

Abstract
Hemophagocytic syndrome or hemophagocytic lymphohistiocytosis (HLH) is a rare but potentially fatal disease caused by dysregulated activation of macrophages against one's blood cells. Major pathologic feature of HLH is hemophagocytosis. We present a case of HLH complicating methotrexate toxicity in a 65-year-old psoriatic patient with history of renal disease. Diagnosis of HLH was established as he fulfilled five out of eight HLH diagnostic criteria. This case report is presented to enlighten clinicians about the clinical entity of HLH and to suspect and recognize this rare and generally fatal disease at the earliest.
Keywords: Cytopenias, hemophagocytic lymphohistiocytosis, hemophagocytic syndrome
Introduction
Hemophagocytic syndrome or hemophagocytic lymphohistiocytosis is a clinical syndrome of hyperinflammation resulting in uncontrolled and ineffective immune response.[1,2] It may be primary or secondary. Primary HLH is a genetic entity. Secondary HLH is associated with infections, malignancies, immune disorders, or drugs.[2,3,4,5] In both types, activated macrophages engulf erythrocytes, leukocytes, platelets, and precursor cells in bone marrow, liver, and lymph nodes.[6] Most severe forms of the disease lead to a sepsis like picture and multiorgan dysfunction. HLH is a disease of rapid progression and has high mortality despite treatment.[3,7,8,9] We report a case of secondary HLH complicating methotrexate toxicity and sepsis.
Case History
A 65-year-old male, a case of chronic plaque psoriasis on intermittent therapy with methotrexate since 15 years and chronic kidney disease since 5 years presented with fever, severe body ache, and ulcerative lesions over lips and trunk. He developed these symptoms 5 days after inadvertent intake of tablet methotrexate 5 mg for 5 consecutive days. Examination showed tender inguinal and axillary lymphadenopathy, hemorrhagic crusting of lips, and necrotic ulcers over psoriatic plaques and as well as normal skin with severe skin tenderness and positive Nikolsky's sign. Since ulcers were present over psoriatic plaques, methotrexate-induced skin necrosis was suspected. Histopathology findings were skin with detached stratum corneum, large subcorneal pustule [Figure 1a], mild spongiosis, marked regenerative atypia, basal vacuolar degeneration, pigment incontinence, and apoptotic keratinocytes [Figure 1b]. Routine investigations showed bicytopenia with hemoglobin of 9.6 g/dl, WBC of 600 cells/mm3 with normal platelet count of 120,000 cells/mm3. His renal function tests were altered with elevated urea (106 mg/dl) and creatinine levels (3.16 mg/dl). The patient was started on antibiotics, leucovorin rescue therapy (folinic acid 10 mg every 6th hourly), and injection Filgrastim (Granulocyte colony-stimulating factor) 300 mg subcutaneously daily. There was worsening pancytopenia and renal function and further developed splenomegaly, features of sepsis. Blood culture was positive for Group B streptococci and methicillin sensitive staphylococcus aureus and procalcitonin levels were elevated (24.5 ng/ml). His antibiotics were upgraded and platelet transfusion given in spite of which patient developed fever, tachycardia, hypotension with further decrease in platelet count to 6,000 cells/mm3 on 5th day. Secondary HLH was suspected and a workup was rapidly initiated based on HLH 2004 diagnostic criteria. Peripheral smear revealed pancytopenia [Figure 2]. Bone marrow aspiration was done which showed megaloblastic erythroid maturation, maturation arrest in the myeloid series, and hemophagocytosis [Figure 3a and b]. Patient had hyperferritinemia (>500 mcg/L) with normal fibrinogen (522 mg/dl) and triglycerides (50 mg/dl) levels. He thus fulfilled 5 out of 8 diagnostic criteria of HLH and was treated as per HLH 2004 protocol with systemic antifungals and injection dexamethasone 10 mg/m2. Despite all supportive measures, patient developed multiorgan dysfunction and succumbed.
Figure 1.
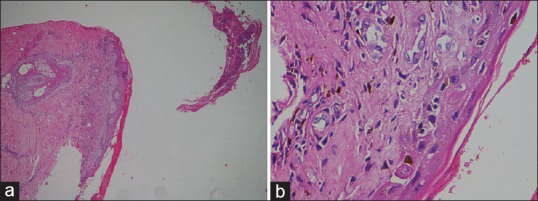
(a) Skin with detached stratum corneum, large subcorneal pustule (H and E, ×4). (b) Mild spongiosis, marked regenerative atypia, basal vacuolar degeneration, pigment incontinence and apoptotic keratinocytes (H and E, ×40)
Figure 2.
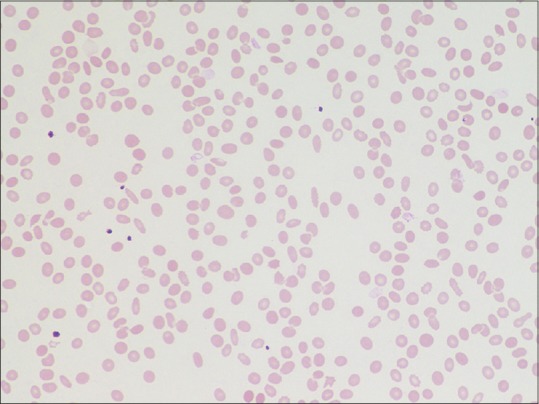
Peripheral smear showing pancytopenia (Giemsa ×40)
Figure 3.
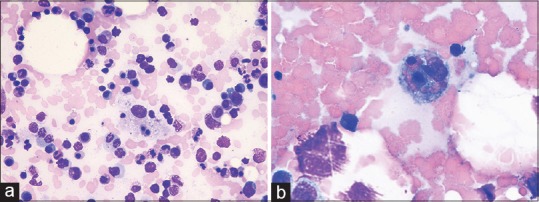
Bone marrow showing hemophagocytosis. (a) Giemsa ×40. (b) Oil immersion ×100)
Discussion
HLH is a rare disease characterized by cytokine overproduction and tissue infiltration with cytotoxic lymphocytes and activated macrophages resulting in fever, hepatosplenomegaly, and cytopenias.[1,2,9] A strict diagnostic criteria of HLH was given in 2004 by Histiocyte Society and is as follows.[9,10] Molecular diagnosis consistent with HLH that is the identification of mutations of PRF1, UNC13D, or STX11 or patient should fulfill five out of the eight criteria: fever (>100.4°F), splenomegaly, cytopenia affecting at least two of three lineages: Hb <9 g/dl (in infants <4 weeks: Hb <10 g/100 ml, platelets <100 × 109/L, Neutrophils <1 × 109/L), hypertriglyceridemia (fasting >265 mg/dl) and or hypofibrinogenemia (≤150 mg/dl), ferritin >500 ng/ml, hemophagocytosis in bone marrow, spleen, or lymph nodes, low or absent NK cell activity, soluble CD25 (soluble IL-2 receptor) >2400 U/ml and in the case of familial HLH there should be no evidence of malignancy.
This patient presented with features of methotrexate toxicity with skin necrosis, bone marrow suppression, and renal failure. His progressive pancytopenia was initially thought to be due to high dose methotrexate. He did not respond to leucovorin rescue therapy or injection Filgrastim and worsened clinically with features suggestive of sepsis due to which antibiotics were upgraded. He was evaluated for HLH as he had progressive pancytopenia unresponsive to treatment and decreased counts in spite of platelet transfusions. The major pathologic feature of HLH is hemophagocytosis as seen in our patient. It is also seen in conditions like sepsis and multiorgan failure.[3] There is considerable clinical and laboratory overlap between sepsis and HLH.[8,9] Diagnosis of HLH was established in our patient as he fulfilled five out of eight diagnostic criteria of HLH. This is a rare presentation of HLH complicating methotrexate toxicity. This case report is presented to enlighten clinicians about HLH inorder to reduce the mortality associated with the disease.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Janka G. Hemophagocytic lymphohistiocytosis: When the immune system runs amok. Klin Padiatr. 2009;221:278–85. doi: 10.1055/s-0029-1237386. [DOI] [PubMed] [Google Scholar]
- 2.Rosado FG, Kim AS. Hemophagocytic lymphocytosis: An update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713–27. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 3.Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: Clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6:137–54. doi: 10.1586/eci.09.58. [DOI] [PubMed] [Google Scholar]
- 4.Mehta RS, Smith RE. Hemophagocytic lymphohistiocytosis (HLH): A review of literature. Med Oncol. 2013;30:740. doi: 10.1007/s12032-013-0740-3. [DOI] [PubMed] [Google Scholar]
- 5.Freeman HR, Ramanan AV. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96:688–93. doi: 10.1136/adc.2009.176610. [DOI] [PubMed] [Google Scholar]
- 6.Favara BE. Hemophagocytic lymphohistiocytosis: A hemophagocytic syndrome. Semin Diagn Pathol. 1992;9:63–74. [PubMed] [Google Scholar]
- 7.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–22. doi: 10.1016/S1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price B, Lines J, Lewis D, Holland N. Haemophagocytic lymphohistiocytosis: A fulminant syndrome associated with multiorgan failure and high mortality that frequently masquerades as sepsis and shock. S Afr Med J. 2014;104:401–6. doi: 10.7196/samj.7810. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 10.Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol. 2004;50:157–74. doi: 10.1016/j.critrevonc.2004.01.002. [DOI] [PubMed] [Google Scholar]