

Abstract
Background
Although immune checkpoint inhibitors have revolutionized cancer treatment, clinical benefit with this class of agents has been limited to a subset of patients. Hence, more effective means to target tumor cells that express immune checkpoint molecules should be developed. For the first time, we report a novel natural killer (NK) cell line, programmed death-ligand 1 (PD-L1) targeting high-affinity natural killer (t-haNK), which was derived from NK-92 and was engineered to express high-affinity CD16, endoplasmic reticulum-retained interleukin (IL)-2, and a PD-L1-specific chimeric antigen receptor (CAR). We show that PD-L1 t-haNK cells also retained the expression of native NK receptors and carried a high content of granzyme and perforin granules.
Methods
NanoString, flow cytometry, and immunofluorescence analyses were performed to characterize the phenotype of irradiated PD-L1 t-haNK cells. In vitro PD-L1 t-haNK cell activity against cancer cell lines and human peripheral blood mononuclear cells (PBMCs) was determined via flow-based and 111In-release killing assays. The antitumor effect of PD-L1 t-haNK cells in vivo was investigated using MDA-MB-231, H460, and HTB1 xenograft models in NOD-scid IL2Rgammanull (NSG) mice. Additionally, the antitumor effect of PD-L1 t-haNK cells, in combination with anti-PD-1 and N-803, an IL-15 superagonist, was evaluated using mouse oral cancer 1 syngeneic model in C57BL/6 mice.
Results
We show that PD-L1 t-haNK cells expressed PD-L1-targeting CAR and CD16, retained the expression of native NK receptors, and carried a high content of granzyme and perforin granules. In vitro, we demonstrate the ability of irradiated PD-L1 t-haNK cells to lyse 20 of the 20 human cancer cell lines tested, including triple negative breast cancer (TNBC) and lung, urogenital, and gastric cancer cells. The cytotoxicity of PD-L1 t-haNK cells was correlated to the PD-L1 expression of the tumor targets and can be improved by pretreating the targets with interferon (IFN)-γ. In vivo, irradiated PD-L1 t-haNK cells inhibited the growth of engrafted TNBC and lung and bladder tumors in NSG mice. The combination of PD-L1 t-haNK cells with N-803 and anti-PD-1 antibody resulted in superior tumor growth control of engrafted oral cavity squamous carcinoma tumors in C57BL/6 mice. In addition, when cocultured with human PBMCs, PD-L1 t-haNK cells preferentially lysed the myeloid-derived suppressor cell population but not other immune cell types.
Conclusion
These studies demonstrate the antitumor efficacy of PD-L1 t-haNK cells and provide a rationale for the potential use of these cells in clinical studies.
Keywords: immunology, oncology, tumors
Background
The discovery and establishment of continuously expanding natural killer (NK) cell lines provide a potential source of allogeneic ‘off-the-shelf’ cellular therapy due to its low risk of causing graft-versus-host disease or other alloimmune toxicities.1 NK-92, which was derived from a non-Hodgkin’s lymphoma patient, is a highly cytotoxic NK shown to have activity against a broad range of leukemic and solid malignancies.2–5 In several clinical studies, adoptive transfer therapy using irradiated NK-92 cells has demonstrated safety even up to doses of 1010 cells/m2 and has shown evidence of antitumor clinical benefit.6–8
High-affinity NK (haNK) cells are NK-92 cells engineered to express high-affinity CD16/FcγRIIIa (158V) allele and endoplasmic reticulum (ER)-retained interleukin (IL)-2.9 10 haNK cells were demonstrated to retain high granular stocks of perforin and granzyme and to have cytotoxic potency against various human tumor cell lines in vitro. Furthermore, the incorporation of the high-affinity CD16 allele allowed haNK cells to lyse cells via antibody-dependent cell-mediated toxicity (ADCC) mediated by tumor-antigen-specific IgG1 antibodies.
The PD-1/programmed death-ligand 1 (PD-L1) pathway has been co-opted by cancer cells as a mechanism of immune escape and represents a key target in cancer immunotherapy.11 12 Currently, there are five anti-PD-1 and anti-PD-L1 monoclonal antibodies that have been approved in several types of solid tumors.13 14 However, only a limited number of non-melanoma patients benefit from this therapy with more than half of patients with positive PD-L1 expression not responding to the treatment15; thus, better ways to target PD-L1 should be investigated.
Recently, NK cells have been developed to express chimeric antigen receptor (CAR) targeting specific tumor-associated antigens.16 17 In this report, we described a PD-L1 targeting high-affinity natural killer (t-haNK) cell line, which was engineered to express high-affinity CD16, ER-retained IL-2 and a CAR specific against PD-L1. We demonstrated that irradiated PD-L1 t-haNK cells were highly cytolytic against various human tumor cell lines in vitro. In vivo, we showed that adoptive transfer of irradiated PD-L1 t-haNK cells resulted in tumor growth inhibition of several xenograft tumor models in NOD-scid IL2Rgammanull (NSG) mice. Furthermore, PD-L1 t-haNK cells lysed human peripheral myeloid-derived suppressor cells (MDSCs) without affecting the T and NK cells. These studies provide rationale for future clinical trials with this novel PD-L1 t-haNK cell platform.
Methods
Cell lines and culture
The PD-L1 t-haNK and haNK cells were provided by NantKwest through a Cooperative Research and Development Agreement (CRADA) with the National Cancer Institute (NCI), National Institutes of Health (NIH). haNK cells were cultured as described9 10 and were irradiated (10 Gy) 24 hours before being used in in vitro tests. PD-L1 t-haNK cells were provided cryopreserved and irradiated (15 Gy) and were thawed on the same day as the experiment. Human cancer cell lines MDA-MB-231, BT549, T47D, SUM149, MCF7, H460, H441, HCC4006, SW480, SW620, DU145, HTB1, IOMM, CaSki, AGS, SNU-1, SNU16, SNU-5 and KATO III were purchased from American Type Culture Collection. CH22 was a kind gift from the Chordoma Foundation. Mouse oral cancer 1 (MOC1) was obtained from R. Uppaluri (Boston, MA) and cultured as described.18 19 PD-L1 null MDA-MB-231 cells were generated by Synthego via CRISPR/Cas9 technology and sorted to >98% knockout purity via fluorescence-activated cell sorting (FACS) using BD FACSAria (BD Biosciences) at the LGI Flow Cytometry Core, NCI, NIH. All cultures were mycoplasma-free and were maintained in the recommended culture media. Human MDSCs were expanded from human PBMCs as previously described20 and were isolated using CD33 magnetic beads (Miltenyi Biotec).
RNA analysis
Total RNA was extracted from PD-L1 t-haNK and haNK cells using the RNeasy Mini kit (Qiagen). NanoString analysis of the isolated RNA was completed with the nCounter Pan-Cancer Immune Profiling Panel (NanoString Technologies), run by the Genomics Laboratory, Frederick National Laboratory for Cancer Research, Frederick, Maryland. Raw data files and reporter library files were uploaded and analyzed using the nSolver analysis software V.4.0.70 (NanoString) with the mRNA counts normalized to the most consistently expressed housekeeping genes and with the haNK cell sample as the categorical reference values. Genes that were deregulated by log2 fold >3 were selected and uploaded into Ingenuity Pathway Analysis (IPA) software V.01–14 (Qiagen). An IPA Core analysis focused on NK cell-related pathways was performed.
Flow cytometric analysis and FACS
CAR expression was detected using biotinylated soluble human PD-L1 protein (Biosystems Acro) as a primary stain followed by PE-conjugated streptavidin (BD Biosciences) as the secondary stain. The following antibodies and stains were also used: CD56-APC (BD Biosciences), CD16-BV510 (BD), granzyme B-PE (BD Biosciences), perforin-BV421 (BD Biosciences), PD-L1-PE (BD Biosciences), and LIVE/DEAD fixable blue dead stain (Thermo Fisher). To identify the 135 immune subsets in the PBMCs (online supplementary table S1), methodology and gating strategies that have been previously described21 22 were used with slight modifications. Antibody panels were slightly modified (online supplementary table S2). Flow cytometry was performed on BD LSRFortessa or BD FACSVerse (BD Biosciences) and analyzed using FlowJo V.9.7.6 or V.10.5.3 (TreeStar). FACS of PD-L1high and PD-L1low MDA-MB-231 was performed using the MA900 (Sony) sorter.
jitc-2019-000450supp001.pdf (241.2KB, pdf)
Immunofluorescence imaging
PDL1 t-haNK, MDA-MB-231 and PD-L1 null MDA-MB-231 cells were stained as described.9 The following were used as primary antibodies: NKG2d (Abcam) and perforin (Abcam), PD-L1 (Abcam). The following were used as secondary antibodies: goat anti-rabbit-AlexFLuor647 (Thermo Fisher), goat anti-mouse-AlexFLuor488 (Thermo Fisher), and goat-anti-rabbit AlexaFluor488 (Thermo Fisher). The samples were mounted using ProLong gold antifade mountant with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher). The samples were imaged on a Leica DMI4000B microscope (Leica Microsystems).
NK killing assays
111In-release assays were performed as previously described.9 For experiments with interferon (IFN)-γ pretreatment, tumor cell lines were incubated with 20 ng/mL IFN-γ for 24 hours prior to 111In-labeling. For ADCC experiments, 1 µg/mL each of cetuximab (Eli Lilly), anti-PD-L1 (provided by NantKwest through a CRADA) and anti-hen egg lysozyme IgG1 isotype control were added to the appropriate samples. For the blocking experiments, PD-L1 t-haNK cells were preincubated for 2 hours with either 50 µg/mL anti-CD16 antibody (eBioscience) or 200 nM concanamycin A (CMA) before being used in lysis assays. For assays evaluating the indirect effect of N-803 on PD-L1 t-haNK cells, C57BL/6 splenocytes were cultured with 0.5, 1.0, or 2.0 µg immobilized N-803 for 18 hours. Supernatants from the cultures were quantified for IFN-γ using a mouse ELISA kit (Life Technologies). MOC1 cells were incubated for 18 hours in supernatant harvested from a splenocyte culture incubated overnight with 1 µg immobilized N-803. Afterwards, the MOC1 cells were evaluated for PD-L1 expression and were used as PD-L1 t-haNK cell target.
For the PDL1high vs PDL1low coculture flow-based killing assay, MDA-MB-231, IFN-γ-treated MDA-MB-231 and PD-L1high flow-sorted MDA-MB-231 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) (BioLegend). BT549, MCF7, T47D, MDA-MB-231, and PD-L1 null MDA-MB-231 were labeled with CellTrace Violet Proliferation dye (Thermo Fisher). Equal numbers of CFSE- and violet-labeled cells were mixed and cocultured with PD-L1 t-haNK cells at 37°C for 18 hours. The samples were then analyzed by flow cytometry. Per cent killing was determined based on the formula 100%×[1–(percentage of viable tumor cells in the presence of NK cells/percentage of viable tumor cells in the absence of NK cells)].
For the killing assays targeting PBMCs, cryopreserved or fresh PBMCs were cocultured with PD-L1 t-haNK cells. For some experiments, PBMC-PD-L1 t-haNK cell contact was blocked using a 3 μm transwell membrane. Following the 16 hours coculture incubation, cells were harvested and examined by flow cytometry. Immune subsets with a potentially biologically relevant change following coincubation with PD-L1 t-haNK cells were defined as those with a p value <0.05, the majority of donors having a >25% change, difference in medians of >0.05% of PBMCs, and a median frequency of >1% of total PBMCs.
Live cell imaging
PD-L1 t-haNK cells were labeled with CellTracker Violet BMQC (Thermo Fisher), and MDA-MB-231 cells were labeled with CellTracker CM-Dil (Thermo Fisher) and were cocultured together in an eight-well glass bottom μ-slide (Ibidi) at 1:2 effector to target (E:T) ratio. Immediately, CellEvent Caspase 3/7 Green (Thermo Fisher) was added to the coculture. The cells were imaged using the Zeiss LSM 780 at the NCI Confocal Microscopy Core Facility.
Western blot
Wild-type (WT) MDA-MB-231 and PD-L1 null MDA-MB-231 lysates (30 µg each) were resolved by SDS-PAGE before electro-transfer onto nitrocellulose membranes before probing with anti-PD-L1 (Abcam) and anti-vinculin (Sigma-Aldrich) antibodies followed by goat anti-rabbit-IRDye 680RD (LI-COR) and goat anti-mouse-IRDye 800CW (LI-COR) antibodies. The probed proteins were visualized using the Odyssey system (LI-COR).
Tumor studies
Female NSG mice 10–16 week old (originally obtained from Jackson Laboratory and bred in-house at NCI, Bethesda, Maryland) were inoculated with a suspension of MDA-MB-231, PD-L1 null MDA-MB-231, H460 or HTB1 cells (5.0×106 per mouse) in 100 µL of phosphate-buffered saline (PBS) admixed with Matrigel 50% (v/v). Tumors grew for 9–10 days before randomizing into untreated control (PBS) or PD-L1 t-haNK cell-treated groups of 8–10 mice per group with an average initial tumor volume of ∼100 mm3. The treated cohorts were injected with 1×107 cells PD-L1 t-haNK cells intraperitoneally one or two times per week. For the combination study, C67BL/6 mice were inoculated with MOC1 cells (5×106 cells/mouse, subcutaneously). Tumors grew for 10 days (such that the average tumor volume was ~80 to 100 mm3) before randomizing into groups of eight. The tumor-bearing mice were administered with PD-1 (200 mg, intraperitoneally), N-803 (1 µg, subcutaneously) or PD-L1 t-haNK cells (107 cells, intraperitoneally) as monotherapy, doublets or triplets on days 10, 17, and 24. For all tumor studies, tumor growth was monitored. The animal studies were terminated within a week after the last treatment or when the tumors reached the ethical limit. Liver and lungs were collected and metastatic tumor nodules were manually counted. Lungs were perfused with India ink to increase contrast.
Statistical analyses
Student’s t-test was used to determine the significance of two-group comparisons. One-way analysis of variance (ANOVA) was used to compare more than two groups. Two-way ANOVA was used to determine the significance of multiple comparisons between two or more groups. Tukey’s or Sidak’s post hoc test was used to correct both one-way and two-way ANOVAs. Pearson’s correlation coefficient was calculated to determine the correlation between two factors, and linear regression was used to calculate the best-fit lines. P values less than 0.05 were considered significant. Error bars represent means±SEM.
Results
PD-L1 t-haNK cells are haNK cells engineered to target PD-L1-expressing tumor cells
We have previously demonstrated that irradiated haNK cells (10 Gy) continued to express NK markers such as NKG2D and CD56 and maintained high levels of granzyme B and perforin.9 10 Here, we characterized PD-L1 t-haNK cells, which are haNK cells that have been engineered to express a CAR receptor composed of an extracellular single-chain variable fragment that binds to PD-L1, a transmembrane domain, and an FcεRIγ intracellular domain for downstream signaling. RNA analysis of irradiated PD-L1 t-haNK and irradiated haNK cells using the nCounter PanCancer Immune Profiling Panel (NanoString) revealed that the two cell types have comparable gene expression of common NK markers, perforin, and granzyme B (figure 1A). Of the genes investigated, 15.4% were deregulated by threefold or more (44 genes upregulated and 72 genes downregulated) in PD-L1 t-haNK cells compared with haNK cells (online supplementary figure S1). IPA focused on NK cell-related pathways revealed that this set of genes is involved in NK activation and migration (online supplementary table S3). Flow cytometric analysis verified that PD-L1 t-haNK and haNK cells have comparable expression of CD16, perforin, and granzyme (figure 1B). More importantly, it demonstrated that PD-L1 t-haNK cells maintained the expression of CAR molecules that are capable of binding to PD-L1 post-irradiation and post-thaw (figure 1B). NKG2D and perforin expression of PD-L1 t-haNK cells was confirmed via immunofluorescence microscopy (figure 1C).
Figure 1.

PD-L1 t-haNK cells are haNK cells engineered to target PD-L1-expressing tumor cells. (A) Immune-related transcriptomes of irradiated PD-L1 t-haNK and haNK cells were analyzed using the nCounter PanCancer immune profiling panel. Heatmap showing select NK-related genes with data presented as fold change values on a scale of −3 (blue) to 3 (red). (B) Representative FACS plots showing the frequencies of anti-PD-L1 CAR+, CD16+, and perforin+granzyme B+ irradiated PD-L1 t-haNK and haNK cells. Results are from two independent experiments and were gated on live cells. Blue histograms represent controls, while red histograms represent samples. (C) Immunofluorescence microscopy data showing perforin (green) and NKG2D (orange) expressions on irradiated PD-L1 t-haNK cells. (D) MDA-MB-231 tumor cell lysis by PD-L1 t-haNK and haNK cells with or without anti-PD-L1 antibody (1 µg/mL) was evaluated via 111In-release assay at 25:1 E:T ratio. Results shown are the means with SEM of triplicate measurement and representative of three independent experiments. (E) Cytolytic capacity of PD-L1 t-haNK cells on CMA and anti-CD16 treatment with MDA-MB-231 as target at 25:1 E:T ratio as evaluated in 111In-release assay. Results shown are the means with SEM of triplicate measurement. (F) PD-L1 t-haNK cell-mediated killing of MDA-MB-231 tracked in real time using live-cell imaging. CellTracker violet BMQC was used to differentiate the PD-L1 t-haNK cells (blue) from the MDA-MB-231 tumor cells (bright field), and CellEvent-caspase 3/7 was used to identify cells undergoing apoptosis. Scale bars on microscopy images (C, F) represent 10 µM. One-way analysis of variance with Tukey’s multiple comparison test was used for statistical analyses. *P<0.05, **P<0.01, ***P<0.005, ****P<0.001. CAR, chimeric antigen receptor; CMA, concanamycin A; E:T, effector to target; FACS, fluorescence-activated cell sorting; haNK, high-affinity NK; NK, natural killer; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer.
Next, we examined the cytolytic function of PD-L1 t-haNK cells. The triple negative breast cancer (TNBC) cell line MDA-MB-231 was used as target because it has been shown to express high levels of PD-L1,23 and we have previously shown that it is susceptible to haNK cell ADCC-mediated lysis.9 10 We observed that anti-PD-L1 antibody (1 µg/mL) augmented haNK cell lytic activity through ADCC mechanisms (p<0.05, figure 1D). PD-L1 t-haNK cells mediated MDA-MB-231 cell lysis at a higher rate compared with haNK cells with anti-PD-L1 antibody (p<0.01). The difference may be due to the complex interactions involved in ADCC versus the integrated extracellular recognition and intracellular signaling with CAR that can accelerate the activation of PD-L1 t-haNK cells.17 Indeed, real-time tracking of cell lysis revealed that PD-L1 t-haNK cells initially killed MDA-MB-231 at a faster rate, but after an extended incubation, the overall killing was comparable between the two NK cell lines (online supplementary figure S2). Anti-PD-L1 antibody decreased the activity of PD-L1 t-haNK cells (figure 1D) likely through competition for PD-L1 binding, implying that this combination may not be a viable option for therapy.
The anti-EGFR antibody cetuximab has previously been shown to elicit ADCC-mediated killing of MDA-MB-231 by haNK cells.9 Although PD-L1 t-haNK cells express high-affinity CD16, their cytolytic activity against MDA-MB-231 was not enhanced by cetuximab nor was it diminished by CD16 blockade (figure 1E). This suggests that PD-L1 t-haNK cell cytotoxicity is more dependent on the CAR pathway than ADCC in the context of PD-L1high cells such as MDA-MB-231. On the other hand, CMA effectively mitigated PD-L1 t-haNK cell-mediated tumor cell lysis (p<0.005; figure 1F), demonstrating perforin and granzyme B release as a critical mechanism of action of PD-L1 t-haNK cell killing.
We employed live cell imaging to observe PD-L1 t-haNK cell-mediated killing of MDA-MB-231 in real time (figure 1F). When a PD-L1 t-haNK cell associated with an MDA-MB-231 cell, it resulted in the activation of the caspase 3/7 pathway in the tumor cell, ultimately triggering the target’s apoptotic cell death. Overall, these data demonstrated that PD-L1 t-haNK cells are specialized haNK cells engineered to target PD-L1 expressing tumor cells via CAR.
PD-L1 t-haNK cells induced the cell lysis of various human cancer cell lines in vitro
Through In111-release assays, we found that PD-L1 t-haNK cells efficiently lysed different human cancer cell lines, including breast (n=5, 3 of which are TNBC), lung (n=3), colon (n=2), urogenital (n=2), ovarian (n=1), chordoma (n=1), meningioma (n=1), and gastric (n=5) cancer cell lines at varying degrees (figure 2A and online supplementary figure S3). To determine the relationship between PD-L1 t-haNK cell-mediated cell lysis and PD-L1 expression, we calculated the Pearson correlation coefficient of % cell lysis with % PD-L1 score, which was calculated by scoring each cell line for % positive cells and for mean fluorescence intensity (MFI) on a quartile scale ranging from 1 to 4. The sum of the two values was considered the PD-L1 score.24 The degree of cell lysis of a cancer cell line as a result of PD-L1 t-haNK cell activity strongly corresponded to its PD-L1 score (figure 2B).
Figure 2.

PD-L1 t-haNK cells induced the lysis of a range of human cancer cell lines in vitro. (A) Cell lysis of MDA-MB-231, BT549, T47D, SUM149, MCF7, H460, H441, HCC4006, SW480, SW620, DU145, HTB1, CaSKi, CH22, and IOMM mediated by PD-L1 t-haNK cells and haNK cells±anti-PD-L1 AB (1 µg) at different E:T ratios evaluated in 111In-release assay. Results shown are the means with SEM of triplicate measurement and representative of at least two independent experiments for each cell line. (B) Correlation between PD-L1 score and % tumor cell lysis (p=0.0133, r=0.6222). The PD-L1 score of each cell line was calculated by scoring the % PD-L1+ cells and the MFI on a quartile scale of 1–4 and then adding the two values. The best-fit lines were determined using linear regression. E:T, effector to target; MFI, mean fluorescence intensity; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer.
As in figure 1D, we also found that at 50:1 E:T ratio, PD-L1 t-haNK cells induced a greater tumor cell killing than haNK cells on all cell lines tested, except SW620, which expressed low PD-L1 (figure 2A). The addition of anti-PD-L1 antibody generally improved haNK cell activity through ADCC mechanisms.9 10 The increased haNK cell-mediated lysis on anti-PD-L1 antibody addition was particularly noticeable in cell lines with high PD-L1 scores.
After establishing the correlation between PD-L1 expression and the % cell lysis induced by PD-L1 t-haNK cells, we investigated whether modulating the cell surface expression of PD-L1 affects PD-L1 t-haNK cell-mediated tumor cell lysis. We pretreated four breast cancer cell lines (MDA-MB-231, BT549, T47D, and MCF7) with IFN-γ, which has been reported to regulate PD-L1 expression,25 and then used these cells as PD-L1 t-haNK cell targets. We observed that IFN-γ treatment upregulated % PD-L1 and/or MFI in the four breast cancer cell lines, which corresponded to a significant improvement in cell lysis induced by PD-L1 t-haNK cells (figure 3).
Figure 3.
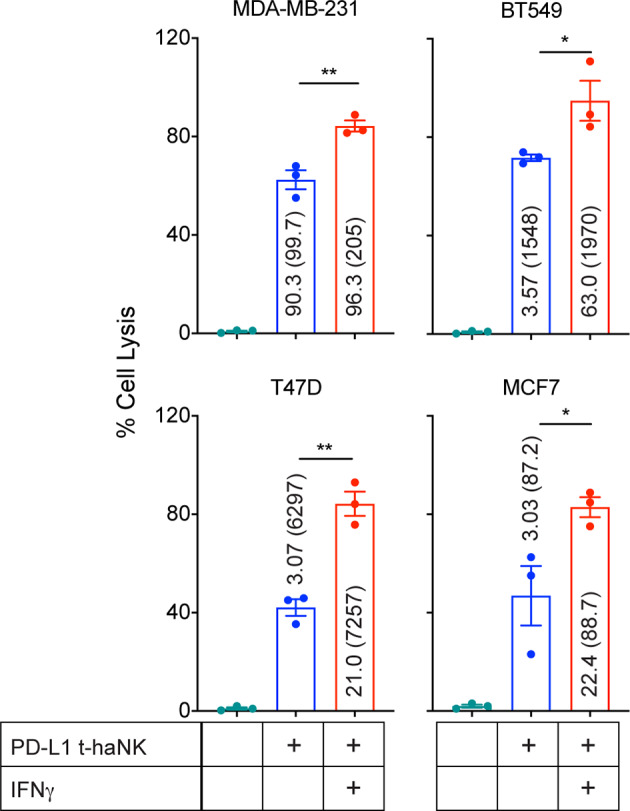
IFN-γ pretreatment improved PD-L1 t-haNK cell targeting of human breast cancer cell lines in vitro. MDA-MB-231, BT549, T47D, and MCF7 were pretreated overnight with IFN-γ prior to being incubated with PD-L1 t-haNK cells. Cell lysis was evaluated in 111In-release assay at 25:1 E:T ratio. Values represent the %PD-L1 (MFI) for each cell treatment. Results shown are the means with SEM of triplicate measurement and representative of two independent experiments. One-way analysis of variance with Tukey’s multiple comparisons test was used for statistical analyses. *P<0.05, **P<0.01. E:T, effector to target; IFN, interferon; MFI, mean fluorescence intensity; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer.
PD-L1 t-haNK cells preferentially lysed PD-L1high over PD-L1low human breast cancer cell line in vitro
After determining that cell lysis by PD-L1 t-haNK cells is correlated to the level of PD-L1 expression on the tumor cell surface, we examined the specificity of PD-L1 t-haNK cells against PD-L1+ cells. To address this, cocultures of CFSE-labeled PD-L1high (MDA-MB-231) and CellTrace Violet-labeled PD-L1low (BT549, MCF7, and T47D) breast cancer cells were coincubated with PD-L1 t-haNK cells at different E:T ratios overnight (figure 4A–F). The numbers of viable residual PD-L1high and PD-L1low breast cancer cells were then analyzed by flow cytometry. The results showed a greater decrease in the PD-L1high population, demonstrating that PD-L1 t-haNK cells preferentially lysed the cells expressing the CAR target.
Figure 4.

PD-L1 t-haNK cells preferentially targeted PD-L1high versus PD-L1low human breast cancer cell line in vitro. Carboxyfluorescein succinimidyl ester (CFSE)-labeled PD-L1high MDA-MB-231 was cocultured with CellTrace Violet-labeled PD-L1low breast cancer cell lines BT549 (A, B), MCF7 (C, D), and T47D (E, F). CellTrace Violet-labeled IFN-γ-treated MDA-MB-231 was cocultured with CFSE-labeled untreated cells (G, H). Flow-sorted PD-L1high and PD-L1low MDA-MB-231 cell populations were labeled with CFSE and CellTrace Violet, respectively (I, J). Irradiated PD-L1 t-haNK cells were incubated with the cocultures at different E:T(PD-L1high):T(PD-L1low) overnight prior to flow cytometric analysis to determine cell lysis. The flow cytometric plots shown have been stratified to live cells and have been downsampled, such that all the plots for each coculture have the same cell count in every E:T:T ratio. The numbers indicate the cell count for each population in the downsampled plots. Data are representative of two independent experiments for each cell line for panels A–F. E:T, effector to target; IFN, interferon; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer.
To ensure that the difference in targeting is not due to the difference in cell type, we treated MDA-MB-231 cells with IFN-γ, which we have shown to increase PD-L1 expression (figure 3), and labeled these cells with CFSE prior to coculture with untreated CellTrace Violet-labeled MDA-MB-231 (figure 4G). We also flow-sorted for PD-L1high and PD-L1low MDA-MB-231 cells and labeled the two cell populations with CFSE and CellTrace Violet, respectively (figure 4I). After coincubation with PD-L1 t-haNK cells, we observed that these effector cells preferentially killed the MDA-MB-231 populations that have higher PD-L1 expression. This confirms that the PD-L1 t-haNK cell activity is robust against tumor cells that express its target.
Next, PD-L1 was knocked out of the MDA-MB-231 via CRISPR/Cas9 (PD-L1 null MDA-MB-231). The PD-L1 knockout was confirmed via flow cytometry (figure 5A), western blot (figure 5B), and immunofluorescence staining (figure 5C). Predictably, the removal of PD-L1 cell surface expression resulted in the markedly diminished ability of PD-L1 t-haNK cells to lyse the PD-L1 null MDA-MB-231 compared with the WT (p<0.005, figure 5D). We also observed that PD-L1 t-haNK cell cytotoxicity against PD-L1 null MDA-MB-231 cells is comparable to the cytotoxicity of haNK cells against WT and PD-L1 null MDA-MB-231 cells (figure 5E), again illustrating the importance of the CAR activity of PD-L1 t-haNK cells. Furthermore, the PD-L1 t-haNK cells preferentially killed the WT MDA-MB-231 over the PD-L1 null MDA-MB-231 at the lower E:T:T ratios (figure 5F). Overall, the data demonstrate that the PD-L1 t-haNK cells preferentially target the PD-L1-expressing tumor cells.
Figure 5.

PD-L1 silencing on MDA-MB-231 diminished the ability of PD-L1 t-haNK cells to target the tumor cells in vitro. PD-L1 null MDA-MB-231 was generated through the CRISPR/Cas9 system. PD-L1 knockout was confirmed via (A) flow cytometry, (B) western blot, and (C) immunofluorescence microscopy. Scale bar=75 µm (D). WT MDA-MB-231 and PD-L1 null MDA-MB-231 tumor cell lysis mediated by PD-L1 t-haNK cells was evaluated via 111In-release assay at different E:T ratios. (E) WT MDA-MB-231 and PD-L1 null MDA-MB-231 tumor cell lysis mediated by PD-L1 t-haNK and haNK cells were evaluated via 111In-release assay at 50:1 E:T ratio. Results for via 111In-release assay shown are the means with SEM of triplicate measurement and representative of two independent experiments. (F) CFSE-labeled WT MDA-MB-231 and CellTrace Violet-labeled PD-L1 null MDA-MB-231 were cocultured together and added with irradiated PD-L1 t-haNK cells at different E:T:T ratios overnight before flow cytometric analysis of cell lysis. The FACS plots shown have been stratified to live cells and downsampled such that each plot has the same cell count in each E:T:T ratio. The numbers indicate the cell count for each population in the downsampled plots. Data are representative of two independent experiments for each cell line. Two-way analysis of variance with Sidak’s multiple comparison test was used for statistical analyses. **P<0.01, ***P=0.09, ****P<0.0001. E:T, effector to target; PD-L1, programmed death-ligand 1; CFSE, carboxyfluoresceinsuccinimidyl ester; DAPI,4′,6-diamidino-2-phenylindole; ns, not significant; t-haNK, targeting high-affinity natural killer; WT, wild type.
PD-L1 t-haNK cells trafficked into PD-L1+ tumors and delayed tumor growth
We first determined whether PD-L1 t-haNK cells traffic into the tumors. Female NSG mice bearing WT or PD-L1 null MDA-MB-231 tumors were injected with 107 111In-labeled irradiated PD-L1 t-haNK cells intravenously or intraperitoneally. 111In signal was found in the WT MDA-MB-231 tumors 24 and 72 hours after injection, indicating successful trafficking of PD-L1 t-haNK cells into the PD-L1-expressing tumor (online supplementary figure S4). Furthermore, PD-L1 t-haNK cell tumor infiltration was greater in the WT MDA-MB-231-bearing mice than in the PD-L1 null MDA-MB-231-bearing cohort 24 hours after treatment.
Next, we investigated the antitumor therapeutic effect of these effector cells. Female NSG mice (n=10/group) bearing WT or PD-L1 null MDA-MB-231 tumors were treated with irradiated PD-L1 t-haNK cells (107 cells/animal intraperitoneally) two times a week for four consecutive weeks. Significant tumor growth control was achieved in PD-L1 t-haNK cell-treated WT MDA-MB-231-bearing mice on day 17 (figure 6A). Meanwhile, significant tumor growth inhibition was achieved in the PD-L1 null MDA-MB-231-bearing mice much later on day 28 (figure 6B). The delayed ability of PD-L1 t-haNK cells to inhibit PD-L1 null MDA-MB-231 in vivo corresponded with the decreased PD-L1 t-haNK cell-mediated cell lysis observed in vitro (figure 5D) and reduced PD-L1 t-haNK cell trafficking (online supplementary figure S4).
Figure 6.

PD-L1 t-haNK cells inhibited primary and metastatic tumor growth in vivo. (A, B) Female NSG mice (10–16 weeks old) were inoculated with WT (A) or PD-L1 null MDA-MB-231 (B) tumors. The tumor-bearing mice (n=10/group) were treated with irradiated PD-L1 t-haNK cells intraperitoneally two times per week for 4 weeks and tumor growth was monitored. Data shown are representative of two independent experiments. (C, D) Macrometastases were counted from liver (C) and lung (D) tissues collected from the WT MDA-MB-231-bearing mice cohorts. (E) PD-L1 t-haNK cell and CMA-treated irradiated PD-L1 t-haNK cells were injected into MDA-MB-231 tumor-bearing female NSG mice (n=10/group) once a week for 4 weeks and tumor growth was monitored. (F, G) HTB1-bearing (F, n=7–8 mice/group) and H460-bearing (G, n=10/group) female NSG mice were treated with PD-L1 t-haNK cells once a week and tumor growth was monitored. Arrows in the growth curves indicate PD-L1 t-haNK cell treatment. Two-way analysis of variance with Tukey’s (E) or Sidak’s (A, B, F, G) multiple comparisons test statistical analyses of the tumor growth curves. *P<0.05, **P<0.01, ***P<0.005, ****P<0.001. Student’s t-test was used for the statistical analyses of the metastatic lesions. *P<0.05. CMA, concanamycin A; NSG, NOD-scid IL2Rgammanull; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer; WT, wild type.
The aggressive nature of MDA-MB-231 can result in the dissemination of the cancer cells leading to the development of metastases in multiple sites.26 27 To investigate the metastatic disease burden, organs were harvested from the WT MDA-MB-231 cohort 2 days after the last treatment and examined.
Significantly more macroscopic metastatic lesions were found in the liver and lungs of the control animals than in the corresponding tissues of the PD-L1 t-haNK cell-treated mice (figure 6C).
Next, we investigated whether PD-L1 t-haNK cells could provide therapeutic effect when given less frequently. Female NSG mice (n=10/group) were implanted with MDA-MB-231 and were injected with PD-L1 t-haNK cells (107 cells/mouse intraperitoneally) once a week (figure 6E). Compared with the control group, tumor growth inhibition was observed in the PD-L1 t-haNK cell-treated animals starting on day 14 (p<0.05). Also, the anti-tumor response was abrogated when the PD-L1 t-haNK cells were pretreated with CMA, indicating the importance of perforin and granzyme B in the activity of PD-L1 t-haNK cells in vivo.
Lastly, we examined whether PD-L1 t-haNK cells could also be effective against other solid tumor xenografts. HTB1 and H460, which are human bladder and lung tumor models, respectively, were established onto female NSG mice (n=7–10/group, figure 6F). PD-L1 t-haNK cells (107 cells/mouse intraperitoneally) were administered to the animals once a week for four consecutive weeks and tumor growth was monitored. For both models, PD-L1 t-haNK cell-treated animals had significantly smaller tumor volumes compared with their control counterparts. Due to the accelerated growth of H460, the animals were only given PD-L1 t-haNK cells two times, and the experiment had to be aborted early before the ethical tumor size limit was reached. Nevertheless, we were able to show the efficacy of PD-L1 t-haNK cells in MDA-MB-231, HTB1, and H460 xenograft models.
PD-L1 t-haNK cells in combination with anti-PD-1 antibody and N-803 resulted in improved tumor growth control
Other studies have reported that N-803 (formerly ALT-803), an IL-15 superagonist complexed with IL-15RαSushi-Fc fusion protein, significantly expanded and activated NK cells in mice.28 29 We investigated whether N-803 could improve the cytolytic activity of PD-L1 t-haNK cells by coincubating the two immunotherapeutic agents with MDA-MB-231 tumor cells (figure 7A). We observed that N-803 had no synergistic effect with PD-L1 t-haNK cells; however, it should be noted that the activity of PD-L1 t-haNK cells alone was already high. In addition, PD-L1 t-haNK cells do not express the IL-15 receptor (not shown). On the other hand, we found that N-803 was able to increase the activity of healthy donor NK cells (figure 7B); thus, while N-803 did not directly enhance PD-L1 t-haNK cells, it is capable of activating endogenous NK cells that can potentially contribute an additional antitumor effect.
Figure 7.

PD-L1 t-haNK cells, α-PD-1, and N-803 combination therapy resulted in superior tumor growth control. (A, B) PD-L1 t-haNK (A) and NK cells isolated from healthy human donor PBMCs (B) were cocultured with MDA-MB-231 cells in the presence of N803 (50 ng/mL), and target cell killing was assessed via 111In-release assay. (C) Splenocytes from C57BL/6 mice were incubated with varying amounts of N-803 for 24 hours. Afterwards, IFN-γ levels in the culture supernatant were quantified via ELISA. (D) MOC1 cells were incubated for 24 hours in supernatants harvested from untreated or N-803-treated (1 μg) splenocyte cultures as described in panel C. As positive control, MOC1 cells were incubated with IFN-γ (20 ng/mL). The MOC1 cells were then analyzed for PD-L1 expression and used as target cells for PD-L1 t-haNK cell-mediated killing via 111In-release assay. (E) Model of indirect enhancement in killing of tumors by N-803 mediated increase of tumor PD-L1. (F) C67BL/6 mice were transplanted with MOC1 cells. The tumor-bearing mice (n=8 mice/group) were treated starting at day 10 (tumor volume ~80 to 100 mm3) with once weekly PD-1, N-803 or PD-L1 t-haNK cells alone or in concurrent combination for a total of three treatments. The arrows below the x-axis of growth plots indicate individual or concurrent treatments. Inset under the legend for each plot is the number of established tumors that rejected with treatment. Two-way analysis of variance with Tukey’s multiple comparison test was used for statistical analyses. ***P=0.09, ****P<0.0001. IFN, interferon; MOC1, mouse oral cancer 1; NK, natural killer; PBMC, peripheral blood mononuclear cell; PD-L1, programmed death-ligand 1; t-haNK, targeting high-affinity natural killer.
Next, we explored whether N-803 could have an indirect effect on PD-L1 t-haNK cell–cell activity. N-803 has been demonstrated to stimulate IFN-γ production in mouse and human immune cells.30 Indeed, we have detected significant IFN-γ levels in the supernatant of murine splenocytes cultured with immobilized N-803 (approximately 2 ng/mL IFN-γ for all N-803 concentrations tested, figure 7C). When MOC1 cells were incubated in the supernatant harvested from N-803-treated splenocyte cultures, PD-L1 t-haNK cell killing of the MOC1 cells was improved and the improvement corresponded with the increased PD-L1 expression on the tumor cells (figure 7D). Therefore, we were able to demonstrate that N-803 could potentially increase PD-L1 t-haNK cell cytolytic activity by inducing other immune cells to produce IFN-γ, which results in elevated PD-L1 expression on tumor cells, thereby making them more targetable by PD-L1 t-haNK cells (figure 7E).
We tested the therapeutic efficacy of PD-L1 t-haNK cells in combination with N-803 and anti-PD-1 antibody in a MOC1 murine tumor model (figure 7F). C57BL/6 mice (n=8/group) were implanted with MOC1 cells and treatment was commenced when the average tumor size reached 80–100 mm3, wherein PD-L1 t-haNK cells (107 cells, intraperitoneally), N-803 (1 µg, subcutaneously on the contralateral flank), and anti-PD-1 antibody (200 mg, intraperitoneally) were administered weekly for 3 weeks. Monotherapy with N-803 or anti-PD-1 antibody was not effective in inhibiting MOC1 tumor growth. On the other hand, weekly administration of PD-L1 t-haNK cells significantly reduced the tumor burden in the mice. Contrary to our in vitro data, the PD-L1 t-haNK cells+N-803 combination did not result in better tumor growth control when compared with PD-L1 t-haNK cell treatment alone. Likewise, the combination of PD-L1 t-haNK cells and anti-PD-1 antibody did not result in improved therapeutic benefit over PD-L1 t-haNK cell treatment alone. However, when PD-L1 t-haNK cells, N-803, and anti-PD-1 were all coadministered, tumor growth was significantly impeded with five out of eight animals completely rejecting the tumor. N-803 and anti-PD-1 each exerts an effect on T cells: N-803 expands and activates T cells while anti-PD-1 maintains T-cell function by minimizing T-cell exhaustion.28 29 31 A phase Ib trial employing N-803 with nivolumab has shown that this combination resulted in increased frequency and activity of CD8+ T cells32; this may explain the modest antitumor activity we observed in our N-803+anti-PD-1 group. Our animal data imply that superior antitumor effect could be achieved by having robust NK and T-cell compartments, and this could be attained through the combination of PD-L1 t-haNK cells with immunomodulators that expand, enhance, and enable the T cells.
PD-L1 t-haNK cells lysed human MDSC cells in vitro
A previous report demonstrated that different immune cell types in PBMCs from patients with cancer have varied PD-L1 expressions.22 We investigated whether PD-L1 t-haNK cells have any adverse effects on human peripheral immune cells. PBMCs from human healthy donors were coincubated with PD-L1 t-haNK cells at different E:T ratios for 24 hours before flow cytometric analysis for different immune subsets (figure 8A). We observed that PD-L1 t-haNK cells had no effect on NK, NK-T, CD4+ T and CD8+ T cells but promoted the expansion of the regulatory T cell (Treg) (p<0.05) and B-cell populations (p<0.001). However, we observed that the peripheral blood MDSC population was drastically decreased by PD-L1 t-haNK cells by up to 90% (p=0.001 for all E:T ratios tested, figure 8A). Furthermore, PD-L1 t-haNK cells were capable of killing both monocytic (in two out of two healthy donors) and granulocytic (in one out of two healthy donors) MDSCs (figure 8B). In addition, killing assays performed using transwell chambers demonstrated that PD-L1 t-haNK cell-mediated killing of MDSCs was contact dependent (figure 8B).
Figure 8.

PD-L1 t-haNK cells decreased human PBMC-derived MDSC population in vitro. (A) PBMCs from healthy HDs were cocultured with PD-L1 t-haNK cells at different E:T ratios overnight and then analyzed for different immune subset populations (n=11). (B) Healthy HD PBMCs and PD-L1 t-haNK cells (0.5:1.0 E:T ratio) were separated by a transwell insert and incubated overnight prior to flow cytometric analysis of MDSC populations. MDSCs were identified as CD11b+ CD33+ HLA-DRlow/− and were either CD14+ or CD15+. (C, D) PBMCs from patients with prostate cancer (C, n=3) and patients with HNSCC (D, n=3) were cocultured with PD-L1 t-haNK cells at different E:T ratios and were analyzed for mMDSC (CD11b+ CD33+ HLA-DRlow/− CD14+ CD15−) and gMDSC (CD11b+ CD33+ HLA-DRlow/− CD14− CD15+) populations after an overnight coincubation. (E) MDSCs were isolated from healthy donor PBMCs using a CD33 isolation kit and were used as PD-L1 t-haNK cell targets. Cell lysis was evaluated through 111In-release assay at 20:1 E:T ratio. **P<0.01. cDC, conventional dendritic cells; E:T, effector to target; gMDSC, granulocytic myeloid-derived suppressor cell; HD, human donor; HNSCC, head and neck squamous cell carcinoma; MDSC, myeloid-derived suppressor cell; mMDSC, monocytic myeloid-derived suppressor cell; NK, natural killer; PBMC, peripheral blood mononuclear cell; PD-L1, programmed death-ligand 1; pDC, plasmacytoid dendritic cells; t-haNK, targeting high-affinity natural killer.
Among the different immune cells in the peripheral blood of patients with cancer, the MDSCs express high levels of PD-L1.22 We sought to determine whether the PD-L1 t-haNK cell-mediated anti-MDSC activity we observed in healthy donor PBMCs also holds true in patients with cancer PBMCs. PBMCs from patients with prostate cancer (n=3, figure 8C) and head and neck squamous cell carcinoma (n=3, figure 8D) were obtained and cocultured with PD-L1 t-haNK cells for 24 hours before the frequency of MDSCs was assessed via flow cytometry. Coincubation of patients with cancer PBMCs with PD-L1 t-haNK cells resulted in the reduction of peripheral MDSCs by more than 60% in both cancer types. To determine whether the MDSC killing was a direct effect of PD-L1 t-haNK cells, we isolated MDSCs from healthy donor PBMCs and performed an 111In-release assay. Figure 8E validates that MDSC lysis by PD-L1 t-haNK cells did not rely on other immune cells but is a direct consequence of the activity of PD-L1 t-haNK cells itself.
Discussion
Immunotherapy is a powerful tool that has transformed cancer treatment. Immune checkpoint inhibitors such as anti-PD-1 and anti-PD-L1 have demonstrated clinical activity and are currently being used as first-line or second-line treatment for different cancer types.13 14 More recently, the Food and Drug Administration approved two CD19 CAR-T cells, axicabtagene ciloleucel and tisagenlecleucel, as therapies for patients with relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma.33 Both immunotherapy approaches are important but are not without challenges. For example, only a small fraction of patients demonstrate response to anti-PD-1/PD-L1 treatment with more than half of the patients with positive PD-L1 expression not benefiting from the treatment.15 Autologous CAR T cells, on the other hand, are time-consuming and expensive to generate, and their success in hematological malignancies has yet to be conveyed in solid tumors.34 These limitations highlight the need to seek and develop alternative immunotherapy options to treat cancer.
Recently, CAR-engineered NK cells have received increasing attention as more evidence shows that these effectors induce potent antitumor activity in preclinical studies (reviewed in Rezvani et al and Zhang et al16 17). The current study investigated the antitumor efficacy of PD-L1 t-haNK cells, which is a novel human, allogeneic NK cell line that has been engineered to express a CAR targeting tumor-associated antigen PD-L1, high-affinity variant (158V) of CD16/FcγRIIIa receptor, and an ER-retained IL-2. These characteristics of the PD-L1 t-haNK cell allow it to target tumor cells in three distinct mechanisms: CAR-mediated killing, ADCC-mediated killing, and native NK receptor-mediated killing.
In vitro, 20 of the 20 tumor cell lines used in this study were shown to be lysed by PD-L1 t-haNK cells in vitro, including breast (three of which are TNBCs), lung, colon, urogenital, ovarian, chordoma, meningioma and gastric cancer cell lines at varying degrees (figure 2A and online supplementary figure S3). The PD-L1 t-haNK cytolytic activity was more robust than the parental haNK cell activity (figures 1D and 2A). However, haNK cell killing could generally be improved by extending the incubation time (online supplementary figure S2) or by promoting ADCC mechanisms via the addition of anti-PD-L1 antibody (figure 2A). PD-L1 expression was correlated to the efficacy of PD-L1 t-haNK cell-mediated lysis (figure 2B), denoting that the PD-L1 t-haNK cell effectively recognizes its cognate tumor-associated antigen via the anti-PD-L1 CAR. In fact, removal of the PD-L1 target reduced the ability of the PD-L1 t-haNK cell to lyse MDA-MB-231 cells to a level that is comparable to that of haNK cells (figure 5D, E). Furthermore, in several cocultures of PD-L1low and PD-L1high breast cancer cell lines, it was observed that PD-L1 t-haNK cells selectively lysed the PD-L1high tumor targets (figure 4). The cytotoxic activity of the PD-L1 t-haNK cell against its tumor targets was found to be dependent on the perforin/granzyme B pathway (figure 1B) and the activation of caspase3/7 (figure 1F). Taken together, the data demonstrated that the engineered CAR promoted the specific activity of the PD-L1 t-haNK cells against PD-L1expressing tumor cells in vitro.
In vivo, we have shown that PD-L1 t-haNK cell treatment resulted in profound growth inhibition of PD-L1-expressing MDA-MB-231, HTB1, and H460 tumors. Moreover, PD-L1 t-haNK cells prevented the development of MDA-MB-231 metastatic lesions in the liver and lungs. For claudin-low breast cancers like MDA-MB-231, PD-L1 expression is induced by the epithelial to mesenchymal (EMT) transition and is important for the maintenance of the EMT status.35 36 PD-L1 is also expressed in the cancer stem cell population of MDA-MB-231 and is important in the process of cell renewal.37 38 Therefore, in addition to killing primary tumors, PD-L1 t-haNK cells may also be targeting premetastatic cells and cancer stem cells that are essential for the dissemination and invasion of tumor cells that lead to the formation of metastatic lesions.
In addition to the PD-L1-directed CAR, the PD-L1 t-haNK cells retained their expression of native activating receptors such as NKp46 and NKG2D (figure 1), which suggests that the PD-L1 t-haNK cells have the ability to exert anticancer activity through mechanisms apart from that dictated by the specificity of the CAR. In line with this, we have observed that PD-L1 t-haNK cells have the capacity to lyse tumor cells that have low-level PD-L1 expression (figure 2). Furthermore, although we have shown that PD-L1 knockout resulted in reduced cytotoxic activity of PD-L1 t-haNK cells, an appreciable magnitude of cell lysis was still observed in the PD-L1 null MDA-MB-231 in vitro (>50% at 25:1 E:T; figure 5D). Our data suggest that against PD-L1 low or null targets, PD-L1 t-haNK cells essentially perform as haNK cells (figure 5E). More notably, we have shown that repeated PD-L1 t-haNK cell treatment resulted in significant, although delayed, tumor growth control of PD-L1 null MDA-MB-231 in NSG mice. The delay in tumor growth control corresponded with the decreased PD-L1 t-haNK cell-mediated cell lysis observed in vitro and reduced PD-L1 t-haNK cell trafficking (online supplementary figure S4). Despite the delayed response, this observation is very critical since the loss or mutation of CAR-targeted antigen is one of the mechanisms of resistance observed in CAR-T-cell therapy.39 The data suggest that innate characteristics of the PD-L1 t-haNK cells have an important antitumor role and could potentially minimize the risk of relapse or resistance to this effector cell.
Although PD-L1 t-haNK cells expressed high-affinity CD16, ADCC-mediated killing of MDA-MB-231 with cetuximab was not observed (figure 1E). haNK cells, which are similar to PD-L1 t-haNK cells but lacking the CAR, have been shown to mediate ADCC through the anti-PD-L1 (avelumab) and anti-EGFR (cetuximab) IgG1 monoclonal antibodies9 10; hence, in principle, the PD-L1 t-haNK cells are capable of killing through this means. The difference in the stoichiometry and steric hindrances in the initiation of CAR signaling and ADCC signaling favors the CAR in terms of signaling rate and strength that the contribution of ADCC to the overall antitumor activity was masked.17 Nevertheless, ADCC is an important aspect of CAR-NK killing and future investigations should characterize this aspect of PD-L1 t-haNK cell killing. PD-L1 t-haNK cell-mediated ADCC might be better observed when targeting tumor cells that have low expression of PD-L1.
In the pathological disease state, tumor cells evade the antitumor response of tumor-specific T cells by inducing cell surface expression of PD-L1 in response to IFN-γ.25 This mechanism of tumor escape could be exploited to enhance the activity of PD-L1 t-haNK cells. Thus, anticancer agents, such as N-803 (figure 7C–F), that can promote IFN-γ production by immune cells and/or PD-L1 expression on cancer cells are rational candidates for strategic combination with PD-L1 t-haNK cells. Therapies such as chemotherapy, radiation, IL-15, and IL-12 that have been reported to have anticancer therapeutic benefits when combined with anti-PD-L1 checkpoint blockade and have also been found to promote NK activity and trafficking are some of the logical choices for combination.28 40–42 In addition to agents that promote NK activity, we have presented that the combination of PD-L1 t-haNK cells with immunomodulators, such as N-803 and anti-PD-1 antibody, which are known to activate and maintain T cell functions, is a viable strategy for cancer treatment (figure 7E28 29 31).
One potential advantage of CAR-NK cells over CAR-T cells is the low occurrence of graft versus host disease and other alloimmune diseases observed with NK adoptive transfer.16 17 However, some immune cells express PD-L1,22 and these populations could potentially be targeted by PD-L1 t-haNK cells. Coculture assays of PD-L1 t-haNK cells with healthy donor PBMCs showed that PD-L1 t-haNK cells did not affect NK, NKT, CD4 T, and CD8 T cells. PD-L1 t-haNK cells stimulated the expansion of B cells and Tregs. The parental haNK cells have been shown to secrete IL-2,10 which plays a key role in Treg and B cell proliferation.43 44 Although IL-2 has an essential lymphoproliferative effect, well-documented toxicities related to this cytokine pose a risk.45 Clinical data showed that haNK cells are generally well tolerated with no treatment-induced ‘cytokine storm’ observed.46–48 In contrast to the effect on Tregs and B cells, PD-L1 t-haNK cells caused the contraction of the peripheral MDSC population in both healthy donors and patients with cancer populations (figure 8). MDSCs have a crucial role in promoting and maintaining the immunosuppressive condition in the tumor microenvironment (TME), including expressing high levels of PD-L1 to promote PD-1/PD-L1 signaling in the TME.20 22 Interventions that could block or deplete MDSCs have been the focus of numerous studies, and it has been shown that a decrease in MDSC population is correlated with increased T-cell activity and clinical outcome.49 Therefore, MDSC killing is an advantageous effect of PD-L1 t-haNK cells and may be one of the mechanisms by which PD-L1 t-haNK cells can exert antitumor activity.
Because the parental cell line of PD-L1 t-haNK cells, NK-92, was derived from a patients with progressing non-Hodgkin’s lymphoma, malignant formation in patients is a concern. In order to prevent this, PD-L1 t-haNK cells will be irradiated prior to infusion into patients to inhibit proliferation while maintaining cytotoxicity. Non-clinical NK-92 studies show that mice injected with irradiated NK-92 cells did not develop detectable tumors.1 Moreover, no engraftment of NK-92 and haNK cells has been reported in clinical studies so far.1 46–48 Irradiation also ensures that the shorter life span of the engineered NK cells will not cause cytopenia, which is one of the side effects of CAR T cells.50 However, this also means that repeated infusions with PD-L1 t-haNK cells will be necessary. Tracking studies demonstrated that PD-L1 t-haNK cells persist in the tumors for up to 72 hours, indicating that PD-L1 t-haNK cells have to be ideally administered two times a week to ensure the maintenance of the cells in the TME.
The data presented in this report and the safety track record of NK-92 and haNK cells render PD-L1 t-haNK cells as a potential new agent for cancer treatment. Clinical studies have to be performed to better assess the efficacy and safety of PD-L1 t-haNK cells. An ongoing phase I monotherapy study in patients with locally advanced or metastatic solid tumor malignancies (NantKwest sponsored) will evaluate the overall safety profile of PD-L1 t-haNK cells and will provide dosing and safety data required for subsequent clinical studies.
Conclusions
We demonstrate that PD-L1 t-haNK cells target various human cancers in vitro. We also show that these cells inhibit tumor growth in several tumor xenograft models. The results provide a rationale for the potential use of these cells in clinical studies.
Acknowledgments
The authors thank the NCI Flow Cytometry Core Facility (Dr Ferenc Livak) and NCI Confocal Microscopy Core Facility (Dr Michael Kruhlak) for their assistance and expertise in flow cytometry and live-cell imaging, respectively. The authors also thank Debra Weingarten for her editorial assistance in the preparation of this paper.
Footnotes
Contributors: KPF, CTA, JHL, SR, PS-S, JS, and JWH conceptualized and designed the research studies. KPF, MRP, RND, KS, YR, and JWH conducted the experiments and acquired the data. KPF, MRP, RND, JS, and JWH analyzed the data. KPF and JWH wrote the manuscript. KPF, MRP, RDN, KS, YR, CTA, JHL, SR, PS-S, JS, and JWH reviewed the manuscript.
Funding: This work was funded by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), and via a Cooperative Research and Development Agreement between the NCI and NantBioScience, Inc.
Disclaimer: The authors from the National Cancer Institute do not have any competing interests to disclose.
Competing interests: JHL is an employee of NantKwest and ImmunityBio; RS is an employee of NantOmics and ImmunityBio; PS-S is a founder of NantKwest, NantOmics and ImmunityBio.
Patient consent for publication: Not required.
Ethics approval: Cryopreserved peripheral blood mononuclear cells were obtained from healthy donors at the NIH Clinical Center Blood Bank (NCT00001846), from patients with prostate cancer enrolled in a phase II study at the NCI (NCT00514072), and from patients with head and neck neoplasms enrolled on a Biospecimen procurement protocol through the National Institute on Deafness and Other Communication Disorders (NCT0342906). All experimental studies were performed under the approval of the NIH Intramural Animal Care and Use Committee. All mice were housed and maintained in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request
References
- 1.Suck G, Odendahl M, Nowakowska P, et al. . NK-92: an 'off-the-shelf therapeutic' for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother 2016;65:485–92. 10.1007/s00262-015-1761-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994;8:652-8. [PubMed] [Google Scholar]
- 3.Yan Y, Steinherz P, Klingemann HG, et al. . Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res 1998;4:2859-68. [PubMed] [Google Scholar]
- 4.Tam YK, Miyagawa B, Ho VC, et al. . Immunotherapy of malignant melanoma in a scid mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother 1999;8:281–90. 10.1089/106161299320316 [DOI] [PubMed] [Google Scholar]
- 5.Maki G, Klingemann HG, Martinson JA, et al. . Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res 2001;10:369–83. 10.1089/152581601750288975 [DOI] [PubMed] [Google Scholar]
- 6.Arai S, Meagher R, Swearingen M, et al. . Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 2008;10:625–32. 10.1080/14653240802301872 [DOI] [PubMed] [Google Scholar]
- 7.Tonn T, Schwabe D, Klingemann HG, et al. . Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013;15:1563–70. 10.1016/j.jcyt.2013.06.017 [DOI] [PubMed] [Google Scholar]
- 8.Williams BA, Law AD, Routy B, et al. . A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017;8 10.18632/oncotarget.19204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jochems C, Hodge JW, Fantini M, et al. . An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016;7:86359-86373. 10.18632/oncotarget.13411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jochems C, Hodge JW, Fantini M, et al. . Adcc employing an NK cell line (haNK) expressing the high affinity CD16 allele with avelumab, an anti-PD-L1 antibody. Int J Cancer 2017;141:583–93. 10.1002/ijc.30767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel SP, Kurzrock R. Pd-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–56. 10.1158/1535-7163.MCT-14-0983 [DOI] [PubMed] [Google Scholar]
- 12.Hamanishi J, Mandai M, Matsumura N, et al. . Pd-1/Pd-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol 2016;21:462–73. 10.1007/s10147-016-0959-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong J, Chehrazi-Raffle A, Reddi S, et al. . Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 2018;6 10.1186/s40425-018-0316-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol 2019;12:92. 10.1186/s13045-019-0779-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Chen W, Xu ZP, et al. . Pd-L1 distribution and perspective for cancer Immunotherapy—Blockade, knockdown, or inhibition. Front Immunol 2019;10 10.3389/fimmu.2019.02022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezvani K, Rouce R, Liu E, et al. . Engineering natural killer cells for cancer immunotherapy. Mol Ther 2017;25:1769–81. 10.1016/j.ymthe.2017.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, Oberoi P, Oelsner S, et al. . Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol 2017;8:533. 10.3389/fimmu.2017.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Judd NP, Winkler AE, Murillo-Sauca O, et al. . Erk1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res 2012;72:365–74. 10.1158/0008-5472.CAN-11-1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onken MD, Winkler AE, Kanchi K-L, et al. . A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res 2014;20:2873–84. 10.1158/1078-0432.CCR-14-0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol 2010;185:2273-84. 10.4049/jimmunol.1000901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lepone LM, Donahue RN, Grenga I, et al. . Analyses of 123 peripheral human immune cell subsets: defining differences with age and between healthy donors and cancer patients not detected in analysis of standard immune cell types. J Circ Biomark 2016;5:5. 10.5772/62322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donahue RN, Lepone LM, Grenga I, et al. . Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J Immunother Cancer 2017;5:20. 10.1186/s40425-017-0220-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittendorf EA, Philips AV, Meric-Bernstam F, et al. . Pd-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2014;2:361–70. 10.1158/2326-6066.CIR-13-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyerinas B, Jochems C, Fantini M, et al. . Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res 2015;3:1148–57. 10.1158/2326-6066.CIR-15-0059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong H, Strome SE, Salomao DR, et al. . Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793-800. 10.1038/nm730 [DOI] [PubMed] [Google Scholar]
- 26.Iorns E, Drews-Elger K, Ward TM, et al. . A new mouse model for the study of human breast cancer metastasis. PLoS One 2012;7:e47995. 10.1371/journal.pone.0047995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puchalapalli M, Zeng X, Mu L, et al. . NSG mice provide a better spontaneous model of breast cancer metastasis than athymic (nude) mice. PLoS One 2016;11:e0163521. 10.1371/journal.pone.0163521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim PS, Kwilas AR, Xu W, et al. . Il-15 superagonist/IL-15RαSushi-Fc fusion complex (IL-15SA/IL-15RαSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016;7:16130–45. 10.18632/oncotarget.7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu B, Jones M, Kong L, et al. . Evaluation of the biological activities of the IL-15 superagonist complex, ALT-803, following intravenous versus subcutaneous administration in murine models. Cytokine 2018;107:105–12. 10.1016/j.cyto.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhode PR, Egan JO, Xu W, et al. . Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models. Cancer Immunol Res 2016;4:49–60. 10.1158/2326-6066.CIR-15-0093-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alsaab HO, Sau S, Alzhrani R, et al. . Pd-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol 2017;8:561. 10.3389/fphar.2017.00561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrangle JM, Velcheti V, Patel MR, et al. . ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1B trial. Lancet Oncol 2018;19:694–704. 10.1016/S1470-2045(18)30148-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter BD, Rogalski M, Jacobson CA. Chimeric antigen receptor T-cell therapy for the treatment of aggressive B-cell non-Hodgkin lymphomas: efficacy, toxicity, and comparative chimeric antigen receptor products. Expert Opin Biol Ther 2019;19:1157–64. 10.1080/14712598.2019.1644316 [DOI] [PubMed] [Google Scholar]
- 34.Newick K, O'Brien S, Moon E, et al. . Car T cell therapy for solid tumors. Annu Rev Med 2017;68:139–52. 10.1146/annurev-med-062315-120245 [DOI] [PubMed] [Google Scholar]
- 35.Alsuliman A, Colak D, Al-Harazi O, et al. . Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: significance in claudin-low breast cancer cells. Mol Cancer 2015;14:149. 10.1186/s12943-015-0421-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noman MZ, Janji B, Abdou A, et al. . The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017;6:e1263412. 10.1080/2162402X.2016.1263412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almozyan S, Colak D, Mansour F, et al. . Pd-L1 promotes Oct4 and Nanog expression in breast cancer stem cells by sustaining PI3K/Akt pathway activation. Int J Cancer 2017;141:1402–12. 10.1002/ijc.30834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu J-M, Xia W, Hsu Y-H, et al. . STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun 2018;9:1908. 10.1038/s41467-018-04313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sotillo E, Barrett DM, Black KL, et al. . Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 2015;5:1282–95. 10.1158/2159-8290.CD-15-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakai H, Kokura S, Ishikawa T, et al. . Effects of anticancer agents on cell viability, proliferative activity and cytokine production of peripheral blood mononuclear cells. J Clin Biochem Nutr 2013;52:64–71. 10.3164/jcbn.12-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujii R, Jochems C, Tritsch SR, et al. . An IL-15 superagonist/IL-15Rα fusion complex protects and rescues NK cell-cytotoxic function from TGF-β1-mediated immunosuppression. Cancer Immunol Immunother 2018;67:675–89. 10.1007/s00262-018-2121-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinhuis KM, Ros W, Kok M, et al. . Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Annals of Oncology 2019;30:219–35. 10.1093/annonc/mdy551 [DOI] [PubMed] [Google Scholar]
- 43.Mingari MC, Gerosa F, Carra G, et al. . Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature 1984;312:641–3. 10.1038/312641a0 [DOI] [PubMed] [Google Scholar]
- 44.Ye C, Brand D, Zheng SG. Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther 2018;3:2. 10.1038/s41392-017-0002-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartz RN, Stover L, Dutcher JP. Managing toxicities of high-dose interleukin-2. Oncology 2002;16:11-20. [PubMed] [Google Scholar]
- 46.Bhatia SBM, Zhang H, Lee T. Adoptive cellular therapy (act) with allogeneic activated natural killer (aNK) cells in patients with advanced Merkel cell carcinoma (MCC): preliminary results of a phase 2 trial. Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD, 2016. [Google Scholar]
- 47.Carlson E, Kistler M, Soon-Shiong P. NANT cancer vaccine an orchestration of immunogenic cell death by overcoming immune suppression and activating NK and T cell therapy in patients with third line or greater TNBC and head & neck SCC (abstr). Society for Immunotherapy of Cancer Annual Meeting, Washington, DC, 2018. [Google Scholar]
- 48.Soon-Shiong P, Lee J, Seery T. NANT cancer vaccine an orchestration of immunogenic cell death by overcoming immune suppression and activating NK and T cell therapy in patients with third line or greater metastatic pancreatic cancer (abstr). Society for Immunotherapy of Cancer Annual Meeting, Washington, DC, 2018. [Google Scholar]
- 49.Tobin RP, Davis D, Jordan KR, et al. . The clinical evidence for targeting human myeloid-derived suppressor cells in cancer patients. J Leukoc Biol 2017;102:381–91. 10.1189/jlb.5VMR1016-449R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016;127:3321–30. 10.1182/blood-2016-04-703751 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2019-000450supp001.pdf (241.2KB, pdf)