

Abstract
Lytic replication of Epstein-Barr virus (EBV) is not only essential for its cell–to–cell spread and host–to–host transmission, but it also contributes to EBV-induced oncogenesis. Thus, blocking EBV lytic replication could be a strategy for managing EBV-associated diseases. Previously, we identified a series of natural lignans isolated from the roots of Saururus chinensis (Asian lizard's tail) that efficiently block EBV lytic replication and virion production with low cytotoxicity. In this study, we attempted to elucidate the molecular mechanism by which these lignans inhibit EBV lytic replication. We found that a representative compound, CSC27 (manassantin B), inhibits EBV lytic replication by suppressing the expression of EBV immediate-early gene BZLF1 via disruption of AP-1 signal transduction. Further analysis revealed that manassantin B specifically blocks the mammalian target of rapamycin complex 2 (mTORC2)-mediated phosphorylation of AKT Ser/Thr protein kinase at Ser-473 and protein kinase Cα (PKCα) at Ser-657. Using phosphoinositide 3-kinase–AKT-specific inhibitors for kinase mapping and shRNA-mediated gene silencing, we validated that manassantin B abrogates EBV lytic replication by inhibiting mTORC2 activity and thereby blocking the mTORC2–PKC/AKT-signaling pathway. These results suggest that mTORC2 may have utility as an antiviral drug target against EBV infections and also reveal that manassantin B has potential therapeutic value for managing cancers that depend on mTORC2 signaling for survival.
Keywords: virology, mTOR complex (mTORC), viral DNA, viral replication, protein kinase C (PKC), inhibition mechanism, inhibitor, EBV reactivation, Epstein-Barr virus (EBV), lignan, manassantin B, mTORC2 inhibitor
Introduction
Epstein-Barr virus (EBV)5 ubiquitously infects more than 90% of the population worldwide and is also an etiological agent of several human diseases and malignancies (1, 2). Like other herpesviruses, the EBV life cycle consists of two phases, latent and lytic. When EBV infects memory B-lymphocytes, it establishes latent infection by default and expresses a few latent genes to maintain latent infection (3, 4). Establishment of latency is a viral strategy to avoid host immune surveillance for lifetime-persistent infection. When latent EBV is reactivated in response to physiological stimuli or chemical assault, lytic replication occurs where the virus expresses almost all the genes in its genome, replicates its genomic DNA, and produces progeny viral particles releasing from cells. EBV lytic replication initiates at the activation of viral immediate-early genes (ZTA and RTA) and proceeds as almost all viral genes are expressed in a cascade fashion that leads to production of progeny virions (5). Lytic replication is essential for cell–to–cell spread as well as host–to–host transmission of the virus. In addition, increasing evidence indicates that EBV lytic replication also contributes to viral pathogenesis and oncogenesis. (i) EBV lytic replication is crucial for efficient dissemination to the sites of disease. (ii) The presence of low numbers of EBV lytically-infected cells could enhance tumor growth through the release of growth factors and immunosuppressive cytokines (6–8). (iii) Elevated antibody titers against EBV lytic antigens and increased viral DNA load in serum/plasma are directly correlated with advanced cancer stages, poor prognosis, or tumor recurrence in nasopharyngeal carcinoma (NPC), Hodgkin's disease, and Burkitt's lymphoma (9). (iv) EBV lytic replication is directly linked to chronic active EBV infection (CAEBV) and oral hairy leukoplakia (10, 11). (v) EBV lytic genes BZLF1, BGLF4, and BGLF5 can induce genome instability (12) and other lytic gene products, such as LF1, LF2, LF3, BILF1, BALF4, and BHLF1, are detected in tumor biopsies (13–15). Therefore, EBV lytic life cycle contributes to EBV-induced oncogenesis, and inhibition of EBV lytic replication can be a strategy for treatment of EBV-associated malignancies.
Reactivation of EBV can be initiated when B cells are differentiated into plasma following B-cell receptor stimulation by antigen (16). In the cell culture system, EBV lytic life cycle can be induced by certain chemicals such as 12-O-tetradecanoylphorbol-13-acetate (TPA) and sodium butyrate (17). These findings indicate that host cell signal transduction and epigenetic regulation control the switch of the virus between latency and lytic replication. TPA is found to trigger EBV reactivation cascade through activating protein kinase C (PKC), leading to stimulation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway (18, 19). As a consequence of the activation of the pathway, c-Fos is accumulated, and c-Jun is phosphorylated, leading to the formation of an active AP-1 complex on the ZTA promoter, activation of the gene of EBV immediate-early transcriptional activator ZTA, and initiation of the lytic cascade of EBV (20). In B-cell receptor–mediated EBV activation, PI3K–AKT-activated bZip transcription factors (ATF/c-Jun/CREB) also bind to the ZII domain and activate the BZLF1 promoter (21).
PI3K/mTOR signaling is a chief mechanism for controlling cell proliferation, survival, and metabolism (22). mTOR exists in two different complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). In mTORC1, mTOR is associated with Raptor, PRAS40, and mLST8/GβL (23), whereas in mTORC2, mTOR complexes with two unique regulatory proteins named Rictor and Sin1 in addition to mLST8/GβL, PROTOR/PRR5, and DEPTOR (24–27). mTORC1 is highly sensitive to rapamycin, whereas mTORC2 is insensitive to short-term treatment of rapamycin. mTORC1 and mTORC2 participate in different pathways and recognize distinct substrates. mTORC1 regulates cell growth by controlling the activity of the S6 kinases 1 (S6K1) and the eIF-4E-binding protein 1 (4E-BP1) (23), whereas mTORC2 modulates cell survival, growth, proliferation, and metabolism by controlling the phosphorylation of AKT Ser-473 (29). Previous studies showed that inhibition of mTORC1 by rapamycin inhibits EBV and KSHV lytic replication (30, 31). In addition, EBV in turn is able to activate or manipulate PI3K/mTOR signaling, which is associated with oncogenesis. PI3K/mTOR is activated in EBV-positive post-transplant lymphoproliferative diseases, Hodgkin's lymphoma, NPC, and gastric carcinoma (32). In this study, we found that EBV reactivation depends on mTORC2 and inhibition of mTORC2 function by a potential mTORC2 inhibitor, CSC27 (manassantin B), and blocks EBV switch from latency to lytic replication. Manassantin B inhibits mTORC2 phosphorylation of AKT and PKCα and, as a consequence, blocks EBV lytic replication.
Results
Manassantin B blocks EBV lytic replication by suppressing ZTA-initiated reactivation cascade
In our previous work, we identified a series of natural lignans isolated from the roots of Saururus chinensis and showed that they efficiently block EBV lytic replication with low cytotoxicity (33). To understand the mechanism underlying their antiviral activities, a representative compound among these lignans, CSC27 (manassantin B, a dineolignan), was chosen for further investigation based on its antiviral potency (Fig. 1A). The effect of manassantin B on EBV lytic replication was analyzed using P3HR-1 cells, an EBV latently-infected Burkitt lymphoma cell line. P3HR-1 cells were induced for reactivation with TPA and sodium butyrate (NaB). Three hours post-induction, the cells were exposed to manassantin B in a wide range of concentrations. Intracellular viral genomic DNA was determined at 48 h post-induction. Encapsidated viral DNA was measured for released virions in the media 5 days post-induction. The half-maximal DNA replication inhibitory concentration (IC50) and the half-maximal antiviral effective concentration (EC50) of manassantin B were determined from the dose-response curves of the intracellular DNA and extracellular virion to be 1.72 and 0.5 μm, respectively (Fig. 1B). The cytotoxicity of the compound was assessed using the trypan blue exclusion method for cell viability at 48 h, and the 50% cytotoxic concentration (CC50) was determined to be >50 μm, leading to a selectivity index (= CC50/EC50) of >100. In addition, the antiviral property of manassantin B was also examined in EBV-carrying Akata-BX1 cells in which EBV reactivation was induced by anti-IgG treatment. Result showed that manassantin B inhibited EBV lytic DNA replication and virion production with IC50 of 12.96 μm and EC50 of 3.06 μm in Akata-BX1 cells (Fig. 1C).
Figure 1.

Effect of manassantin on EBV lytic replication. A, structure of manassantin B (CSC27). B, P3HR-1 cells were treated with a wide range concentration of manassantin B 3 h after being induced by TPA and sodium butyrate (NaB) for EBV lytic replication. Intracellular EBV DNA replication (blue), extracellular EBV virion production (green), and cell viability (orange) were determined as described under “Experimental procedures.” The mean values from three independent experiments and standard deviations are presented on the y axis of dose-response curves. Calculated IC50, EC50, and CC50 values are presented in the table. C, Akata-BX1 cells were treated with manassantin B in a wide range concentration 3 h after being induced by anti-IgG antibody for EBV lytic replication. Intracellular EBV DNA replication (blue), extracellular EBV virion production (green), and cell viability (orange) were determined and presented as IC50, EC50, and CC50 values in the table. D, cytotoxicity of manassantin B to PBMC was assessed after 48 h of treatment by trypan blue staining. E, effect of manassantin B on ZTA and RTA mRNA levels in TPA/NaB-induced P3HR-1 cells were determined using qRT-PCR at 24 h post-induction. F, expression of ZTA and EA–D were detected by Western blot analysis at 24 h post-induction.
Cytotoxicity of manassantin B to primary lymphocytes was assessed with human peripheral blood mononuclear cells (PBMCs) after 48 h of treatment, and the result demonstrated low cytotoxicity of manassantin B to primary lymphocytes (Fig. 1D).
Transition from latency to lytic state is controlled by two viral transcription factors, namely ZTA and RTA, encoded by BZLF1 and BRLF1. Both ZTA and RTA are essential for viral lytic gene expression and ori-Lyt–dependent DNA replication. The expression of BZLF1 and BRLF1 in induced P3HR-1 cells was determined 48 h post-treatment by qRT-PCR and Western blot analysis. Results showed significant reductions of both BZLF1 and BRLF1 gene expression in the cells treated with manassantin B in both mRNA and protein levels (ZTA) in a dose-dependent manner (Fig. 1, E and F). As a consequence, the expression of downstream gene, EA–D protein, was also reduced (Fig. 1F). We conclude that manassantin B inhibits EBV lytic replication by blocking ZTA- and RTA-initiated viral reactivation cascade.
Manassantin B inhibits AP-1–signaling pathway
EBV reactivation can be initiated through activating protein kinase C (PKC). As a consequence, c-Fos is accumulated and c-Jun is phosphorylated, leading to the formation of an active AP-1 complex on the ZII domain of the BZLF1 promoter and activation of ZTA gene (20). In addition, other cellular transcription factors, such as CREB and ATF, are also reportedly associated with the promoter in response to PI3K–AKT signaling (21). We asked whether manassantin B inhibits EBV ZTA activation through disrupting the AP-1–signaling pathway. To this end, we first employed an electrophoretic mobility shift assay (EMSA) to study the effects of the lignan on the AP-1 DNA-binding property. Two dsDNA fragments, one containing a consensus AP-1–binding motif and the other harboring the ZII element of the ZTA promoter, were reacted with the nuclear extract of TPA-induced P3HR-1 cells that have been treated with manassantin B in various concentrations. Results showed that a DNA–protein complex was detected with the AP-1–binding oligonucleotide, and the binding was reduced with the extracts of the cells treated with manassantin B (Fig. 2A). Similarly, a DNA–protein complex that appeared only with TPA-induced P3HR-1 nuclear extract on the ZII element was also diminished in response to manassantin B in a dose-dependent manner (Fig. 2B), indicating that manassantin B prevents the TPA-induced transcription factor (AP-1 or CREB/c-Jun/ATF) from binding to the ZTA promoter. However, manassantin B did not block the transcription factors in the nuclear extract of nonmanassantin B–treated cells binding to AP-1–responsive promoters (Fig. 2, C and D), suggesting that manassantin B acts at the pathway upstream of AP-1 activation rather than blocking activated AP-1 binding to DNA. Second, the effect of manassantin B on the transcription activity of AP-1–responsive promoter was analyzed using a promoter–reporter assay. 293T cells were transfected with the AP-1 promoter–luciferase reporter plasmids. Six hours post-transfection, cells were treated with TPA to activate AP-1 signaling. Manassantin B in different concentrations was added into the cell culture. The activation of the AP-1–responsive promoter in the absence and presence of manassantin B was measured through the luciferase activities 36 h post-induction. Manassantin B was found to be able to block TPA-induced transcription activity of the AP-1–responsive promoter in a dose-dependent manner (Fig. 2E). Taken together, manassantin B inhibits the AP-1–signaling pathway upstream of ZTA, thereby blocking EBV reactivation.
Figure 2.

Manassantin B blocks AP-1 binding to AP-1–responsive promoter and the BZLF1 promoter. A, Cy5-labeled double-stranded oligonucleotide containing a consensus AP-1–binding motif; B, double-stranded oligonucleotides of the ZII element of the BZLF1 promoter were incubated with the nuclear extracts (NE) prepared from TPA/NaB-induced P3HR-1 cells that were treated with manassantin B in various concentrations. Protein DNA complexes were resolved by EMSA. Unlabeled DNA duplexes were used to determine binding specificity. C and D, nuclear extracts prepared from TPA/NaB-induced P3HR-1 cells that were not treated with manassantin B were incubated with the same double-stranded oligonucleotides above and manassantin B in different concentrations. Protein DNA complexes were resolved by EMSA. E, 293T cells were co-transfected with pAP-1–promotor–luciferase vector and pRL-TK control vector, followed by induction with TPA. Manassantin B in a wide range of concentrations was added to the culture medium. The promoter activities were measured after 36 h post-treatment using a luciferase assay, normalized by Renilla luciferase activity.
Kinase mapping: mTOR is required for EBV lytic replication
To identify the target of manassantin B, we continued to trace upstream pathways that are responsible for EBV lytic replication and targeted by manassantin B. PI3K/mTOR and the Ras–ERK pathway are two main upstream pathways of AP-1 transduction signaling (34, 35). To understand whether these two pathways are involved in EBV reactivation in P3HR-1 cells, we employed a whole panel of pharmacological inhibitors of the PI3K/mTOR and ERK pathways for a kinase mapping. The effect of each inhibitor on EBV lytic DNA replication was determined, and results showed that the inhibitors to ERK (U0126), PI3K subunits (LY294002, HS173, TGX221, and AS252424 CAL-101), and AKT1/2/3 (AT7867) in the concentrations of 10× their IC50 exhibited little effect on EBV lytic replication. In contrast, mTORC1 inhibitor rapamycin gave a moderate inhibition, and PI3K/mTOR dual inhibitor BEZ235 led to an efficient inhibition of EBV DNA synthesis (Fig. 3, A and B). The cytotoxicity of these inhibitors to P3HR-1 cells in these concentrations was determined (Fig. 3C). Furthermore, the effects of rapamycin and BEZ235 in a wide range of concentrations on EBV ZTA expression and viral DNA replication were further examined. Rapamycin exhibited partial inhibition on ZTA expression and EBV DNA replication even at the concentration of 1000× IC50 (Fig. 3E). In contrast, BEZ235 was able to block ZTA expression and EBV DNA replication in P3HR-1 cells (Fig. 3F).
Figure 3.

mTOR is critical for EBV lytic replication. A, list of PI3K/AKT/mTOR inhibitors used in this experiment and their targets, IC50 and 10 × IC50. B, each inhibitor listed in A was added to TPA/NaB-induced P3HR-1 cells at a concentration of approximately 10 × IC50. EBV lytic DNA replication was determined by real-time PCR. C, associated cytotoxicity of each inhibitor to P3HR-1 cells was assessed by trypan blue staining. D, effect of each inhibitor on EBV ZTA expression was determined by Western blotting 24 h after treatment. E, wide range concentration of rapamycin (Rapa) was used to examine the effect of the mTORC1 inhibitor on EBV gene expression (ZTA and EA–D) as well as EBV lytic DNA replication. F, wide range concentration of BEZ235 was used to analyze the effect of the dual inhibitor on EBV gene expression (ZTA and EA–D) as well as EBV lytic DNA replication.
Manassantin B inhibits mTORC2, AKT, and PKC signaling
Given that mTOR signaling is required for EBV reactivation, we wondered whether manassantin B inhibits EBV lytic replication by targeting mTOR signaling. mTOR assembles into two distinct complexes, designated mTORC1 and mTORC2. To gain insight into how manassantin B acts on mTOR pathways, we examined the effect of manassantin B on the activation status of the components in mTOR pathways, including AKT, PKC, MEK, ERK, and p70S6K using Western blot analysis 24 h post-induction. AKT activation is tightly controlled by PI3K/mTOR signaling and involves the phosphorylation of Thr-308 in the activation loop or the phosphorylation of Ser-473 in the hydrophobic motif. As shown in Fig. 4, manassantin B specifically blocked the phosphorylation of AKT at Ser-473 but not Thr-308. It is known that phosphorylation of Thr-308 residue in AKT is accomplished by PDK1, whereas phosphorylation of Ser-473 residue is mediated by mTORC2 (29). Furthermore, phosphorylation of another mTORC2 substrate, PKCα, was also inhibited in the presence of manassantin B. In contrast, manassantin B showed little effect on mTORC1 activity as the phosphorylation of p70S6K was not decreased or even slightly increased by this lignan. Therefore, manassantin B appears to target mTORC2 and inhibit its kinase activity.
Figure 4.
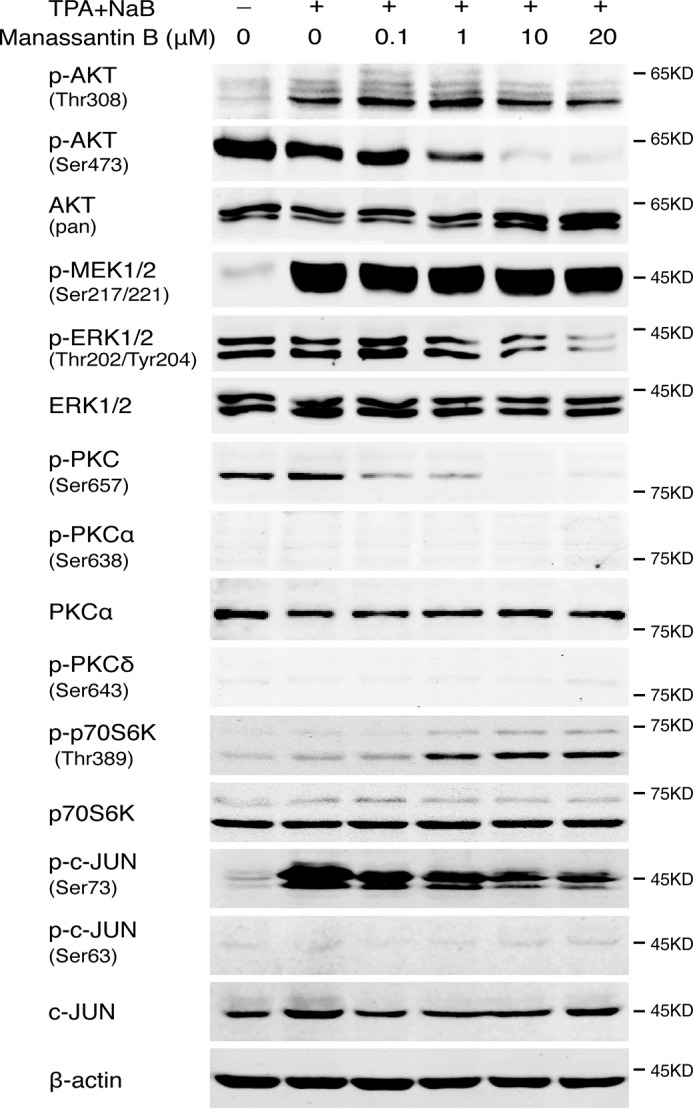
Manassantin B inhibits mTORC2–PKC-signaling pathways. The expression and phosphorylation of AKT, PKC, MEK, ERK, p70S6K, and c-Jun were examined by Western blot analysis at 24 h post-induction in the absence and presence of manassantin B.
The ERKs1/2 are ubiquitous Ser/Thr kinases that phosphorylate hundreds of substrates. Manassantin B inhibits the phosphorylation of ERK1/2. However, the phosphorylation of MEK1/2 was not affected by manassantin B.
As the end effector of the signal transduction, the phosphorylation of AP-1 (c-Jun) in response to manassantin B treatment was examined. Results showed that the phosphorylation of c-Jun at Ser-73 increased in the cells induced with TPA/butyrate, and the induced phosphorylation of c-Jun was inhibited in the presence of manassantin B in a dose-dependent manner (Fig. 4).
Knockdown of mTORC2 completely blocks EBV lytic replication
To further confirm that mTORC2 is critically required for EBV lytic replication and serves as the target of manassantin B, we silenced the expression of the genes for mTOR, Raptor, and Rictor using an shRNA knockdown approach. mTOR is a shared component of mTORC1 and mTORC2, whereas Raptor and Rictor are specific components of mTORC1 and mTORC2, respectively. Knockdown of mTOR, which destroyed both mTORC1 and mTORC2, resulted in a complete shutdown of ZTA expression (Fig. 5A), EBV lytic DNA replication (Fig. 5D), and virion production in TPA-induced P3HR-1 cells (Fig. 5E). Silencing Rictor, which down-regulates mTORC2, also led to dramatic reduction in ZTA expression (Fig. 5B), viral DNA replication (Fig. 5D), and virion production (Fig. 5E). In contrast, knockdown of Raptor expression, which down-regulates mTORC1, only reduced ZTA expression to a certain extent (Fig. 5C) and led to partial inhibition of viral DNA replication (Fig. 5D) and virion production (Fig. 5E). These results, together with manassantin B inhibiting the phosphorylation of Akt-473 and PKCα-657, led to the conclusion that mTORC2 is crucial for EBV lytic replication, and manassantin B blocks the EBV reactivation through inhibiting mTORC2 signaling. A model for the function of mTORC2 in EBV lytic replication and action of manassantin B (CSC27) is illustrated in Fig. 6.
Figure 5.

mTORC2 is critical for EBV lytic replication. shRNA lentivirus targeting mTOR (A), Rictor (B), and Raptor (C), along with a nontargeting control shRNA lentivirus (shCtr), were transduced into P3HR-1 cells. The effects of the shRNA on each target gene and EBV ZTA gene expression were determined by Western blotting. D, cells stably expressing mTOR, Raptor, Rictor, and control shRNAs were induced by TPA and NaB for lytic viral replication. EBV lytic DNA replication was evaluated at 48 h post-induction by qRT-PCR as described. E, EBV virion production was assayed 5 days after induction as described under “Experimental procedures.”
Figure 6.

Signaling network of mTORC2 that leads to activation of PKC pathway and reactivation of latent EBV. The mTORC2–PKC–AP-1–ZTA as well as mTORC2–AKT–ATF/CREB–ZTA axes are critical for EBV reactivation, and manassantin B (CSC27) blocks EBV reactivation by inhibiting mTORC2 kinase activity.
Discussion
A class of lignans isolated from the roots of S. chinensis was found to be able to effectively block EBV lytic replication (33). In this study, we investigated the pharmacological property and antiviral mechanism of a representative compound, namely manassantin B (CSC27). The study showed that manassantin B blocks EBV reactivation via preventing the viral switch gene ZTA from being activated by cellular signaling pathways. Interestingly, manassantin B targets and disrupts mTOR signaling to achieve the inhibitory effect on EBV reactivation. Revelation of the pharmacology of manassantin B paved the way for exploring the application value of this natural lignan in antiviral drug development. Our findings have shown additional salient features as follows.
(i) This study demonstrated essential roles of mTORC2 signaling in EBV reactivation. EBV can be induced to enter lytic replication by either phorbol ester (TPA) treatment or B-cell receptor engagement at the B-cell surface (17, 19, 37). This study revealed the involvement of mTORC2 in TPA-mediated EBV lytic replication, which can be blocked by manassantin B (Fig. 1B). We also showed that manassantin B can inhibit anti-IgG–induced EBV lytic replication in Akata cells (Fig. 1C), suggesting that both TPA and B-cell receptor engagement-mediated EBV reactivation are likely to share or merge into the same pathway, mTORC2 signaling, and ultimately lead to EBV lytic replication. It has been reported that B-cell receptor-mediated EBV lytic replication in Akata cells is regulated by mTORC2 (38). Therefore, the study on manassantin B pharmacology revealed a crucial role of mTORC2 signaling in EBV lytic replication as follows. (a) Stimuli activate mTORC2 that phosphorylates AKT at Ser-473 and PKCα at Ser-657. (b) Active PKC–AP-1 signaling leads to activation of ZTA and RTA, which initiates EBV lytic replication cascade (Fig. 6). On the other side, manassantin B inhibits mTORC2 kinase activity and blocks the mTORC2–PKC–AP-1–ZTA pathway, thereby halting the EBV reactivation from latency.
(ii) The findings that the mTORC2–PKC–AP-1 axis is absolutely required for EBV reactivation and that manassantin B can block EBV reactivation validated mTORC2 as an effective drug target for EBV-associated diseases. EBV is an important pathogen responsible for a number of human diseases and cancers. Although much of the EBV-induced pathology has been attributed to viral latency, the importance of EBV lytic replication in viral pathogenicity has been increasingly recognized. Evidence is emerging that reactivation of lytic virus and the lytic activator protein ZTA is associated with tumorigenesis and autoimmune diseases (13, 14, 39, 40). EBV lytic replication is directly linked to chronic active EBV infection (CAEBV), oral hairy leukoplakia in immunosuppressed individuals, and an increased risk of EBV-associated nasopharyngeal carcinoma (41–46). Therefore, inhibition of EBV lytic replication may be a strategy for treatment of some EBV-associated diseases. Although a few drugs have been developed against α- and β-herpesviruses (47, 48), there are no effective drugs available to treat γ-herpesvirus (KSHV and EBV) infections. Therefore, efficacious drugs against EBV are very much needed. Identification of the natural lignin manassantin B as an effective antiviral agent that inhibits EBV reactivation with minimal cytotoxicity provides a candidate compound or lead for further development of a new drug to treat EBV-associated diseases.
(iii) mTOR signaling is known as a master regulator of homeostasis, which controls most anabolic and catabolic processes in response to nutrients and growth factors. Two distinct complexes, mTORC1 and mTORC2, correspond two major branches of signal output. mTORC1 has been well-characterized in terms of its structure, regulation, and function, whereas mTORC2 is less studied. mTOR has been a validated drug target for cancer treatment. Rapamycin (mTORC1 inhibitor) and derivatives were approved for treatment of certain cancers but have been less successful than anticipated (22). Several PI3K/mTOR dual inhibitors, such as NVP-BEZ235 and VS-5584, are under clinical trial for cancer therapy, including lymphoma treatment (49, 50). These drugs have also shown some promise in preclinical and early clinical trial data but have raised concerns over dose-limiting toxicities (22). Deregulation of mTORC2, particularly hyperactivation, has been observed in many types of human cancers (51, 52). Recent studies in cancer biology indicate that mTORC2 activity is essential for the transformation and vitality of a number of cancer cell types, but is less essential in normal cells (28, 36, 53). Thus, the mTORC2-specific inhibitor has special value for cancer treatment. Our recent study showed that mTORC2 is involved in NPC cancer stem cell formation, and manassantin B is able to inhibit NPC growth in a xenograft mouse model.6 Thus, manassantin B exhibits a unique advantage as an mTORC2 inhibitor to treat EBV-associated cancers by targeting multiple layers of pathogenesis, i.e. EBV reactivation and tumorigenesis and exhibiting low cytotoxicity.
Experimental procedures
Cell culture
P3HR-1 cells, an EBV-positive Burkitt's lymphoma cell line, were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS, Gibco-BRL). Akata-BX1 cells that carry a recombinant EBV were cultured in RPMI 1640 medium supplemented with 10% FBS. PBMCs were cultured in RPMI 1640 medium supplemented with 10% FBS. The PBMCs were isolated from human peripheral blood buffy coat samples of healthy volunteers, obtained from Guangzhou Blood Centre. Human embryonic kidney HEK293T cells were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS. All cultures contained penicillin/streptomycin (100 units/ml).
Chemicals and reagents
Manassantin B (CSC27) was isolated from the roots of S. chinensis (33). LY294002, HS173, TGX211, AS252424, CAL101, AT7867, and BEZ235 were purchased from Medchem Express. These compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted to different concentrations before being added to P3HR-1, 293T cell cultures. The final DMSO concentration in the culture medium was maintained below 0.5%. TPA and NaB were purchased from Sigma-Aldrich. Goat anti-human IgG was purchased from Hua Yang Zheng Long. mTOR pathway antibody sampler kit, phospho-Akt pathway antibody sampler kit, and antibodies against phospho-p44/42 MAPK (ERK1/2) (Thr-202/Tyr-204), p44/42 MAPK (ERK1/2), phospho-MEK1/2 (Ser-217/221), phospho-p70S6K (Thr-389), p70S6K, and phospho-PKCδ (Ser-643) were obtained from Cell Signaling Technology. Antibody against β-actin was purchased from Sigma-Aldrich. Antibodies against PKCα, phospho-PKCα (Ser-657), phospho-PKCα (Ser-638), EBV ZTA, and EBV EA–D were obtained from Santa Cruz Biotechnology.
Quantification of intracellular EBV genomic DNA
P3HR-1 cells were induced with 20 ng/ml TPA and 0.3 mm NaB for EBV reactivation. Akata–BX1 cells were induced with 8 μg/ml IgG. Three hours post-induction, CSC27 or other compounds in dilutions were added to cells. Cells were harvested at 48 h post-induction, and total DNAs were purified using Qiagen DNeasy kit. EBV genomic DNA copy number was quantified by real-time PCR on a LightCycler 480 instrument (Roche Applied Science) using primers specified for EBNA1 (forward primer 5′-CATTGAGTCGTCTCCCCTTTGGAAT-3′ and reverse primer 5′-TCATAACAAGGTCCTTAATCGCATC-3′). The intracellular viral genomic DNA in each sample was normalized to GAPDH using primers directed to GAPDH (forward 5′-ACATCATCCCTGCCTCTAC-3′ and reverse 5′-TCAAAGGTGGAGGAGTGG-3′). The half-maximal inhibitory concentration (IC50) values of compounds were determined from a dose-response curve of EBV DNA content values from TPA-induced and chemically-treated cells. The viral DNA contents with those of uninduced cells subtracted were divided by those of the control cells with no drug treatment and then represented on the y axes of dose-response curves: y axis value = (TPAX − noTPAX)/(TPA0 − noTPA0), where X is any concentration of the drug, and 0 represents nondrug treatment. The IC50 on viral DNA synthesis for each compound was calculated with the aid of GraphPad Prism software.
Quantification of extracellular EBV genomic DNA
Five days post-induction, P3HR-1 and Akata-BX1 culture media were collected, and extracellular virions were pelleted from the medium supernatant. To remove contaminating DNA outside viral particles, the concentrated viruses were treated with turbo DNase I (Ambion) at 37 °C for 1 h, followed by proteinase K digestion. Virion DNA was extracted with phenol/chloroform, precipitated with ice-cold ethanol, and then dissolved in Tris-EDTA (TE) buffer. The EBV genomic copy numbers were determined by real-time PCR, and the values were corrected as described above. The half-maximal antiviral effective concentration (EC50) values were calculated from dose-response curves with GraphPad Prism software.
Cytotoxicity assay
P3HR-1, Akata-BX1, and PBMCs were treated with CSC27 in a wide range of concentration for 2 days. The viability of cells was assessed by counting trypan blue–stained cells using a Countstar instrument. The half-maximal CC50 was calculated from dose-response curves with Graph-Pad Prism software.
Western blotting assay
Cells were lysed with lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% NP-40, 10% glycerol, 40 mm β-glycerophosphate, 30 mm sodium fluoride, 5 mm EDTA, protease inhibitor mixture (Roche Applied Science), 1 mm sodium orthovanadate). Lysates were subsequently denatured, resolved by SDS-PAGE, and transferred to nitrocellulose membranes. Membranes were reacted with the primary antibodies. Anti-IR Dye 800 or Dye 680 anti-rabbit or anti-mouse IgGs were used as the secondary antibodies. The images were visualized using the LI-COR Odyssey system. All immunoblottings were repeated at least twice, and representative images are shown.
EMSA
Two double-stranded oligonucleotides were synthesized, one containing consensus AP-1–binding site (5′-CGCTTGATGACTCAGCCGGAA-3′) and the other harboring the ZII region of the ZTA promoter (5′-ACGTCCCAAACCATGACATCACAGAGGAGGCT-3′). Oligonucleotides were end-labeled with 3′Cy5 before being annealed to form dsDNA fragments. Nuclear protein extracts were prepared using low-salt buffer (10 mm HEPES-KOH, pH 7.5, 0.05% NP40, 10 mm KCl, 1.5 mm MgCl2, 0.1 mm EDTA, 0.5 mm DTT, 1 mm PMSF, protease inhibitor mixture, 1 mm NaF, 1 mm NaNO3) and high-salt extraction buffer (20 mm HEPES-KOH pH 7.5, 500 mm KCl, 1.5 mm MgCl2, 0.2 mm EDTA, 10% glycerol, 0.5 mm DTT, 1 mm PMSF, protease inhibitor mixture, 1 mm NaF, 1 mm NaNO3). EMSAs were carried out in EMSA reaction buffer (10 mm Tris-HCl, pH 7.5, 50 mm KCl, 0.5 mm MgCl2, 1 mm DTT, 2.5% glycerol) for 30 min at 15 °C. The DNA–protein complexes were resolved on 5.5% native polyacrylamide gels at 4 °C, 120 V for 1 h, and then scanned with an Odyssey imager (LI-COR). Unlabeled DNA fragments of ZII and AP-1 were used to determine binding specificity.
Luciferase assay
The promoter–reporter plasmid pAP-1-luc was provided by Dr. Ersheng Kuang at Sun Yat-sen University. 293T cells grown in 48-well plates were co-transfected with 50 ng of pAP-1-luc and 5 ng of pRL-TK using Lipofectamine 2000 reagent (Life Technologies, Inc.). 6 h after transfection, cells were treated with TPA. The pRL-TK plasmid expressing Renilla luciferase was used as an internal control. 36 h post-induction, the luciferase assay was performed with Promega's dual-luciferase assay kit. Each sample was duplicated, and each experiment was repeated at least three times.
shRNA-mediated gene silencing
The shRNA lentiviral vectors targeting mTOR (Clone ID: NM_004958.2-5477s1c1; NM_004858.2-7897s1c1), Raptor (Clone ID: NM_020761.1-4689s1c1; NM_020761.1-4325s1c1), and Rictor (Clone ID: NM_152756.2-2620s1c1; NM_152756.3-5273s21c1) were purchased from Sigma-Aldrich. Lentiviral particles were prepared by transfecting HEK293T cells with pLKO.1-shRNA plasmid (or pLKO.1-shControl plasmid), miR8.2 packing plasmid, and pCMV–VSV-G plasmid in the ratio of 4:3:1. Media containing lentiviruses were harvested at 48 and 72 h and subjected to ultracentrifugation to concentrate the lentiviruses. Then the concentrated lentiviruses were used to transduce P3HR-1 cells, followed by selection under 2 μg/ml puromycin for a week. The knockdown efficiencies were verified by Western blot analysis.
Data availability
All data described are contained within the article.
Author contributions
Q. W., N. Z., Y. W., and Y. Y. data curation; Q. W. formal analysis; Q. W. and N. Z. investigation; Q. W., N. Z., and J. H. methodology; Q. W. writing-original draft; Y. W. and Y. Y. funding acquisition; J. X., Q. G., and P. M. L. resources; Y. Y. conceptualization; Y. Y. supervision; Y. Y. writing-review and editing.
Acknowledgments
We thank the members in Yuan Lab for discussion and constructive suggestions. We are grateful to Musheng Zeng at Sun Yat-sen University Cancer Center and Ersheng Kuang at Sun Yat-sen University for providing cell lines and reagents.
This work was supported by National Institutes of Health Grant P01CA174439 and National Natural Science Foundation of China Grants 81530069 and 81772177. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
N. Zhu and Y. Yuan, unpublished data.
- EBV
- Epstein-Barr virus
- PBMC
- peripheral blood mononuclear cell
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- EMSA
- electrophoretic mobility shift assay
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex
- PI3K
- phosphoinositide 3-kinase
- qRT
- quantitative RT
- CREB
- cAMP-response element-binding protein
- FBS
- fetal bovine serum
- PMSF
- phenylmethylsulfonyl fluoride
- PKCα
- protein kinase Cα
- NPC
- nasopharyngeal carcinoma
- CAEBV
- chronic active EBV infection
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- KSHV
- Kaposi's sarcoma–associated herpesvirus
- NaB
- sodium butyrate.
References
- 1. Young L. S., and Rickinson A. B.. 2004) Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768 10.1038/nrc1452 [DOI] [PubMed] [Google Scholar]
- 2. Longnecker R. M., Kieff E., and Cohen J. I.. 2013) Epstein-Barr virus. In Fields Virology (Knipe D. M., Howley P. M., Cohen J. I., Griffin D. E., Lamb R. A., Martin M. A., Racaniello V. R., and Roizman B., eds) pp. 1898–1959, 6th Ed., Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Nilsson K., Klein G., Henle W., and Henle G.. 1971) The establishment of lymphoblastoid cell lines from adult and from foetal human lymphoid tissue and its dependence on EBV. Int. J. Cancer 8, 443–450 10.1002/ijc.2910080312 [DOI] [PubMed] [Google Scholar]
- 4. Thorley-Lawson D. A., Miyashita E. M., and Khan G.. 1996) Epstein-Barr virus and the B cell: that's all it takes. Trends Microbiol. 4, 204–208 10.1016/S0966-842X(96)90020-7 [DOI] [PubMed] [Google Scholar]
- 5. Kenney S. C. 2007) in Reactivation and Lytic Replication of EBV (Arvin A., Campadelli-Fiume G., Mocarski E., Moore P. S., Roizman B., Whitley R., and Yamanishi K., eds) pp. 403–433, Cambridge University Press, Cambridge, UK: [PubMed] [Google Scholar]
- 6. Hong G. K., Gulley M. L., Feng W. H., Delecluse H. J., Holley-Guthrie E., and Kenney S. C.. 2005) Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 79, 13993–14003 10.1128/JVI.79.22.13993-14003.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma S. D., Hegde S., Young K. H., Sullivan R., Rajesh D., Zhou Y., Jankowska-Gan E., Burlingham W. J., Sun X., Gulley M. L., Tang W., Gumperz J. E., and Kenney S. C.. 2011) A new model of Epstein-Barr virus infection reveals an important role for early Lytic viral protein expression in the development of lymphomas. J. Virol. 85, 165–177 10.1128/JVI.01512-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato Y., Kamura T., Shirata N., Murata T., Kudoh A., Iwahori S., Nakayama S., Isomura H., Nishiyama Y., and Tsurumi T.. 2009) Degradation of phosphorylated p53 by viral protein–ECS E3 ligase complex. PLoS Pathog. 5, e1000530 10.1371/journal.ppat.1000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gulley M. L., and Tang W.. 2008) Laboratory assays for Epstein-Barr virus-related disease. J. Mol. Diagn. 10, 279–292 10.2353/jmoldx.2008.080023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dardari R., Khyatti M., Benider A., Jouhadi H., Kahlain A., Cochet C., Mansouri A., El Gueddari B., Benslimane A., and Joab I.. 2000) Antibodies to the Epstein-Barr virus transactivator protein(ZEBRA) as a valuable biomarker in young patients with nasopharyngeal carcinoma. Int. J. Cancer 86, 71–75 [DOI] [PubMed] [Google Scholar]
- 11. Kimura H., and Cohen J. I.. 2017) Chronic active Epstein-Barr virus disease. Front. Immunol. 8, 1867 10.3389/fimmu.2017.01867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang Y. H., Lee C. P., Su M. T., Wang J. T., Chen J. Y., Lin S. F., Tsai C. H., Hsieh M. J., Takada K., and Chen M. R.. 2012) Epstein-Barr virus BGLF4 kinase retards cellular S-Phase progression and induces chromosomal abnormality. PLoS ONE 7, e39217 10.1371/journal.pone.0039217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tierney R. J., Shannon-Lowe C. D., Fitzsimmons L., Bell A. I., and Rowe M.. 2015) Unexpected patterns of Epstein–Barr virus transcription revealed by a high throughput PCR array for absolute quantification of viral mRNA. Virology 474, 117–130 10.1016/j.virol.2014.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin Z., Xu G., Deng N., Taylor C., Zhu D., and Flemington E. K.. 2010) Quantitative and qualitative RNA-Seq–based evaluation of Epstein-Barr virus transcription in type I latency Burkitt's lymphoma cells. J. Virol. 84, 13053–13058 10.1128/JVI.01521-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu L., Lin Z., Wu Y., Dong J., Zhao B., Cheng Y., Huang P., Xu L., Xia T., Xiong D., Wang H., Li M., Guo L., Kieff E., Zeng Y., Zhong Q., and Zeng M.. 2016) Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 10, 61–75 10.1007/s11684-016-0436-0 [DOI] [PubMed] [Google Scholar]
- 16. Laichalk L. L., and Thorley-Lawson D. A.. 2005) Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79, 1296–1307 10.1128/JVI.79.2.1296-1307.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. zur Hausen H., O'Neill F. J., Freese U. K., and Hecker E.. 1978) Persisting oncogenic herpesvirus induced by the tumour promoter TPA. Nature 272, 373–375 10.1038/272373a0 [DOI] [PubMed] [Google Scholar]
- 18. Liu S., Liu P., Borras A., Chatila T., and Speck S. H.. 1997) Cyclosporin A-sensitive induction of the Epstein–Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. EMBO J. 16, 143–153 10.1093/emboj/16.1.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies A. H., Grand R. J., Evans F. J., and Rickinson A. B.. 1991) Induction of Epstein-Barr virus Lytic cycle by tumor-promoting and non-tumor–promoting phorbol esters requires active protein kinase C. J. Virol. 65, 6838–6844 10.1128/JVI.65.12.6838-6844.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flemington E., and Speck S. H.. 1990) Identification of phorbol ester response elements in the promoter of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64, 1217–1226 10.1128/JVI.64.3.1217-1226.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iwakiri D., and Takada K.. 2004) Phosphatidylinositol 3-kinase is a determinant of responsiveness to B cell antigen receptor-mediated Epstein-Barr virus activation. J. Immunol. 172, 1561–1566 10.4049/jimmunol.172.3.1561 [DOI] [PubMed] [Google Scholar]
- 22. Saxton R. A., and Sabatini D. M.. 2017) mTOR signaling in growth. Cell 168, 960–976 10.1016/j.cell.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M.. 2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 10.1016/S0092-8674(02)00808-5 [DOI] [PubMed] [Google Scholar]
- 24. Frias M. A., Thoreen C. C., Jaffe J. D., Schroder W., Sculley T., Carr S. A., and Sabatini D. M.. 2006) mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 16, 1865–1870 10.1016/j.cub.2006.08.001 [DOI] [PubMed] [Google Scholar]
- 25. Pearce L. R., Huang X., Boudeau J., Pawłowski R., Wullschleger S., Deak M., Ibrahim A. F., Gourlay R., Magnuson M. A., and Alessi D. R.. 2007) Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 405, 513–522 10.1042/BJ20070540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M.. 2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 10.1016/j.cub.2004.06.054 [DOI] [PubMed] [Google Scholar]
- 27. Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., and Hall M. N.. 2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128 10.1038/ncb1183 [DOI] [PubMed] [Google Scholar]
- 28. Hietakangas V., and Cohen S. M.. 2008) TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer 8, 282 10.1186/1471-2407-8-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M.. 2005) Phosphorylation and regulation of Akt/PKB by the Rictor–mTOR complex. Science 307, 1098–1101 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 30. Nichols L. A., Adang L. A., and Kedes D. H.. 2011) Rapamycin blocks production of KSHV/HHV8: insights into the anti-tumor activity of an immunosuppressant drug. PLoS ONE 6, e14535 10.1371/journal.pone.0014535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adamson A. L., Le B. T., and Siedenburg B. D.. 2014) Inhibition of mTORC1 inhibits lytic replication of Epstein-Barr virus in a cell-type specific manner. Virol. J. 11, 110 10.1186/1743-422X-11-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alsayed Y., Leleu X., Leontovich A., Oton A. B., Melhem M., George D., and Ghobrial I. M.. 2008) Proteomics analysis in post-transplant lymphoproliferative disorders. Eur. J. Haematol. 81, 298–303 10.1111/j.1600-0609.2008.01106.x [DOI] [PubMed] [Google Scholar]
- 33. Cui H., Xu B., Wu T., Xu J., Yuan Y., and Gu Q.. 2014) Potential antiviral lignans from the roots of Saururus chinensis with activity against Epstein-Barr virus lytic replication. J. Nat. Prod. 77, 100–110 10.1021/np400757k [DOI] [PubMed] [Google Scholar]
- 34. Vivanco I., and Sawyers C. L.. 2002) The phosphatidylinositol 3-kinase–Akt pathway in human cancer. Nat. Rev. Cancer 2, 489–501 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 35. Karin M. 1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486 10.1074/jbc.270.28.16483 [DOI] [PubMed] [Google Scholar]
- 36. Sparks C. A., and Guertin D. A.. 2010) Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 29, 3733–3744 10.1038/onc.2010.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takada K., and Ono Y.. 1989) Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J. Virol. 63, 445–449 10.1128/JVI.63.1.445-449.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kosowicz J. G., Lee J., Peiffer B., Guo Z., Chen J., Liao G., Hayward S. D., Liu J. O., and Ambinder R. F.. 2017) Drug modulators of B cell signaling pathways and Epstein-Barr virus lytic activation. J. Virol. 91, e00747–17 10.1128/JVI.00747-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chien Y. C., Chen J. Y., Liu M. Y., Yang H. I., Hsu M. M., Chen C. J., and Yang C. S.. 2001) Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N. Engl. J. Med. 345, 1877–1882 10.1056/NEJMoa011610 [DOI] [PubMed] [Google Scholar]
- 40. Tsai M. H., Raykova A., Klinke O., Bernhardt K., Gärtner K., Leung C. S., Geletneky K., Sertel S., Münz C., Feederle R., and Delecluse H. J.. 2013) Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 5, 458–470 10.1016/j.celrep.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 41. Lau R., Middeldorp J., and Farrell P. J.. 1993) Epstein Barr virus gene expression in oral hairy leukoplakia. Virology 195, 463–474 10.1006/viro.1993.1397 [DOI] [PubMed] [Google Scholar]
- 42. Kimura H., Hoshino Y., Kanegane H., Tsuge I., Okamura T., Kawa K., and Morishima T.. 2001) Clinical and virologic characteristics of chronic active Epstein-Barr virus infection. Blood 98, 280–286 10.1182/blood.V98.2.280 [DOI] [PubMed] [Google Scholar]
- 43. Lei K. I., Chan L. Y., Chan W. Y., Johnson P. J., and Lo Y. M.. 2000) Quantitative analysis of circulating cell-free Epstein-Barr virus (EBV) DNA levels in patients with EBV-associated lymphoid malignancies. Br. J. Haematol. 111, 239–246 10.1046/j.1365-2141.2000.02344.x [DOI] [PubMed] [Google Scholar]
- 44. Lo Y. M. 2001) Quantitative analysis of Epstein-Barr virus DNA in plasma and serum: applications to tumor detection and monitoring. Ann. N.Y. Acad. Sci. 945, 68–72 10.1111/j.1749-6632.2001.tb03865.x [DOI] [PubMed] [Google Scholar]
- 45. Geser A., de Thé G., Lenoir G., Day N. E., and Williams E. H.. 1982) Final case reporting from the Ugandan prospective study of the relationship between EBV and Burkitt's lymphoma. Int. J. Cancer 29, 397–400 10.1002/ijc.2910290406 [DOI] [PubMed] [Google Scholar]
- 46. Lin J. C., Wang W. Y., Chen K. Y., Wei Y. H., Liang W. M., Jan J. S., and Jiang R. S.. 2004) Quantification of plasma Epstein–Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N. Engl. J. Med. 350, 2461–2470 10.1056/NEJMoa032260 [DOI] [PubMed] [Google Scholar]
- 47. Andrei G., and Snoeck R.. 2011) Emerging drugs for varicella zoster virus infections. Expert Opin. Emerg. Drugs 16, 507–535 10.1517/14728214.2011.591786 [DOI] [PubMed] [Google Scholar]
- 48. Whitley R. J., Alford C. A., Hirsch M. S., Schooley R. T., Luby J. P., Aoki F. Y., Hanley D., Nahmias A. J., and Soong S. J.. 1986) Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N. Engl. J. Med. 314, 144–149 10.1056/NEJM198601163140303 [DOI] [PubMed] [Google Scholar]
- 49. Serra V., Markman B., Scaltriti M., Eichhorn P. J., Valero V., Guzman M., Botero M. L., Llonch E., Atzori F., Di Cosimo S., Maira M., Garcia-Echeverria C., Parra J. L., Arribas J., and Baselga J.. 2008) NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 68, 8022–8030 10.1158/0008-5472.CAN-08-1385 [DOI] [PubMed] [Google Scholar]
- 50. Kolev V. N., Wright Q. G., Vidal C. M., Ring J. E., Shapiro I. M., Ricono J., Weaver D. T., Padval M. V., Pachter J. A., and Xu Q.. 2015) PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 75, 446–455 10.1158/0008-5472.CAN-14-1223 [DOI] [PubMed] [Google Scholar]
- 51. Grabiner B. C., Nardi V., Birsoy K., Possemato R., Shen K., Sinha S., Jordan A., Beck A. H., and Sabatini D. M.. 2014) A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 4, 554–563 10.1158/2159-8290.CD-13-0929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gkountakos A., Pilotto S., Mafficini A., Vicentini C., Simbolo M., Milella M., Tortora G., Scarpa A., Bria E., and Corbo V.. 2018) Unmasking the impact of Rictor in cancer: novel insights of mTORC2 complex. Carcinogenesis 39, 971–980 10.1093/carcin/bgy086 [DOI] [PubMed] [Google Scholar]
- 53. Guertin D. A., Stevens D. M., Saitoh M., Kinkel S., Crosby K., Sheen J. H., Mullholland D. J., Magnuson M. A., Wu H., and Sabatini D. M.. 2009) mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 10.1016/j.ccr.2008.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data described are contained within the article.