Samavati L. Aspergillus lung disease in patients with sarcoidosis: a case series and review of the literature. Lung. 2011 Apr;189(2):167–72. doi: 10.1007/s00408-011-9280-9. [DOI] [PubMed] [Google Scholar]
- 38.Judson MA, Boan AD, Lackland DT. The clinical course of sarcoidosis: presentation, diagnosis, and treatment in a large white and black cohort in the United States. Sarcoidosis Vasc Diffuse Lung Dis. 2012 Oct;29(2):119–27. [PubMed] [Google Scholar]
- 39.Minces LR, Bhama JK, Abdel-Massih R, et al. Successful double lung transplantation in a patient with bilateral pulmonary and sinus aspergillomas. Transpl Infect Dis. 2011 Oct;13(5):485–8. doi: 10.1111/j.1399-3062.2011.00615.x. [DOI] [PubMed] [Google Scholar]
- 40.Panselinas E, Judson MA. Acute pulmonary exacerbation of sarcoidosis. In Pulmonary Sarcoidosis: A Guide for the Practicing Clinician, Respiratory Medicine 17, Ed Judson M. 2014:65–78. [Google Scholar]
- 41.Moller DR, Ho L. Pulmonary sarcoidosis. In: Clinical Handbook of Interstitial Lung Disease. In: Thillai M, Moller DR, Meyer KC, editors. Boca Raton: CRC Press, Taylor & Francis Group; 2018. pp. 257–270. [Google Scholar]
- 42.Schutt AC, Bullington WM, Judson MA. Pharmacotherapy for pulmonary sarcoidosis: a Delphi consensus study. Respir Med. 2010 May;104(5):717–23. [Google Scholar]
- 43.Korsten P, Strohmayer K, Baughman RP, Sweiss NJ. Refractory pulmonary sarcoidosis - proposal of a definition and recommendations for the diagnostic and therapeutic approach. Clin Pulm Med. 2016 Mar;23(2):67–75. doi: 10.1097/CPM.0000000000000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baughman RP, Drent M. The treatment of pulmonary sarcoidosis. In Pulmonary Sarcoidosis: A Guide for the Practicing Clinician, Respiratory Medicine 17, Ed Judson M. 2014:41–64. [Google Scholar]
- 45.Grutters JC, van den Bosch JM. Corticosteroid treatment in sarcoidosis. Eur Respir J. 2006 Sep;28(3):627–36. doi: 10.1183/09031936.06.00105805. [DOI] [PubMed] [Google Scholar]
- 46.Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006 Oct 1;174(7):795–802. doi: 10.1164/rccm.200603-402OC. [DOI] [PubMed] [Google Scholar]
- 47.Baughman RP, Engel PJ, Meyer CA, Barrett AB, Lower EE. Pulmonary hypertension in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2006 Jun;23(2):108–16. [PubMed] [Google Scholar]
- 48.Valeyre D, Nunes H, Bernaudin JF. Advanced pulmonary sarcoidosis. Curr Opin Pulm Med. 2014 Sep;20(5):488–95. doi: 10.1097/MCP.0000000000000075. [DOI] [PubMed] [Google Scholar]
- 49.Kirkil G, Lower EE, Baughman RP. Predictors of Mortality in Pulmonary Sarcoidosis. Chest. 2018 Jan;153(1):105–113. doi: 10.1016/j.chest.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 50.Judson MA. Strategies for identifying pulmonary sarcoidosis patients at risk for severe or chronic disease. Expert Rev Respir Med. 2017 Feb;11(2):111–118. doi: 10.1080/17476348.2017.1281745. [DOI] [PubMed] [Google Scholar]
- 51.Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015 Jan;34(1):1–15. doi: 10.1016/j.healun.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 52.Eberlein M, Garrity ER, Orens JB. Lung allocation in the United States. Clin Chest Med. 2011 Jun;32(2):213–22. doi: 10.1016/j.ccm.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Egan TM, Edwards LB. Effect of the lung allocation score on lung transplantation in the United States. J Heart Lung Transplant. 2016 Apr;35(4):433–9. doi: 10.1016/j.healun.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 54.Lamas DJ, Lederer DJ. Lung transplantation for idiopathic pulmonary fibrosis. In; Idiopathic Pulmonary Fibrosis: A Comprehensive Clinical Guide. In: Eds Meyer KC, Nathan SD, editors. New York: Humana Press, Springer; 2014. pp. 363–377. [Google Scholar]
- 55.Shlobin OA, Nathan SD. Management of end-stage sarcoidosis: pulmonary hypertension and lung transplantation. Eur Respir J. 2012 Jun;39(6):1520–33. doi: 10.1183/09031936.00175511. [DOI] [PubMed] [Google Scholar]
- 56.Hadjiliadis D, Sporn TA, Perfect JR, Tapson VF, Davis RD, Palmer SM. Outcome of lung transplantation in patients with mycetomas. Chest. 2002 Jan;121(1):128–34. doi: 10.1378/chest.121.1.128. [DOI] [PubMed] [Google Scholar]
- 57.Minces LR, Bhama JK, Abdel-Massih R, et al. Successful double lung transplantation in a patient with bilateral pulmonary and sinus aspergillomas. Transpl Infect Dis. 2011 Oct;13(5):485–8. doi: 10.1111/j.1399-3062.2011.00615.x. [DOI] [PubMed] [Google Scholar]
- 58.Julliard WA, Meyer KC, De Oliveira NC, Osaki S, Cornwell RC, Sonetti DA, Maloney JD. The presence or severity of pulmonary hypertension does not affect outcomes for single-lung transplantation. Thorax. 2016 May;71(5):478–80. doi: 10.1136/thoraxjnl-2015-207354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nathan SD, Shlobin OA, Ahmad S, Burton NA, Barnett SD, Edwards E. Comparison of wait times and mortality for idiopathic pulmonary fibrosis patients listed for single or bilateral lung transplantation. J Heart Lung Transplant. 2010 Oct;29(10):1165–71. doi: 10.1016/j.healun.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 60.Jaksch P, Koinig H, Klepetko W. Critical care management. In: Lung Transplantation. In: Vigneswaran WT, Garrity ER Jr, editors. Lung Biology in Health and Disease. Vol. 243. London: Informa; 2010. pp. 224–236. [Google Scholar]
- 61.Meyer KC. Lung transplantation: chronic complications and management. In: Lung Transplantation. In: Vigneswaran WT, Garrity ER Jr, editors. Lung Biology in Health and Disease. Vol. 243. London: Informa; 2010. pp. 357–374. [Google Scholar]
- 62.Shah RJ, Diamond JM, Cantu E, et al. Latent class analysis identifies distinct phenotypes of primary graft dysfunction after lung transplantation. Chest. 2013;144(2):616–622. doi: 10.1378/chest.12-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morrison MI, Pither TL, Fisher AJ. Pathophysiology and classification of primary graft dysfunction after lung transplantation. J Thorac Dis. 2017 Oct;9(10):4084–4097. doi: 10.21037/jtd.2017.09.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brugière O, Suberbielle C, Thabut G, et al. Lung transplantation in patients with pretransplantation donor-specifi c antibodies detected by Luminex assay. Transplantation. 2013;95(5):761–765. doi: 10.1097/TP.0b013e31827afb0f. [DOI] [PubMed] [Google Scholar]
- 65.Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J. 2014 Dec;44(6):1479–503. doi: 10.1183/09031936.00107514. [DOI] [PubMed] [Google Scholar]
- 66.Verleden GM, Raghu G, Meyer KC, Glanville AR, Corris P. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant. 2014 Feb;33(2):127–33. doi: 10.1016/j.healun.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 67.Benden C, Haughton M, Leonard S, Huber LC. Therapy options for chronic lung allograft dysfunction-bronchiolitis obliterans syndrome following first-line immunosuppressive strategies: A systematic review. J Heart Lung Transplant. 2017 Sep;36(9):921–933. doi: 10.1016/j.healun.2017.05.030. [DOI] [PubMed] [Google Scholar]
- 68.Meyer KC. Diagnosis and management of bronchiolitis obliterans syndrome following lung or hematopoietic cell transplantation. Expert Rev Respir Med. 2016 Jun;10(6):599–602. doi: 10.1586/17476348.2016.1162717. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Liu Y, Su L, Jiang SJ. Recipient-related clinical risk factors for primary graft dysfunction after lung transplantation: a systematic review and meta-analysis. PLoS One. 2014 Mar 21;9(3):e92773. doi: 10.1371/journal.pone.0092773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong A, King CS, Brown AW, Ahmad S, Shlobin OA, Khandhar S, Bogar L, Rongione A, Nathan SD. Hemothorax following lung transplantation: incidence, risk factors, and effect on morbidity and mortality. Multidiscip Respir Med. 2016 Nov 15;11:40. doi: 10.1186/s40248-016-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wille KM, Gaggar A, Hajari AS, et al. Bronchiolitis obliterans syndrome and survival following lung transplantation for patients with sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2008;25(02):117–124. [PubMed] [Google Scholar]
- 72.Walker S, Mikhail G, Banner N, Partridge J, Khaghani A, Burke M, Yacoub M. Medium term results of lung transplantation for end stage pulmonary sarcoidosis. Thorax. 1998 Apr;53(4):281–4. doi: 10.1136/thx.53.4.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nunley DR, Hattler B, Keenan RJ, Iacono AT, Yousem S, Ohori NP, Dauber JH. Lung transplantation for end-stage pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999 Mar;16(1):93–100. [PubMed] [Google Scholar]
- 74.Collins J, Hartman MJ, Warner TF, Müller NL, Kazerooni EA, McAdams HP, Slone RM, Parker LA. Frequency and CT findings of recurrent disease after lung transplantation. Radiology. 2001 May;219(2):503–9. doi: 10.1148/radiology.219.2.r01ma12503. [DOI] [PubMed] [Google Scholar]
- 75.Ionescu DN, Hunt JL, Lomago D, Yousem SA. Recurrent sarcoidosis in lung transplant allografts: granulomas are of recipient origin. Diagn Mol Pathol. 2005 Sep;14(3):140–5. doi: 10.1097/01.pas.0000176765.26047.6f. [DOI] [PubMed] [Google Scholar]
- 76.Schultz HH, Andersen CB, Steinbruuchel D, Perch M, Carlsen J, Iversen M. Recurrence of sarcoid granulomas in lung transplant recipients is common and does not affect overall survival. Sarcoidosis Vasc Diffuse Lung Dis. 2014 Jul 8;31(2):149–53. [PubMed] [Google Scholar]
- 77.Banga A, Sahoo D, Lane CR, Farver CF, Budev MM. Disease Recurrence and Acute Cellular Rejection Episodes During the First Year After Lung Transplantation Among Patients With Sarcoidosis. Transplantation. 2015 Sep;99(9):1940–5. doi: 10.1097/TP.0000000000000673. [DOI] [PubMed] [Google Scholar]
- 78.Bjørtuft O, Foerster A, Boe J, Geiran O. Single lung transplantation as treatment for end-stage pulmonary sarcoidosis: recurrence of sarcoidosis in two different lung allografts in one patient. J Heart Lung Transplant. 1994 Jan-Feb;13(1 Pt 1):24–9. [PubMed] [Google Scholar]
- 79.Yserbyt J, Wuyts WA, Verleden SE, Verleden GM, Van Raemdonck DE, Verbeken EK, Vanaudenaerde BM, Vos R. Solid Organ Transplantation in Sarcoidosis. Semin Respir Crit Care Med. 2017 Aug;38(4):538–545. doi: 10.1055/s-0037-1602383. [DOI] [PubMed] [Google Scholar]