

Abstract
G protein–coupled receptors (GPCRs) use a series of conserved microswitches to transmit signals across the cell membrane via an allosteric network encompassing the ligand-binding site and the G protein-binding site. Crystal structures of GPCRs provide snapshots of their inactive and active states, but poorly describe the conformational dynamics of the allosteric network that underlies GPCR activation. Here, we analyzed the correlation between ligand binding and receptor conformation of the α1A-adrenoreceptor, a GPCR that stimulates smooth muscle contraction in response to binding noradrenaline. NMR of [13CϵH3]methionine-labeled α1A-adrenoreceptor variants, each exhibiting differing signaling capacities, revealed how different classes of ligands modulate the conformational equilibria of this receptor. [13CϵH3]Methionine residues near the microswitches exhibited distinct states that correlated with ligand efficacies, supporting a conformational selection mechanism. We propose that allosteric coupling among the microswitches controls the conformation of the α1A-adrenoreceptor and underlies the mechanism of ligand modulation of GPCR signaling in cells.
Keywords: G protein-coupled receptor (GPCR), ligand-binding protein, conformational change, nuclear magnetic resonance (NMR), adrenergic receptor, allosteric coupling, binding mechanism, conformational equilibrium, microswitch, solution structure
Introduction
G protein–coupled receptors (GPCRs)3 are integral membrane proteins sharing a common seven-helix transmembrane domain (TMD). Conformational changes to the TMD are required to transmit the extracellular stimuli intracellularly to activate signaling pathways. Over the past 20 years X-ray crystal structures, and more recently cryo-EM structures, have revealed a plethora of structural details on how functionally different ligands interact with GPCRs and the conformational changes they induce. Most structures solved to date are of GPCRs in inactive states, bound to inverse agonists or antagonists (1). A few have been crystalized with agonist alone (1), with resultant structures similar to antagonist-bound inactive states. Complexes of GPCRs with active state-stabilizing nanobodies, engineered mini G proteins, Gα C-terminal peptide, or heterotrimeric G proteins appear necessary to stabilize agonist-bound GPCRs in active states for X-ray and cryoelectron microscopy structure determination (2). Using these tools, several active state GPCR structures have been solved (1–3), revealing conserved conformational changes that occur upon receptor activation. These include rearrangements in the ligand-binding site and a large outward movement at the cytoplasmic side of transmembrane (TM) helix 6 (TM6) to accommodate G protein binding. Although providing a wealth of structural detail of static receptor conformations, these structures generally do not provide insight into GPCR signaling complexities such as basal receptor activity, partial agonism, and biased agonism.
To address this shortfall, spectroscopic techniques, supported by molecular dynamic simulations, have given insight into the conformational dynamics that underlie the activity of a few diffusible ligand-activated GPCRs including β2-adrenergic receptor (β2-AR) (4–12), β1-adrenergic receptor (β1-AR) (13, 14), adenosine A2A receptor (A2AR) (15–18), μ-opioid receptor (μOR) (19, 20), leukotriene B4 receptor (BLT2) (21), and the M2 muscarinic acetylcholine receptor (M2R) (22). By far the most studied receptor in this regard is β2-AR, for which [13CϵH3]methionine labeling NMR (5, 8, 9), 19F NMR (6, 7, 10, 11), and electron paramagnetic resonance (10) have been applied to characterize the conformational signatures of this receptor when bound to various ligands and a G protein mimetic nanobody. These studies reveal that GPCRs are highly dynamic, sampling inactive and active conformational states, and are thought to predominantly function via a conformational selection mechanism (15, 23). Such a mechanism posits that a GPCR constantly samples various inactive and active conformations, all existing in equilibrium. Ligands preferentially bind to particular receptor states, depending on their pharmacological characteristics, thus shifting the conformational equilibrium toward these preferred states and modulating the signaling output of the system. The extracellular orthosteric ligand-binding site in adrenoceptors is connected to the intracellular G protein-binding site through a series of conserved microswitches (24–27) (Fig. 1): a central transmission switch (also called the connector region, CWXP motif or PIF motif (28)), the NPXXY switch, and the intracellular G protein-binding site, characterized by the DRY motif (or switch). How these microswitches coordinate the transmission of the extracellular signal is not clear, but molecular dynamics (MD) simulations and NMR data have led to a mechanistic description of “loose allosteric coupling” (28).
Figure 1.

Methionine residues in α1A-AR. A, the location of six methionines on a cartoon representation of α1A-AR. Methionine side chains are highlighted as red sticks. Bound adrenaline and G protein are colored in green and purple, respectively. B–D, homology models of α1A-AR-A4 in the inactive state (blue; modeled on the X-ray crystal structure of inactive β2-AR, PDB ID 5JQH) and active state (pink; modeled on the X-ray crystal structure of active β2-AR, PDB ID 3SN6) are superimposed showing inferred conformational changes that occur in the ligand-binding pocket (B), transmission switch (C), and G protein-binding site (D).
This mechanism refers to each microswitch as conformationally independent from the others, which as an active DRY motif state is not significantly dependent on an active state in the transmission switch. That said, an active state in the transmission switch does increase the probability of the DRY motif (and thus the receptor) to sample active states (thus, loose allosteric coupling) (28). Put simply, the conformational changes that occur in the microswitches are thought to drive the overall equilibrium state of the receptor system. Despite recent work, it is not well-understood how the binding of ligands such as inverse agonists influence the microswitch state equilibria to decrease basal receptor activity.
α1-Adrenoceptors (α1-ARs) comprise three Gq-coupled GPCR subtypes (α1A-, α1B-, and α1D-AR) that bind and sense the endogenous catecholamines, adrenaline and noradrenaline, to modulate a range of physiological processes. In the periphery, postsynaptic α1-AR stimulation by catecholamines mediates smooth muscle contraction, thus α1-AR antagonists and inverse agonists are clinically prescribed to treat hypertension and benign prostatic hyperplasia (BPH) (29). α1-ARs are also widely expressed in the central nervous system, but the lack of subtype-selective antibodies and ligands limits the understanding of their role in neuroplasticity and neurodegeneration (30). Currently there are no available crystal structures of an α1-AR family member, which limits the rational design of more selective compounds to probe the physiological role of α1A-adrenoreceptor (α1A-AR) in the central nervous system.
Recombinant α1A-AR expresses poorly and the resultant protein is particularly unstable when purified in detergent (31), which has hindered biochemical studies of this GPCR. Recently, we engineered an α1A-AR variant, α1A-AR-A4, that can be expressed in Escherichia coli and exhibits improved stability when purified in detergents (32). When expressed in COS-7 cells α1A-AR-A4 exhibits no signaling efficacy in response to adrenaline stimulation (32). In the present study, α1A-AR-A4 was labeled with [13CϵH3]methionine at the five naturally occurring methionine residues, providing NMR probes to assess how inverse agonists, partial agonists, and full agonists influence receptor conformational equilibria. Three of these methionines are excellent probes of the ligand-binding site and the microswitches proposed to be markers of signal transmission: Met-2926.55 (superscript denotes GPCRdb numbering (33)) is located in the ligand-binding site; Met-1153.41 is proximal to the transmission switch (Ile-1143.40, Pro-1965.50, Leu-1975.51, Phe-2816.44, and Trp-2856.48); and Met-2035.57 sits above the tyrosine of the DRY motif (Asp-1303.49, Arg-1313.50, and Tyr-1323.51). Using the inactive α1A-AR variant, α1A-AR-A4, and by reverse mutation to an active receptor (α1A-AR-A4–active) we show that for Met-1153.41 and Met-2035.57 the chemical shifts and line widths of the 13CϵH3 groups are dependent on ligand efficacy (from strong inverse agonist to full agonist), suggesting that α1A-AR activation proceeds primarily through a conformational selection mechanism.
Results
13CϵH3 Methionine labeling and NMR signal assignment
α1A-AR-A4 is a thermostabilized variant of the human α1A-AR that contains 15 amino acid substitutions over wildtype (WT) human α1A-AR (Fig. S1). Excluding Met-1, α1A-AR-A4 possesses six methionine residues, five of which are naturally occurring (Met-1153.41, Met-1454.44, Met-2035.57, Met-248ICL3, and Met-2926.55) and one, Met-802.58, is a thermostabilizing mutation previously selected for using directed evolution for detergent stability (32) (Fig. S1). Homology models of α1A-AR (Fig. 1) built on in inactive and active states of X-ray structures of β2-AR show that three of these methionines were particularly interesting as conformational probes as they are located either within the adrenaline-binding site (Met-2926.55), immediately adjacent to the highly conserved Ile-1143.40 of the transmission switch (Met-1153.41), or sitting above Tyr-1253.51 of the DRY motif within the G protein-binding site (Met-2035.57). These homology models of α1A-AR suggest that each of these regions undergo significant local rearrangements between inactive to active conformations (Fig. 1).
α1A-AR-A4 was expressed and labeled with [13CϵH3]methionine using an adapted E. coli methionine biosynthesis pathway inhibition protocol that we have previously used to generate [13CϵH3]methionine-labeled neurotensin receptor 1 (NTS1) samples labeled with 96% incorporation efficiency (34, 35). Using this method α1A-AR-A4 expressed well and could be purified, solubilized in n-dodecyl-β-d-maltopyranoside (DDM), with a yield of (0.5–1 mg/liter of culture). 40–60 μm samples of [13CϵH3]methionine-labeled α1A-AR-A4 were subsequently used to record 2D 1H-13C SOFAST-heteronuclear multiple quantum coherence (HMQC) spectra in the apo state, and in the bound states for prazosin (full inverse agonist), WB-4101 (partial inverse agonist), phentolamine (partial inverse agonist), silodosin (or KMD-3213, neutral antagonist), oxymetazoline (partial agonist), and adrenaline (full agonist) (Fig. 2 and Fig. S2). Individual [13CϵH3]methionine resonances were assigned by expressing and analyzing α1A-AR-A4 M80L, α1A-AR-A4 M115I, α1A-AR-A4 M203L, α1A-AR-A4 M248I, and α1A-AR-A4 M292I mutants in the same way. 1H-13C SOFAST-HMQC spectra enabled clear assignment of mutated methionines as the remaining five resonances in these spectra showed only small chemical shift differences in the presence of the mutation (Fig. S3). The [13CϵH3]methionine of the apo state of α1A-AR-A4 showed clear single resonances for each methyl with no significant heterogeneity (Fig. 2), in contrast to many previously studied GPCRs (5, 8, 14, 20–22). Met-1454.44 and Met-248ICL3 exhibited intense signals with 1H and 13C chemical shifts of the methyl group indicative of solvent-exposed, unrestrained methyl groups. Met-248ICL3, located within ICL3 (Fig. 1A), showed strong signal intensity most likely due to the mobility of this loop and exposure to the bulk solvent. Met-1454.44 is at the C-terminal intracellular end of TM4, predicted to be exposed on the surface of the helix (Fig. 1A) and thus also highly mobile. Met-802.58 was not unambiguously assigned (Fig. S3, A and F) as it either is significantly broadened and difficult to resolve in all receptor states or may overlap with Met-1454.44 and under some conditions with Met-2926.55 (Fig. S3E). The remaining methionines, Met-1153.41, Met-2035.57, and Met-2926.55, were readily assigned (Fig. S3, B, C, E, G, H, and J) and exhibited resolved chemical shifts for the 13CϵH3 that were sensitive to the bound ligand (Fig. 2, B–D).
Figure 2.
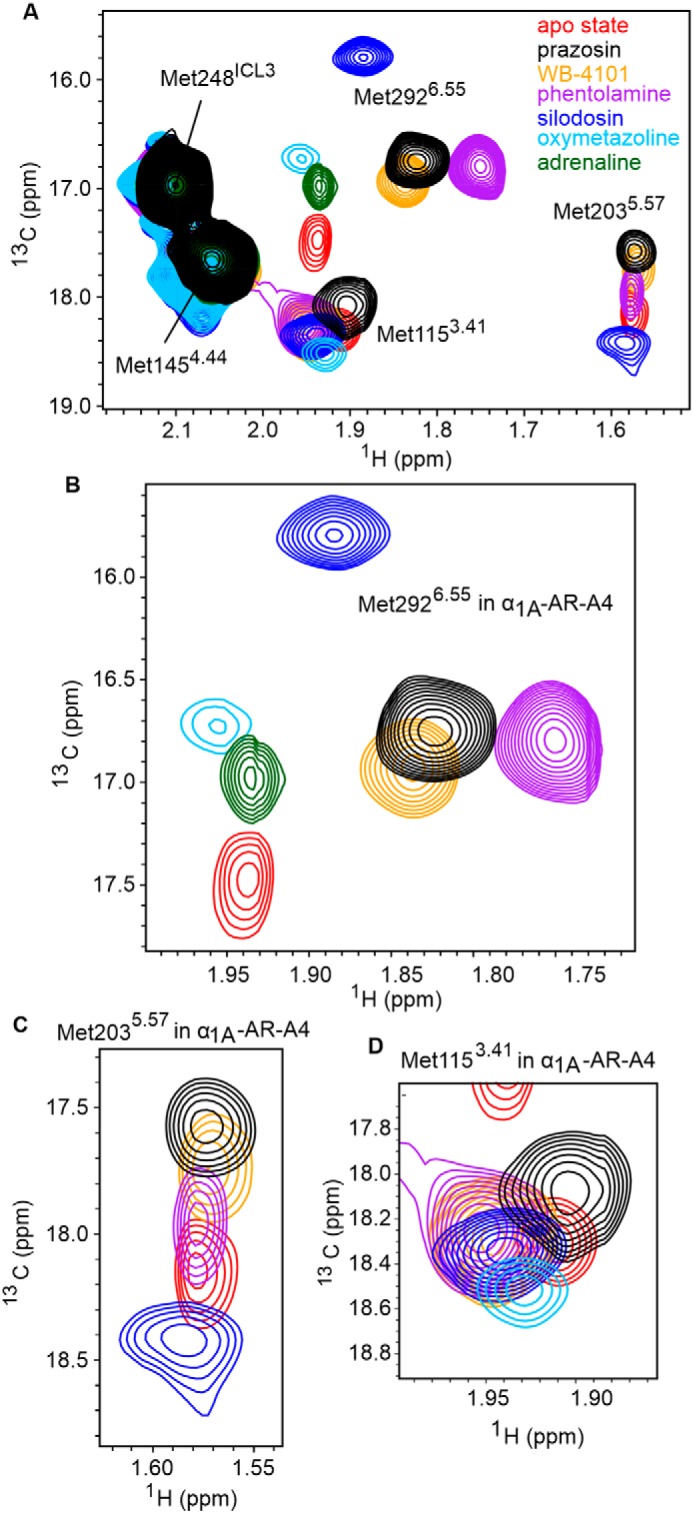
1H-13C SOFAST-HMQC spectra of α1A-AR-A4. A, overlay of 2D 1H-13C SOFAST-HMQC spectra for ([13CϵH3]Met-) α1A-AR-A4 collected in the apo state (red) and bound to prazosin (black, inverse agonist), WB-4101 (orange, inverse agonist), phentolamine (purple, inverse agonist), silodosin (blue, neutral antagonist), oxymetazoline (cyan, partial agonist), and adrenaline (green, full agonist). B, close-up of the Met-2926.55 resonance. C, close-up of the Met-2035.57 resonance. D, close-up of the Met-1153.41 resonance. Spectra were acquired on ∼50 μm α1A-AR-A4 dissolved in 0.02–0.1% DDM micelle, pH 7.5, and 25 °C.
Based on homology models, Met-2926.55 projects into the orthosteric ligand-binding pocket (Fig. 1A) and mutational studies support a role for this residue in ligand binding (36). Thus, the 13CϵH3 chemical shifts of Met-2926.55 likely reflect a direct interaction with chemical groups of each ligand. Interestingly, the resonance intensities of Met-2926.55 increased in the presence of antagonists and inverse agonists relative to the apo state (Fig. 2B), indicating that binding of these ligands reduces conformational dynamics in the orthosteric binding site. Met-1153.41 and Met-2035.57 are distant from the orthosteric site, but both the chemical shifts and line widths of their 13CϵH3 groups were sensitive to ligand binding (Fig. 2, C and D), likely reflecting receptor conformational changes in the transmission switch and G protein-binding site, respectively (Fig. 1, C and D). The 1H chemical shift of the methyl of Met-2035.57 was shifted upfield from typical small-peptide positions (2.1 ppm) to 1.58 ppm in agreement with our models, which predict ring-current induced effects from Tyr-1253.51 of the DRY motif (Fig. S4). Met-2035.57 therefore serves as a probe of conformational change within this region. Indeed, the resonances of the 13CϵH3 of Met-2035.57 exhibited a significant linear chemical shift change depending on which ligand was bound, demonstrating that allosteric coupling between the ligand-binding site and the G protein-binding site is retained in the inactive α1A-AR-A4 in solution. Such a linear chemical shift change is also expected for ligands modulating the receptor state via conformational selection. We postulate that the Met-2035.57 signal reflects the average, equilibrium signal, between inactive and active states undergoing fast exchange. Inverse agonists preferentially bound to inactive states, shifting the Met-2035.57 equilibrium to an upfield position (inactive state) compared with the apo state receptor, which can sample active-like states to a certain degree.
We were interested to see if an opposite trend could be observed for receptor agonists, which we hypothesized would shift the position of the Met-2035.57 resonance downfield. For α1A-AR-A4, however, the binding of the full agonist adrenaline to α1A-AR-A4 resulted in complete line broadening of the Met-1153.41 and Met-2035.57 resonances despite the promotion of a distinct chemical shift for Met2926.55 in the binding site. Binding of the partial agonist oxymetazoline resulted in substantial broadening of Met-2035.57 and Met-2926.55, but not Met-1153.41. The loss of these chemical shifts upon agonist binding was likely due to the significantly weaker agonist affinities at α1A-AR-A4 compared with unmutated, WT α1A-AR, as a result of the F312L-stabilizing mutation (32). Thus, NMR experiments were repeated on α1A-AR-A4 (L312F), for which agonist affinities were largely restored to that of WT α1A-AR (Fig. S5 and Table S1) (32).
Agonist-induced chemical shifts of Met-1153.41 and Met-2035.57 resonances
Despite the reduced thermostability of α1A-AR-A4 (L312F) (32), we were able to [13CϵH3]methionine label and record 1H-13C SOFAST-HMQC spectra for this receptor in the apo state and bound to adrenaline (full agonist), phenylephrine (full agonist), A-61603 (full agonist), and oxymetazoline (partial agonist) in addition to the inverse agonists and neutral antagonists tested on α1A-AR-A4 (Fig. 3A). Overall, the 1H-13C SOFAST-HMQC spectra of the apo, antagonist, and inverse agonist-bound states of α1A-AR-A4 (L312F) were similar to those of α1A-AR-A4. Again, single resonances for the [13CϵH3]methionine groups of α1A-AR-A4 (L312F) were observed for all ligands. The chemical shifts of Met-2926.55 induced by each ligand in α1A-AR-A4 (L312F) were slightly different to those of α1A-AR-A4, most likely due to orthosteric binding site changes after the L312F reversion. Inverse agonist binding increased the intensity of the Met-2926.55 resonance in α1A-AR-A4 (L312F), as was seen with α1A-AR-A4; whereas the neutral antagonist silodosin significantly decreased the peak intensity and the partial agonist oxymetazoline and full agonist A-61603 highly broadened the resonance of Met-2926.55 in α1A-AR-A4 (L312F) (Fig. S6).
Figure 3.
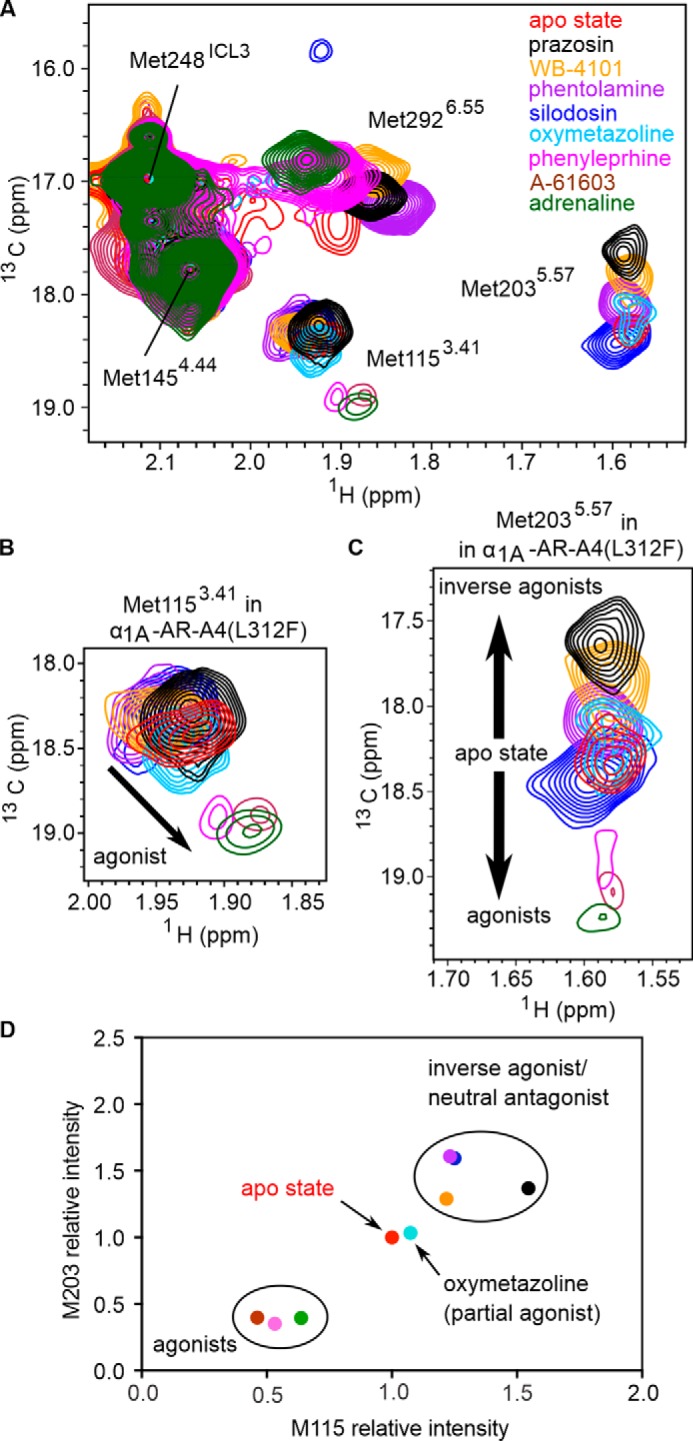
Ligand efficacy-dependent chemical shifts of Met-1153.41 and Met-2035.57 resonances. A, overlay of 2D 1H-13C SOFAST-HMQC spectra for ([13CϵH3]Met-) α1A-AR-A4 (L312F) in the apo state (red) and bound to ligands: prazosin (black, inverse agonist), WB-4101 (orange, inverse agonist), phentolamine (purple, inverse agonist), silodosin (blue, neutral antagonist), oxymetazoline (cyan, partial agonist), phenylephrine (magenta, full agonist), A-61603 (maroon, full agonist), and adrenaline (green, full agonist). B, close-up of the Met-1153.41 resonance in α1A-AR-A4 (L312F). C, close-up of the Met-2035.57 resonance in α1A-AR-A4 (L312F). The spectra for adrenaline, A-61603, and phenylephrine are plotted at a level 1.8-times lower than the main figure. D, normalized peak intensities of Met-1153.41 and Met-2035.57 of α1A-AR-A4 (L312F) show differences between agonists, antagonists, and partial agonists. Ligands are colored as listed above. Spectra were acquired on ∼50 μm α1A-AR-A4 (L312F) dissolved in 0.02–0.1% DDM micelle, pH 7.5, and 25 °C.
The recovered agonist affinity for α1A-AR-A4 (L312F) allowed the measurement of 1H-13C SOFAST-HMQC spectra where we were confident of full receptor-agonist saturation. Binding of the full agonist adrenaline to α1A-AR-A4 (L312F) produced a similar Met-2926.55 chemical shift to that seen with α1A-AR-A4 (Fig. 3A), and also weak peaks were now observed for Met-1153.41 and Met-2035.57 (Fig. 3, B and C), which were completely broadened in adrenaline-bound α1A-AR-A4. Importantly, the binding of all agonists, adrenaline, phenylephrine, and A-61603 to α1A-AR-A4 (L312F) induced distinct chemical shift and line broadening changes to Met-1153.41 and Met-2035.57 compared with neutral antagonists and inverse agonists (Fig. 3, B and C). The agonist-induced Met-1153.41 resonances cluster together as potentially indicative of an active transmission switch conformation (Fig. 3B). Binding of the partial agonist oxymetazoline induced a chemical shift of Met-1153.41 falling between the inverse agonist and full agonist clusters, consistent with partial agonists promoting a weaker shift in the inactive-active transmission switch state equilibrium. The linear change in the Met-2035.57 chemical shift position upon inverse agonist binding seen with α1A-AR-A4 was retained in α1A-AR-A4 (L312F), but as hypothesized, agonist binding promoted opposite, downfield resonance shifts in the 13C dimension along the same vector (Fig. 3C). This change in [13CϵH3]methionine chemical shift in the 13C dimension reflects a change in the χ3 dihedral angle. The 13C chemical shift dependence of this angle is about 19 ppm for trans and 16 ppm for ±gauche (37). For the apo and antagonist states the chemical shift of 18.25 to 18.5 ppm suggests a trend toward trans, whereas in the full-inverse agonist state the resonance shifts up-field to 17.7 ppm, indicative of an averaging between gauche and trans. Consistent with our homology models (Fig. S4) for full agonist a further downfield shift between 19 and 19.25 ppm infers an increase in the trans conformer. Interestingly, the partial agonist oxymetazoline induced a small upfield 13Cϵ shift of Met-2035.57, similar to the inverse agonist phentolamine. The fact that full agonists induced Met-2035.57 chemical shifts to move in the opposite direction to inverse agonists suggests an equilibrium shift away from inactive to active conformational states of the DRY motif. Furthermore, the resonance intensities of both Met-1153.41 and Met-2035.57 in α1A-AR-A4 (L312F), relative to the ligand-insensitive Met-1454.44 resonance, were weakened upon agonist binding compared with the intensity increases seen with antagonists and inverse agonists (Fig. 3D). The intensities of Met-1153.41 and Met-2035.57 upon binding of the partial agonist oxymetazoline fell between the antagonist- and agonist-induced intensities. The behavior of the 13CϵH3 of Met-1153.41 and Met-2035.57 is consistent with the current concept that agonists increase conformational heterogeneity in GPCRs, where agonists increase microsecond timescale transitions to active receptor states, to increase the probability of engaging and activating effector proteins (5, 8, 10, 14, 15, 23).
Chemical shift changes of Met-2035.57 correlate with ligand efficacy
In mammalian cells, α1A-AR exhibits basal activity in the absence of bound ligands (38). Such basal activity is unaffected by the binding of neutral antagonists but is reduced by the binding of inverse agonists to the receptor. In the case of α1A-AR, by probing the ability of various antagonists to reduce the signaling of a constitutively active receptor mutant, the rank order of inverse agonist efficacies has been found to be: prazosin (strongest); WB-4101, phentolamine (weakest), and silodosin being a neutral antagonist (38). To understand how the NMR signals of Met-2035.57 in α1A-AR-A4 (L312F) relate to receptor conformational equilibria, the changes to the chemical shifts for the 13CϵH3 of Met-2035.57 were plotted against the previously published relative efficacy values for the inverse agonists, revealing a strong linear correlation (R2 = 0.99, Fig. 4A). To test if this correlation is retained when probing inverse agonism at the WT α1A-AR, we determined the relative inverse agonist efficacies of these ligands using a NanoBiT split luciferase assay (39). In this assay the 18-kDa Large BiT (LgBiT) fragment was fused to the N terminus of Gαq and the 1.3-kDa Small BiT (SmBiT) was fused to the N terminus of Gγ2. When co-expressed with Gβ1, the formation of a Gαq(LgBiT)-Gβ1-Gγ2(SmBiT) heterotrimer results in bright luminescence. GPCR-induced stimulation of this G protein complex causes dissociation of the heterotrimer and thus reduction in luminescence output, whereas inhibition of basal GPCR activation would be predicted to increase luminescence output. COS-7 African green monkey kidney cells stably expressing WT α1A-AR were transfected with Gαq(LgBiT), Gβ1, and Gγ2(SmBiT) encoding expression plasmids, incubated with luminescence substrate, and then treated with various α1A-AR ligands, while monitoring cellular luminescence. A-61603 induced α1A-AR activation led to heterotrimer dissociation of the Gαq(LgBiT)-Gβ1-Gγ2(SmBiT) complex and thus a reduction in luminescence output (Fig. 4B). Inverse agonists on the other hand reduced basal activation of α1A-AR, maintaining the Gαq(LgBiT)-Gβ1-Gγ2(SmBiT) complex leading to increased luminescence output from the cells (Fig. 4B). The specificity of these responses was probed by conducting the same experiments on COS-7 cells not expressing α1A-AR (Fig. S7, A–C). The observed changes in luminescence after α1-AR ligand treatments were specific to α1A-AR expressing cells except for WB-4101, which induced a short (5 min) increase in luminescence in the control cells (Fig. S7A). To exclude this nonspecific effect the net luminescence change for each sample group was calculated as the area under the luminescence curves between 5 and 10 min after ligand addition. A strong linear correlation was found between the chemical shift changes for the 13CϵH3 of Met-2035.57 in α1A-AR-A4 (L312F) and the net luminescence increase generated by each inverse agonist over the 5-min period in the WT α1A-AR-expressing cells (R2 = 0.72, Fig. 4C). Importantly, the Met-2035.57 chemical shift positions of α1A-AR-A4 (L312F) did not correlate with the affinity of these antagonists for α1A-AR (Fig. 4D), demonstrating that the differences in chemical shift were not due to varying receptor occupancy. Furthermore, no correlation was seen between the [13CϵH3]Met-2035.57 chemical shift changes of α1A-AR-A4 (L312F) and the net luminescence changes in COS-7 cells not expressing WT α1A-AR (Fig. S7B). Critically, the correlation between chemical shift changes of [13CϵH3]Met-2035.57 in α1A-AR-A4 (L312F) and WT α1A-AR–specific luminescence increases in the NanoBiT assay remained when the analysis window was extended to include the full 10 min after ligand addition (Fig. S7D).
Figure 4.
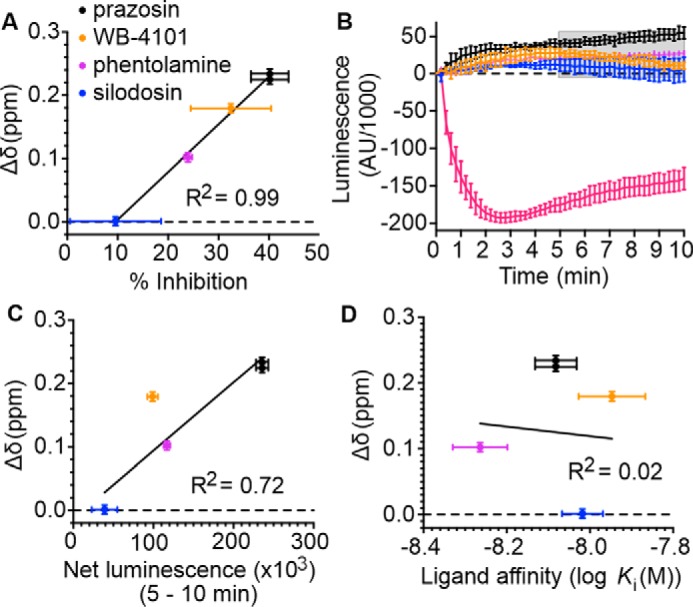
Correlation between the chemical shift positions of the 13CϵH3 in Met-2035.57 and inverse agonists efficacy. A, linear regression analysis of the average chemical shift differences (Δδ) for the 13CϵH3 of Met-2035.57 in α1A-AR-A4 (L312F) when bound to prazosin (black circles), WB-4101 (orange circles), and phentolamine (purple circles) compared with silodosin (blue circles) and the published efficacy of each ligand in reducing the signaling of a constitutively active mutant of α1A-AR (38). Published data were extracted using WebPlotDigitizer (RRID: SCR_013996). Testing the resultant equation against the null hypothesis of a slope of zero resulted in a p value of <0.0001. B, NanoBit G protein activity assay demonstrating inverse agonism of prazosin, WB-4101, phentolamine, and silodosin at WT α1A-AR–expressing COS-7 cells. Each of these inverse agonist experiments were repeated in three independent biological replicate experiments, with the mean ± S.E. of the resultant luminescence plotted for each time point. To demonstrate the response from an agonist, A-61603 treatment was performed in two independent biological replicate experiments. Each biological replicate comprised three technical replicates measured in parallel. The gray shaded region indicates where the area under each biological replicate curve was calculated for C. C, linear regression analysis of the average chemical shift differences (Δδ) for the 13CϵH3 of Met-2035.57 in α1A-AR-A4 (L312F) and the increase in luminescence seen in the NanoBit assay for each inverse agonist and neutral antagonist. Ligands are colored as listed above and the p value testing against a slope of 0 was 0.011. D, linear regression analysis of the average chemical shift differences (Δδ) for the 13CϵH3 of Met-2035.57 in α1A-AR-A4 (L312F) and the affinities of each inverse agonist and neutral antagonist. Ligands are colored as listed above and the p value testing against a slope of 0 was 0.89. In A, C, and D, Δδ is plotted for two independent titrations of prazosin and silodosin, and single experiments for WB-4101 and phentolamine. Average chemical shift differences (Δδ) were normalized using the equation Δδ = [(Δδ1H)2 + (Δδ13C/3.5)2]0.5 and error was calculated by the formula |[Δδ1H × R1H + Δδ13C × R13C/(3.5)2]|/Δδ, where R1H and R13C are the digital resolutions in ppm in the 1H and 13C dimensions, respectively (5).
Improving signaling competency in α1A-AR-A4
When expressed in COS-7 cells, α1A-AR-A4 is incapable of stimulating cellular increases in inositol phosphate 1 (IP1) in response to adrenaline binding, or activation of a cAMP response element (CRE) reporter gene after treatment with another α1-AR agonist, phenylephrine (32). To ensure biological relevance of our NMR studies we thus sought α1A-AR-A4 back-mutants that were able to stimulate canonical signaling pathways in mammalian cells upon agonist treatment. Seven thermostabilizing mutations within the TMD of α1A-AR-A4 were back-mutated (Y67N, M80L, A127G, F151W, K322N, L327P, and Y329S) as single changes or in combinations, and screened for phenylephrine- and oxymetazoline-induced signaling with an IP1 assay in transfected COS-7 cells (Fig. S8A). Although the back-mutant, α1A-AR-A4 (Y67N, M80L, K322N, L327P, and Y329S) was able to facilitate significant oxymetazoline-induced cellular accumulation of IP1 compared with α1A-AR-A4 and WT α1A-AR (Fig. S8A) it expressed poorly in bacteria. The back-mutant α1A-AR-A4 (Y67N, K322N), termed α1A-AR-A4–active, however, was able to stimulate IP1 accumulation in response to both phenylephrine and oxymetazoline treatment (Fig. S8A) and it expressed well in bacteria. Importantly, α1A-AR-A4 contains the N322K-stabilizing mutation in the NPXXY switch(25), which is hypothesized to form a stabilizing salt bridge with Asp-722.50 to lock the NPXXY switch in an inactive and stable state. We thus expected that reversion of this mutation (K322N) would restore the function of the NPXXY switch and the signaling activity of α1A-AR-A4. Interestingly, the Y67N mutation was required on top of K322N to restore signaling activity in α1A-AR-A4–active. Asn-672.45 is distant from the NPXXY switch and its importance is not clear.
Using an intracellular calcium mobilization assay, α1A-AR-A4–active was able to be activated by the full agonists adrenaline and A-61603, as well as the partial agonists oxymetazoline and PF-3774076 (Fig. S8, B–E). The affinity of QAPB for α1A-AR-A4–active was retained upon purification of the receptor in DDM (Fig. S9A). Competition binding assays revealed, however, that the affinities of agonists for α1A-AR-A4–active (Fig. S9B and Table S1) were weaker than WT α1A-AR due to the F312L stabilizing mutation, but stronger than at α1A-AR-A4 (32). When purified in DDM α1A-AR-A4–active was significantly less stable than α1A-AR-A4 (Fig. S9C) and thus back-mutation of F312L to recover agonist potency was not pursued as it was deemed unlikely that the resultant receptor would be stable enough for NMR experiments.
α1A-AR-A4–active was labeled with [13CϵH3]methionine and 2D 1H-13C SOFAST-HMQC spectra were acquired as above (Fig. 5, A and D). Overall the ligand-perturbed chemical shifts of the [13CϵH3]methionine resonances in α1A-AR-A4–active were similar to those in α1A-AR-A4 and α1A-AR-A4 (L312F), except for several key differences with the Met-1153.41 and Met-2035.57 resonances. We acquired spectra of four independent preparations of apo α1A-AR-A4–active (four biological replicates) and found that in the absence of bound ligand the data were not easily reproduced (Fig. S10A). The well-resolved Met-2035.57 varied between an intense peak, two peaks of similar intensity, or a peak of weak intensity. Met-1153.41 persisted as a split peak, although the two components varied in intensity. Importantly, in the presence of the most potent inverse agonist, prazosin, the resonances of Met-1153.41 and Met-2035.57 were single peaks, and regardless of sample preparation, exhibited the same chemical shifts. The [13CϵH3]Met-1153.41 resonance, as perturbed by prazosin, aligned approximately with the upfield component of the resonance for apo α1A-AR-A4–active (Fig. S10B). In contrast, upon titration with the neutral antagonist, silodosin, the peaks of Met-1153.41 also collapsed to a single resonance with identical chemical shifts, but now aligned best with the downfield component of apo α1A-AR-A4–active (Fig. S10B). For the two partial inverse agonists, WB-4101 and phentolamine, the 13CϵH3 resonance of Met-1153.41 was a single resonance, positioned midway between the “prazosin” (upfield) and “silodosin” (downfield) peaks (Fig. 5E). These trends for ligand efficacy were present in α1A-AR-A4 and α1A-AR-A4 (L312F), but were not as distinct as now observed for α1A-AR-A4–active, and notably the apo states for α1A-AR-A4 and α1A-AR-A4 (L312F) did not show two discrete peaks for Met-1153.41. Such apo state sample-to-sample heterogeneity may suggest the presence of misfolded contaminants, but upon the addition of prazosin or silodosin each of these samples gave identical spectra (Fig. S10A), supporting the binding competency of the α1A-AR-A4–active samples. The diversity of apo state spectra likely reflects diversity of conformational states of similar free energy. The addition of agonist again resulted in a single resonance for [13CϵH3]Met-1153.41 that shifts upfield in 1H and downfield in 13C (Fig. 5E). The trend in shifts of these resonances, however, suggests they follow in a linear manner evolving from the downfield (basal) signal of the apo state and reflects the selection of the active-like state.
Figure 5.

1H-13C SOFAST-HMQC spectra of α1A-AR-A4–active. A, Overlay of 2D 1H-13C SOFAST-HMQC spectra of ([13CϵH3]Met-) α1A-AR-A4–active bound to prazosin (black, inverse agonist), WB-4101 (orange, inverse agonist), phentolamine (purple, inverse agonist), and silodosin (blue, neutral antagonist). B, close-up of the Met-2035.57 resonance. C, linear regression analysis of the average chemical shift differences (Δδ) for the 13CϵH3 of Met-2035.57 in α1A-AR-A4–active when bound to prazosin (black circles), WB-4101 (orange circles), and phentolamine (purple circles) compared with silodosin (blue circles) and the increase in luminescence seen in the NanoBit assay with α1A-AR-A4–active expressing COS-7 cells treated with the same antagonist (from Fig. S11A). Testing the resultant equation against the null hypothesis of a slope of zero resulted in a p value of 0.0041. D, overlay of 2D 1H-13C SOFAST-HMQC spectra of ([13CϵH3]Met-) α1A-AR-A4–active bound to prazosin (black, inverse agonist), silodosin (blue, neutral antagonist), oxymetazoline (cyan, partial agonist), PF-3774076 (magenta, partial agonist), A-61603 (maroon, full agonist), and adrenaline (green, full agonist). E, close-up of the Met-1153.41 resonance. F, linear regression analysis of the average chemical shift differences (Δδ) for the 13CϵH3 of Met-1153.41 in α1A-AR-A4–active when bound to oxymetazoline (cyan circles), PF-3774076 (pink circles), A-61603 (dark red circles), adrenaline (green circles), and silodosin (blue circles) and the efficacy of each agonist in triggering Ca2+ mobilization in α1A-AR-A4–active expressing COS-7 cells (from Fig. S8, B–E). Testing the resultant equation against the null hypothesis of a slope of zero resulted in a p value of 0.0154. C and F, Δδ are plotted for two independent titrations of prazosin, silodosin, and oxymetazoline, and single experiments for other ligands. In A, B, D, and E, spectra were acquired on ∼50 μm α1A-AR-A4–active dissolved in 0.02–0.1% DDM micelle, pH 7.5, and 25 °C. Average chemical shift differences (Δδ) were normalized using the equation Δδ = [(Δδ1H)2 + (Δδ13C/3.5)2]0.5 and errors were calculated by the formula |[Δδ1H × R1H + Δδ13C × R13C/(3.5)2]|/Δδ, where R1H and R13C are the digital resolutions in ppm in the 1H and 13C dimensions respectively (5).
A major difference between the spectra of α1A-AR-A4 and α1A-AR-A4–active, however, was significantly increased line broadening of the Met-2035.57 signal of α1A-AR-A4–active in the apo state (Fig. S10A) and when bound to antagonists (Fig. 5B). This broadening suggests that the DRY motif near the G protein-binding site of α1A-AR-A4–active is more dynamic compared with α1A-AR-A4, consistent with a receptor that more readily transitions between inactive and active-receptor states. Importantly, similar to α1A-AR-A4 and α1A-AR-A4 (L312F) variants, the 13CϵH3 of Met-2035.57 shows an efficacy-dependent linear 13CϵH3 chemical shift change in the presence of inverse agonist and neutral antagonist, trending to an upfield 13C position (χ3 of ±gauche) for the more potent inverse agonist (Fig. 5, A and B). Unexpectedly, the addition of silodosin (neutral antagonist) resulted in a significant 13C downfield shift to near 19.5 ppm consistent with a trans χ3 angle for Met-2035.57. Furthermore, similar to α1A-AR-A4, the Met-2035.57 13CϵH3 resonance of α1A-AR-A4–active was near completely broadened in the presence of agonists.
NanoBiT G protein activity assays were performed on COS-7 cells expressing α1A-AR-A4–active to determine relative inverse agonist efficacies. The inverse agonists reduced basal Gq activity in α1A-AR-A4–active in a similar way as WT α1A-AR expressing cells (Fig. 4A and Fig. S11A). The net luminescence change induced by each inverse agonist at α1A-AR-A4–active expressing cells correlated well, in a linear fashion, with the 13CϵH3 chemical shift changes of Met-2035.57 that each ligand induced at purified α1A-AR-A4–active, when analyzed over two separate time periods (Fig. 5C and Fig. S11B). Interestingly, the agonist-induced chemical shift changes of [13CϵH3]Met-1153.41 showed a linear correlation with the efficacy of each agonist in Ca2+ mobilization assays on α1A-AR-A4–active expressing COS-7 cells (Fig. 5F) although the partial agonist PF-3774076 was a notable outlier. Overall, these cell-based assays with the ligand efficacy-correlated chemical shift changes of Met-1153.41 and Met-2035.57 clearly demonstrate that a conformational selection mechanism underlies receptor function in cells.
Discussion
Recent spectroscopic studies have demonstrated that different classes of GPCR ligands distinctly alter the population of receptor states within the GPCR conformational equilibrium (23). GPCR conformational changes are driven by defined structural changes in the microswitches (24–26, 28) (Fig. 1) and, thus, how particular ligands affect the GPCR microswitch states likely underlies their pharmacological output as inverse, partial, full, or biased agonists. Observing these effects, however, remains challenging. α1A-AR was one of the first GPCRs to be cloned and pharmacologically characterized (40) and is clinically targeted with agonists as nasal decongestants and antagonists for hypertension and BPH. Despite the importance of this receptor there are currently no three-dimensional structures of α1A-AR, reflecting the inherent instability of this protein. Here, we demonstrate that prototypical ligands modulate the conformational equilibrium, as measured at the microswitches, of α1A-AR in defined and predictable ways by [13CϵH3]methionine labeling α1A-AR variants and monitoring the 1H and 13C chemical shifts of these methyl resonances in the presence of ligands of different efficacy.
It is well-accepted that the NMR signals of methionine methyl groups are sensitive to the local environment and the conformation of the methionine side chain (5, 8). Many of the conclusions made in this study rely on Met-1153.41, a probe for the conformation of the transmission switch, and Met-2035.57 as a probe of the DRY motif that signifies intracellular TMD rearrangements for G-protein binding. In our model of α1A-AR, Met-2035.57 sits over Tyr-1253.51 of the DRY motif but is distant from Arg-1243.50, which is expected to undergo significant rotameric changes within this motif (2) (Fig. 1D). Met-1153.41 is sequential to the Ile-1143.40 in TM3 but it points away and is distant to the transmission switch residue Phe-2816.44 located on TM6, which is expected to undergo significant rotameric changes (Fig. 1C). Although, in the thermostabilized inactive α1A-AR-A4 mutant the residues of the transmission switch and DRY motif are retained, the asparagine of a third microswitch, the NPXXY motif, is mutated to lysine, which likely forms a salt bridge with Asp-722.50 to lock this switch in an inactive state. The transmission switch and NPXXY motif are proximal to each other and therefore Met-1153.41, whereas distant to NPXXY, is likely to be sensitive to conformational changes involving both switches. In the reported active-state GPCR structures, three conserved residues (Arg3.50 of the DRY motif, Tyr7.53 of the NPXXY motif, and Tyr5.58) adopt near identical positions and connect these microswitches through water-mediated hydrogen bonds (1, 2). Furthermore, in our model of active α1A-AR, which is based on structures of β2-AR, Arg-1243.50 of the DRY motif is in contact with Tyr-3267.53 of the NPXXY motif (Fig. 1D).
In our NMR experiments for all ligands the 13CϵH3 group of both Met-1153.41 and Met-2035.57 show significant directional chemical shift and line width changes that are correlated with ligand efficacy, not affinity. As a distinct peak is observed for the addition of each ligand the chemical shift likely reflects an average population exchanging on a fast to intermediate timescale. The chemical shift differences, however, reflect a shift in the equilibrium, and specifically for the 13CϵH3 of Met-2035.57, from a χ3 of a gauche-trans average (inverse agonist) toward a trans (agonist) average (Fig. 6A). An NMR study using 15N-labeled, thermostabilized β1-AR observed substantial ligand efficacy-correlated backbone chemical shift changes for Val-2265.57, which is in the same position as Met-2035.57 in α1A-AR (13). The authors speculated that these changes were caused by TM5 bending toward the active receptor state (13), an idea that may also apply to α1A-AR and other GPCRs. Here, the linear chemical shift changes of Met-2035.57, and to a lesser degree Met-1153.41, in response to ligands of different efficacy is strong evidence that agonists activate α1A-AR via a conformational selection mechanism. The line broadening of Met-1153.41 and Met-2035.57 upon agonist binding supports an efficacy-driven shift in dynamics, and thereby the equilibrium of conformational states, communicated allosterically by the microswitches and sensed by these methionine residues (Fig. 6).
Figure 6.

How ligands modulate the conformational landscape of the α1A-AR microswitches. A, cartoon representations of α1A-AR in the inverse agonist-bound, apo, and agonist-bound states. The three probe methionines, Met-2926.55 (binding site), Met-1153.41 (transmission switch), and Met-2035.57 (DRY microswitch) are highlighted with red sticks and the labeled methyl group in green. The arrows labeled χ3 illustrate the ligand-induced changes to the equilibrium between the trans (t) and gauche (g) χ3 dihedral angle of Met-2035.57. Other arrows indicate how different ligands alter the conformation equilibria of Met-2926.55, Met-1153.41, and TM6. Hypothetical free energy landscape diagrams of the three microswitches in (b) the inactive α1A-AR-A4 receptor compared with (C) α1A-AR-A4–active. (a) indicates the proposed inactive states, (b) represents basal states, and (c) represents active states of the microswitches.
For our signaling incompetent receptors, α1A-AR-A4 and α1A-AR-A4 (L312F), the NPXXY microswitch has been mutated (N322K) in a way that would bias the NPXXY switch toward inactive states (Lys-322–Asp-72 salt bridge). A consequence of this mutation is that for α1A-AR-A4 (L312F) the DRY motif probe, Met-2035.57, gives relatively intense chemical shifts in the apo and antagonist-bound states (conformational equilibrium biased toward inactive states) (Fig. 6B). On restoration of the NPXXY microswitch in α1A-AR-A4–active, however, the Met-2035.57 chemical shifts broaden and shift toward the agonist-bound position (as defined for α1A-AR-A4 (L312F)), even with neutral antagonist bound. Therefore, restoring the NPXXY microswitch enables the DRY motif of α1A-AR-A4–active to more readily sample active-like states. The full trans (19.5 ppm in 13C) populated by silodosin indicates that we may observe the full-active state, although previous studies showed that the full-active state is only populated in the presence of both agonist and nanobody (8, 10, 14, 19, 22). Our two methionine probes, Met-1153.41 and Met-2035.57 retain similar ligand-induced behavior in α1A-AR-A4–active compared with α1A-AR-A4 and α1A-AR-A4 (L312F), where the latter are both essentially inactive. The chemical shift and line-broadening trends of Met-1153.41 and Met-2035.57 suggest that the transmission switch and DRY motif, in the presence of an inactive NPXXY motif, can independently adopt conformations representative of active and inactive states. To fully adopt the conformational signatures of an active receptor, a functional NPXXY motif is required (in α1A-AR-A4–active) thus increasing the dynamics of the transmission switch and DRY motif, suggesting that the interdependence of the three microswitches is a consequence of their dynamic nature, and that this is required for full receptor function.
Although the striking linear chemical shift dependence for the 13CϵH3 of Met-2035.57 on ligand efficacy is consistent with a smooth change in equilibria from inactive to active, linear trends for 13CϵH3 of Met-1153.41 were less clear. In the inactive variants α1A-AR-A4 and α1A-AR-A4 (L312F) the resonance for apo, inverse agonist, and neutral antagonist shows little variation, but clear chemical shift changes and broadening are observed for agonists. The most distinct changes for the 13CϵH3 of Met-1153.41 were for α1A-AR-A4–active, where in the apo state two peaks were consistently observed, suggesting slow exchange (> millisecond) between two distinct states, to which we attribute to restoring the NPXXY microswitch. On the basis of chemical shift, we propose that the upfield peak of apo α1A-AR-A4–active represents fully inactive receptor (state A in Fig. 6), expected for full inverse agonists, and the downfield peak with a basal state receptor that is stabilized by the neutral antagonist (state B in Fig. 6). This downfield “basal” peak shows approximate linear efficacy-dependent chemical shift changes with agonist titrations. Therefore, the 13CϵH3 of Met-1153.41 reflects three states, inactive (inverse agonist), an intermediate (basal), and active states (state(s) C in Fig. 6), where the latter progressively shift from partial to full agonist states. Interestingly, in a [13CϵH3]methionine-labeled study on the M2R, Met-1123.41, which is equivalent to Met-1153.41 in α1A-AR, did not display efficacy-dependent chemical shift changes. In the presence of ligands, however, M2R Met-1123.41 was resolved as two separate resonances, consistent with a slow exchanging microswitch (22) and may highlight some differences between how different rhodopsin family GPCRs function.
In this NMR study, by starting with a signaling incompetent variant of α1A-AR and subsequently restoring signaling activity through back-mutations, we were able to study the functional dynamics of the key GPCR microswitches and how different ligands modulate this. Our NMR data for the transmission and DRY microswitches revealed ligand efficacy-dependent changes to the microswitch conformational equilibria, supporting a conformational selection mechanism for α1A-AR modulation. This and the agonist-driven line broadening for both microswitches suggest similar mechanistic actions on the different microswitches, supporting ligand-driven allosteric communication between the microswitches. MD simulations (41) of β2-AR suggest that these microswitches behaved independently of each other, with only loose allosteric coupling. Although this may be true over the relatively short timescales of MD, we believe that over the course of an NMR experiment such loose allosteric coupling culminates in significant coupled shifts to the microswitch conformations and that this is likely how ligands modulate GPCR signaling in cells.
Experimental procedures
α1A-AR constructs
The α1A-AR-A4 variant is a thermostabilized human α1A-AR, containing 15 stabilizing mutations (32). Met-802.58 in the spectra was introduced through the stabilization process in lieu of the naturally occurring amino acid leucine. As compared with WT, the C termini of the α1A-AR-A4 variant was modified by truncation at Ser-351 and addition of a deca-His tag to facilitate purification (Fig. S1). For expression, the α1A-AR variants sequences were subcloned into the pQE30-derived vector, pDS15, with a maltose-binding protein and a methionine-free monomeric ultrastable GFP (−Met-muGFP) (42) attached, respectively, to the N and C termini of the receptor via HRV 3C protease-cleavage sites. For the purpose of KingFisher-binding assays, α1A-AR variants were subcloned into a similar vector, pDS11, in which muGFP was replaced with mCherry because the excitation and emission wavelengths of fluorescent QAPB were overlapping with those of GFP. The final sequence of α1A-AR-A4 after purification (with residues left from HRV 3C cleavage) is: GPGSVFLSGNASDSSNSIQPPAPVNISKAILLGVILGGIILFGVLGNILVILSVACHRHLHSVTHYYVVYLAVADLLLTSTVMPFSAIYEVLGYWAFGRVFCNIWAAVDVLCCTASIMGLCIISIDRYIAVSYPLRYPTIVTQRRALMALLCVFALSLVISIGPLFGWRQPAPVDETICQINEEPGYVLFSALGSFYLPLAIILVMYCRVYVVAKRESRGLKSGLKTDKSDSEQVTLRIHRKNAPAGGSGMASAKTKTHFSVRLLKFSREKKAAKTLGIVVGCFVLCWLPFFLVMPIGSFFPDFKPSETVFKIVLWLGYLNSCIKPIIYLCYSQEFKKAFQNVLRIQCLCRKQSASHHHHHHHHHHGTRSLRGGLEVLFQ.
In the α1A-AR-A4 (L312F) variant one stabilizing mutation Leu-312 was reversed to phenylalanine to improve the affinity of ligands compared with α1A-AR-A4. α1A-AR-A4–active (Y67N, K322N) is a signaling competent variant in which the two stabilizing mutations Tyr-67 and Lys-322 were reverted to the WT asparagines. α1A-AR-A4 was used for NMR assignment, where each methionine was substituted to either leucine or isoleucine. All mutations were introduced through site-directed mutagenesis using PrimeSTAR DNA polymerase (TaKaRa).
α1A-AR expression
All α1A-AR variants were expressed in E. coli C43 (DE3) cells (Lucigen, Middleton, WI). For [13CϵH3]methionine-labeled expressions, 5 ml of LB pre-culture containing 100 mg/liter of ampicillin and 1% (w/v) glucose was inoculated with a single colony of C43 cells freshly transformed with the expression plasmid and incubated at 37 °C, 225 rpm for approximately 8 h. 2 ml of the LB day culture was centrifuged (1700 × g, 22 °C, 5 min) and the pellet was used to inoculate 100 ml of a defined minimal medium (M1 medium) (34) as overnight pre-culture. 10 ml of the overnight pre-culture was used to inoculate 500 ml of M1 medium in 2-liter flasks. The expression cultures were incubated at 37 °C, 225 rpm to reach OD600 of 0.6, at which point 50 mg/liter of [13CϵH3]methionine (Cambridge Stable Isotopes) was added along with 100 mg/liter of each lysine, threonine, phenylalanine, and 50 mg/liter of each leucine, isoleucine, and valine. The flasks were transferred to 20 °C and left shaking for 15 min prior to inducing protein expression with 250 μm isopropyl β-d-1-thiogalactopyranoside. After overnight expression (15–18 h, 20 °C, 225 rpm), the culture was harvested by centrifugation (2600 × g, 4 °C, 15 min). The final pellet was snap frozen in liquid nitrogen and stored at −80 °C. For unlabeled expression 5 ml of LB pre-culture was used to inoculate 500 ml of 2YT medium containing 100 mg/liter of ampicillin and 0.4% (w/v) glucose. At OD600 of 0.6 the culture was chilled on ice for 2 min prior to inducing protein expression with 250 μm isopropyl β-d-1-thiogalactopyranoside overnight and harvesting was carried out as described above.
α1A-AR purification
The frozen cell pellet was thawed at room temperature for 30 min. A 10-ml pellet was gently resuspended in 40 ml of ice-cold solubilization buffer (25 mm HEPES, pH 7.5, 200 mm NaCl, 10% glycerol, 1% DDM (Anatrace), 0.12% cholesterol hemisuccinate (Anatrace), 0.6% CHAPS (Sigma), 50 mg of lysozyme, 5 mg of DNase, one tablet of EDTA-free complete protease inhibitor mixture (Roche Applied Science), 0.2–0.4 mm phenylmethylsulfonyl fluoride, and incubated on a turning wheel for 30 min at 4 °C. The cell membranes were then disrupted by a sonication device (Diagenode Bioruptor Plus, high power, 10 s on/20 s off for 30 cycles) followed by another 1-h incubation at 4 °C on the turning wheel. The cell debris was removed by centrifugation (12,000 × g, 4 °C, 40 min) and the supernatant was filtered using a 45-μm Durapore syringe filter (Merck Millipore). The cleared cell lysate was incubated with 3 ml of Talon metal affinity resin pre-equilibrated with 45 ml of equilibrium buffer (20 mm HEPES, pH 7.5, 300 mm NaCl, 10% glycerol, 0.05% DDM). After 1.5 h incubation at 4 °C, the resin retaining the receptor was washed three times with washing buffer 1 (20 mm HEPES, pH 7.5, 500 mm NaCl, 10% glycerol, 0.05% DDM) and then the full-length protein was eluted by 30 ml of elution buffer (20 mm HEPES, pH 7.5, 300 mm NaCl, 10% glycerol, 0.05% DDM, 250 mm imidazole). The eluate was concentrated down to 0.5–1 ml using a 100-kDa cut-off centrifugal filter device (Amicon Ultra, Millipore). Imidazole was removed by using a PD10 desalting column (GE Healthcare). Cleavage of fusion proteins from the receptor was carried out overnight at 4 °C by adding 100 mm Na2SO4, 1 mm tris(2-carboxyethyl)phosphine, and 300 pmol of glutathione S-transferase-tagged HRV 3C protease (made in house).
The cleaved mixture was incubated for 1 h with 2 ml of pre-equilibrated Talon resin. The resin was washed using 30 ml of washing buffer 2 (20 mm HEPES, pH 7.5, 300 mm NaCl, 10% glycerol, 0.05% DDM, 30 mm imidazole) and the receptor was eluted by 20 ml of elution buffer. The eluate was concentrated down to 450 μl by a 30-kDa cut-off centrifugal filter device (Amicon Ultra, Millipore) and was loaded onto a Superdex 200 10/300 increase column (GE Healthcare) equilibrated with size exclusion chromatography buffer (50 mm sodium phosphate, pH 7.5, 100 mm NaCl, 0.02% DDM). Size exclusion chromatography was carried out at a flow rate of 0.5 ml/min. The peak fractions containing receptor were pooled and concentrated down to 100 μl using a 30-kDa cut-off centrifugal filter device (Amicon Ultra, Millipore). The sample buffer was exchanged twice to NMR buffer (50 mm sodium phosphate, pH 7.5, 100 mm NaCl, 99.9% D2O). Yields were generally between 0.5 and 1 mg of receptor/liter of expression culture. Protein concentration was measured by BCA protein assay (Pierce, ThermoFisher).
NMR spectroscopy
NMR samples were prepared to 130 μl at 40–60 μm receptor in 3-mm Shigemi NMR tubes (Shigemi Inc., Allison Park, PA). Ligands were added at saturating concentrations that were 2 mm adrenaline for α1A-AR-A4 and α1A-AR-A4 (active), 400 μm prazosin, and 1 mm of other ligands to all mutants (Table S1). Adrenaline was supplemented with 1 mm of the antioxidant ascorbic acid. Samples containing low affinity agonists (phenylephrine and adrenaline) were recycled via competition with high affinity ligands, exchange was judged via the chemical shift of the Met-2926.55 resonance. Experiments on α1A-AR-A4 and α1A-AR-A4-L312F, apo, and all ligands were performed at least twice on independent receptor samples (biological replicates), except WB-4101 and phentolamine, which were acquired once. Experiments on apo α1A-AR-A4–active and bound to prazosin, silodosin, and oxymetazoline were performed at least twice on independent receptor samples, and for other ligands were performed once.
All NMR spectra were collected at 25 °C on an 800-MHz Bruker Avance II spectrometer equipped with a triple resonance cryoprobe. 2D 1H-13C SOFAST-HMQC (43) spectra were recorded by excitation with a 2.25-ms PC9 120° 1H pulse and refocusing with a 1-ms r-SNOB shaped 180° 1H pulse. The spectral widths were set to 12 and 25 ppm for 1H and 13C dimensions, respectively. For the spectra recorded for α1A-AR-A4 variant (Fig. 2 and Fig. S3), 1024 × 128 complex points were recorded with a 25% Poisson-gap sampling schedule and 2048 scans; an acquisition time of 8.5 h. For the other spectra, 1024 × 200 complex points were recorded with either traditional or 60% Poisson-gap sampling schedule and 368 scans resulting in acquisition times of 10 and 6 h, respectively. Spectra were reconstructed with compressed sensing using qMDD and processed using NMRpipe (44) where data were multiplied by cosine bell functions and zero-filled once in each dimension. Spectra were analyzed in NMRFAM-Sparky (45) (Goddard, T. D. and Kneller, D. G., University of California, San Francisco).
The average chemical shift differences, Δδ, were normalized using the equation Δδ = [(Δδ1H)2 + (Δδ13C/3.5)2]0.5. The error values were calculated by the formula |[Δδ1H × R1H + Δδ13C × R13C/(3.5)2]|/Δδ, where R1H and R13C are the digital resolutions in ppm in the 1H and 13C dimensions, respectively (5).
Saturation and competition-binding assays
1 nmol of purified full-length α1A-AR variant (mCherry attached) was resuspended in 10 ml of assay buffer (20 mm HEPES, pH 7.5, 100 mm NaCl, 0.02% DDM) and immobilized onto 200 μl of Dynabeads (Streptavidin T1) for 30 min at 4 °C. 100 μl of the suspension-containing beads with immobilized receptor were aliquoted to a 96-Deep Well plate from which the beads were transferred to another 96-Deep Well plate containing 100 μl of ligand solution using a KingFisher Flex magnetic particle processor. For saturation binding, immobilized receptors in each well were incubated with 100 μl of assay buffer containing increased concentrations (0, 3.125, 6.25, 12.5, 25, 50, 100, and 200 nm) of QAPB (quinazoline piperazine BODIPY) for 2 h at 22 °C. Nonspecific binding was determined by repeating the experiment in the presence of 10 μm prazosin. For competition binding, immobilized receptors were incubated with 100 μl of assay buffer containing 10 nm QAPB with the addition of ligands at various concentrations, as shown in supporting Figs. S5 and S8, for 2 h at 22 °C. Immobilized receptors were subsequently washed with 200 μl of assay buffer and resuspended in 100 μl of assay buffer. 90 μl of the final beads solution was transferred to a 96-well Greiner Bio-One nonbinding black plate. Fluorescence of bound QAPB was measured using a POLARstar OMEGA plate reader (BMG Labtech, Ortenburg, Germany) and normalized to mCherry fluorescence, which was detected simultaneously. Data represent the mean ± S.D. of three independent biological replicate experiments each performed in duplicate technical measurements. To compare ligand-binding affinities at α1A-AR-A4 (L312F) and α1A-AR-A4–active of α1A-AR-A4, raw data from our previously published paper (32), were reanalyzed and presented in Table S1.
Thermostability assay
1 nm purified full-length α1A-AR-A4 or α1A-AR-A4–active (mCherry attached) was prepared in base buffer (20 mm HEPES, 100 mm NaCl, 0.1% DDM). To measure thermostability of receptors in the apo state, 100 μl of receptor solution was aliquoted into 24 wells of a 96-well PCR plate. 10 of the 12 duplicates were heated in gradient temperatures for 30 min and the two remaining duplicates were left at 4 °C for normalization. After thermo-treatment, the receptors were transferred to a KingFisher 96-Deep Well plate containing 2 μl of paramagnetic Dynabeads per well (streptavidin T1, ThermoFisher Scientific). The following few steps were automatically performed by using a KingFisher 96 magnetic particle processor. The receptor was first incubated with magnetic beads for 30 min at 4 °C. Then, magnetic beads were transferred to another 96-Deep Well plate containing 100 μl of ligand solution (20 mm HEPES, 100 mm NaCl, 0.1% DDM, 100 nm QAPB). The nonspecific binding was determined by competing QAPB with 100 μm of prazosin. After 1.5 h incubation, immobilized receptors were subsequently washed with 200 μl of assay buffer and resuspended in 100 μl of assay buffer. 90 μl of the final beads solution was transferred to a 96-well Greiner Bio-One nonbinding black plate. Fluorescence of bound QAPB was measured using a POLARstar OMEGA plate reader (BMG Labtech, Ortenburg, Germany) and normalized to mCherry fluorescence, which was detected simultaneously. To measure the thermostability of α1A-AR variants in the presence of ligand, receptors were preincubated with 100 nm QAPB for 1 h on ice prior to being heated at varying temperatures. The remaining steps were carried out as described for apo state thermostability assay. Data represent the mean ± S.D. of three independent biological replicate experiments each performed in duplicate technical replicate measurements.
IP1 assay
Gαq/11 signaling assays were carried out using the IP-One HTRF® Assay Kit (Cisbio Bioassays, France) measuring IP1 using the manufacturer's protocol. COS-7 cells were seeded at 25,000 cells per well in a 96-well plate and incubated overnight at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS) (Scientifix Life, Melbourne, Australia), 1% l-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco). Cells were transfected with pcDNA3.1 constructs of WT or mutant α1A-ARs using Lipofectamine 2000 (Invitrogen) at 0.25 μg of DNA/well. 24 h later, cells were stimulated with ligands for 2 h at 37 °C in 40 μl of Stimulation Buffer, then frozen at −80 °C. 14 μl of thawed sample were transferred to a white HTRF® 384-well Optiplate (PerkinElmer Life Sciences), incubated with development reagents in the dark for 1 h with shaking, and analyzed by time-resolved fluorescence using a POLARstar OMEGA plate reader (BMG Labtech, Ortenburg, Germany). Data were analyzed against the kit's standard curve. Data represent mean ± S.D. of three independent biological replicate experiments (except for some samples in the screening assay in Fig. S8, where some receptors were measured only once or twice). For each biological replicate samples were measured in triplicate technical replicate measurements, unless otherwise stated in the figure legends (as in Fig. S8).
NanoBiT G protein activity assay
COS-7 cells grown in 10% FBS, 1% l-glutamine, 1% penicillin/streptomycin, DMEM were seeded at 250,000 cells/well on a 6-well–plate. Cells were then transiently co-transfected in the 6-well–plate, with 0.1 μg of Gαq-LgBiT (GNAQ-11S) DNA, 0.5 μg of Gβ-untagged (GNB1) DNA, 0.5 μg of Gγ-SmBiT (114-GnG2) DNA, 0.2 μg of Guanine Release Factor (RIC8A) DNA, and 0.5 μg of α1A-AR (or respective AR mutants) DNA using Lipofectamine 2000 transfection reagent as per the manufacturer's instructions. The next day the cells were resuspended in phenol red-free DMEM containing 10% FBS, 1% l-glutamine, 1% penicillin/streptomycin, 25 mm HEPES and seeded at 50,000 cells/well to a white 96-well–plate and incubated overnight. On the day of the assay, plates were preincubated with 10 μm furimazine for 1 h. Following incubation, raw luminescence counts in each well were measured every 12 s over the course of the assay using a POLARstar Omega plate reader (BMG Labtech). Cells were treated with either vehicle or a saturating concentration of each ligand (50 nm for A-61603 and 100 nm for antagonists). Luminescence counts were plotted against time, with the final preincubation reading assigned as the zero-time point (time of vehicle/ligand addition). A baseline correction was then performed by subtracting the luminescence counts in the vehicle-treated samples from the ligand-treated samples, which resulted in a time course plot of ligand-induced luminescence counts. Initial raw luminescence counts were used as a readout of G protein expression levels. Data represent the mean ± S.E. of three independent biological replicate experiments each performed in triplicate technical replicate measurements, unless otherwise stated in the figure legends.
Intracellular Ca2+ mobilization assays
COS-7 cells were seeded in 10-cm culture dishes at 3 × 106 cells/dish and allowed to grow overnight at 37 °C, 5% CO2 in DMEM supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin (Life Technologies). The next day the cells were transfected with 30 μg of receptor DNA construct (pcDNA3.1 expression vector containing WT or mutant α1A-ARs) using 60 μl of Lipofectamine 2000 (Invitrogen) transfecting reagent per dish. The following day, cells were transferred to 96-well–culture plates (5 × 104 cells/well) and allowed to grow overnight. On the day of the experiment cells were washed twice with Ca2+ assay buffer (150 mm NaCl, 2.6 mm KCl, 1.2 mm MgCl2, 10 mm d-glucose, 10 mm HEPES, 2.2 mm CaCl2, 0.5% (w/v) BSA, and 4 mm probenecid, pH 7.4) and incubated in Ca2+ assay buffer containing 1 mm Fluo-4-AM for 1 h in the dark at 37 °C and 5% CO2. After two washes with Ca2+ assay buffer, fluorescence was measured for 1.5 min upon the addition of ligands in a Flexstation 3 (Molecular Devices, Sunnyvale, CA) using an excitation wavelength of 485 nm and emission wavelength of 520 nm. Data were normalized to the peak response elicited by 3 μm ionomycin (Life Technologies). Data represent the mean ± S.D. of three independent biological replicate experiments each performed in triplicate technical replicate measurements, unless otherwise stated in the figure legends.
Homology modeling
Homology models of inactive and active state α1A-AR were built with I-TASSER (46) using crystal structures of β2-AR as the templates, which show 23% identity and 39% similarity. For inactive state models, the structure of β2-AR bound to the antagonist carazolol and the inactive state stabilizing nanobody, Nb60 (PDB ID 5JQH) (47), was used as a template. For the active state models, the crystal structure of a β2-AR–Gs protein complex bound to the agonist BI-167107 (PDB ID 3SN6) (48) was used as a template. The N- and C-terminal regions as well as the ICL3 regions, which have no sequence similarity to the template, were deleted from the model. Energy minimization was performed using Minimize tool in Maestro version 11.7.012 (Schrödinger, Inc.) under OPLS 2005 (49) forcefield.
Data availability
All data generated or analyzed during this study are included in the published article (and supporting information).
Author contributions
F.-J. W., A. A.-R., R. A. B., D. J. S., and P. R. G. data curation; F.-J. W. formal analysis; F.-J. W., L. M. W., A. A.-R., A. G., T. M. V., M. K., A. R. W., M. D. G., and P. R. G. investigation; F.-J. W. methodology; F.-J. W. writing-original draft; F.-J. W., L. M. W., A. A.-R., A. G., T. M. V., M. K., A. R. W., M. D. G., R. A. B., D. J. S., and P. R. G. writing-review and editing; A. A.-R., A. R. W., M. D. G., R. A. B., D. J. S., and P. R. G. conceptualization; A. A.-R., M. D. G., R. A. B., D. J. S., and P. R. G. resources; A. A.-R., A. R. W., M. D. G., R. A. B., D. J. S., and P. R. G. supervision; A. A.-R., M. D. G., R. A. B., D. J. S., and P. R. G. funding acquisition; A. A.-R., R. A. B., D. J. S., and P. R. G. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Fabian Bumbak (The Florey Institute of Neuroscience and Mental Health) for assistance with optimizing the expression and purification of receptor samples; Prof. Dmitry Veprintsev (University of Nottingham) and Dr. Franziska Heydenreich (Stanford University) for suggesting back-mutations for the generation of α1A-AR-A4–active; Prof. Asuka Inoue (Tohoku University) for supplying plasmids for the G protein activity assays; Sharon Layfield (The Florey Institute of Neuroscience and Mental Health) and Dr. Ashish Sethi (The University of Melbourne) for assistance with cell-based assays and NMR data analysis, respectively; Dr. David Chalmers (Monash University) and the Monash University Medicinal Chemistry Computational Chemistry Facility for the assistance with computational modeling; and The Bio21 NMR facility for access to spectrometers.
This work was supported by National Health and Medical Research Council (NHMRC) project Grants 1081801 (to D. J. S.), 1081844 (to P. R. G., D. J. S., and R. A. D. B.), and 1141034 (to D. J. S., P. R. G., and R. A. D. B.), and a NHMRC Boosting Dementia Research Leadership Fellowship (to D. J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S11 and Table S1.
- GPCR
- G protein–coupled receptors
- β2-AR
- β2-adrenergic receptor
- μ-OR
- μ-opioid receptor
- BPH
- benign prostatic hyperplasia
- DDM
- n-dodecyl-β-d-maltopyranoside
- HMQC
- heteronuclear multiple quantum coherence
- MD
- molecular dynamics
- TMD
- transmembrane domain
- IP
- inositol phosphate
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- QAPB
- quinazoline piperazine BODIPY
- PDB
- Protein Data Bank
- α1A-AR
- α1A-adrenoreceptor.
References
- 1. Manglik A., and Kruse A. C.. 2017) Structural basis for G protein-coupled receptor activation. Biochemistry 56, 5628–5634 10.1021/acs.biochem.7b00747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carpenter B., and Tate C. G.. 2017) Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr. Opin. Struct. Biol. 45, 124–132 10.1016/j.sbi.2017.04.010 [DOI] [PubMed] [Google Scholar]
- 3. García-Nafría J., and Tate C. G.. 2019) Cryo-EM structures of GPCRs coupled to Gs, Gi, and Go. Mol. Cell. Endocrinol. 488, 1–13 10.1016/j.mce.2019.02.006 [DOI] [PubMed] [Google Scholar]
- 4. Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., and Kobilka B. K.. 2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–112 10.1038/nature08650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kofuku Y., Ueda T., Okude J., Shiraishi Y., Kondo K., Maeda M., Tsujishita H., and Shimada I.. 2012) Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 3, 1045 10.1038/ncomms2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J. J., Horst R., Katritch V, Stevens R. C., Wüthrich K.. 2012) Biased signaling pathways in β2-AR characterized by 19F-NMR. Science 335, 1106–1110 10.1126/science.1215802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horst R., Liu J., Stevens R., and Wüthrich K.. 2013) β2-adrenergic receptor activation by agonists studied with 19F NMR spectroscopy. Angew. Chem. 125, 10962–10965 10.1002/ange.201305286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., and Kobilka B. K.. 2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 10.1016/j.cell.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kofuku Y., Ueda T., Okude J., Shiraishi Y., Kondo K., Mizumura T., Suzuki S., and Shimada I.. 2014) Functional dynamics of deuterated β2-adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 53, 13376–13379 10.1002/anie.201406603 [DOI] [PubMed] [Google Scholar]
- 10. Manglik A., Kim T. H., Masureel M., Altenbach C., Yang Z., Hilger D., Lerch M. T., Kobilka T. S., Thian F. S., Hubbell W. L., Prosser R. S., and Kobilka B. K.. 2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111 10.1016/j.cell.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eddy M. T., Didenko T., Stevens R. C., and Wüthrich K.. 2016) β2-Adrenergic receptor conformational response to fusion protein in the third intracellular loop. Structure 24, 2190–2197 10.1016/j.str.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kobilka B., Gether U., Seifert R., Lin S., and Ghanouni P.. 1999) Characterization of ligand-induced conformational states in the β2-adrenergic receptor. J. Recept. Signal. Transduct. Res. 19, 293–300 10.3109/10799899909036652 [DOI] [PubMed] [Google Scholar]
- 13. Isogai S., Deupi X., Opitz C., Heydenreich F. M., Tsai C. J., Brueckner F., Schertler G. F., Veprintsev D. B., and Grzesiek S.. 2016) Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 530, 237–241 10.1038/nature16577 [DOI] [PubMed] [Google Scholar]
- 14. Solt A. S., Bostock M. J., Shrestha B., Kumar P., Warne T., Tate C. G., and Nietlispach D.. 2017) Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound β1-adrenergic receptor. Nat. Commun. 8, 1795 10.1038/s41467-017-02008-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ye L., Van Eps N., Zimmer M., Ernst O. P., and Prosser R. S.. 2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 10.1038/nature17668 [DOI] [PubMed] [Google Scholar]
- 16. Clark L. D., Dikiy I., Chapman K., Rödström K. E., Aramini J., LeVine M. V., Khelashvili G., Rasmussen S. G., Gardner K. H., and Rosenbaum D. M.. 2017) Ligand modulation of sidechain dynamics in a wild-type human GPCR. Elife 6, e28505 10.7554/eLife.28505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye L., Neale C., Sljoka A., Lyda B., Pichugin D., Tsuchimura N., Larda S. T., Pomès R., García A. E., Ernst O. P., Sunahara R. K., and Prosser R. S.. 2018) Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9, 1372 10.1038/s41467-018-03314-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eddy M. T., Lee M. Y., Gao Z. G., White K. L., Didenko T., Horst R., Audet M., Stanczak P., McClary K. M., Han G. W., Jacobson K. A., Stevens R. C., and Wüthrich K.. 2018) Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 172, 68–80.e12 10.1016/j.cell.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sounier R., Mas C., Steyaert J., Laeremans T., Manglik A., Huang W., Kobilka B. K., Démené H., and Granier S.. 2015) Propagation of conformational changes during μ-opioid receptor activation. Nature 524, 375–378 10.1038/nature14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okude J., Ueda T., Kofuku Y., Sato M., Nobuyama N., Kondo K., Shiraishi Y., Mizumura T., Onishi K., Natsume M., Maeda M., Tsujishita H., Kuranaga T., Inoue M., and Shimada I.. 2015) Identification of a conformational equilibrium that determines the efficacy and functional selectivity of the μ-opioid receptor. Angew. Chem. Int. Ed. Engl. 54, 15771–15776 10.1002/anie.201508794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Casiraghi M., Damian M., Lescop E., Point E., Moncoq K., Morellet N., Levy D., Marie J., Guittet E., Banères J. L., and Catoire L. J.. 2016) Functional modulation of a G protein-coupled receptor conformational landscape in a lipid bilayer. J. Am. Chem. Soc. 138, 11170–11175 10.1021/jacs.6b04432 [DOI] [PubMed] [Google Scholar]
- 22. Xu J., Hu Y., Kaindl J., Risel P., Hübner H., Maeda S., Niu X., Li H., Gmeiner P., Jin C., and Kobilka B. K.. 2019) Conformational complexity and dynamics in a muscarinic receptor revealed by NMR spectroscopy. Mol. Cell 75, 53–65.e7 10.1016/j.molcel.2019.04.028 [DOI] [PubMed] [Google Scholar]
- 23. Shimada I., Ueda T., Kofuku Y., Eddy M. T., and Wüthrich K.. 2019) GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 18, 59–82 10.1038/nrd.2018.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deupi X., and Standfuss J.. 2011) Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr. Opin. Struct. Biol. 21, 541–551 10.1016/j.sbi.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 25. Trzaskowski B., Latek D., Yuan S., Ghoshdastider U., Debinski A., and Filipek S.. 2012) Action of molecular switches in GPCRs: theoretical and experimental studies. Curr. Med. Chem. 19, 1090–1109 10.2174/092986712799320556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ahuja S., and Smith S. O.. 2009) Multiple switches in G protein-coupled receptor activation. Trends Pharmacol. Sci. 30, 494–502 10.1016/j.tips.2009.06.003 [DOI] [PubMed] [Google Scholar]
- 27. Bhattacharya S., and Vaidehi N.. 2014) Differences in allosteric communication pipelines in the inactive and active states of a GPCR. Biophys. J. 107, 422–434 10.1016/j.bpj.2014.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Latorraca N. R., Venkatakrishnan A. J., and Dror R. O.. 2017) GPCR dynamics: structures in motion. Chem. Rev. 117, 139–155 10.1021/acs.chemrev.6b00177 [DOI] [PubMed] [Google Scholar]
- 29. Akinaga J., García-Saínz J. A., and S. Pupo A.. 2019) Updates in the function and regulation of α1-adrenoceptors. Br. J. Pharmacol. 176, 2343–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez D. M., and Doze V.. 2011) Cardiac and neuroprotection regulated by α1-adrenergic receptor subtypes. J. Recept. Signal Transduct. Res. 31, 98–110 10.3109/10799893.2010.550008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scott D. J., and Plückthun A.. 2013) Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells. J. Mol. Biol. 425, 662–677 10.1016/j.jmb.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 32. Yong K. J., Vaid T. M., Shilling P. J., Wu F. J., Williams L. M., Deluigi M., Plückthun A., Bathgate R. A. D., Gooley P. R., and Scott D. J.. 2018) Determinants of ligand subtype-selectivity at α1A-adrenoceptor revealed using saturation transfer difference (STD) NMR. ACS Chem. Biol. 13, 1090–1102 10.1021/acschembio.8b00191 [DOI] [PubMed] [Google Scholar]
- 33. Isberg V., de Graaf C., Bortolato A., Cherezov V., Katritch V., Marshall F. H., Mordalski S., Pin J.-P., Stevens R. C., Vriend G., and Gloriam D.. 2015) Generic GPCR residue numbers: aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 36, 22–31 10.1016/j.tips.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bumbak F., Keen A. C., Gunn N. J., Gooley P. R., Bathgate R. A. D., and Scott D. J.. 2018) Optimization and 13CH3 methionine labeling of a signaling competent neurotensin receptor 1 variant for NMR studies. Biochim. Biophys. Acta 1860, 1372–1383 10.1016/j.bbamem.2018.03.020 [DOI] [PubMed] [Google Scholar]
- 35. Bumbak F., Bathgate R. A. D., Scott D. J., and Gooley P. R.. 2019) Expression and purification of a functional E. coli 13CH3-methionine-labeled thermostable neurotensin receptor 1 variant for solution NMR studies. Methods Mol. Biol. 1947, 31–55 10.1007/978-1-4939-9121-1_3 [DOI] [PubMed] [Google Scholar]
- 36. Hwa J., Graham R. M., and Perez D. M.. 1995) Identification of critical determinants of α1-adrenergic receptor subtype selective agonist binding. J. Biol. Chem. 270, 23189–23195 10.1074/jbc.270.39.23189 [DOI] [PubMed] [Google Scholar]
- 37. Butterfoss G. L., DeRose E. F., Gabel S. A., Perera L., Krahn J. M., Mueller G. A., Zheng X., and London R. E.. 2010) Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J. Biomol. NMR 48, 31–47 10.1007/s10858-010-9436-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu J., Taniguchi T., Takauji R., Suzuki F., Tanaka T., and Muramatsu I.. 2000) Inverse agonism and neutral antagonism at a constitutively active α-1A adrenoceptor. Br. J. Pharmacol. 131, 546–552 10.1038/sj.bjp.0703584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inoue A., Raimondi F., Kadji F. M. N., Singh G., Kishi T., Uwamizu A., Ono Y., Shinjo Y., Ishida S., Arang N., Kawakami K., Gutkind J. S., Aoki J., and Russell R. B.. 2019) Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e25 10.1016/j.cell.2019.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cotecchia S., Schwinn D. A., Randall R. R., Lefkowitz R. J., Caron M. G., and Kobilka B. K.. 1988) Molecular cloning and expression of the cDNA for the hamster α1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 85, 7159–7163 10.1073/pnas.85.19.7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dror R. O., Arlow D. H., Maragakis P., Mildorf T. J., Pan A. C., Xu H., Borhani D. W., and Shaw D. E.. 2011) Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18684–18689 10.1073/pnas.1110499108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scott D. J., Gunn N. J., Yong K. J., Wimmer V. C., Veldhuis N. A., Challis L. M., Haidar M., Petrou S., Bathgate R. A. D., and Griffin M. D. W.. 2018) A novel ultra-stable, monomeric green fluorescent protein for direct volumetric imaging of whole organs using CLARITY. Sci. Rep. 8, 667 10.1038/s41598-017-18045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schanda P., Kupce E., and Brutscher B.. 2005) SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 33, 199–211 10.1007/s10858-005-4425-x [DOI] [PubMed] [Google Scholar]
- 44. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A.. 1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 45. Lee W., Tonelli M., and Markley J. L.. 2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J., Yang J., Jang R., and Zhang Y.. 2015) GPCR-I-TASSER: a hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure 23, 1538–1549 10.1016/j.str.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Staus D. P., Strachan R. T., Manglik A., Pani B., Kahsai A. W., Kim T. H., Wingler L. M., Ahn S., Chatterjee A., Masoudi A., Kruse A. C., Pardon E., Steyaert J., Weis W. I., Prosser R. S., et al. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452 10.1038/nature18636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siu S. W., Pluhackova K., and Böckmann R. A.. 2012) Optimization of the OPLS-AA force field for long hydrocarbons. J. Chem. Theory Comput. 8, 1459–1470 10.1021/ct200908r [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the published article (and supporting information).