

Abstract
Polycomb repressive complex 1 (PRC1) is critical for mediating gene expression during development. Five chromobox (CBX) homolog proteins, CBX2,4,6,7,8, are incorporated into PRC1 complexes, where they mediate targeting to trimethylated lysine 27 of histone H3 (H3K27me3) via the N-terminal chromodomain (ChD). Individual CBX paralogs have been implicated as drug targets in cancer; however, high similarity in sequence and structure among the CBX ChDs provide a major obstacle in developing selective CBX ChD inhibitors. Here we report the selection of small, focused, DNA-encoded libraries (DELs) against multiple homologous ChDs to identify modifications to a parental ligand that confer both selectivity and potency for the ChD of CBX8. This on-DNA, medicinal chemistry approach enabled the development of SW2_110A, a selective, cell-permeable inhibitor of the CBX8 ChD. SW2_110A binds CBX8 ChD with a Kd of 800 nM, with minimal 5-fold selectivity for CBX8 ChD over all other CBX paralogs in vitro. SW2_110A specifically inhibits the association of CBX8 with chromatin in cells and inhibits the proliferation of THP1 leukemia cells driven by the MLL-AF9 translocation. In THP1 cells, SW2_110A treatment results in the significant decrease in the expression of MLL-AF9 target genes, including HOXA9, validating the previously established role for CBX8 in MLL-AF9 transcriptional activation, and defining the ChD as necessary for this function. The success of SW2_110A provides great promise for the development of highly selective and cell permeable probes for the full CBX family. In addition, the approach taken provides a proof-of-principle demonstration of how DELs can be used iteratively for optimization of both ligand potency and selectivity.
Keywords: DNA-encoded chemical library, Polycomb Chromobox homolog (CBX) proteins, Affinity selection assays, Chromodomain, MLL-AF9 Leukemogenesis
INTRODUCTION
Polycomb group (PcG) proteins are transcriptional repressors required for proper body segmentation in Drosophila1 and for maintaining progenitor cell populations in mammals.2 PcG proteins are part of two distinct complexes, polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2) (Figure 1A).3 Canonical polycomb function, as defined in Drosophila, begins with PRC2-mediated trimethylation of lysine 27 of histone 3 (H3K27me3), which recruits PRC1 via the chromodomain (ChD) of the chromobox homolog (CBX) subunit. PRC1 then compacts chromatin and ubiquitinates lysine 119 on histone H2A to promote transcriptional repression.4,5
Figure 1.

(A) (left) Canonical polycomb-mediated gene repression. Trimethyllysine marks installed by PRC2 on H3K27 are recognized by ChDs in the CBX subunit of PRC1. This is followed by monoubiquitination at H2AK119, chromatin compaction, and transcriptional repression. (right) Paralogous subunits assemble combinatorially to produce distinct PRC1 complexes with unknown differential function. (B) Preparation and selection of DNA-encoded chemical libraries for CBX ChD ligands. Positional scanning libraries were prepared by parallel chemical modification of an amine-modified oligonucleotide immobilized on DEAE Sepharose as a solid support. Subsequent encoding was performed by parallel PCRs with unique templates. Libraries were pooled, split, and selected against immobilized CBX ChDs. Enriched libraries were then pooled for DNA sequencing.
In mammals, PRC1 subunits are represented by multiple, mutually exclusive paralogs that combinatorially assemble into dozens of distinct PRC1 complexes (Figure 1A).6,7 The CBX subunit is represented by five paralogs (CBX2, CBX4, CBX6, CBX7, CBX8) that shift in expression during development,8,9,10,11,12 as well as cancer progression.13,14 Studies in multiple cell types show that individual paralogs, even when expressed simultaneously, have unique and non-overlapping functions in development and disease.8,10 In particular, CBX8 has recently emerged as a potential oncogenic target in multiple malignancies. It drives growth in lymphoma,15 hepatocellular carcinoma,16 breast cancer,17 and leukemia with MLL translocations.18 While a dependency on CBX8 has been defined at the genetic level, potential druggable sites on the protein have not been explored, and chemical probes specifically targeting CBX8 have not been developed.
The high flexibility of the apo structures, the shallow, extended nature of the peptide binding site, and the high sequence similarity among paralogs present significant challenges for development of potent and selective inhibitors to CBX ChDs. Computational analysis of the peptide-bound polycomb CBX ChD structures suggests that the aromatic cage forming the trimethyllysine binding site is “druggable” based on binding site volume, enclosure, and hydrophobicity.19 Yet, prior work has shown this site to be particularly difficult to target with traditional small molecules. Two reported small molecule ChD inhibitors target CBX7,20,21 yet these molecules display weak (~20 µM) affinity for CBX7 and over 10-fold weaker affinity for CBX8. Larger molecular weight trimethyllysine-containing peptidomimetics (5–6-mers) developed for CBX4,22 CBX6,23 and CBX722,24 ChDs display much greater affinity (<1 µM); however, they have limited cell permeability.
An additional challenge lies in developing ChD inhibitors with specificity for one paralog over another. There is high sequence similarity among the CBX ChDs, particularly among the polycomb CBX ChDs (CBX 2,4,6,7,8) that recognize H3K27me3 (>67% conserved residues).25 Moderate selectivity has been achieved for CBX6 ChD,23 and CBX7/CBX4 ChDs.22,24 No ligand has been developed with selectivity for CBX8 ChD.
To address these challenges, we have employed DNA-encoded chemical libraries, which have numerous advantages over conventional ligand optimization approaches.26,27 In a previous study, we described an approach for synthesizing and selecting small DNA-encoded libraries (DELs) of peptidic compounds against a panel of targets (see Figure 1B for overview). We used previously published CBX7 ChD ligands with over 10-fold selectivity over CBX8 ChD to develop quantitative metrics for affinity selection assays of DNA-encoded libraries against CBX ChDs.22 We demonstrated that selection assays are capable of faithfully replicating known SAR of CBX7 and CBX8 ChD ligands and identified 5 monomers that increased affinity and selectivity to CBX8 ChD.28 In this manuscript, we utilize DNA-encoding and affinity selection with on-DNA medicinal chemistry optimization to obtain CBX8 ChD inhibitors with high affinity (3–800 nM), selectivity (>5–20-fold over other paralogs), and cell permeability. We used these ligands as chemical probes to define the CBX8 ChD as a therapeutic target in MLL-AF9 leukemia.
RESULTS AND DISCUSSION
In Vitro Selection Assays of Ligands to CBX Chromodomains via DNA-Encoded Positional Scanning Library (PSL)
First-generation DNA-Encoded Positional Scanning Library (PSL1)
We performed selections of a DNA-encoded positional scanning library (PSL1) reported in Denton et al.28 against all five ChDs of the polycomb CBX paralogs and the ChD of CBX5, an HP1 protein (SI 1). As observed previously with selections against CBX8 and CBX7 ChDs, position –2 (P(−2), see SI 1 for overview) was the most critical position for determining selectivity for binding to CBX paralogs due to differences in the size of a hydrophobic binding pocket lined by two valines and a leucine in CBX4 and CBX7, but by a valine, leucine, and alanine in CBX2 and CBX8.22–29 We synthesized two off-DNA ligands (KED97 and KED98) composed of monomers that gave improved affinity and selectivity for CBX8 over CBX7. Both peptides are highly selective for CBX8 over CBX7, with KED98 showing the best CBX8 ChD selectivity over all other CBX ChDs (SI 2A–C). Unfortunately, a diethyllysine variant of KED97, KED97L, displayed no activity in published CBX8-dependent assays of cell viability,18 transcription,30 and chromatin binding (SI 2D–F).31 Although the diethyllysine substitution has shown to increase cellular permeability without compromising binding affinity for CBX ChDs,15 this ligand still displays poor cellular permeability (SI 2G) in the chloroalkane penetration assay (CAPA).32 While this is the first reported CBX8-specific ligand, the low selectivity and cell permeability severely limit its utility.
Second-generation DNA-encoded Positional Scanning Library (PSL2)
To identify probes of CBX8 ChD with improved affinity, selectivity, and cellular permeability, we designed and synthesized a second-generation positional scanning library (PSL2) around KED98, the most selective CBX8 ligand derived from PSL1. For PSL2, we again varied the four positions to the N-terminal side of the trimethyllysine. Several monomers were chosen to expand upon the structure-activity relationships (SAR) observed with PSL1.28 In particular, we explored a number of P(−2) side chains with sizes between the valine and the cyclopentyl group to optimize binding in the ChD hydrophobic pocket. Based on the methyl isoxazole hit, a number of 5-substitued isoxazoles were included at the P(−4) cap along with additional heterocyclic aromatics.17 To facilitate the discovery of cell permeable molecules, we sought to reduce the number of amide bonds and molecular weight through truncation and incorporation of dipeptide mimetics or simple linkers (both rigid and flexible) to bridge side chain binding sites. Therefore, we included a number of monomers that might substitute for two sequential monomers from the parental molecule (either [–1 + –2], [–2 + –3], or [–3 + –4]) (Figure 2, see Figure SI3 for full monomer set).
Figure 2. Select enrichment data from affinity-based selection of 192 compounds (PSL2) against 6 CBX ChD isoforms by DNA sequencing.

(A) Compound KED98 was utilized as the parental ligand for PSL2. Unique monomers (188) were tested in total at four positions along with four replicates of the parental peptide in (B) PSL2A and (C) PSL2B. Parallel selections against all five polycomb CBX ChD and CBX5 ChD using PSL2 library were performed, and selected DNA pools sequenced. A color map designates the enrichment of each library member to a particular CBX ChD. For each position, representative synthons are ordered by enrichment of CBX8 in decreasing order. Enrichment was calculated as the fold change of sequencing reads from selected DNA over sequencing reads of input DNA and was normalized to a non-ligand control.
Using 96 unique 140-mer dsDNA constructs, we prepared the 192-membered PSL2 in two sets: PSL2A and PSL2B. PSL2A incorporated 32 synthons each for positions –1 and –2 and 16 synthons each for combined [–1 + –2] and [–2 + –3] positions. Likewise, PSL2B included 32 synthons each for positions –3 and –4, as well as 16 synthons for combined [–3 + –4] positions and 16 amino acid synthons for position –4 together with an acetate cap.
For position –1 (PSL2A_1–32), a wide variety of lipophilic amino acids were enriched for all paralogs, with the exception of non-α-amino acids (e.g. PSL2A_2 and PSL2A_30) and phenylglycine derivatives (e.g. PSL2A_14). In general, there was little indication that modification of the P(–1) residue could increase selectivity of the parental ligand for CBX8, or any paralog, with the notable exception of compound PSL2A_5, where a naphthyl Phe derivative decreased binding to all paralogs except CBX4 (Figure 2B, SI 3). For position –2 (PSL2A_49–80), synthons with small hydrophobic groups were well tolerated by all polycomb (Pc) CBXs, while non-α-amino acids were not tolerated by any CBX paralogs at this position. The tolerance of larger side chains at P(–2) by CBX8 in particular was reiterated, and synthons as large as phenylglycine (PSL2A_75) were tolerated for CBX8. For position –3, Phe derivatives (e.g. PSL2B_10, PSL2B_13, PSL2B_15) similar to parental synthon Cl-Tyrosine were all favored for binding. For position –4, the substituted isoxazole derivatives, as well as particular additional heterocyclic aromatics were favored or well tolerated by the polycomb CBX paralogs (Figure 2C, SI 3).
Monomers included to substitute for both P[–1 + –2] positions (compounds PSL2A_33–48) were not tolerated, with the exception of gamma-amino butyric acid (PSL2A_35), which could not be confirmed in off-DNA follow up studies (SI 4B). Similarly, monomers intended to substitute for both P[–2 + –3] residues (PSL2A_81–96) were not tolerated. Gratifyingly, two monomers among those included to substitute for both P[–3 + –4] monomers (PSL2B_33–48) were tolerated without a large loss in CBX8 binding. Specifically, ligands with a biphenylcarboxylic acid (PSL2B_42) and phenoxybenzoic acid (PSL2B_48) acyl caps at the [–3 + –4] position demonstrated retained affinity and improved selectivity towards CBX8 (Figure 2C, SI 3).
Several trends in the PSL2 data sets suggested that selection conditions were not sufficiently stringent to yield differential enrichments among high affinity ligands. In prior work with PSL1-derived compounds KED97 and KED98, we observed greater enrichments in the PSL1 selections and greater affinity in displacement assays of valine over cyclopentyl glycine at P(–2) for all Pc ChDs (Figures SI 1 and 2), yet enrichments observed for PSL2A_49 and PSL2A_50 were similar. Additionally, the majority of the acyl cap monomers at P(−4) displayed similarly high levels of enrichment for all the Pc CBX ChDs despite their varied structures (SI 3). In order to more effectively differentiate the top binders, we performed selections against CBX8 a second time using more stringent conditions.
Optimized High Stringency (HS) selections against CBX8 ChD
To increase the stringency of the affinity selection assay, we increased the number and time of bead wash cycles, and further reduced on-bead protein concentration (Figure 3). Under these conditions, valine at P(–2) (PSL2A_50) now showed higher enrichment than cyclopentylglycine (PSL2A_49), consistent with prior PSL1 selections and off-DNA competitive FP assays (Figures SI 1, SI 2A). Overall, few substitutions to the parental ligand at either the P(–1) or P(–3) positions suggested that significant gains in affinity could be achieved (Figure SI 5). Several substitutions at the P(−2) and P(−4) positions gave increased enrichment over the parental ligand (Figure 3).
Figure 3.

Increased stringency selections of PSL2 against CBX8 ChD. Two on-bead effective protein concentrations (0.01x: ~0.5 μM, 0.05x: ~2.5 μM, calculated based on bead capacity), and two washing stringencies (HS: high stringency, increased washing cycles and time; NS: normal stringency) were applied in the selection of PSL2 against CBX8 ChD. Select enrichment data for library molecules with various building blocks in (A) PSL2A at P(−2), (B) PSL2B at P(−4), and (C) PSL2B with capping at P(−3). A color map indicates the enrichment of selected library members to CBX8 ChD under the indicated conditions (increasing stringency bottom to top). Values are ordered by CBX8 enrichment at the highest stringency in decreasing order left to right. (D) For select compounds, enrichment values from the normal stringency (NS 0.05x) CBX8 selections were divided by the enrichments of 4 additional ChDs under the same conditions and were normalized using the ratio of enrichments for the parental ligand. (E) For select compounds, enrichment values from the high stringency (HS 0.01x) selections to CBX8 were divided by the enrichment of the parental ligand under the same conditions. Enrichment was calculated as fold change of sequencing reads from selected DNA over sequencing reads of input DNA, normalized to the enrichment of a non-ligand control.
Among additional substitutions at the –2 position, we now observe the highest enrichment of cyclopropylglycine (PSL2A_52), 3-thienylglycine (PSL2A_57), and L-alanine (PSL2A_76) (Figure 3A). Enrichment of other monomers at P(–2) decreased roughly with increasing side chain size. Similar enrichment was observed for cyclobutaneacetic acid (PSL2A_55), propargylglycine (PSL2A_63), and allylglycine (PSL2A_64), and decreased enrichment was found for allo-isoleucine (PSL2A_51), isoleucine (PSL2A_60), norleucine (PSL2A_53), and phenylglycine (PSL2A_75) compared to the parental compound.
As with P(–2), the higher stringency (HS) selection results for modification at P(–4) showed greater differentiation of high affinity ligands (Figure 3B). Several 3-substituted isoxazole carboxylic acids with various substituents (PSL2B_53, 54, 56, 57) gave improved enrichment with enrichment decreasing roughly with the size of the substituent. In agreement with this SAR was the low enrichment of the desmethyl isoxazole (PSL2B_55). In addition, 5-bromo-2-pyrazinecarboxylic acid (PSL2B_63), 5-bromopyridine-2-carboxylic acid (PSL2B_65), and isoquinoline-3-carboxylic acid (PSL2B_66) gave higher enrichment to CBX8 than the parental ligand, while 2-thiopheneacetic acid (PSL2B_50) and 1H-imidazole-4-carboxylic acid (PSL2B_59) showed lower enrichment. These results were consistent with both published and PSL1 SAR of ligands to CBX8 (SI 1), which have shown increased affinity for benzoyl caps with lipophilic para substituents.28
For the single building blocks intended to substitute for both P[–3 + –4] monomers in the parental ligand (PSL2B_33–48), the increased stringency selections showed a marked decrease in enrichment (Figure 3C). This was particularly the case for the ligands with phenoxybenzoic acid (PSL2B_48) or biphenylcarboxylic acid (PSL2B_42) acyl caps at the [–3 + –4] position, which showed high enrichment under the normal stringency selection conditions.
Selection, Validation, and Structural Optimization of Off-DNA Ligands
To facilitate decision making for off-DNA follow up, we used results from the normal stringency selections for all Pc ChDs as indicators of selectivity and the high stringency selections to CBX8 as indicators of affinity (Figure 3D and 3E). To assess improvements in selectivity, we plotted the ratio of CBX8 enrichment to the enrichment for each of the other isoforms and normalized this value to the same ratio observed for each isoform with the parental compound. Compounds with the P(−3) capping with phenoxybenzoic acid (PSL2B_48) or biphenylcarboxylic acid (PSL2B_42) showed the largest gains in CBX8 selectivity. PSL2B_42 showed improved selectivity against all isoforms, and PSL2B_48 showed improvements in selectivity over CBX4 and CBX6. Compound PSL2A_75 with phenylglycine at P(−2) showed more modest selectivity gains but for all isoforms.
Among the P(−2) substitutions that indicated increased affinity, all showed lower selectivity for CBX 4, 6, and 7. These decreases roughly tracked with decreasing size of the side chain with P(−2) alanine (PSL2A_76) demonstrating the largest decrease in selectivity. For selectivity over CBX4 and 7, this observation is consistent with the structural differences in the binding pocket for this side chain.23 Also, prior work has shown high affinity binding of ligands with L-alanine at this position for all Pc CBX ChDs.23,28 For the P(−4) substitutions that showed high enrichment (3-isopropyl isoxazole was selected as an example) only modest, if any, changes in selectivity were observed.
To facilitate comparison of affinity across the two library selections, which were conducted separately, we plotted the ratio of the enrichment of a compound to the enrichment of the parental ligand under the highest stringency conditions (Figure 3E). For the molecules that indicated improved selectivity, decreased enrichment relative to the parental compound was observed at varying levels, with the greatest decrease due to the P(−2) phenylglycine (PSL2A_75). Similar ratios were observed for the improved affinity P(−2) substitutions, as well as the exemplary isopropyl isoxazole.
Based on this analysis, we selected five molecules for off-DNA synthesis on solid phase for subsequent determination of IC50 values using a competition fluorescence polarization (FP) assay against CBX6, 7, and 8 (Table 1).23 Consistent with sequencing results, both cyclopropylglycine (SW2_90) and 3-thienylglycine (SW2_101E) substitutions at P(−2) have greater affinity to CBX8 than the cyclopentylglycine parental compound (Table 1A). Selectivity for CBX8 with these compounds suffers, particularly against CBX6, however. The larger phenylglycine at this position was tolerated by CBX8 and not CBX7, but this compound (SW2_49B) shows a large decrease in affinity. For the P(−3) capped compounds PSL2B_48 and PSL2B_42 with high selectivity in the sequencing data, this selectivity was confirmed with the off-DNA compounds. The phenoxyphenyl compound SW2_101B shows very high selectivity over both CBX6 and CBX7. The biphenyl compound CBX2_89 shows high selectivity over CBX6. For both of these compounds, affinity to CBX8 is largely unchanged from the parental compound.
Table 1.
IC50 values for off-DNA ligands in a ligand displacement fluorescence polarization assay. (A) Ligands containing trimethyllysine (B) Ligands containing diethyllysine. IC50 values for each ligand against CBX6, CBX7, and CBX8 ChDs, were measured using 100 nM fluorescent probe and 1 µM CBX6, 0.4 µM CBX7, 4 µM CBX8 ChDs. Reported values are the average of quadruplicates ± s.d. NB: No binding observed. ND: Value not determined. In some cases, full curves could not be determined due to IC50’s >100 μM or compound aggregation (Aggreg.). Full binding curves are shown in SI 6 and SI 4.
 |
IC50 (µM) | |||
|---|---|---|---|---|
| Compound | R = | CBX6 | CBX7 | CBX8 |
| KED98 (Parental) |
 |
34 ± 1 | 83 ± 2 | 14 ± 1 |
| SW2_90 (PSL2A_52) |
 |
0.36 ± 0.05 | 0.8 ± 0.1 | 2.1 ± 0.5 |
| SW2_101E (PSL2A_57) |
 |
4.8 ± 0.6 | 83 ± 1 | 3.1 ± 0.8 |
| SW2_49B (PSL2A_75) |
 |
ND | ND (> 100) |
44 ± 8 |
| SW2_101B (PSL2B_48) |
 |
NB | ND (> 100) |
12 ± 1 |
| SW2_89 (PSL2B_42) |
 |
ND (Aggreg.) |
22 ± 1 | 13 ± 1 |
| SW2_101F |  |
ND (> 100) |
1.6 ± 0.3 | 4.0 ± 1 |
| SW2_101A |  |
98 ± 26 | 35 ± 9 | 40 ± 8 |
 |
IC50 (µM) | |||
|---|---|---|---|---|
| Compound | R = | CBX6 | CBX7 | CBX8 |
| SW2_110A | 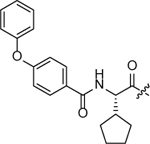 |
ND (Aggreg.) |
ND (Aggreg.) |
>7.0 (Aggreg.) |
| SW2_104B |  |
ND (Aggreg.) |
ND (Aggreg.) |
>15 (Aggreg.) |
| SW2_110B |  |
ND (Aggreg.) |
ND (Aggreg.) |
>5.7 (Aggreg.) |
| SW2_104A |  |
2.7 ± 0.7 | 4.8 ± 0.9 | 1.6 ± 0.6 |
In addition, we combined monomers that were able to substitute for both the –3 and –4 positions (either the phenoxybenzyl or biphenyl) together with the high affinity monomers identified for the –2 position (cyclopropyl, thienyl) (Table 1). SW2_101F (P(−2) cyclopropyl, P(−3) phenoxylphenyl) had increased affinity for CBX8, but gave decreased selectivity for CBX8 over CBX7. Interestingly, the biphenyl cap gave significantly reduced affinity for both CBX8 and CBX7 when paired with the P(–2) cyclopropyl glycine (SW2_101A), while in the context of the P(–2) cyclopentyl glycine (SW2_89), there was little change in affinity. For diethyl compounds SW2_110A, SW2_110B, and SW2_104B, full IC50 curves could not be obtained due to aggregation issues with the fluorescein-containing probe at high compound concentrations. Lastly, we also combined the 5-isopropyl-3-isoxazole at P(–4) with the 3-thienylglycine at P(–2) in compound SW2_104A. Tight binding to CBX8 ChD was observed, albeit with low selectivity.
To measure Kd values, fluorescence polarization assays of fluorescein conjugates were conducted (Table 2) with titration of each of the Pc ChDs. The Kd of SW2_110A-FL for the CBX8 ChD was determined to be ~800 nM, which was similar to the ~700 nM obtained by microscale thermophoresis (MST) (SI 8) and the ~500 nM value obtained with underivatized SW2_110A in a thermal shift assay (SI 9). The affinity of SW2_110A-FL to CBX8 is decreased slightly compared to the KED97L-FL; however, SW2_110A-FL displayed dramatic improvements in selectivity. This compound is completely selective for CBX8 over CBX4 and CBX6, while maintaining 20-fold selectivity over CBX7 and 5-fold selectivity over CBX2. In contrast, the selectivity of SW2_104A for CBX8 ChD was only modestly improved over KED97L while the affinity to all paralogs was increased significantly. The Kd of SW2_104A is 2.9 nM for CBX8 (5.4 nM by MST, SI 8), making it the tightest binding CBX ChD ligand reported to date. Intriguingly, combining the phenoxyphenyl cap with the thienylglycine at P(–2) in compound SW2_110B-FL did not increase binding compared to SW2_110A-FL with cyclopentyl at P(−2), which differs from the difference observed within the 6-mer ligand context (Table 1), suggesting potential crosstalk between the P(−2) pocket and the biphenyl binding region.
Table 2.
Kd values of fluorescein-conjugated ligands to PcG CBX ChDs. Values were determined by direct fluorescence polarization and are displayed as the average of n=4 ± s.d. NB: No binding. ND: Not determined due to an inability to acquire full binding curves due to low affinity or compound aggregation. MST: MicroScale Thermophoresis. TSA: Thermal Shift Assay. *The non-derivatized ligand was used for for Kd determination by TSA. Full binding curves are provided in SI 7 for FP data, SI 8 for MST data, and SI 9 for TSA data.

| ||||||
|---|---|---|---|---|---|---|
| CBX Protein Chromodomain (Kd (µM)) | ||||||
| Compound | R = | CBX2 | CBX4 | CBX6 | CBX7 | CBX8 |
| KED97L-FL |  |
1.1 ± 0.4 | 2.7 ± 0.4 | 0.3 ± 0.1 | 4.7 ± 0.9 | 0.24 ± 0.03 |
| SW2_110A-FL |  |
4.6 ± 0.9 | NB | NB | >16 ± 5 | 0.8 ± 0.2 (MST: 0.7 ± 0.1) (TSA*: 0.5 ± 0.3) |
| SW2_104A-FL |  |
0.05 ± 0.04 | 0.17 ± 0.04 | 0.014 ± 0.004 | 0.8 ± 0.4 | 0.0029 ± 0.0008 (MST: 0.005 ± 0.001) |
| SW2_110B-FL |  |
ND | NB | NB | NB | ND (MST: 0.8 ± 0.1) |
| SW2_90L-FL |  |
0.79 ± 0.01 | 0.17 ± 0.01 | 0.030 ± 0.001 | 0.140 ± 0.008 | 0.110 ± 0.008 |
For moving forward towards an ideal CBX8 chemical probe, we selected the phenoxyphenyl compound SW2_110A. While the fluorescein-conjugate of this compound displayed modest affinity for CBX8 (800 nM Kd), the selectivity profile was far more favorable compared to the high affinity SW2_104A (3 nM Kd). In addition, SW2_110A includes one less amino acid, which will improve the physiochemical properties of this compound as a probe. As SW2_110A, in particular, demonstrated some aggregation effects in the displacement FP assays (Table 1), we tested the solubility of both SW2_110A and SW2_104A in a shake-flask equilibrium assay.33 With 24-hour incubation in PBS, SW2_110A showed solubility at 130 µM, and SW2_104A was soluble to 41 µM.
Structural Basis of SW2_110A Association with CBX8 ChD
To investigate the structural basis of inhibitor binding, we utilized NMR spectroscopy. We collected a series of 1H,15N heteronuclear single quantum coherence (15N-HSQC) spectra on 15N-labeled CBX8 ChD upon the titration of SW2_110A. Addition of the inhibitor led to substantial changes in the CBX8 ChD spectrum, including chemical shift perturbations (CSPs) and disappearance of resonances, indicating binding (Figure 4A). Mapping these CSPs onto the solved structure of the ChD-H3K9me3 complex reveals a cluster of residues with significant CSPs cluster in and around the canonical histone binding pocket, indicating that the inhibitor can directly compete with histone tail binding (Figure 4B and Figure 4C), which was confirmed using a competitive fluorescence polarization of SW_110A_L-FL with H3K27me3 peptides (SI 10A). Note that a ChD-H3K27me3 structure is currently unavailable, but that we have previously demonstrated that H3K9me3 and H3K27me3 bind in the same pocket.34 Importantly, the limited solubility of the inhibitor at high concentrations required introduction of DMSO. A control titration with DMSO only showed minimal CSPs, suggesting that DMSO alone does not significantly alter the ChD structure (SI 10B).
Figure 4.

Structural basis of SW2_110A binding. (A) Overlay of 15N-HSQC spectra of CBX8 CD upon addition of increasing concentration of SW2_110A. Molar ratios are color coded as indicated in the legend inset. (B) Chemical shift perturbations (CSPs) as a function of CBX8 CD residue. Residues for which resonances disappear upon binding are denoted by a blue sphere, and residues missing resonances entirely are denoted with gray spheres. Perturbations were considered significant if they were greater than the average plus one standard deviation (denoted by the blue line), not including the highest 10% of CSP values. (C) Residues with significant CSPs or for which resonances broadened to the point of disappearing upon binding are colored blue on a cartoon representation of CBX8 in complex with H3K9me3 (PDB ID 3I91). The peptide is shown as white sticks, and the aromatic cage residues important for coordinating the methyllysine are shown as gray sticks.
We have previously determined the structural basis of H3K27me3 and H3K9me3 binding using NMR spectroscopy.34 Comparing the CSPs induced upon addition of SW2_110A to those of an H3K27me3 peptide (residues 23–34) reveals the largest differences are in resonances corresponding to E43, N47, and I48, as well as D50, L53, and L54 (SI 10C). Both subsets of resonances are significantly more perturbed upon inhibitor binding as compared to H3K27me3 and were not significantly perturbed upon addition of DMSO alone (SI 10B and SI 10C). Residues E43, N47, and I48 are in the expected location of the P(–1) and P(–2) side-chains, which are Arg(P–1) and Ala(P–2) in the H3K27me3 peptide. In addition, residues D50, L53, and L54 lie where the phenoxyphenyl group is expected to bind. Notably, these residues are in the hydrophobic pocket of the CBX8 ChD, which is a key determinant for CBX8 ChD specificity for H3K27me3. A number of resonances (corresponding to V10, F11, A12, E14, A15, K33, G34, T41, and L49) disappear upon binding (Figure 4B). These residues lie in the β1 strand and the β1-β2 loop, which contain the aromatic cage residues. Based on the crystal structures of the apo CBX8 ChD and the ChD in complex with H3K9me3, the N-terminal portion of the β1-strand is stabilized upon histone tail binding.25 The disappearance of these peaks upon addition of SW2_110A suggests that, in contrast, inhibitor binding may not fully stabilize this region instead leading to conformational exchange in the bound state on the intermediate NMR timescale. Together, the NMR analysis reveals the structural basis of SW2_110A binding and suggests the determinants of histone binding inhibition.
Interestingly, although the inhibitor is selective for CBX8, it associates with regions of the ChD that are conserved between homologues. Indeed, most of the residues that differ between the ChD homologues do not exhibit CSPs upon SW2_110A binding with two exceptions: A15, which is a Ser in CBX6 and CBX7, and S36, which is an Ala in CBX6 and a Pro in CBX7. Neither residue, however, is expected to make direct contact with the inhibitor. To gain further insight into the potential source of selectivity, we investigated binding to two compounds, which demonstrated low selectivity between homologues. Titration of KED97L or KED98L led to very similar subsets of CSPs as compared to SW2_110A, revealing a similar binding pocket (SI 10D). The non-selective KED97L and KED98L, however, led to far fewer disappearance of resonances as compared to SW2_110A, suggesting that both KED97L and KED98L lead to greater stabilization of the CBX8 ChD. This is especially evident in the β1 strand. Together, these data suggest that rather than differences in the manner in which each CD directly coordinates the inhibitor, selectivity likely arises from a difference in the accessible conformational ensemble available to each CBX ChD. This could be modulated by small differences in the ChD sequence ultimately leading to differences in the size and nature of chemical groups that can be accommodated.
Molecular Dynamics Simulations of Ligand Association with CBX8 ChD and CBX6 ChD
To gain insight into the ligand selectivity for CBX8 over CBX6, we carried out MD simulations of CBX8 ChD and CBX6 ChD with the trimethyllysine version of SW2_110A (SW2_101B). Due to the increased selectivity of cyclopentyl glycine at –2 position for CBX8 over CBX6 in off-DNA validations (SI 2A), we, firstly, investigated the interaction of cyclopentyl ring in the –2 position. The closest residue difference within this region is a leucine (CBX8)/isoleucine (CBX6) at the bottom of the –2 binding pocket. We evaluated RMSD traces (Figure 5A) for two sets of five residues at the bottom of the –2 binding pocket in CBX6 (orange) and CBX8 (teal) (residues V30, A13, L53, L16, I48 for CBX8 and V30, A13, L53, I16, I48 for CBX6). This area is directly involved in enclosing the cyclopentyl ring within the pocket (Figure 5C). The RMSDs presented in Figure 5A indicate persistent flexibility of the CBX6-ligand system, and a relative binding stability for CBX8-ligand complex. The increased flexibility in the CBX6 –2 pocket leads to higher cyclopentyl mobility and a coupled disruption of the hydrogen bonding network (illustrated by larger variance of the hydrogen bond distances presented in Figure 5B).
Figure 5.

Molecular dynamics simulations of inhibitor (SW2_101B) binding to CBX8 and CBX6 ChDs. (A) Root Mean Square Deviation (RMSD) plots of surface area portrayed residues at bottom of –2 binding pocket in CBX6 (orange) and CBX8 (teal). RMSDs were taken relative to highest clustered pose taken from the 100 ns trajectories. (B) Hydrogen bonding map for CBX8. H-bonds labeled to their respective panel. A-C hydrogen bonding distances contributed by cyclopentyl alanine and the N-terminus diphenyl ether residues on CBX6 and CBX8. (C) The cyclopentyl ring in the (–2) pocket. The differing residue at the bottom of the (−2) pocket (leucine (CBX8), isoleucine (CBX6)) gives a slight change in pocket shape.
The diaryl ether group determined experimentally to promote SW2_110A selectivity for CBX8 does not appear to preferentially interact with either isoform in the MD simulation data. The terminal phenyl ether group oscillates back and forth to either side of the beta groove for both CBX8 and CBX6, as described via the calculated distances of the terminal phenyl ring to the side of beta-groove region (SI 11D). The intermediate phenyl ring, however, adopts distinct orientations emerging from under the clasp; this likely carries a steric penalty, as well as further contribution to the destabilization of the hydrogen bond network discussed above. Despite the instability below the clasp, the hydrophobic clasps at the –1 position for both isoforms are closed around the ligand in both simulations. The binding geometries in this region are highly similar for CBX6 and CBX8, as shown in the distances between residues V10 and L49 (SI 11B).
Overall, the MD data present a complex binding motif for SW2_110A to CBX6 and CBX8, which is complicated by the collective motion of the two isoforms. However, decreased stability of the binding configuration around the –2 pocket (including the collective effects of binding of the cyclopentyl ring, the hydrogen bonding network and steric effects in the accommodation of the diphenyl ether group) appears to disfavor CBX6 complexation of the SW2_110A ligand.
Cellular Selectivity Studies
Chemoprecipitations
The CBX ChDs demonstrate significant structural flexibility in vitro, making it necessary to determine whether recombinant CBX ChDs accurately recapitulate binding properties of ChDs found within the context of fully formed PRC1. Therefore, we utilized biotinylated derivative SW2_104A-B and SW2_110A-B to enrich CBX8 and its paralogs from mouse embryonic fibroblast (MEF) lysates (Figure 6A-left) and HEK293T lysates (Figure 6A-right) using affinity purification. Both SW2_104A-B and SW2_110A-B robustly enrich CBX8 in support of the in vitro results. Further, enrichment of CBX4, CBX6 and CBX7 is higher for SW2_104A-B than SW110A-B, supporting the in vitro FP assay results with recombinant CBX ChDs that suggest better CBX8 selectivity for SW2_110A. In contrast to the results from both in vitro assays, however, we did observe some enrichment of CBX6 with SW2_110A-B. Considering the high homology between these two ChDs (86% sequence identity), this isn’t surprising, but indicates that in vitro properties for the ChDs may not completely be recapitulated in a cellular environment.
Figure 6:

Cellular activity of CBX8 ChD ligands. (A) Chemoprecipitations from Mouse Embryonic Fibroblast (MEF) (left) and HEK293T (right) nuclear lysates using biotin-labeled SW2_104A (SW2_104A-B) and SW2_110A (SW2_110A-B) were analyzed using immunoblot analysis. (B) Chromatin immunoprecipitation (ChIP) followed by quantitative PCR of genomic regions with CBX8 and CBX7 binding in Hs68 fibroblast cell line. ChIP-qPCR was used to evaluate the ability of SW2_110A to disrupt endogenous CBX protein associations with chromatin in cells. Cells were treated with 100 µM SW2_110A for 4h prior to harvest. ChIP-qPCR of CBX7 and CBX8 at LMNB2 (negative locus), RUNX3, GATA6, and CCND2. For all qPCR, error bars represent SEM n=3 biological replicates, p-values were calculated using two-tailed Student’s t-test, * = p < 0.05, ** = p <0.01, *** = p < 0.001, **** = p <0.0001. (C) Relative cytosolic access of chloroalkane-modified ligands, SW2_110A-CA, SW2_104A-CA and KED97L-CA, was evaluated by chloroalkane penetration assay (CAPA). (D) Dose-dependent cytosolic access of SW2_110A-CA, SW2_104A-CA and KED97L-CA was assessed by preincubation of designated concentrations of CA-molecules, followed by HT-TAMRA dye. CP50 of CA-molecules were evaluated and averaged from three independent curve fits.
ChIP-qPCR
In order to evaluate the ability of SW2_110A to disrupt CBX8 association with chromatin, we used chromatin immunoprecipitation (ChIP) followed by quantitative PCR (ChIP-qPCR) (Figure 6B). Using ChIP-Seq datasets for CBX6, CBX7 and CBX8 in Hs68 fibroblast cells,31 we selected representative target loci with detectable enrichment of CBX7 and CBX8 using ChIP-qPCR.34 Upon incubation of cells with SW2_110A, we observe significant reduction of CBX8 binding at these sites, while CBX7 binding was unaffected or even increased upon treatment with SW2_110A, a phenomenon similarly observed with CBX8 knockdown (SI 12B). Since CBX paralogs primarily localize together at H3K27me3 sites,8,31 it is possible that selective reduction of CBX8 at a genomic locus could increase the enrichment of other paralogs at the same site. To confirm that SW2_110A can disrupt global CBX8 chromatin binding, we also performed sequential salt extraction (SSE), which can determine the impact of chromodomain inhibition on the bulk chromatin binding affinity of CBX paralogs.34,35,36 We confirmed that SW2_110A abrogates CBX8, but not CBX7, binding to bulk chromatin (SI 12B).
Evaluation of Cytosolic Access using ChloroAlkane Penetration Assay (CAPA)
The ChIP-qPCR results imply that SW2_110A has increased cell permeability compared to KED97L. To confirm this difference, we used CAPA, a cell penetration assay recently developed by the Kritzer Lab.32 CAPA is easy to perform, quantitative, and measures compound availability in the cytosol without interference from molecules trapped in endosomes. CAPA utilizes a HeLa cell line stably transfected with a cytosolic HaloTag protein37 in a pulse-chase experiment. Cells are incubated with ligands conjugated to a chloroalkane (CA) (pulse), which will covalently react with the HaloTag protein when/if the ligand reaches the cytoplasm. The cells are then treated with chloroalkane-TAMRA dye (chase), which reacts with any remaining, unblocked HaloTag. The red fluorescence is quantified using flow cytometry, which is inversely proportional to the CA-molecule cytosolic concentration. SW2_110A and SW2_104A were conjugated to the chloroalkane (denoted SW2_110A-CA and SW2_104A-CA) and compared to KED97L-CA. A significant increase in permeability was observed for both SW2_110A-CA and SW2_104A-CA with CP50 values of 26 ± 2 µM and 22 ± 1 µM respectively, compared to CP50 > 100 µM for KED97L-CA (Figure 6C,D).
Cellular Activity in MLL-AF9 Transformed Leukemia
We next wished to evaluate the activity of SW2_110A in a CBX8-dependent cell line. Previous studies identified that CBX8 is required for leukemogenesis in an MLL-AF9 mouse model of leukemia, as well as for the viability of human leukemia cell lines with MLL-AF9 translocations.18 Supporting a requirement for CBX8 in the viability of MLL-AF9 transformed cell lines, we were unable to isolate a CBX8 knockout in the THP1 leukemia cell line using CRISPR-Cas9. As an alternative approach, we used lentiviral-mediated shRNA knockdown of CBX8 and CBX7 and confirmed that CBX8, but not CBX7, is required for maintaining the growth of THP1 cells (Figure 7A). In contrast, the viability of K562 leukemia cells driven by a BCR-ABL translocation38 was not affected by either CBX7 or CBX8 knockdown. To determine whether CBX8 ChD inhibitors can inhibit MLL-AF9 mediated oncogenesis, we measured the viability of THP1 (MLL-AF9 translocation) and K562 (BCR-ABL translocation) leukemia cells cultured with SW2_110A. We observed a significant decrease in the proliferation of THP1 cells starting at 3 days, while the growth of control leukemia cell line K562 was not affected (Figure 7B). Similar proliferation effects were observed with SW2_104A (SI 13A) and the IC50 of the two inhibitors after 12 days of THP1 treatment were similar with IC50 = 26 µM for SW2_110A, and IC50 = 25 µM for SW2_104A (SI 13B).
Figure 7: Dependency on CBX8 for MLL-AF9 Mediated Cell Proliferation and Gene Transcription.

(A) Cell viability of THP1 and K562 cells upon CBX8 and CBX7 knockdown. (B) SW2_110A treatment (100 μM) inhibits proliferation of THP1 cells but not K562 cells. Cells were split at 1:4 on day 6 to avoid over-confluency. s.d. was represented by error bars (n=3, three biological replicates with three technical replicates for each biological replicate). (C) qRT-PCR analysis of CBX8, CBX7, CBX6 and HOXA9 gene expression in THP1 cells with knockdown of CBX8 and CBX7. (D) qRT-PCR analysis of gene expression for MLL-AF9 target genes (HOXA9, CDK6, MYB, RUNX2, and RUNX3) and controls (ACTB and BCR-ABL) in THP1 and K562 cells after 48 hours of 100 μM SW2_110A treatment. (E) qRT-PCR analysis of HOXA9 gene expression in THP1 cells after 48 hours of SW2_110A treatment at the indicated dose. (F) qRT-PCR analysis of MLL-AF9 target gene expression in THP1 cells treated with SW2_110A (100 μM for 24h) after CBX8 knockdown. (G) CBX8 and AF9 genome-wide localization analysis using publicly available ChIP-Seq datasets from K562 cells.51 (H) Genome-wide analysis of CBX8, CBX2 and H3K27me3 peak overlaps in K562 cells using published ChIP-Seq datasets.51 Numbers indicate number of called peaks, overlap percent of total CBX8 peaks. (I) Previously published ChIP-Seq tracks of H3K4me3 and H3K27me3 enrichment at HOXA locus in hematopoetic stem cell (HSC) (top) in HSCs that have undergone differentiation to granulocyte monocyte progenitors (GMP) (middle) and in GMP cells with exogenous MLL-AF9 expression (bottom). Data obtained from Bernt et al.52 and visualized using UCSC genome browser. (J) ChIP-qPCR analysis of AF9, CBX8, and CBX7 enrichment at sites upstream of the HOXA9 transcription start site. Inhibitor treatment was 100 µM for 24h. Error bars represent s.e.m. (n=3) and p-values were calculated using Student’s two-tailed t-test: * = p < 0.05, ** = p < 0.01,*** = p < 0.001, **** = p < 0.0001.
Gene Expression Analysis
The MLL-AF9 translocation is a fusion of the N-terminus of the MLL H3K4 histone methyltransferase with the C terminus of AF9, a subunit of the Super Elongation Complex (SEC). The MLL portion is missing methyltransferase activity and is primarily responsible for targeting, while the AF9 portion drives transcriptional activation.39,40,41 Previous studies identified a paralog-specific interaction between CBX8 and AF9 (or its paralog ENL), which is conserved in the MLL-AF9 (or MLL-ENL) translocation product.18,42,43 MLL-AF9 mediated oncogenesis is mediated primarily through aberrant gene activation,44,45,46 most notably the activation of HOXA9, which drives “de-differentiation” to a hematopoetic stem cell-like state.47,48 Previous work defined CBX8 as required for HOXA9 activation by MLL-AF9,18 which we confirmed using qRT-PCR analysis of HOXA9 gene expression in THP1 cells with knockdown of CBX8 (Figure 7C).
To define whether CBX8 ChD inhibition results in a decrease in the transcription of MLL-AF9 target genes, we performed qRT-PCR for HOXA9, as well as CDK6, MYB, RUNX2, and RUNX3, which have been defined as MLL-AF9 transcriptional targets in THP1 cells.49 All MLL-AF9 targets genes displayed significantly decreased expression after 48 hours of SW2_110A treatment, while non-MLL-AF9 target genes B2M and ACTB were unchanged in the presence of compound. Neither these gene targets, nor BCR-ABL, were altered upon compound treatment in the K562 cell line, consistent with their different oncogenic drivers (Figure 7D). To demonstrate the on-target activity, as well as the kinetic control afforded by the inhibitor, SW2_110A treatment displayed significantly decreased HOXA9 expression with increased inhibitor concentration (Figure 7E). To confirm that the expression changes for MLL-AF9 targets observed with compound treatment are in fact mediated through CBX8, we further analyzed gene expression of MLL-AF9 target genes in THP1 cells treated with SW2_110A after CBX8 knockdown. We found that while both compound treatment and CBX8 knockdown reduced the expression of MLL-AF9 target gene expression, there was no additive effect, which supports that compound effects on gene expression are mediated through CBX8 (Figure 7F).
AF9 and CBX8 genome-wide localization
The specific association between CBX8 and the AF9/ENL proteins is well characterized18,42,43; however, it is still unclear how CBX8, a known transcriptional repressor,50 mediates AF9-mediated gene activation. Additionally confounding is how the ChD of CBX8 specifically is involved in gene activation, as the ChD’s established role is binding H3K27me3, a mark associated with gene repression.6 Using publicly available ChIP-Seq datasets from K562 cells,51 we confirmed that CBX8 is almost exclusively localized at sites with H3K27me3, and that ENL is almost exclusively localized at sites with H3K4me3, a mark associated with gene activation (Figure 7G). Further, ENL binding displays almost no overlap with sites of H3K27me3 or CBX8 binding, providing evidence that CBX8 association does not affect ENL chromatin targeting. In addition, CBX8 binding almost completely overlaps with that of CBX2, a paralog that does not associate with AF9/ENL, providing evidence that AF9/ENL association conversely does not affect CBX8 targeting (Figure 7H).
CBX8 Binding at the HOXA9 Locus
During hematopoetic stem cell (HSC) differentiation to granulocyte monocyte progenitors (GMPs), HOXA9 expression decreases and the repressive H3K27me3 mark spreads towards the HOXA9 coding region (Figure 7I, visualization of data from Bernt et al).52 Transformation of GMPs using exogenous MLL-AF9 re-establishes H3K4me3 and pushes the H3K27me3 boundary back upstream to allow for HOXA9 re-activation (Figure 7I-bottom). Using ChIP-qPCR in THP1 cells, we confirmed robust enrichment of DNA across the HOXA9 locus with AF9 IP, but with the CBX8 IP, no significant enrichment of DNA was observed until ~8.5 kb upstream of the HOXA9 start site where the H3K27me3 boundary begins. This enrichment is reduced upon 24h treatment with SW2_110A, which also reduces AF9 enrichment at the boundary region (Figure 7J). Previous studies have identified an antagonistic relationship for CBX8 and AF9/ENL where overexpression of CBX8 represses AF9 target gene expression,30 and overexpression of ENL activates CBX8 repressed genes.53 This supports a model by which the direct association between AF9 and CBX8 allows for MLL-AF9 to antagonize CBX8 and prevent repressive chromatin from spreading into the HOXA9 locus. Release of CBX8 from the boundary region using SW2_110A likely allows other CBX paralogs to bind, which we observed using ChIP-qPCR of CBX7 (Figure 7J). CBX7 cannot be antagonized by MLL-AF9 so the increase in CBX7 binding can facilitate repressive memory and further deposition of H3K27me3 through the HOXA locus, a function observed for CBX7 in other cell types.54
Conclusions
Using two generations of directed DNA-encoded chemical libraries, we identified selective, cell-permeable, peptidomimetic ligands for the CBX8 ChD. While increased potency and partial selectivity for CBX8 were achieved after the selection in PSL1, a subsequent library led to compounds with further increases in potency, cell permeability, and selectivity. While DELs are often used for hit generation to initiate traditional optimization efforts, this work highlights the ability to use DNA-encoded chemistry within the design-make-test-analyze cycles (DMTA cycles) of medicinal chemistry.55 Compared with traditional synthesis, purification, and discreet screening of individual molecules, we found this DNA-encoded approach to be lower cost and less labor-intensive for the identification and optimization of ligands. These benefits largely arise from the nature of the in vitro selection assay. The ease of this assay allowed concurrent optimization of affinity and selectivity against multiple protein targets. In this assay, it is the concentration of the protein target (not the synthetic ligand) that drives the binding event. Thus, the concentration of the DNA-encoded molecules is insignificant and can be very low, which allows synthesis on a very small scale, permitting the incorporation of monomers that would be too expensive to include using traditional approaches. Several studies have shown how enrichment values from selection assays can correlate to ligand affinity.28,56,57,58 An additional advantage is the low requirement for purity of the synthetic ligands using this approach. While ligand purity can complicate the relationship between the observed enrichment of a molecule and its affinity to the protein target, this can be addressed by performing selections at multiple concentrations, as performed here.59 Likewise, the scale of this approach requires only small amounts of the protein target for the selection assays.
Using this methodology, we have identified the tightest binding and most selective ligands to date for the CBX8 ChD, indicating that new combinations of monomers can improve the affinity of ligands for this target class. Of particular note, we found that a truncated scaffold lacking the position –4 monomer retains affinity to CBX8 and displays improved selectivity. SW2_110A, a ligand containing the truncated phenoxyphenyl cap, is the first CBX ChD ligand to demonstrate complete selectivity over certain isoforms; however, the structural basis for this selectivity is not straightforward, as indicated by both NMR and MD analysis. These structural analyses suggest that unique conformations of the ChDs that are accessible for ligand binding are largely responsible for selectivity, highlighting the difficultly in applying structure-based design for optimizing ligands to these highly dynamic domains.
Importantly, the removal of a single amino acid reduced the molecular weight, reduced the number of rotatable bonds, increased hydrophobicity, and showed improved cell permeability compared to the parental peptidomimetic. A major hurdle for developing peptidomimetics as chemical probes is cell permeability. Identification of SW2_110A as a selective and cell-permeable CBX8 ligand indicates that further improvements can still be made to increase “druglikeness”. The chemical tractability of the phenoxyphenyl group will facilitate further improvements in probe properties as will additional minimization of the ligand through removal of amide bonds.
While there are examples of redundant repressive functions for CBX paralogs,60,61 there are also examples of individual paralogs acting in non-redundant roles.8,9,10,11,12 In particular, CBX8 has been implicated in transcriptional activation in both development and disease.18,62 Previous studies have demonstrated the significance of HOXA9 gene activation for MLL-AF9 leukemogenesis, and the requirement for CBX8 in HOXA9 gene activation; however, the mechanism by which CBX8 activates gene expression has been elusive. In fact, it has been suggested to be through a PRC1-independent function,18,15 which may or may not require the chromodomain. Using our cell permeable CBX8 ChD inhibitors, we have determined that the ChD of CBX8 is required for CBX8-mediated HOXA9 gene activation in MLL-AF9 leukemia and likely works through canonical binding of CBX8 at H3K27me3 to maintain chromatin boundaries via interaction with AF9.52 Based on this model, the gene repression observed upon CBX8 inhibition requires the activity of other CBX paralogs, highlighting the necessity for highly selective CBX inhibitors for determining paralog-specific function.
METHODS
Materials and Methods
Oligonucleotides were purchased from IDT (Coralville, IA) or Bioneer (Alameda, CA) and used as provided. Analytical high-performance liquid chromatography (HPLC) separations were completed using an Agilent 1100 system with detection at 260 nm using a water/MeCN gradient containing 100 mM triethylammonium acetate, pH 5.5. Preparative HPLC separations were completed using a Varian ProStar system with detection at 260 and 280 nm using a water/MeOH gradient containing 0.75% hexafluoroisopropanol, 0.0035% triethylamine, pH 7.0. Reagents and solvents were used as received from commercial sources.
Preparation of 96 Single 140-Mer dsDNA Constructs
The integrated polymerase chain assembly (PCA)–PCR experiments were used to generate 96 single-gene barcode DNA constructs using a modified procedure.63 For each reaction, six pairs of complementary 40-mer DNA oligonucleotides were used.64 Six 40-mer oligos were pooled and used as templates for PCA. Each 5.0 μL PCA reaction contained 0.2 μM of each template 40-mer, with the following: 1.0 mM dNTPs, 0.1 U/μL of Vent DNA polymerase in 1x DNA polymerase buffer (NEB). All thermocycle procedures were as follows: 3 min at 94 ℃, then cycling for denaturation at 94 ℃ for 15 s, annealing at 58 ℃ for 15 s, extension at 72 ℃ for 30 s, and a final extension of 72 ℃ for 5 min after 20 cycles. Each 50 μL PCR reaction contained 5 μL of PCA product, 0.2 mM each dNTP, 0.4 μM of each end primer (ZA and ZD’), and 0.025 U/μL DreamTaq DNA polymerase in 1X DreamTaq buffer (Thermo Fisher). The successive PCR went for 20 cycles using the same thermocycling conditions as PCA. Following PCR, each reaction was purified using SeraMag Carboxylate-Modified Magnetic SpeedBeads (GE Healthcare, Pittsburgh, PA) as previously reported65 and quantified by UV absorbance at 260 nm.
CBX ChD Protein Expression and Purification
CBX chromodomain constructs (addgene plasmid #25158 (CBX2), #25237 (CBX4), #25296 (CBX6), #25241 (CBX7) and #62514 (CBX8, provided by Cheryl Arrowsmith)25 were transformed into chemically competent BL21 CodonPlus RIL E. coli cells (Stratagene, La Jolla, CA) as N-terminal His6–tagged proteins. Bacterial growth was completed at 37 ℃ in LB media to OD600 = 2.0, followed by reducing the temperature to 16 ℃ over 30–60 min and induced with 1 mM IPTG for 16 h. Cells were collected by centrifugation at 6000 rpm for 20 min and resuspended in ChD binding buffer (20 mM Tris, pH 8, 150 mM NaCl, 0.01% Tween 20, 20 mM imidazole) with 1.0 mM PMSF. Bacteria pellets were stored at –80 ℃ until needed. Pellets were thawed on ice for 10 min in ChD binding buffer, and shaked at 4 ℃ supplemented with 100 μg/mL lysozyme, 1 mg/mL CHAPS and 1 mM PMSF, for 30 min. Cells were subsequently lysed by sonication (2x: 15W for 30s on, 30 s off, followed by 1x: 20W for 1 min). The solubilized fraction was collected by centrifugation at 15000 rpm for 40 min at 4 ℃. Meanwhile, Ni-NTA Agarose resin (QIAGEN, Venlo, Netherlands) was washed with H2O and equilibrated with ChD binding buffer. The soluble fraction was incubated with the prewashed Ni-NTA agarose resin at 4 ℃ for 2 h. The resin was then washed 3X with ChD purification buffer (20 mM Tris, pH 8, 150 mM NaCl, 0.01% Tween 20, 1 mM PMSF). Proteins were eluted by the addition of 0.5 M imidazole to ChD purification buffer. The elution was diluted with 30% glycerol, flash frozen, and stored at –80 ℃ until needed. Protein purity was assessed by SDS-PAGE and concentration was determined by the Pierce 660 kit (Thermo Scientific).
Preparation of Kme3-Ser-CPF
The first two residues of the CBX consensus sequence were synthesized in bulk as previously described.28 Briefly, 150 nmol of NH2-5′-CPF in DEAE binding buffer (10 mM HOAc and 0.005% Triton X-100) was split between 6 cartridges. Each contained 220 µL 50% DEAE Sepharose slurry in 50% ethanol and was pre-washed with DEAE binding buffer. The DNA-loaded cartridges were washed 3x 3 mL MeOH. Fmoc-amino acid coupling was achieved by incubating the cartridges in 1 mL of 50 mM Fmoc-amino acid, 50 mM EDC-HCl, and 5 mM HOAt in 40% DMF/ 60% MeOH for 30 minutes at RT, with double couplings. After couplings, the cartridges were washed 3x 3 mL MeOH followed by 3x 3 mL DMF. Fmoc deprotection was achieved by incubating the cartridges in 1 mL of 20% piperidine in DMF for 30 minutes at RT, then washed with 3x 3 mL DMF, 3x 3 mL MeOH, and 1 mL DEAE binding buffer after the final coupling. The DNA was eluted and collected by passing 1 mL of DEAE Elution Buffer (1.5 M NaCl and 0.005% Triton X-100) through each cartridge. The crude conjugate was desalted and concentrated to dryness.
Positional scanning library synthesis.
The purified Kme3-Ser-CPF conjugate was suspended in 4.8 mL DEAE binding buffer. To 96 wells in a 384-well filter plate, 20 µL DEAE Sepharose was added and washed 3x 90 µL DEAE binding buffer. To each well, 50 µL of Kme3-Ser-CFF solution (approximately 1 nmol conjugate per well) was added and washed 3x 90 µL MeOH. Briefly, Fmoc-amino acids were coupled using 50 mM Fmoc-amino acid, 50 mM EDC-HCl, 5 mM HOAt in 40% DMF/60% MeOH for 30 minutes at RT with double coupling and deprotected by 20% piperidine in DMF for 30 minutes at RT. Wells were washed 3x 90 µL MeOH and 3x 90 µL DMF between each step. Following the final chemistry step, wells were washed 3x 90 µL DMF, 3x 90 µL MeOH, and 90 µL DEAE binding buffer. DNA-conjugates were eluted by incubating 2x 40 µL DEAE Elution Buffer in each well for 5 minutes at RT and then collected by centrifugation. Each conjugate was then attached to a unique 140-mer dsDNA template sequence by PCR individually (1X DreamTaq Buffer, 0.5 µM CFF-conjugate (PSL library member), 0.5 µM CPR, 0.2 mM dNTPs, 0.05 ng/µL template, and 0.025 U/µL). All PCRs were pooled and purified by SPRI and quantified by UV absorbance at 260 nm.
Positional scanning library selection against PcG CBX ChDs.
A frozen pellet from an induced 5 mL E. coli culture with CBX-His6 ChD was suspended in 300 μL of ice-cold lysis buffer (20 mM Tris, pH 8, 150 mM NaCl, 100 μg/mL, 1 mg/mL CHAPS, 0.02% Tween-20, 1 mM PMSF) and lysed by sonication for 2 min (3 s on, 3 s off) at 30% power while on ice. The lysate was collected after centrifugation at 4000g at 4 ℃. Meanwhile, 21 μL of His Mag Sepharose Ni (Ni-NTA-Sepharose–MBs) were pre-washed with 3X 21 μL of purification buffer (20 mM Tris, pH 8.0, 150mM NaCl, 20 mM imidazole, 1 mM PMSF, 0.02% Tween- 20). The soluble lysate was then combined with 12 μL of pre-washed Ni-NTA-Sepharose-MBs and incubated at 4 ℃ for 1 h. The MBs were separated and washed in 5X 11 μL of purification buffer. After the last wash, the MBs were suspended in 12 μL of purification buffer. The CBX-bound MBs were split and diluted to yield 10 μL of 1X (~50 μM CBX), 10 μL of 1/10X (~5 μM CBX), and 10 μL of 1/20X (~2.5 μM CBX). The MBs were separated and 10 μL of the DNA pre-mix (50 nM benzylamide (Bz) on DNA construct 97 (nonligand), 0.5 nM 4-BrBA-F-A-I-Kme3-S on DNA construct 98 (high-affinity ligand), and 50 nM CBX positional scanning library-DNA conjugates (approximately 0.5 nM of each library member) in 20 mM Tris, pH 8, 150 mM NaCl, 10 mM MgCl2 , 0.02% Tween-20, 1 mg/mL BSA, 1 mg/mL sheared salmon sperm DNA) was added to all four samples (mock [no protein/MBs only], 50 μM CBX, 5 μM CBX, and 2.5 μM CBX) and allowed to incubate at RT for 1 h. The MBs were then separated and washed 5X in 10 μL of the above buffer. DNA conjugates and protein were eluted by incubating the MBs for 5 min at RT in the above buffer with 0.5 M imidazole. Each elution was collected and prepared for PCR and next-generation sequencing (NGS). The above procedure was applied to selections against all PcG CBX paralog.
Solid phase peptide synthesis (SPPS).
Off-DNA peptides were prepared using traditional SPPS methods. All couplings and deprotections were monitored by ninhydrin tests. Briefly, 50 mg of Rink Amide MBHA resin was swelled for 20 minutes in 1,2-dichloroethane and 20 minutes in DMF before suspension in 20% piperidine in DMF for the initial Fmoc deprotection for 30 minutes at RT. Couplings were completed using 5.0 eq. (relative to the capacity of the resin) of Fmoc-AA (or carboxylic acid), 5.0 eq. HOAt, and 5.0 eq. DIC in DMF (approximately 0.1 M) and pre-activated for 20 minutes at RT before being added to the resin. Fmoc deprotections were achieved by incubating the resin for 30 minutes at RT in 20% piperidine in DMF. Peptides were cleaved and deprotected by incubating in 95% TFA, 2.5% triisopropylsilane, and 2.5% H2O for 3 hours at RT. The crude peptide was collected by precipitation out of ice-cold diethyl ether and then suspended in 50% MeOH/50% H2O and concentrated to dryness. The residue was dissolved in DMSO and purified on a semi-prep HPLC using a H2O/MeOH + 0.1% TFA gradient with detection at 215 nm and 254 nm. Yield was determined by mass of the dried, purified peptide as the TFA salt relative to the equivalents as determined by the mass of resin used. Purity was confirmed to be > 95% by HPLC.
Synthesis of diethyllysine derivatives
Crude peptides were synthesized as described above. After purification of the peptide, reductive amination was accomplished via dissolvation of peptides in 80% MeOH, and 20% DMSO, followed with 100 eq. acetaldehyde and 50 eq. NaCNBH3 (final peptide conc. 0.1M) and incubated at 37 ºC overnight. The mixture was concentrated and HPLC purified as described above.
Synthesis of C-terminal alkyne peptide
Modified methods from previously reported procedure66 were used to synthesize C-terminal alkyne peptides. Polystyrene-linked aldehyde resin (FMPB AM resin, 100 mg, 1.08 mmol/g) was added to a round bottom flask and gently stirred for 30 minutes at RT in DCM. DCM was gently evaporated and 5 mL of DMF with 5 mL of MeOH was added to the resin. To this, 10 eq. glacial AcOH was added with 10 eq. propargyl amine and 10 eq. NaCNBH3 and gently stirred under light reflux for 3 hours at 80 °C. The mixture was cooled and washed with MeOH, DCM, and DMF and re-swelled for 30 mins in 1,2-dichloroethane prior to the first acylation. To the resin, 5.0 eq. Fmoc-Ser(OtBu)-OH with 5.0 eq. DIC, 8.0 eq. HOAt in DMF was added and incubated at 37 °C overnight. The remaining synthesis, purification, and reductive amination were completed as described above. Purity was confirmed to be > 95% by HPLC.
Synthesis of 5-/6-FAM.
To 10.0 mg of 5-/6-FAM NHS ester (ThermoFisher), 422 μL of THF was added. Once dissolved, 7.2 mg (3.0 eq.) of 4-azido-1-aminobutane was added and mixed vigorously. A precipitate initially formed but dissolved upon mixing and then the reaction was incubated at RT, protected from light, and incubated at RT for 16 hours. The reaction was then concentrated and purified by semi-prep HPLC with H2O/MeOH 0.1% TFA gradient.
Synthesis of FAM-peptide conjugates.
To a 150 μL of 100 mM alkyne peptides in DMSO, 13.1 mg of a single isomer of 4-Azido-5/6-FAM (0.5 eq.) was added. To this, 5.0 μL of 2 M TEAA, pH 5.5, and 10 μL 0.1 M aminoguanidinium-HCl was added. Separately, 25 μL of CuBr-saturated DMSO was suspended in 50 μL of 50 mM THPTA and then added to the azide/alkyne mixture. The mixture was incubated at RT overnight and then 10 μL of 0.5 M EDTA, pH 8 was added to the mixture. The FAM-peptide conjugate was purified as described above. Purity was confirmed to be > 95% by HPLC.
Synthesis of Biotin-peptide conjugate
To a 150 μL of 100 mM alkyne peptides in DMSO, 13.1 mg of Biotin-PEG3-azide (2.0 eq.) was added. To this, 5.0 μL of 2 M TEAA, pH 5.5, and 10 μL 0.1 M aminoguanidinium-HCl was added. Separately, 25 μL of CuBr saturated DMSO was suspended in 50 μL of 50 mM THPTA and then added to the azide/alkyne mixture. The mixture was incubated at RT for overnight and then 10 μL of 0.5M EDTA, pH 8 was added. The peptide-biotin conjugate was purified as described above. Purity was confirmed to be > 95% by HPLC.
Synthesis of Chloroalkane Linker
2-(2-azidoethoxy)ethanol was prepared as previously described.67
Competitive Fluorescence polarization (FP) assay of PSL hits against CBX6 ChD, CBX7 ChD and CBX8 ChD.
FP was measured by titration of CBX ChDs to a FITC-labelled probe as previously reported.24 Binding and competition FP assays were performed in black 384-well plates with optical bottoms. Buffer used in FP assays consists of 20 mM Tris, pH 8, 150 mM NaCl, 0.01% Tween 20. The FITC-labeled probe was kept constant at 100 nM with 1 µM CBX6 ChD, 0.4 µM CBX7 ChD or 4 µM CBX8 ChD, concentrations selected based on the reported relative affinity of the CBX ChD protein for the FITC probes. Two-fold dilutions of ligand were used, starting with 500 µM as highest peptide concentration to 0.488 µM as the lowest. Four replicates were tested at each concentration. Raw data were analyzed using GraphPad Prism 7 following a “one site-Fit logIC50” competition model with any outliers (95 % confidence interval) being excluded.
Competitive Fluorescence polarization (FP) assay with H3K27me3
FP assays were performed in black 384-well plates with 100 nM FAM-labelled SW2_110A (SW2_110AL-FL) and 3 µM CBX8 ChD CBX8. Two-fold dilutions of H3K27me3(21–44)) peptide (Active Motif 81052) were added starting at 1 mM as highest peptide concentration. Raw data were analyzed using GraphPad Prism 7 following a “one site-Fit logIC50” competition model.
Direct fluorescence polarization (FP) binding assay of PSL hits against PcG CBX ChDs
FP assays were conducted as above with slight modifications. The FAM-labeled peptide was kept constant at 100 nM except for high-affinity ligands (SW2_104A-FL), where the concentration was reduced to 10 nM. The CBX ChD proteins were titrated by 2-fold series dilutions in the assays with varying protein concentrations, depending on the binding affinity of the ligands. Four replicates were used for each ligand. Raw data were analyzed for determinations of Kd, using GraphPad Prism 7 following a “one-site” total binding model with any outliers (95% confidence interval) being excluded.
Thermal Shift Assay (TSA)
The TSA was performed according to a previous reported protocol.68 In brief, the reaction was run in 20 µL using a standard qPCR machine with a ROX filter (Applied Biosystems). The reaction was run in the following reaction buffer: 10 mM HEPES 7.0, 150 mM NaCl, 8X SYPRO Orange S6651 Invitrogen (5000X stock), 0.2 mg/mL CBX8 ChD, 5% DMSO containing SW2_110A at designated concentrations. Melt curves were obtained using a temperature gradient of 25–75 °C in 40 minutes with readings every 0.5 °C. Melt curves for CBX8 ChD were obtained for four replicates at each ligand concentration and the Tm values were calculated using nonlinear least squares fit on Prism 8. The approximate Kd was calculated from Tm values using nonlinear least squares fit on Prism 8.
Solubility assay
Aqueous solubility assays (Eurofins, ITEM 435) involved assessment of solubility (from 10 mM DMSO stock solution) using a shake-flask method with UV-Vis detection.33 The DMSO stock solution was diluted to 200 μM with phosphate buffered saline (pH 7.4) at room temperature and shaken for 24 h. After centrifugation, the concentration of corresponding soluble compound was determined by HPLC-UV, comparing the peak area obtained with that from a series of reference compounds in DMSO. The results presented are the average of duplicate measurements.
NMR analysis of SW2_110A binding.
The CBX8 CD construct was a gift from Cheryl Arrowsmith (Addgene plasmid #62514). GST-tagged CBX8 CD was created using Infusion and the pGSTag vector, provided by Gerald Crabtree. The CBX8 CD were expressed in BL21 (DE3) pLysS E. coli cells. Cells were grown in LB media at 37 °C to an A600 OD of ~1.0. Cells were pelleted using centrifugation at 4,000 rpm and 18 °C for 10 minutes. Cells were then resuspended in M9 minimal media (4L LB cells per 1L M9) supplemented with 15N-NH4Cl. Cells were allowed to recover at 18 °C and 210 rpm for up to one hour before induction with 1 mM IPTG for 16–18 hours. Cells were pelleted via centrifugation at 6,000 rpm and 18°C for 20 minutes. Cells were resuspended in 40 mL Low Salt Buffer (25 mM Tris-HCl (pH-7.5), 50 mM NaCl) with DNase I and a protease inhibitor tablet. Resuspended pellets were lysed using the Emulsiflex. Cell lysate was cleared at 15,000 rpm and 4°C for one hour.
15N-CBX8 CD was purified according to the following protocol. Clarified cell lysate containing GST-tagged 15N-CBX8 CD was rocked with glutathione agarose resin for one hour at 4°C. GST-tagged 15N-CBX8 CD bound beads were purified using a gravity flow column. Bound beads were rinsed thoroughly with High Salt Buffer (25 mM Tris-HCl (pH-7.5), 1 M NaCl), followed by Low Salt Buffer. GST-tagged 15N-CBX8 CD was eluted from the beads using 50 mM glutathione in Low Salt Buffer, adjusted to a pH of 7.5. GST-tagged CBX8 CD was concentrated to a volume of 2 mL using a 10,000 MWCO filter. The GST-tag was cleaved using TEV protease at room temperature (25 °C) for three hours. The cleaved 15N-CBX8 CD was then purified using cation exchange chromatography and size exclusion (Superdex S75, 300/10). All 15N-CBX8 CD were stored in a final buffer containing 40 mM NaPi (pH-6.8) and 100 mM NaCl.
SDS-PAGE was used to confirm the identity and purity of 15N-CBX8 CD samples. Quantification of 15N-CBX8 CD was performed using the calculated extinction coefficient (ε= 19,480 M−1cm−1) and measured A280 value. All 15N-CBX8 CD samples were concentrated to 25–50 μM for NMR and flash frozen in liquid nitrogen for long-term storage at –80 °C. Prior to collecting HSQC data, 15N-CBX8 CD samples were thawed overnight at 4 °C.
15N-HSQC spectra were collected on 25–50 μM 15N-CBX8 CD at 25 °C on a Bruker Avance II 800 MHz spectrometer equipped with a cryogenic probe. Titration with DMSO was performed by subsequently adding 1%, 2%, and 5% (v/v%) DMSO. Titration with SW2_110A was performed by addition of 0.5 (1% DMSO), 1.0 (2% DMSO), 2.5 (5% DMSO) or 11 (6% DMSO) molar ratios of compound to protein. All spectra were processed using NMRPipe and ccpNmr.
Normalized chemical shift perturbation values (Δδ) were calculated for the DMSO and SW2_110A in Excel using the following equation:
where Δδ is the chemical shift perturbation in in parts per million (ppm). Δδ values were considered significant when greater than the average plus one standard deviation after trimming the 10% of residues with the largest Δδ value.
MD Simulation
Molecular dynamics using the Amber16 suite69 were carried out for CBX6 (PDB: 3GV6)25 and CBX8 (PDB: 3i91)25 hosts bound to compound SW2 in the presence of approximately 8000 TIP3PBOX waters along with charge neutralizing chloride ions using the ff14S forcefield.70 Simulations were minimized, heated to 300K over 200 ps, and then equilibrated for 100 ns to yield the data presented in MD related figures. Equilibration trajectories were done using a 2 fs time step at 300K under NPT conditions. Interaction cutoffs were set to 8 Å and SHAKE hydrogen constraints were applied. Initial pose generation for MD was done using AutoDock Vina71 in combination with UCSF Chimera.72 Parameterization of non-standard residues was in part done using Gaussian09 (Gaussian, Inc., Wallingford CT)73 and the AmberTools1669 suite of preparatory programs.
File Preparation and Simulation Setup:
Non-standard residue parameterization was done by construction in Avogadro with C and N terminal caps. This includes the diphenyl ether n-terminus, the cyclopentyl alanine, and the trimethyllysine. Residue structures were minimized in Gaussian09 at the HF 6–31G* level of theory. AmberTools16 residuegen utility was then used to construct the preparatory files for use in tLeAP molecular dynamics preparation environment. The SW2_101B ligand was also loaded into UCSF Chimera to generate pdbqt files required for AutoDock Vina. Host coordinates were taken from the crystal structures of PDB 3i91 and 3GV6 by stripping extraneous atoms such as ligand, solvent, and ions.
500 ligand poses for both CBX6 and CBX8 were generated using AutoDock Vina (Exhaustiveness 7) on an MD generated ensemble of 50 host configurations. Top ten docked poses were then selected for molecular dynamics. This process was done iteratively until converged structures were found. RMSD clustering was done on the final replicate trajectories to provide a starting pose for the 100 ns production trajectories.
Computational Resources:
Each 10-pose iteration required 12 hours on 280 cores (10 nodes with 2 Intel Xeon E5–2680 v4 processors each). 100 ns trajectories required approximately 120 hours using 28 cores each. Total simulation time for both systems including starting pose generation was approximately 180 hours.
Cell Culture
HEK293T cells were cultured in Dubecco’s Modified Essential Media (DMEM), 10% fetal bovine serum (FBS, JR Scientific), 1% glutagro (Corning), 1% penicillin/streptomycin (Corning), 1% sodium pyruvate (Corning). Human THP1 cells were cultured in RPMI (Gibco), 10% FBS (J R Scientific), 1% Sodium pyruvate (Invitrogen), 1% Pen/Strep(Invitrogen), 1% Glutamax (Thermo Scientific), 0.1% 2-mercaptoethanol. Human K562 cells were cultured in RPMI (Gibco), 10% FBS (JR Scientific), 1% Sodium pyruvate (Invitrogen), 1% Pen/Strep (Invitrogen), 1% Glutamax (Thermo Scientific). Halo-GFP-Mito Hela Cell were cultured in DMEM (Gibco), 10% FBS (JR Scientific), 1% Sodium pyruvate (Invitrogen), 1% Pen/Strep (Invitrogen), 1% Glutamax (Thermo Scientific), 1 µg/mL puromycin. Human Hs68 cells were cultured in DMEM (Gibco), 10% FBS (JR Scientific), 1% Sodium pyruvate (Invitrogen), 1% Pen/Strep(Invitrogen), 1% Glutamax (Thermo Scientific). Human G401 cells were cultured in McCoy’s (Corning), 10% FBS (Thermo Scientific), 1% Pen/Strep (Invitrogen), 1% Glutamax (Thermo Scientific), 1% MEM NEAA (Invitrogen). Human LNCaP cells were cultured in RPMI (Gibco), 10% FBS (J R Scientific), 1% Pen/Strep (Invitrogen), 1% Glutamax (Thermo Scientific). All cells were grown at 37 °C and 5% CO2. For generation of CBX8 and control CRISPR knockout lines, 200,000 THP1 cells were plated in 6-well 24 hrs prior to transfection. The respective vector (3.3 µg) was co-transfected with 13 µL of Fugene 6 (Promega). Media was changed 24 hrs post-transfection. Transfected cells underwent puromycin selection (2 µg/mL) for 3 days, 48 hrs post-transfection.
Lentiviral transduction
HEK293T cells were co-transfected with pLKO.1 constructs and viral packaging vectors (pMD2.G and psPAX2). Short hairpin constructs for knockdown are below: CBX8 (TRCN0000021896), CBX7 (TRCN0000019144). Viral supernatant was harvested 72 hrs after transfection and concentrated by ultracentrifugation at 17,300 rpm for 2h. Virus was resuspended in 100 µL PBS and 5 µL was added to K562 or THP1 cells in a minimal volume of media for 1h on an orbital shaker. Additional media was added to bring cells to 0.1 × 106 cells/mL and cells were grown at 37 °C and 5% CO2. Twenty-four hours after infection, cells were selected with puromycin (2 µg/mL) for 48 hrs.
Cell proliferation assays
Leukemia cell lines were seeded at 0.1×106 cells/mL in 24-well flat bottom cell culture plate (#353047, Corning). Peptides SW2_110A and SW2_104A (100 μM in DMSO) were added to the cells. Cells were grown for 3 days, the cells were counted and the media was exchanged with fresh media containing DMSO, SW2_110A or SW2_104A. At day 6, cells in each well were split 1:4 and transferred to new wells to avoid over confluency, and media was replaced with fresh compounds in new media. The cells were counted at 12 days, with additional cell counting and fresh compound replenishment at day 3, 6, 9.
CellTiter-Glo luminescent dose-dependent cell viability assay
The effect of SW2_110A and SW2_104A on cell viability was determined using a CellTiter-Glo ATP detection system (#G7573, Promega). THP1 cells were seeded in 0.1 × 106 cells/mL density in 96-well clear bottom white microplate (#655098, Greiner Bio-One). Cells were treated with compounds SW2_110A and SW2_104A for 12 days, with fresh compounds replenishment at day 3, 6, 9. For dose-response studies, IC50 was derived from an eight-point 2-fold titration ranging from 100 μM to 1.56 μM of SW2_110A or SW2_104A. CellTiter-Glo reagent was added to cells, and incubated with gentle shake for 15 minutes in dim light at R.T. Luminescence was read on a GloMax® microplate reader. Luminescence was normalized to DMSO-treated groups. The IC50 was calculated using the “log[inhibitor] vs. the normalized response-variable slope” equation in GraphPad Prism 7.
Sequential Salt Extraction (SSE)
SSE was performed as previously described.35 2.5 × 106 293T cells were seeded in 10 cm cell culture dish (#353003, Corning) overnight. Next day, media was removed and cells were washed with PBS. Then, cells were pretreated with peptides SW2_110A or DMSO on plate (100 μM, 1% DMSO) in 3 mL media for 4 hours in 37 ºC. After the 4-hour pretreatment, media was removed and cells were washed again with PBS. Cells were harvested and washed with PBS. (Note: it is critical to balance the cell numbers the same in the peptide treated group and the DMSO treated control group.) Cells were resuspended in 1 mL Buffer A (25 mM HEPES, 25 mM KCl, 5 mM MgCl2, 0.1% NP-40, 10% Glycerol, 0.05 M EDTA, pH 7.8, plus protease inhibitor) was added to the cell pellet from centrifuge and rotated at 4 ℃ for 10 minutes Cells were spun down at 6500 × g for 5 min at 4 ºC. Supernatant was removed and cell pellet was resuspended with 500 μL of mRIPA (Modified Radioimmunoprecipitation Assay) Buffer (50 mM Tris, 1% Nonidet P-40, 0.25% Sodium deoxycholate, plus protease inhibitors) by pipetting up and down 15 times and incubated on ice for 5 mintes. (Note: it is critical to maintain the consistence for all washing steps with the subsequent NaCl containing mRIPA buffers) Then the sample was centrifuged for 3 min at 6500 × g. The supernatant was saved in a separate tube, labeled as “0 mM fraction”- 0mM sequential salt extraction washing supernatant. (Notes: DMSO or the peptide SW2_110A was also added to the mRIPA washing buffers, in order to maintain the peptide in binding to the protein through the assay) The pellet was sequentially resuspended in 500 μl of mRIPA Buffer with increasing NaCl concentrations (100, 200, 300, 400, 500 mM). The procedures for 0 mM was repeated for each salt concentrations in the subsequent washes. All washing supernatants were saved and labeled. 4× Bolt LDS Sample Buffer (Invitrogen) with 10% β-mercaptoethanol (AMRESCO LLC, Solon, OH) was added to each sample, and 50 μl of each fraction was loaded onto a 4–12% gradient gel (Invitrogen) for immunoblotting analysis of the proteins of interest. ImageJ was employed to quantitate the protein bands.
Chemoprecipitation (peptide pull-down)
Cells were grown to confluency in a 15 cm cell culture dish and washed with PBS. Cells were scraped into with 2 ml Buffer A (25 mM HEPES, 5 mM KCl, 25 mM MgCl2, 0.05 mM EDTA, 10% glycerol, 0.1% NP-40, plus protease inhibitors) and lysed on ice for 15 minutes. The nuclei were pelleted and re-suspended in 2 mL peptide pulldown buffer (20 mM HEPES pH 7.9, 250 mM NaCl, 0.6% NP-40, protease inhibitor and 0.5 mM DTT). Benzonase (200U) was added and the lysate was incubated at 37 ℃ for 10 minutes and room temperature for 10 minutes with agitation. The samples were centrifuged at 13,000 rpm at 4 ℃ for 10 min and lysates were transferred to separate tubes. Meanwhile, 30 µL streptavidin M-270 Dynabeads (Solulink, San Diego, CA) was washed three times with peptide pulldown buffer. Biotinylated ligands in DMSO (0.3 µL of 10 mM stock), were incubated with pre-equilibrated beads at room temperature for an hour. Extra unbound ligands were removed by washing 2× with peptide pulldown buffer and 300 μL (~300 µg) of nuclear lysate supernatant was added to immobilized biotinylated peptide (SW2_110A-B, SW2_104A-B) or beads alone. The mixture was rotated at 4 ºC overnight, the depleted lysate was removed, and the beads were washed with peptide pulldown buffer 3×5 minutes at room temperature. The bound proteins were eluted from the beads with 1× Bolt LDS Sample Buffer (Invitrogen) with 10% β-mercaptoethanol (AMRESCO LLC, Solon, OH). The samples, along with 10% input samples were heated at 95℃ for 5 min and loaded onto a 4–12% gradient gel (Invitrogen) for immunoblotting analysis of the proteins of interest. ImageJ was employed to quantitate the protein bands.
Immunoblot and antibodies
Lysates were boiled and loaded on a 4–12% SDS-page gel (Invitrogen). Gels were transferred to PDVF membranes (Millipore) and incubated in 5% bovine serum albumin (BSA) in PBS-t (PBS with 0.1% Tween-20) prior to primary antibody. Blots were incubated at 4 °C overnight in primary antibody. Blots were washed with PBS-t and incubated for an hour at room temperature in goat anti-rabbit or mouse conjugated to IRDye® 800CW or IRDye® 680 (LI-COR) secondary antibody. Blots were imaged on the Licor Odyessy®. Primary antibodies used: CBX8 (rabbit, 1:1000, Bethyl Cat# A300–882A), CBX4 (rabbit, 1:500, Bethyl Cat# A302–355A), CBX7 (rabbit, 1:1000, Bethyl Cat# A302–525A), CBX2 (mouse, 1:400, Santa Cruz Cat# sc-136387), CBX6 (mouse, 1:400, Santa Cruz Cat# sc-86354), TBP (mouse, 1:1000, Abcam Cat# ab818).
Chromatin Immunoprecipitation-qPCR (ChIP-qPCR)
ChIP: Cells were grown to confluency in 100 mm cell culture plates (~1–2 × 107 cells). Hs68 cells were treated with 100 µM SW2–110A or DMSO for four hours and THP1 cells were treated with 100 µM SW2–110A or DMSO for 24h. ChIP was performed as previously described.34 Briefly, cells were washed with PBS and fixed with 1% formaldehyde in PBS for 10 min at room temperature. Crosslinking was quenched with 0.125 M glycine for 5 min at 4 °C. Cells were washed once with PBS and resuspended in CiA NP Rinse buffer 1 (50 mM HEPES pH 8.0, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X) for 10 min. Cells were pelleted at 400 x g for 5 min at 4 °C. The supernatant was removed, and the cells were resuspended in CiA NP rinse buffer 2 (10 mM Tris pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl). Cells were collected by centrifugation, 400 x g for 5 min. Supernatant was removed and the cells were washed twice with shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris HCl, pH 8.0). Cells were resuspended in 1 mL shearing buffer and sonicated with a probe sonicator (Branson) to obtain ~300 bp DNA fragments. Lysate was centrifuged at 21,000 x g for 15 min to remove debris. Supernatant was collected and pre-cleared 2h with Protein A Dynabeads (Thermo Fisher). For immunoprecipitation, 250 µL pre-cleared cell lysate was incubated with 2 µg of antibody overnight and 10% input was saved. The IPs were washed 3 x 3 min at RT with IP buffer (50 mM HEPES/KOH pH 7.5, 300 mM NaCl, 1 mM EDTA, 1% Triton X, 0.1% DOC, 0.1% SDS) followed by 1x 3 min with DOC (10 mM Tris pH 8.0, 0.25M LiCl, 0.5% NP-40, 0.5% DOC, 1 mM EDTA) and 1x with TE. Protein was eluted from beads with 300 µL elution buffer (1% SDS, 0.1 M NaHCO3) for 20 minutes at RT with agitation. Samples, including saved input, were treated with RNase A (100U) at 37 °C for 30 min followed by proteinase K (2 µg) digestion for 3 hours at 55 °C. Samples were reverse-crosslinked overnight at 65°C, and extracted with phenol chloroform followed by isopropanol precipitation. The isolated DNA was resuspended in 80 µL water for qPCR analysis (4 µL used per well). Antibodies used for IP were CBX8 (Bethyl Cat# A300–882A, rabbit), IgG (CST Cat# 2729, rabbit), CBX7 (Bethyl Cat# A302–525A, rabbit), AF9 (Bethyl Cat# A300–595A, rabbit).
Genome-wide data analysis:
Published annotated datasets were downloaded from Encyclopedia of DNA Elements (ENCODE) or Gene Expression Omnibus (GEO) as BED files.74,75 Peaks for CBX8, CBX2, H3K4me3, H3K27me3, and ENL (MLLT1) were called in reference to GrCh38. All data sets were imported into R Studio. Peak overlaps were determined using the ChIPpeakAnno package.76,77 Overlaps were defined as being within 150 base pairs of each other and on the same strand. Accession numbers for CBX8: GSM830987, GSM1295078, GSM1295089, ENCFF001SYX; H3K27me3: GSM1295084, GSM1295094, ENCFF001SZF; H3K4me3: GSM1295085, GSM1295095, ENCFF001SZJ; MLLT1: GSM2423844, GSM2423845, ENCSR675LRO
ChIP-Seq visualization:
Published ChIP-Seq coordinate datasets from Bernt et al52 (GSE29130) were uploaded to the UCSC genome browser in reference to mm8 genome for visualization. Accession numbers: GSM721213 GSM721214 GSM721215 GSM721216 GSM721217 GSM721218
qPCR:
qPCR was performed on the isolated ChIP DNA (4 µL) using SYBR master mix (Thermo) and run on the BioRad CFX thermo cycler. Three biological replicates were performed in technical triplicate. Enrichment was determined as percent of input DNA.
| Gene name | Forward primer (5’−3’) | Reverse primer (5’−3’) |
|---|---|---|
| LMNB2 | CCGAATCTCTGAAATGAAAGTCCATGC | TTAAAGATCTGAGGGACTCCTCAGTC |
| CCND2 (Pemberton et al., 2016) | ACTGTCTGAAATGAAGGTGAAGC | GATTTGATGGACACTTGGTTTGT |
| GATA6 (Pemberton et al., 2016) | GCCTCTCCATTCCAGAGTTTT | TCCAGAAACCGTTCTCATCC |
| RUNX3 (Pemberton et al., 2016) | TCAAAAGGCATCCGCCTCTCCGT | AAGGATGCACCTGCCGGGAATTG |
| HOXA9 (−0.5 kb) | AAGTCGGAAACGACCAACAGA | TTACAGGGAGCTCGCCAAC |
| HOXA9 (−2.5 kb) | ACAAAGCTGCAGCGAATGTC | ATGATCACGACCGGATGGC |
| HOXA9 (−4.5 kb) | GCAAAATAACCGGCCTCTGC | CCTATAGCCCTGGTGCCGTA |
| HOXA9 (−6.5 kb) | GACGCTGCGGCTAATCTCTA | AAGAGTGGTCGGAAGAAGCG |
| HOXA9 (−8.5 kb) | CTGATGAGCGAGTCGACCAA | GGAAACTCTGGCTCGGGATT |
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
1× 106 THP1 and K562 cells were treated with SW2_110A at 100 μM or DMSO (1%) for 24 h. Cells were harvested after 24 h for RNA extraction. After homogenization of THP1 or K562 cells using TRIzol reagent (Thermo Scientific), RNA was extracted from the aqueous phase in the phase separation step. RNA pellet was washed with 75% ethanol and concentrated for subsequent reverse transcription. 2 μg RNA was then converted into cDNA using Verso cDNA synthesis kit (Thermo Scientific). SYBR Green Mastermix (Thermo Scientific) was used for quantitative PCR. Primers below are used in the qPCR.
| Gene name | Forward primer (5’−3’) | Reverse primer (5’−3’) |
|---|---|---|
| HOXA9 | GGCCCAGGACCGAGATACTT | CGCTCACGGACAATCTAGTTGT |
| B2M | TGCTGTCTCCATGTTTGATGTATCT | TCTCTGCTCCCCACCTCTAAGT |
| ACTB | GCACCACACCTTCTACAATGA | GTCATCTTCTCGCGGTTGGC |
| MYB | TCAGGAAACTTCTTCTGCTCACA | AGGTTCCCAGGTACTGCT |
| CDK6 | TGGAGACCTTCGAGCACC | CACTCCAGGCTCTGGAACTT |
| RUNX2 | TCTTAGAACAAATTCTGCCCTTT | TGCTTTGGTCTTGAAATCACA |
| RUNX3 | GTTCAACGACCTTCGCTTC | GTCCACGGTCACCTTGATG |
| BCR-ABL | ACTCCAGACTGTCCACAGCA | TTGGGGTCATTTTCACTGG |
| CBX6 | AAACGGCGGATCCGAAAGGGAC | GCTGCAATGAGCCGCGAGTC |
| CBX7 | CGTCATGGCCTACGAGGA | TGGGTTTCGGACCTCTCTT |
| CBX8 | CAACATGGAGCTTTCAGCGG | GTGCTGTACTTCTGCGACCA |
Chloroalkane Penetration Assay (CAPA)
CAPA is a recently developed cell penetration assay for measuring relative cytosolic access without interference from endosomally trapped peptides.32 Halo-GFP-Mito Hela cells were cultured and seeded at a 1× 105 cells/well in a 24- or 48-well plate the day before experiments. Cells were rinsed by PBS and treated with chloroalkane conjugated CBX8 peptidomimetic ligands SW2_110A-CA or KED97L-CA in acidified Opti-MEM (0.15% 6N HCl) for 4 h. Next, media was removed, and cells were washed by phenol red-free Opti-MEM for 30 minutes, followed by incubation with 5 μM HT-TAMRA (HTag-TMR, Promega) for another 30 minutes. Then, cells were washed for 15 minutes by phenol red-free DMEM +10% FBS +1% pen/strep, followed with PBS wash and trypsin incubation. Cells were transferred to a new microcentrifuge tube and pelleted by centrifuge, with two times of PBS washes. Cell pellets were resuspended in 250 μL PBS, and 200 μL was used for flow cytometry analysis. Live cells were gated and 10000 cells were measured per sample. Mean fluorescence intensity was calculated from raw data, and these values were normalized to the samples with no dye (0% red signal) and with dye but no HT-molecule (100% red signal).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the lab of J. Kritzer for providing Halo-GFP-Mito Hela cell lines, as well as advice on CAPA assay. We thank A. Alpsoy for helpful discussions on pulldown assays, and A. Dhiman for technical assistance in flow cytometry. We thank B. Peters and C. Maschinot for their help in NMR. We thank Y. Sun for assistance with off-DNA hit synthesis, purification, and LC-MS characterization. Support for this research was provided by the Purdue Center for Cancer Research, NIH grant P30 CA023168 in the form of the Genomics Core and the Pilot Grant Program. K.E.C. is supported by Indiana Clinical and Translational Science Institute Predoctoral Fellowship [UL1TR001108 to A.S., P.I., 9/26/2013–4/30/2018] and Purdue University Bilsland Dissertation Fellowship Aug. 2018-Dec. 2018; N.M. is supported by fellowships from the WestCoast Ride to Live and PCFBC. F.H. is supported by CCSRI (IG703789). Funding for the computational studies was provided by the National Science and Engineering Research Council of Canada, the Canada Foundation for Innovation, the British Columbia Knowledge Development Fund and the University of Victoria. This research was performed in part using the Compute Canada and WestGrid computing resources. C.A.M. is supported by National Science Foundation [CAREER-1452411] and National Institutes of Health [GM128705]. C.J.K is supported by NIH grants (R35GM128894, 1NS101535). E.C.D is supported by NIH grants (CA207532) and the V Foundation for Cancer Research (V2014–004 and D2016–030).
Footnotes
ASSOCIATED CONTENTS
Supplementary Information.
This material is available free of charge via the Internet at http://pubs.acs.org
Supplemental figures (Figures SI1-SI14) and key resources information
Full selection results with all building block structures, off-DNA hit validation including FP assays, MST and TSA, NMR spectra, molecular dynamic simulations, cellular activity data, and LCMS chromatograms
Declaration of Interests
The authors declare no conflicts.
REFERENCES
- (1).Kennison JA (1995) The Polycomb and Trithorax Group Proteins of Drosophila : Trans-Regulators of Homeotic Gene Function. Annu. Rev. Genet 29, 289–303. [DOI] [PubMed] [Google Scholar]
- (2).Sauvageau M; Sauvageau G Polycomb Group Proteins: Multi-Faceted Regulators of Somatic Stem Cells and Cancer. (2010) Cell Stem Cell 7, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Di Croce L; Helin K (2013) Transcriptional Regulation by Polycomb Group Proteins. Nat. Struct. Mol. Biol 20, 1147–1155. [DOI] [PubMed] [Google Scholar]
- (4).Francis NJ; Kingston RE; Woodcock CL (2004) Chromatin Compaction by a Polycomb Group Protein Complex. Science 306, 1574–1577. [DOI] [PubMed] [Google Scholar]
- (5).Lavigne M; Francis NJ; King IFG; Kingston RE (2004) Propagation of Silencing; Recruitment and Repression of Naive Chromatin in Trans by Polycomb Repressed Chromatin. Mol. Cell 13, 415–425. [DOI] [PubMed] [Google Scholar]
- (6).Connelly KE; Dykhuizen EC (2017) Compositional and Functional Diversity of Canonical PRC1 Complexes in Mammals. Biochim. Biophys. Acta, Gene Regul. Mech 1860, 233–245. [DOI] [PubMed] [Google Scholar]
- (7).Gil J; O’Loghlen A (2014) PRC1 Complex Diversity: Where Is It Taking Us? Trends Cell Biol 24, 632–641. [DOI] [PubMed] [Google Scholar]
- (8).Klauke K; Radulović V; Broekhuis M; Weersing E; Zwart E; Olthof S; Ritsema M; Bruggeman S; Wu X; Helin K; Bystrykh L; de Haan G (2013) Polycomb Cbx Family Members Mediate the Balance between Haematopoietic Stem Cell Self-Renewal and Differentiation. Nat. Cell Biol 15, 353–362. [DOI] [PubMed] [Google Scholar]
- (9).Kloet SL; Makowski MM; Baymaz HI; Van Voorthuijsen L; Karemaker ID; Santanach A; Jansen PWTC; Di Croce L; Vermeulen M (2016) The Dynamic Interactome and Genomic Targets of Polycomb Complexes during Stem-Cell Differentiation. Nat. Struct. Mol. Biol 23, 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Morey L; Pascual G; Cozzuto L; Roma G; Wutz A; Benitah SA; Di Croce L (2012) Nonoverlapping Functions of the Polycomb Group Cbx Family of Proteins in Embryonic Stem Cells. Cell Stem Cell 10, 47–62. [DOI] [PubMed] [Google Scholar]
- (11).Morey L; Santanach A; Blanco E; Aloia L; Nora EP; Bruneau BG; Di Croce L (2015) Polycomb Regulates Mesoderm Cell Fate-Specification in Embryonic Stem Cells through Activation and Repression Mechanisms. Cell Stem Cell 17, 300–315. [DOI] [PubMed] [Google Scholar]
- (12).O’Loghlen A; Muñoz-Cabello AM; Gaspar-Maia A; Wu HA; Banito A; Kunowska N; Racek T; Pemberton HN; Beolchi P; Lavial F; Masui O; Vermeulen M; Carroll T; Graumann J; Heard E; Dillon N; Azuara V; Snijders AP; Peters G; Bernstein E; Gil J (2012) MicroRNA Regulation of Cbx7 Mediates a Switch of Polycomb Orthologs during ESC Differentiation. Cell Stem Cell 10, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mills AA (2010) Throwing the Cancer Switch: Reciprocal Roles of Polycomb and Trithorax Proteins. Nat. Rev. Cancer 10, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Koppens M; Van Lohuizen M (2016) Context-Dependent Actions of Polycomb Repressors in Cancer. Oncogene 35, 1341–1352. [DOI] [PubMed] [Google Scholar]
- (15).Béguelin W; Teater M; Gearhart MD; Calvo Fernández MT; Goldstein RL; Cárdenas MG; Hatzi K; Rosen M; Shen H; Corcoran CM; Hamline MY; Gascoyne RD; Levine RL; Abdel-Wahab O; Licht JD; Shaknovich R; Elemento O; Bardwell VJ; Melnick AM (2016) EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 30, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang CZ; Chen SL; Wang CH; He YF; Yang X; Xie D; Yun JP (2018) CBX8 Exhibits Oncogenic Activity via AKT/ b-Catenin Activation in Hepatocellular Carcinoma. Cancer Res 78, 51–63. [DOI] [PubMed] [Google Scholar]
- (17).Chung CY; Sun Z; Mullokandov G; Bosch A; Qadeer ZA; Cihan E; Rapp Z; Parsons R; Aguirre-Ghiso JA; Farias EF; Brown BD; Gaspar-Maia A; Bernstein E (2016) Cbx8 Acts Non-Canonically with Wdr5 to Promote Mammary Tumorigenesis. Cell Rep 16, 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tan J; Jones M; Koseki H; Nakayama M; Muntean AG; Maillard I; Hess JL (2011) CBX8, a Polycomb Group Protein, Is Essential for MLL-AF9-Induced Leukemogenesis. Cancer Cell 20, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Santiago C; Nguyen K; Schapira M (2011) Druggability of Methyl-Lysine Binding Sites. J. Comput. Aided. Mol. Des 25, 1171–1178. [DOI] [PubMed] [Google Scholar]
- (20).Ren C; Morohashi K; Plotnikov AN; Jakoncic J; Smith SG; Li J; Zeng L; Rodriguez Y; Stojanoff V; Walsh M; Zhou M (2015) Small-Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain. Chem. Biol 22, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ren C; Smith SG; Yap K; Li S; Li J; Mezei M; Rodriguez Y; Vincek A; Aguilo F; Walsh MJ; Zhou M (2016) Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Med. Chem. Lett 7, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Stuckey JI; Dickson BM; Cheng N; Liu Y; Norris JL; Cholensky SH; Tempel W; Qin S; Huber KG; Sagum C; Black K; Li F; Huang X; Roth BL; Baughman BM; Senisterra G; Pattenden SG; Vedadi M; Brown PJ; Bedford MT; Min J; Arrowsmith CH; James LI; Frye SV (2016) A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat. Chem. Biol 12, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Milosevich N; Gignac MC; McFarlane J; Simhadri C; Horvath S; Daze KD; Croft CS; Dheri A; Quon TTH; Douglas SF; Wulff JE; Paci I; Hof F; (2016) Selective Inhibition of CBX6: A Methyllysine Reader Protein in the Polycomb Family. ACS Med. Chem. Lett 7, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Simhadri C; Daze KD; Douglas SF; Quon TTH; Dev A; Gignac MC; Peng F; Heller M; Boulanger MJ; Wulff JE; Hof F (2014) Chromodomain Antagonists That Target the Polycomb-Group Methyllysine Reader Protein Chromobox Homolog 7 (CBX7). J. Med. Chem 57, 2874–2883. [DOI] [PubMed] [Google Scholar]
- (25).Kaustov L; Ouyang H; Amaya M; Lemak A; Nady N; Duan S; Wasney GA; Li Z; Vedadi M; Schapira M; Min J; Arrowsmith CH (2011) Recognition and Specificity Determinants of the Human Cbx Chromodomains. J. Biol. Chem 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Goodnow RA; Dumelin CE; Keefe AD (2017) DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discovery 16, 131–147. [DOI] [PubMed] [Google Scholar]
- (27).Neri D; Lerner RA (2018) DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem 87, 479–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Denton KE; Wang S; Gignac MC; Milosevich N; Hof F; Dykhuizen EC; Krusemark CJ (2018) Robustness of In Vitro Selection Assays of DNA-Encoded Peptidomimetic Ligands to CBX7 and CBX8. SLAS Discov 23, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tardat M; Albert M; Kunzmann R; Liu Z; Kaustov L; Thierry R; Duan S; Brykczynska U; Arrowsmith CH; Peters AHFM (2015) Cbx2 Targets PRC1 to Constitutive Heterochromatin in Mouse Zygotes in a Parent-of-Origin-Dependent Manner. Mol. Cell 58, 157–171. [DOI] [PubMed] [Google Scholar]
- (30).Malik B; Hemenway CS (2013) CBX8, a Component of the Polycomb PRC1 Complex, Modulates DOT1L-Mediated Gene Expression through AF9/MLLT3. FEBS Lett 587, 3038–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pemberton H; Anderton E; Patel H; Brookes S; Chandler H; Palermo R; Stock J; Rodriguez-Niedenführ M; Racek T; de Breed L; Stewart A; Matthews N; Peters G (2014) Genome-Wide Co-Localization of Polycomb Orthologs and Their Effects on Gene Expression in Human Fibroblasts. Genome Biol 15, R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Peraro L; Zou Z; Makwana KM; Cummings AE; Ball HL; Yu H; Lin YS; Levine B; Kritzer JA (2017) Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy. J. Am. Chem. Soc 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ (1997) Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev 23, 3–25. [DOI] [PubMed] [Google Scholar]
- (34).Connelly KE; Weaver TM; Alpsoy A; Gu BX; Musselman CA; Dykhuizen EC (2018) Engagement of DNA and H3K27me3 by the CBX8 Chromodomain Drives Chromatin Association. Nucleic Acids Res 47, 2289–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Porter EG; Dykhuizen EC (2017) Individual Bromodomains of Polybromo-1 Contribute to Chromatin Association and Tumor Suppression in Clear Cell Renal Carcinoma. J. Biol. Chem 292, 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Marian CA; Stoszko M; Wang L; Leighty MW; de Crignis E; Maschinot CA; Gatchalian J; Carter BC; Chowdhury B; Hargreaves DC; Duvall JR; Crabtree GR; Mahmoudi T; Dykhuizen EC (2018) Small Molecule Targeting of Specific BAF (MSWI/SNF) Complexes for HIV Latency Reversal. Cell Chem. Biol 25, 1443–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M; Dan S; Jacqui M; Kris Z; Paul O; Gediminas V; Ji Z; Aldis D; Dieter HK; Robert FB; Keith VW (2008) HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- (38).de Groot RP; Raaijmakers JA; Lammers JW; Jove R; Koenderman L STAT5 Activation by BCR-Abl Contributes to Transformation of K562 Leukemia Cells. Blood 94, 1108–1112. [PubMed] [Google Scholar]
- (39).Zeisig DT; Bittner CB; Zeisig BB; García-Cuéllar M-P; Hess JL; Slany RK (2005) The Eleven-Nineteen-Leukemia Protein ENL Connects Nuclear MLL Fusion Partners with Chromatin. Oncogene 24, 5525–5532. [DOI] [PubMed] [Google Scholar]
- (40).Monroe SC; Jo SY; Sanders DS; Basrur V; Elenitoba-Johnson KS; Slany RK; Hess JL (2011) MLL-AF9 and MLL-ENL Alter the Dynamic Association of Transcriptional Regulators with Genes Critical for Leukemia. Exp. Hematol 39, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mueller D; Bach C; Zeisig D; Garcia-Cuellar MP; Monroe S; Sreekumar A; Zhou R; Nesvizhskii A; Chinnaiyan A; Hess JL; Slany RK (2007) A Role for the MLL Fusion Partner ENL in Transcriptional Elongation and Chromatin Modification. Blood 110, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).García-Cuéllar MP; Zilles O; Schreiner SA; Birke M; Winkler TH; Slany RK (2001) The ENL Moiety of the Childhood Leukemia-Associated MLL-ENL Oncoprotein Recruits Human Polycomb 3. Oncogene 20, 411–419. [DOI] [PubMed] [Google Scholar]
- (43).Hemenway CS; De Erkenez AC; Gould GCD (2001) The Polycomb Protein MPc3 Interacts with AF9, an MLL Fusion Partner in t(9;11)(P22;Q23) Acute Leukemias. Oncogene 20, 3798–3805. [DOI] [PubMed] [Google Scholar]
- (44).Sitwala KV; Dandekar MN; Hess JL (2008) HOX Proteins and Leukemia. Int. J. Clin. Exp. Pathol 1, 461–474. [PMC free article] [PubMed] [Google Scholar]
- (45).Yokoyama A; Cleary ML (2008) Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Armstrong SA; Staunton JE; Silverman LB; Pieters R; den Boer ML; Minden MD; Sallan SE; Lander ES; Golub TR; Korsmeyer SJ (2002) MLL Translocations Specify a Distinct Gene Expression Profile That Distinguishes a Unique Leukemia. Nat. Genet 30, 41–47. [DOI] [PubMed] [Google Scholar]
- (47).Ayton PM; Cleary ML (2003) Transformation of Myeloid Progenitors by MLL Oncoproteins Is Dependent on Hoxa7 and Hoxa9. Genes Dev 17, 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kumar AR; Hudson WA; Chen W; Nishiuchi R; Yao Q; Kersey JH (2004) Hoxa9 Influences the Phenotype but Not the Incidence of Mll-AF9 Fusion Gene Leukemia. Blood 103, 1823–1828. [DOI] [PubMed] [Google Scholar]
- (49).Prange KHM; Mandoli A; Kuznetsova T; Wang SY; Sotoca AM; Marneth AE; Van Der Reijden BA; Stunnenberg HG; Martens JHA (2017) MLL-AF9 and MLL-AF4 Oncofusion Proteins Bind a Distinct Enhancer Repertoire and Target the RUNX1 Program in 11q23 Acute Myeloid Leukemia. Oncogene 36, 3346–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dietrich N; Bracken AP; Trinh E; Schjerling CK; Koseki H; Rappsilber J; Helin K; Hansen KH (2007) Bypass of Senescence by the Polycomb Group Protein CBX8 through Direct Binding to the INK4A-ARF Locus. EMBO J 26, 1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ram O; Goren A; Amit I; Shoresh N; Yosef N; Ernst J; Kellis M; Gymrek M; Issner R; Coyne M; Durham T; Zhang X;Donaghey J; Epstein CB; Regev A; Bernstein BE (2011) Combinatorial Patterning of Chromatin Regulators Uncovered by Genome-Wide Location Analysis in Human Cells. Cell 147, 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bernt KM; Zhu N; Sinha AU; Vempati S; Faber J; Krivtsov AV; Feng Z; Punt N; Daigle A; Bullinger L; Pollock RM; Richon VM; Kung AL; Armstrong SA (2011) MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Maethner E; Garcia-Cuellar MP; Breitinger C; Takacova S; Divoky V; Hess JL; Slany RK (2013) MLL-ENL Inhibits Polycomb Repressive Complex 1 to Achieve Efficient Transformation of Hematopoietic Cells. Cell Rep 3, 1553–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Moussa HF; Bsteh D; Yelagandula R; Pribitzer C; Stecher K; Bartalska K; Michetti L; Wang J; Zepeda-Martinez JA; Elling U; Stuckey JI; James LI; Frye SV; Bell O (2019) Canonical PRC1 Controls Sequence-Independent Propagation of Polycomb-Mediated Gene Silencing. Nat. Commun 10, 1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wesolowski SS; Brown DG (2016) The Strategies and Politics of Successful Design, Make, Test, and Analyze (DMTA) Cycles in Lead Generation. In Lead Generation (Holenz J, Ed.), pp 487–512, Wiley-VCH, Weinheim. [Google Scholar]
- (56).Franzini RM; Ekblad T; Zhong N; Wichert M; Decurtins W; Nauer A; Zimmermann M; Samain F; Scheuermann J; Brown PJ; Hall J; Gräslund S; Schüler H; Neri D (2015) Identification of Structure–Activity Relationships from Screening a Structurally Compact DNA-Encoded Chemical Library. Angew. Chemie 54, 3927–3931. [DOI] [PubMed] [Google Scholar]
- (57).Rogers JM; Passioura T; Suga H (2018) Nonproteinogenic Deep Mutational Scanning of Linear and Cyclic Peptides. Proc. Natl. Acad. Sci 115, 10959–10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Jenson JM; Xue V; Stretz L; Mandal T; Reich L “Luther”; Keating AE (2018) Peptide Design by Optimization on a Data-Parameterized Protein Interaction Landscape. Proc. Natl. Acad. Sci 115, E10342–E10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Satz AL (2015) DNA Encoded Library Selections and Insights Provided by Computational Simulations. ACS Chem. Biol 10, 2237–2245. [DOI] [PubMed] [Google Scholar]
- (60).Bracken AP; Kleine-Kohlbrecher D; Dietrich N; Pasini D; Gargiulo G; Beekman C; Theilgaard-Mönch K; Minucci S; Porse BT; Marine J-C; Hansen KH; Helin K (2007) The Polycomb Group Proteins Bind throughout the INK4A-ARF Locus and Are Disassociated in Senescent Cells. Genes Dev 21, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Maertens GN; El Messaoudi-Aubert S; Racek JK; Nicholls J; Rodriguez-Niedenführ M; Gil J; Peters G (2009) Several Distinct Polycomb Complexes Regulate and Co-Localize on the INK4a Tumor Suppressor Locus. PLoS One 4, e6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Creppe C; Palau A; Malinverni R; Valero V; Buschbeck MA (2014) Cbx8-Containing Polycomb Complex Facilitates the Transition to Gene Activation during ES Cell Differentiation. PLoS Genet 10, e1004851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).TerMaat JR; Pienaar E; Whitney SE; Mamedov TG; Subramanian A (2009) Gene Synthesis by Integrated Polymerase Chain Assembly and PCR Amplification Using a High-Speed Thermocycler. J. Microbiol. Methods 79, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Giaever G; Chu AM; Ni L; Connelly C; Riles L; Véronneau S; Dow S; Lucau-Danila A; Anderson K; André B; Arkin AP; Astromoff A; El-Bakkoury M; Bangham R; Benito R; Brachat S; Campanaro S; Curtiss M; Davis K; Deutschbauer A; Entian K-D; Flaherty P; Foury F; Garfinkel DJ; Gerstein M; Gotte D; Güldener U; Hegemann JH; Hempel S; Herman Z; Jaramillo DF; Kelly DE; Kelly SL; Kötter P; LaBonte D; Lamb DC; Lan N; Liang H; Liao H; Liu L; Luo C; Lussier M; Mao R; Menard P; Ooi SL; Revuelta JL; Roberts CJ; Rose M; Ross-Macdonald P; Scherens B; Schimmack G; Shafer B; Shoemaker DD; Sookhai-Mahadeo S; Storms RK; Strathern JN; Valle G; Voet M; Volckaert G; Wang C; Ward TR; Wilhelmy J; Winzeler E. a; Yang Y; Yen G; Youngman E; Yu K; Bussey H; Boeke JD; Snyder M; Philippsen P; Davis RW; Johnston M (2002) Functional Profiling of the Saccharomyces Cerevisiae Genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- (65).Jetson RR; Krusemark CJ (2016) Sensing Enzymatic Activity by Exposure and Selection of DNA-Encoded Probes. Angew. Chemie - Int. Ed 55, 9562–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ten Brink HT; Meijer JT; Geel RV; Damen M; Löwik DWPM; Van Hest JCM (2006) Solid-Phase Synthesis of C-Terminally Modified Peptides. J. Pept. Sci 12, 686–692. [DOI] [PubMed] [Google Scholar]
- (67).Cai B; Kim D; Akhand S; Sun Y; Cassell RJ; Alpsoy A; Dykhuizen EC; Van Rijn RM; Wendt MK; Krusemark CJ (2019) Selection of DNA-Encoded Libraries to Protein Targets within and on Living Cells. J. Am. Chem. Soc 141, 17057–17061. 10.1021/jacs.9b08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Vivoli M; Novak HR; Littlechild JA; Harmer NJ (2014) Determination of Protein-Ligand Interactions Using Differential Scanning Fluorimetry. J. Vis. Exp 91, e51809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Draughn GL; Milton ME; Feldmann EA; Bobay BG; Roth BM; Olson AL; Thompson RJ; Actis LA; Davies C; Cavanagh J (2018) The Structure of the Biofilm-Controlling Response Regulator BfmR from Acinetobacter Baumannii Reveals Details of Its DNA-Binding Mechanism. J. Mol. Biol 430, 806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C (2015) Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Trott O; Olson AJ (2010) Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE (2004) UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (73).Gaussian 09, Revision A.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, and Fox DJ, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- (74).Sloan CA; Chan ET; Davidson JM; Malladi VS; Strattan JS; Hitz BC; Gabdank I; Narayanan AK; Ho M; Lee BT; Rowe LD; Dreszer TR; Roe G; Podduturi NR; Tanaka F, Hong EL; Cherry JM (2016) ENCODE Data at the ENCODE Portal. Nucleic Acids Res 44 , D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).ENCODE Project Consortium. (2012) An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zhu LJ (2013) Integrative Analysis of ChIP-Chip and ChIP-Seq Dataset. Tiling Arrays 1067, 105–124. [DOI] [PubMed] [Google Scholar]
- (77).Zhu LJ; Gazin C; Lawson ND; Pagès H; Lin SM; Lapointe DS; Green MR (2010) ChIPpeakAnno: A Bioconductor Package to Annotate ChIP-Seq and ChIP-Chip Data. BMC Bioinf 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.