

Dear Editor,
A 16-year-old girl presented with an asymptomatic localized pigmented scaly lesion on her back since birth. Clinical examination revealed brown adherent thick scales and verrucous papules involving the right scapular region in a Blashkoid distribution measuring approximately 5 cm × 5 cm [Figures 1 and 2]. The patient was born to a nonconsanguineous marriage, and there was no family history of similar complaints. Histopathological examination revealed prominent hyperkeratosis, hypergranulosis, acanthosis, and areas of vacuolar epidermal disintegration, especially involving the upper Malphigian layer [Figure 3]. Closer view of the degenerate areas showed keratinocytes with perinuclear vacuoles and large clumped keratohyalin granules [Figure 4]. Based on the clinical and histological features, a diagnosis of mosaic form of epidermolytic ichthyosis was established.
Figure 1.
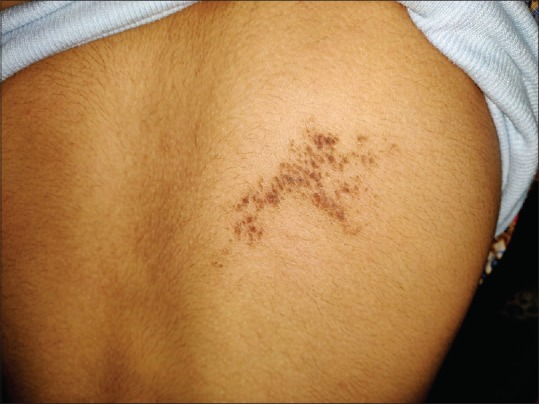
Brown adherent scaly plaque about 5 cm × 5 cm involving the right scapular area in a Blashkoid distribution
Figure 2.
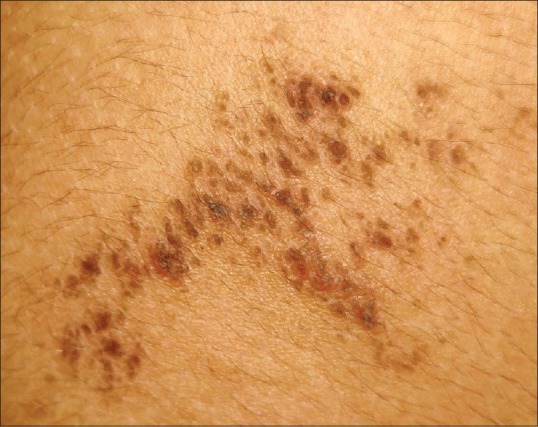
Close-up view of the lesion showing verrucous papules and adherent brown scales
Figure 3.
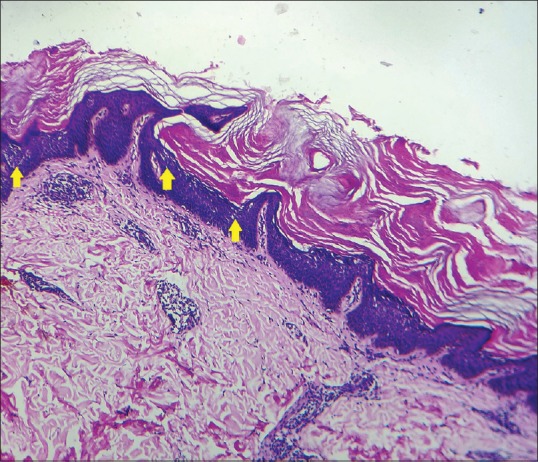
Prominent hyperkeratosis, hypergranulosis, and acanthosis are seen with foci of upper epidermal disintegration (yellow arrows). [H and E, Original magnification ×10]
Figure 4.
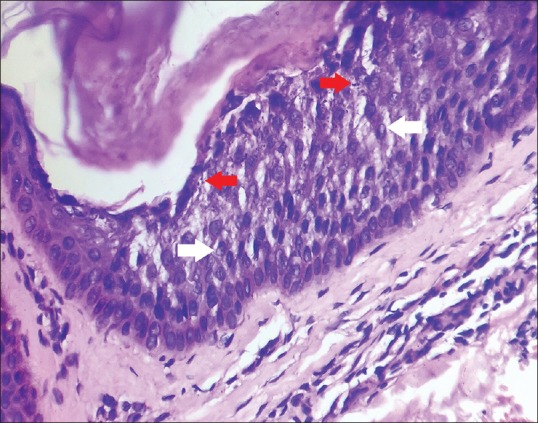
High power view showing vacuolar degeneration of the keratinocytes, perinuclear vacuoles (white arrows), and prominent intracellular keratohyalin granules (red arrows). [H and E, Original magnification x40]
Epidermolytic ichthyosis is an autosomal dominant disorder of keratinization due to mutation in the genes KRT1 and KRT10 encoding cytokeratins 1 and 10. The disease manifests at birth with skin fragility and is characterized by generalized erythema, blistering, and erosions (hence previously known as congenital bullous ichthyosiform erythroderma). With age, the disease tends to localize predominantly at the flexures and sites of trauma as hyperkeratotic yellow-brown plaques or corrugated thick scales with palmoplantar keratoderma. Histopathology is characterized by prominent hyperkeratosis together with disintegration of the epidermis (epidermolytic hyperkeratosis) with the cells typically exhibiting perinuclear vacuoles and clumped keratohyalin granules.[1,2]
Mosaic forms of the disease can occur due to post-zygotic somatic mutations and are referred to as “epidermolytic nevi” due to resemblance with linear epidermal nevi. Type 1 mosaicism, wherein one allele of the gene is mutated produces the classical mosaic form of the disease as in this case. Type 2 mosaicism affects the heterozygous carriers (having a milder generalized form of the disease) and is manifested as a more severe and early-onset disease. The diagnosis is established by typical histological features as described above.[1,2]
Although the mosaic forms (especially like the ones described in this instance) do not pose any major management issues, they are of important clinical relevance when the mosaicism also involves the gonadal cells. In such circumstances, the genetic defect can be transmitted to the offspring who will manifest a full blown disease.[3,4,5,6] The case discussed here is a more localized and limited form of the disease in comparison to the ones described in the literature.[3,4,5,6] Nonetheless, it may be well advised to histologically analyze all the cases of linear epidermal nevi routinely to look for associated epidermolytic hyperkeratosis and the clinical implications associated with such a finding, especially in widespread lesions.[4]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his/her consent for his images and other clinical information to be reported in the journal. The patient understands that his/her names and initials will not be published and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Mendes MS, Kouzak SS, Aquino TA, Takano GH, Lima Ade P. Mosaic epidermolytic ichthyosis - Case report. An Bras Dermatol. 2013;88:116–9. doi: 10.1590/abd1806-4841.20132203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judge MR, McLean WHI, Munro CS. Disorders of Keratinization. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 8th ed. Oxford: Wiley-Blackwell; 2010. pp. 19.1–19.112. [Google Scholar]
- 3.Akhyani M, Kiavash K, Kamyab K. Bullous ichthyosiform erythroderma in a child born to a parent with systematized linear epidermolytic hyperkeratosis. Int J Dermatol. 2009;48:215–7. doi: 10.1111/j.1365-4632.2009.03569.x. [DOI] [PubMed] [Google Scholar]
- 4.Chassaing N, Kanitakis J, Sportich S, Cordier-Alex MP, Titeux M, Calvas P, et al. Generalized epidermolytic hyperkeratosis in two unrelated children from parents with localized linear form, and prenatal diagnosis. J Invest Dermatol. 2006;126:2715–7. doi: 10.1038/sj.jid.5700553. [DOI] [PubMed] [Google Scholar]
- 5.Nomura K, Umeki K, Hatayama I, Kuronuma T. Phenotypic heterogeneity in bullous congenital ichthyosiform erythroderma: Possible somatic mosaicism for keratin gene mutation in the mildly affected mother of the proband. Arch Dermatol. 2001;137:1192–5. doi: 10.1001/archderm.137.9.1192. [DOI] [PubMed] [Google Scholar]
- 6.Nazzaro V, Ermacora E, Santucci B, Caputo R. Epidermolytic hyperkeratosis: Generalized form in children from parents with systematized linear form. Br J Dermatol. 1990;122:417–22. doi: 10.1111/j.1365-2133.1990.tb08292.x. [DOI] [PubMed] [Google Scholar]