

Abstract
Angiokeratomas are variable sized hyperkeratotic vascular papules that are characterized histologically by superficial dilated capillaries in papillary dermis with epidermal proliferation. They can occur as a single lesion to a generalized form (angiokeratoma corporis diffusum). Angiokeratoma corporis diffusum though initially synonymous with Anderson Fabry disease, is now known to occur in a variety of lysosomal enzyme deficiencies. We report a case of 22 year old male with angiokeratoma corporis diffusum associated with acroparesthesias, febrile episodes, sensorineural hearing loss and renal involvement. Histopathological evaluation showed characteristic ectatic blood vessels with vacuolated endothelial cells in papillary dermis. Based on the clinical evaluation and available investigations, we suspected him to be having to Anderson fabry disease. Resource constraints limited our ability to confirm our diagnosis with enzyme assay and electron microscopy. We report this unusual case in desire of re emphasizing the importance of clinical evaluation for reaching a diagnosis in a resource poor setting.
Keywords: Angiokeratoma, Fabry disease, systemic involvement
Introduction
Angiokeratomas are variable sized hyperkeratotic vascular papules that are characterized on histology by superficial dilated capillaries with epidermal proliferation.[1] They can occur singly or in a generalized form and have many different clinical presentations. One of the types is angiokeratoma corporis diffusum (ACD), which most typically involves the “bathing trunk” area of the body, i.e., lower region of the trunk, buttocks, and thighs.[2] For many years, this term had been used synonymously for Fabry disease, a systemic disease caused by a deficiency of α-galactosidase A enzyme.[3,4,5,6,7,8] However, ACD has recently been found to occur in several other enzyme deficiencies also.[9,10,11,12,13,14] We report a case of ACD that we suspected to be associated with Fabry disease according to the clinical evaluation and available investigations.
Case Report
A 22–year-old male with normal intelligence came to our hospital with complain of pinhead sized reddish raised skin lesions over body since 15 years. The skin lesions first appeared on knees and gradually appeared all over the body. There was a history of bleeding from the lesions on scratching. Since last 2 months, patient had a history of shooting pains along lower limbs, which started from the back and radiated downward. The patient also complained of episodes of low-grade fever and generalized weakness for 1 month. There was no history visual disturbance, seizures, or family history of similar lesions. On examination, facies of patient were normal. Multiple discrete as well as grouped non blanchable angiomatous papules of variable size were present over trunk, extremities, and genitalia in a symmetrical pattern [Figures 1 and 2a-d]. Some lesions had a hyperkeratotic surface. Lesions were most clustered over lumbar area, buttocks, penis, and knees. Multiple petechiae were present over oral mucosa. Conjunctival and genital mucosae were normal. Multiple discrete, firm, mobile, and non tender lymph nodes were palpable in the cervical (1.5 cm) and inguinal region (3 cm). Alhough the patient did not complain of hearing loss, pure tone audiometry revealed bilateral sensorineural hearing loss. Ophthalmological examination including slit lamp evaluation did not reveal any abnormality. Hematological investigations revealed microcytic hypochromic anemia and decreased platelets. Renal and liver function tests were normal. On routine urine examination, proteinuria was present, which was later confirmed by evaluation of 24 h urinary protein (1.8 g/day). ECG and ECHO did not show any cardiovascular abnormality. The ultrasonography of the abdomen revealed multiple enlarged lymph nodes in the periumbilical region and right inguinal fossa. There was no organomegaly seen on ultrasonography. X-ray skull was done, which did not reveal any abnormalities.
Figure 1.
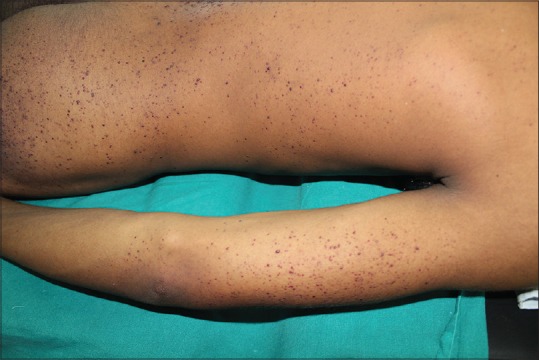
Discrete angiomatous keratotic papules over the trunk and upper limb
Figure 2.
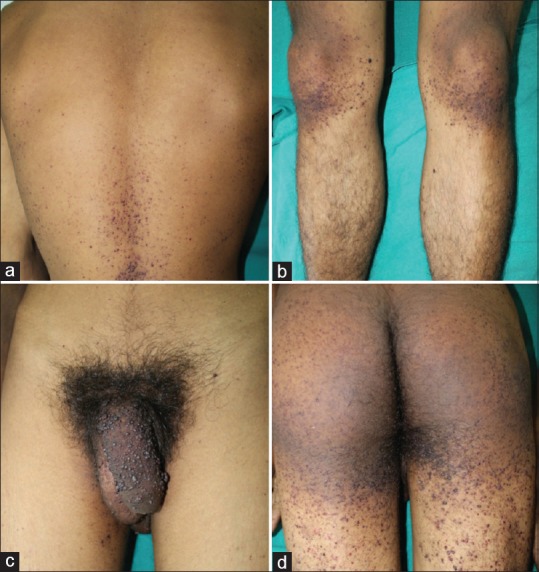
Clustered angiokeratomas around (a) lower back. (b) knees. (c) penis. (d) buttocks
Histopathological evaluation from a papule over back showed the presence of ectatic thin walled capillaries in papillary dermis with a variable degree of hyperkeratosis and elongation of rite ridges [Figures 3 and 4]. A typical finding was the presence of vacuolated endothelial cells, which may be seen in Fabry's disease because of the presence of lipids. Enzyme assays for confirmation of diagnosis could not be done owing to financial and resource constraints. The patient was managed on a multidisciplinary approach. The larger angiokeratomas were removed with electrocautery, and the family was counseled about the possible appearance of new lesions.
Figure 3.
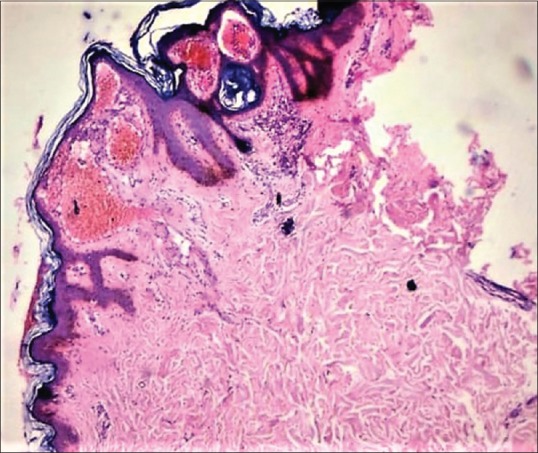
Hyperkeratosis and ectatic blood vessels seen in the papillary dermis (H and E 40×)
Figure 4.
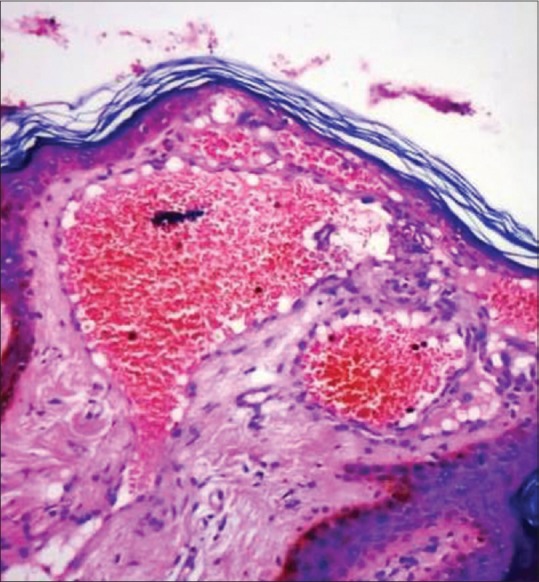
Presence of dilated capillaries lined with vacuolated endothelial cells in the papillary dermis (H and E 100×)
Discussion
ACD was first described in 1898 and was used as a synonym for Anderson Fabry disease.[4,6] It is now known to be associated with other lysosomal disorders also including fucosidosis, sialidosis, mannosidosis, GM1 gangliosidosis, and Kanzaki disease.[9,10,11,12,13,14] However, it has been reported in patients with normal enzyme activity also.[15,16,17,18] The common features seen in all these disorders are outlined in Table 1. Anderson Fabry's disease is an X-linked recessive disorder with a deficiency of α-galactosidase A enzyme. This enzyme is involved in the catabolism of glycosphingo lipids, and its deficiency results in accumulation of these glycosphingo lipids in various cells leading to systemic manifestations. Although each and every cell of the body having lysosomes can be affected, three main organ systems having significant involvement are the heart, kidneys, and nervous system.[6] In Anderson Fabry disease, angiokeratomas start to appear before the onset of puberty and gradually progress in number. Acroparesthesias may be the earliest presenting feature, but their intermittent and transient nature may delay the diagnosis by decades. They are often precipitated by a febrile illness, emotional stress, exercise, or exposure to heat or cold.[19] Anhydrosis or hypohidrosis can be an associated feature and it usually starts in the third decade. Renal impairment is a common finding in males with Fabry disease. Some studies have reported that 50% of patients have proteinuria by age 35 years and develop the end-stage renal disease by age 47 years.[20,21] Cardiac involvement in the form of arrhythmias and conduction defects is a common manifestation of Fabry disease and may produce symptoms as early as the second decade of life.[22]
Table 1.
Enzyme deficiency disorders with angiokeratoma corporis diffusum
| Disease | Enzyme affected | Cutaneous | CNS | PNS | Cardiac | Renal | Eye | Ear | Others |
|---|---|---|---|---|---|---|---|---|---|
| Fabry | Alpha-galactosidase A | ACD | - | Acroparesthesias Hypohydrosis | Arrythmias, conduction defects | Proteinuria, End-stage renal disease | Corneaverticillata, Tortuous retinal vessels | Hearing loss | Lymphedema |
| Fucosidosis | Alpha-L-fucosidase | ACD Coarse facies | Develop-mental delay | Hypohydrosis | - | - | - | - | Recurrent respiratory infections |
| Kanzaki | Alpha-N- acetylgalctosaminidase | ACD coarse facies | Neuro degeneration | - | - | - | - | Hearing loss | - |
| Galacto- sialidosis | Beta-galactosidase and neuraminidase | ACD Dwarfism Coarse facies | Neurodegeneration | - | - | - | Corneal clouding | Hearing loss | Seizures |
| Aspartyl- glycosaminuria | Aspertylglucosaminidase | ACD, coarse facies | Mental retardation | - | Valve involvement | - | - | - | Organomegaly |
| GM1 gangliosidosis | Beta-galactosidase | ACD Facial dysmorphism | Neurodegeneration, dystonia | - | - | - | - | - | Organomegaly |
| Beta- Mannosidosis | Beta-mannosidase | ACD | Learning difficulty | - | - | - | - | Hearing loss | Recurrent infections |
ACD=Angiokeratoma Corporis Diffusum; CNS=Central Nervous System ; PNS=Peripheral Nervous System
With the exception of Fabry disease, as all the enzyme deficiency disorders are usually associated with some impairment of intelligence, we considered a possibility of Fabry disease in our patient. Other features to support our diagnosis included the presence of acroparesthesias, low-grade febrile episodes, sensorineural hearing loss on audiometry, and renal involvement with proteinuria. The presence of vacuolated endothelial cells in a typical angiokeratoma lesson also strengthened our clinical diagnosis. In today's era of investigations being available so easily, clinical judgment sometimes takes a back seat. We report this unusual case just to re-emphasize the reliable value of proper clinical evaluation and use of simple investigations for reaching a diagnosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Weedon D. Skin Pathology. New York, NY: Churchill Livingstone Inc; 2002. Vascular tumors; pp. 1007–8. [Google Scholar]
- 2.Schiller PI, Itin PH. Angiokeratomas: An update. Dermatology. 1996;193:275–82. doi: 10.1159/000246270. [DOI] [PubMed] [Google Scholar]
- 3.Fabry H. Angiokeratoma corporis diffusum—Fabry disease: Historical review from the original description to the introduction of enzyme replacement therapy. Acta Paediatr Suppl. 2002;91:3–5. doi: 10.1111/j.1651-2227.2002.tb03102.x. [DOI] [PubMed] [Google Scholar]
- 4.Anderson W. A case of angeio-keratoma. Br J Dermatol. 1898;10:113–7. [Google Scholar]
- 5.Fabry J. Ein BeitragzurKenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagicaHebrae) Arch Dermatol Syph Berlin. 1898;43:187–200. [Google Scholar]
- 6.Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138:338–46. doi: 10.7326/0003-4819-138-4-200302180-00014. [DOI] [PubMed] [Google Scholar]
- 7.Möhrenschlager M, Braun-Falco M, Ring J, Abeck D. Fabry disease: Recognition and management of cutaneous manifestations. Am J Clin Dermatol. 2003;4:189–96. doi: 10.2165/00128071-200304030-00005. [DOI] [PubMed] [Google Scholar]
- 8.Breunig F, Weidemann F, Beer M, Eggert A, Krane V, Spindler M, et al. Fabry disease: Diagnosis and treatment. Kidney Int Suppl. 2003:S181–5. doi: 10.1046/j.1523-1755.63.s84.5.x. [DOI] [PubMed] [Google Scholar]
- 9.Fleming C, Rennie A, Fallowfield M, McHenry PM. Cutaneous manifestations of fucosidosis. Br J Dermatol. 1997;136:594–7. [PubMed] [Google Scholar]
- 10.Rodríguez-Serna M, Botella-Estrada R, Chabás A, Coll MJ, Oliver V, Febrer MI, et al. Angiokeratoma corporis diffusum associated with beta-mannosidase deficiency. Arch Dermatol. 1996;132:1219–22. doi: 10.1001/archderm.1996.03890340083013. [DOI] [PubMed] [Google Scholar]
- 11.Kanzaki T, Yokota M, Irie F, Hirabayashi Y, Wang AM, Desnick RJ. Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal alpha- N-acetylgalactosaminidase activity: Clinical, morphologic, and biochemical studies. Arch Dermatol. 1993;129:460–5. [PubMed] [Google Scholar]
- 12.Kanzaki T, Yokota M, Mizuno N, Matsumoto Y, Hirabayashi Y. Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum. Lancet. 1989;1:875–7. doi: 10.1016/s0140-6736(89)92867-5. [DOI] [PubMed] [Google Scholar]
- 13.Kanzaki T. Lysosomal storage diseases with angiokeratoma corporis diffusum. Nihon Rinsho. 1995;53:3062–7. [PubMed] [Google Scholar]
- 14.Kodama K, Kobayashi H, Abe R, Ohkawara A, Yoshii N, Yotsumoto S, et al. A new case of alpha-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Meniere's syndrome and without mental retardation. Br J Dermatol. 2001;144:363–8. doi: 10.1046/j.1365-2133.2001.04028.x. [DOI] [PubMed] [Google Scholar]
- 15.Holmes RC, Fensom AH, McKee P, Cairns RJ, Black MM. Angiokeratoma corporis diffusum in a patient with normal enzyme activities. J Am Acad Dermatol. 1984;10:384–7. doi: 10.1016/s0190-9622(84)80012-2. [DOI] [PubMed] [Google Scholar]
- 16.Crovato F, Rebora A. Angiokeratoma corporis diffusum and normal enzyme activities. J Am Acad Dermatol. 1985;12:885–6. doi: 10.1016/s0190-9622(85)80123-7. [DOI] [PubMed] [Google Scholar]
- 17.Marsden J, Allen R. Widespread angiokeratomas without evidence of metabolic disease. Arch Dermatol. 1987;123:1125–7. [PubMed] [Google Scholar]
- 18.Lu YY, Lu CC, Wu CS, Wu CH. Familial angiokeratoma corporis diffusum without identified enzyme defect. Indian J Dermatol Venereol Leprol. 2015;81:46–9. doi: 10.4103/0378-6323.148568. [DOI] [PubMed] [Google Scholar]
- 19.Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. 2004;34:236–42. doi: 10.1111/j.1365-2362.2004.01309.x. [DOI] [PubMed] [Google Scholar]
- 20.Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, et al. Natural history of Fabry renal disease: Influence of alpha-galactosidase aactivity and genetic mutations on clinical course. Medicine (Baltimore) 2002;81:122–38. doi: 10.1097/00005792-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Sessa A, Meroni M, Battini G, Righetti M, Maglio A, Tosoni A, et al. Renal involvement in Anderson-Fabry disease. J Nephrol. 2003;16:310–3. [PubMed] [Google Scholar]
- 22.Linhart A, Magage S, Palecek T, Bultas J. Cardiac involvement in Fabry disease. Acta Paediatr Suppl. 2002;91:15–20. doi: 10.1111/j.1651-2227.2002.tb03104.x. [DOI] [PubMed] [Google Scholar]