

Abstract
Pyrin is an inflammasome sensor in phagocytes that is activated in response to bacterial toxins and effectors that modify RhoA. Pathogen effector-triggered pyrin activation is analogous to an indirect guard mechanism in plants. Pyrin activation appears to be triggered when RhoA GTPases in a host cell are prevented from binding downstream signaling proteins (transducers). RhoA transducers that control this response include PRK kinases, which negatively regulate pyrin by phosphorylation and binding of 14–3-3 proteins. Microtubules regulate pyrin at different levels and may serve as a platform for inflammasome nucleation. Pyrin increases inflammation in the lung, gut or systemically during infection or intoxication in mouse models and protects against systemic infection by decreasing bacterial loads. Pathogenic Yersinia spp. overcome this protective response using effectors that inhibit the pyrin inflammasome. Gain of function mutations in MEFV, the gene encoding pyrin, cause the autoinflammatory disease Familial Mediterranean Fever. Yersinia pestis may have selected for gain of function MEFV mutations in the human population.
Graphical Abstract
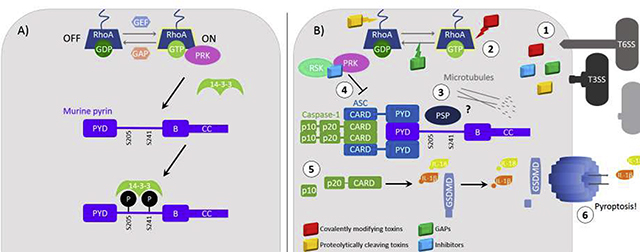
A) In steady-state conditions, RhoA is constantly being turned on and off by GEFs and GAPs that exchange GDP for GTP. RhoA regulates the pyrin inflammasome through interactions with transducers of RhoA signaling, PRKs, that phosphorylate pyrin creating a 14–3-3 binding site. Phosphorylated and 14–3-3 bound pyrin is locked in an off state where its conformation prevents interaction with inflammasome components. B) (1) Bacteria deliver toxins or effecter proteins to host cells using T3SS, T6SS, secretion via ECVs or other general secretion mechanisms. There are several classes of RhoA modifying toxins including, those that covalently modify RhoA, those that proteolytically cleave RhoA and GAPs. (2) Toxins and effectors inactivate RhoA stopping PRK signaling to pyrin. (3) The lack of negative signal by PRKs and possible action of a phosphatase leads to the dephosphorylation of pyrin on its regulatory serine residues and loss of 14–3-3 binding. (4) Another class of toxins and effectors are those that inhibit pyrin inflammasome formation. YopM, an inhibitor of pyrin inflammasome formation represented in blue, recruits kinases RSK and PRK to pyrin to keep it phosphorylated. Other inhibitors of pyrin inflammasome formation work to prevent pyrin dephosphorylation by constitutively activating RhoA, Cnf, or inhibiting inflammatory signaling through MAPKK, YopJ. (5) When pyrin is dephosphorylated it then binds to the ASC adapter protein which binds and dimerizes pro-caspase-1 forming the inflammasome complex. In a mechanism not well understood microtubule polymerization is required for inflammasome formation. Upon dimerization, pro-caspase-1 undergoes autoproteolytic cleavage fully activating the enzyme. Mature caspase-1 then cleaves pro-IL-1β, pro-IL-18 and GSDMD to their mature forms. (6) Gasdermin-D then oligomerizes and forms pores in the plasma membrane which allows secretion of IL-1β and IL-18. If enough pores in the plasma membrane form the cell will undergo cell death via lysis known as pyroptosis.
How to represent inhibitors on this figure? show YopM mechanism?
A) RhoA regulation of pyrin. B) Inactivation of RhoA by bacterial effectors and pyrin inflammasome activation.
Introduction
Inflammasomes function to sense exposure of host cell cytosol to pathogen molecules and mount protective immune defenses [1], [2]. The pattern recognition receptors or sensors that regulate caspase-1 inflammasome assembly are activated by different mechanisms. Once activated, the sensors recruit, dimerize and promote auto-cleavage of pro-caspase-1 into its mature form. Some sensors bind directly to caspase-1, while others oligomerize the adaptor ASC (apoptosis-associated speck-like protein) to recruit caspase-1. Mature caspase-1 in turn generates a proteolytic fragment of gasdermin-D, which oligomerizes to form pores in plasma membranes. Caspase-1 also cleaves pro-IL-1β and pro-IL-18, and the mature forms of these proinflammatory cytokines are released through gasdermin-D pores [1], [2].
Several human monogenic autoinflammatory diseases are caused by mutations in inflammasome sensor genes [3], [4]. The gene responsible for the autoinflammatory disease Familial Mediterranean Fever (FMF) was mapped in 1997 and designated MEFV [3], [4]. MEFV was shown to be selectively expressed in phagocytes such as neutrophils and monocytes [3]. The majority of FMF mutations were mapped to the C-terminal B30.2 domain of human pyrin (Fig. 1), the product of MEFV. These mutations were initially thought to be loss of function, since FMF was considered an autosomal-recessive genetic disease. More than a decade of work was required before it was conceived that FMF variants of pyrin are gain of function and the recessive nature of the disease is due to a gene dosage effect [3], [4]. This concept is consistent with the autoinflammatory phenotype of mice homozygous for Mefv alleles in which human B30.2 domains with FMF mutations were “knocked-in” to murine pyrin, which lacks this domain (Fig. 1) [5].
Figure 1.

The structure of human and murine pyrin. The inflammasome sensor, pyrin, is made up of an N-terminal PYD domain that binds to the PYD domain in the ASC adaptor protein that allows CARD-CARD interactions for caspase-1 inflammasome formation. The linker region following the PYD domain contains 2 serine resides, S208 and S242 of human pyrin, S205 and S241 of murine pyrin. These serine residues are phosphorylated and create a binding site for a 14-3-3 dimer. Following the linker region is a B-box (B) domain and a coiled-coil (CC) domain that mediate protein-protein interactions. In human pyrin, the C-terminus is made up of a B30.2 domain. This domain contains the highest frequency of mutations that lead to FMF or carriage. It is proposed that this domain provides a layer of pyrin regulation, but the mechanism is unknown. Murine pyrin contains a short amino acid sequence following the CC domain but it is unknown if it is involved in pyrin regulation in mice.
Abbreviations: pyrin domain (PYD); apoptosis-associated speck-like protein (ASC); caspase activation and recruitment domain (CARD)
Studies using ectopic protein expression in HEK-293 cells indicated that human pyrin exists as a trimer and interacts with ASC to activate caspase-1 in an inflammasome [6], [7]. The partial dependence of MEFV expression for release of IL-1β in response to exogenous LPS stimulation in human monocytes and THP-1 cells was early evidence that the pyrin inflammasome can be activated in response to a microbial molecule [8]. However, human monocytes have other inflammasomes that are activated in response to exogenous LPS and an alternative NLRP3-dependent mechanism appears to be a dominant pathway [9]. Pyrin was shown to contribute to cleavage of caspase-1 and release of IL-1β in human monocytes infected with Francisella novicida or Burkholderia cenocepacia [10], [11]. The pyrin-dependent response to B. cenocepacia required phagocytosis of the bacteria and a functional T6SS [11]. These results suggested that pyrin can act as a sensor to activate caspase-1 inflammasomes in response to an uncharacterized T6SS effector that is secreted by B. cenocepacia [11]. Activation of the pyrin inflammasome could thus be considered an effector-triggered immune response [12]. Structurally, pyrin is unique among inflammasome sensors (Fig. 1). It has an N-terminal pyrin domain (PYD) found in some members of the NLR (nucleotide-binding domain (NBD) and leucine-rich repeat (LRR)) superfamily [13] followed by B-box, coiled-coil and B30.2 domains found in tripartite motif (TRIM) proteins.
Following reports that Clostridium difficile TcdB could activate an ASC-dependent inflammasome in THP-1 cells or murine macrophages or dendritic cells [14]–[16], Xu et al. demonstrated that pyrin was required for this response [17]. In addition to TcdB, several additional bacterial toxins that covalently inactivate small GTPases the Rho family, RhoA, RhoB or RhoC, trigger the pyrin inflammasome [17]. As there appears to be functional redundancy between RhoA, RhoB or RhoC for controlling pyrin [17], for simplicity this review will refer to all three collectively as RhoA. The toxins that trigger the pyrin inflammasome, C. difficile TcdA and TcdB, C. botulinum C3, Vibrio parahaemolyticus VopS and Histophilus somni IbpA [17], covalently modify RhoA within or near the border of switch 1 (Table 1). The B. cenocepacia T6SS-dependent pyrin inflammasome response was shown to be caused by deamidation of RhoA N41, the same residue that is ADP-ribosylated by C3 (TecA, the effector responsible is described below) [17]. Direct interaction between RhoA and pyrin in cells could not be detected [17]. Thus, pyrin does not directly detect modified RhoA, but functions in an “indirect guard” mechanism [17] as proposed for certain NLRs in the plant innate immunity field [13]. Two mechanisms of pyrin regulation have recently been uncovered. Pyrin is negatively regulated by phosphorylation of two serines located between the PYD and B-box [18]–[20] (Fig. 1). These sites are phosphorylated by PRK (protein kinase C-related kinase, also known as PKN) kinases [18], [21], which bind to and are activated by RhoA. Components of the cytoskeleton, including microtubules, appear to regulate pyrin upstream and downstream of phosphorylation [18], [19], [22].
Table 1. Effectors and toxins that interact with the pyrin inflammasome.
This table lists all known or toxins and effectors that have been shown to or are predicted to either trigger pyrin activation or inhibit pyrin inflammasome formation. Effectors and toxins that target RhoA may do so by several different mechanisms. First, many effectors and toxins inactivate RhoA through covalent modification that prevents loading of GTP or binding to transducers. The amino acid residues modified in RhoA, Rac1 and Cdc42 are noted for those targets that are known. Effectors can also function as GAPs that shift the majority of RhoA in the host cell from the GTP to the GDP-bound state effectively preventing interactions with transducers. Finally, Yersinia YopT cleaves RhoA from the membrane resulting in mislocalization and loss of interaction with transducers. There are also two Yersinia effectors that inhibit the pyrin inflammasome. YopM binds to pyrin and hijacks PRKs to keep pyrin phosphorylated and 14–3-3 bound. YopJ limits inflammasome function by acetylating MAPKKs, which may directly reduce activation of pyrin or decrease expression of inflammasome components such as IL-1β. Purified commercially-available Cnf toxin has been shown to inhibit the pyrin inflammasome by deamidation of RhoA, leading to its constitutive activation. The context in which the interaction of each effector or toxin with the pyrin inflammasome has been studied is described.
| Pathogen and toxin/effector | Mechanism | Targets | Inflammasome outcome | Context | Reference |
|---|---|---|---|---|---|
| Burkholderia cenocepacia TecA | Deamidation | RhoA (N41), Rac1 (N39), Cdc42 (?) | Activator | Ex vivo, in vivo infections | [17], [24] |
| Clostridium difficile TcdA/B | Glucosylation | RhoA (T37), Rac1, Cdc42 (T32) | Activator | Ex vivo intoxications and infections | [17] |
| Clostridium botulinum C3 | ADP Ribosylation | RhoA (N41) | Activator | Ex vivo intoxications | [17] |
| Staphylococcus aureus EDIN-B | ADP Ribosylation | RhoA (N41) | Activator | Predicted | NA |
| Bordetella pertussis Ptx | ADP Ribosylation | Gαi, Gα0 (C351) | Activator | In vivo intoxication | [23] |
| Vibrio parahaemolyticus VopS | Adenylylation | RhoA (T37), Rac1 & Cdc42 (T35) | Activator | Ex vivo ectopic intoxication (LFn) | [17] |
| Histophilus somni IbpA Fic1/2 | Adenylylation | RhoA (Y34), Rac1 & Cdc42 (Y32) | Activator | Ex vivo ectopic intoxication (LFn) | [17] |
| Pseudomonas aeruginosa ExoS GAP | GAP | RhoA, Rac1 & Cdc42 | Activator | Predicted | NA |
| Pseudomonas aeruginosa ExoT GAP | GAP | RhoA, Rac1 & Cdc42 | Activator | Predicted | NA |
| Yersinia spp. YopE | GAP | RhoA, Rac1 & Cdc42 | Activator | Ex vivo, in vivo infections | [21], [25], [26] |
| Yersinia spp. YopT | Cysteine protease | RhoA, Rac1 & Cdc42 | Activator | Ex vivo, in vivo infections | [21], [26] |
| Yersinia spp. YopM | Hijack PRKs to phosphorylate pyrin | Pyrin, PRKs, RSKs | Inhibitor | Ex vivo, in vivo infections | [21], [25] |
| Yersinia spp. YopJ | Acetylation | MAPKKs and TAK1 | Inhibitor | Ex vivo, in vivo infections | [55], [54] |
| Cnf (commercial) | Deamidation | RhoA (Q63) | Inhibitor | Ex vivo intoxication | [18] |
Understanding how toxins and effectors trigger the pyrin inflammasome, and the impact of this innate immune response on bacterial infections, remain important goals in the field of host-microbe interactions. We review new findings that show a multistep process for regulation of the pyrin inflammasome. Studies that have investigate the role of the pyrin inflammasome in host-microbe interactions ex vivo and in vivo [21]–[27] are also described. Ex vivo experiments use cells that natively express pyrin such as human peripheral blood mononuclear cells (PBMCs), purified monocytes or monocyte-like lines (e.g. THP-1), can be induced to express pyrin such as LPS-primed murine bone marrow-derived macrophages (BMDMs) or transfected to ectopically express pyrin (e.g. HEK-293). Regulation of pyrin in the context of FMF has recently been reviewed [3], [4] and is not a focus of this chapter. Additional roles for pyrin in other cellular processes such as autophagy [28], tumor suppression [29] and sensing of microbiota metabolites [30], are emerging but are not covered due to space limitation.
Regulation of the pyrin inflammasome
Toxins and effectors that prevent RhoA from interacting with transducers trigger pyrin activation
RhoA is geranylgeranylated at a C-terminal Cys residue and localizes to membranes [31]. With the help of regulatory proteins RhoA switches between a GDP-bound off conformation and GTP-bound on conformation. Guanine nucleotide exchange factors (GEFs) stimulate the release of GDP from RhoA. The cellular concentration of GTP is higher than GDP, favoring GTP binding. In the GTP-bound conformation the switch I and switch II regions of RhoA bind to and activate signaling proteins such as the PRKs. These signaling proteins are usually referred to as effectors, but here they are referred to as transducers, to avoid confusion with bacterial effectors. GTPase activating proteins (GAPs) accelerate the slow intrinsic rate of GTP hydrolysis, to switch RhoA back to the GDP-bound off conformation. Guanine nucleotide dissociation inhibitors (GDIs) bind to RhoA-GDP and prevent membrane localization.
The toxins and effectors that trigger pyrin activation have one thing in common: they all prevent RhoA from interacting with transducers (Table 1). The toxins and effectors that covalently modify switch I of RhoA prevent transducer binding by steric hindrance (glucosylation of T37 or adenylylation of T37 or Y34) or by driving RhoA-GDP into a complex with GDI in cytosol (ADP ribosylation or deamidation of N41) (Table 1). Experimental approaches that maintain the ability of RhoA to interact with transducers in the presence of a toxin or effector prevent pyrin activation. Aubert et al. showed that overexpression of a deamidation-resistant N41L RhoA and pyrin in a murine dendritic cell line prevented caspase-1 cleavage in response to B. cenocepacia infection [24]. When equivalent mutants of Rac1 and Cdc42 (N39L) were similarity tested, they did not prevent B. cenocepacia-triggered caspase-1 activation, further demonstrating the importance of RhoA. Cytotoxic necrotizing factor (Cnf) deamidates RhoA Q63 in switch II, which blocks GTP hydrolysis and locks RhoA in an active conformation (Table 1). Park et al. found that treatment with purified commercially-available Cnf strongly reduced IL-1β release in C3-intoxicated BMDMs [18] indicating that Cnf can override ADP ribosylation of N41 to maintain interaction of RhoA with transducers at membranes. Some Y. pseudotuberculosis strains encode a Cnf toxin that activates RhoA and it is an important virulence factor in mouse infection models [32]. However, it is not known if the Yersinia Cnf inhibits the pyrin inflammasome in vivo.
Toxins and effectors that modify RhoA without covalently modifying switch I also trigger activation of pyrin (Table 1). Several bacterial effectors have GAP domains that use an arginine finger motif to accelerate conversion of RhoA-GTP to RhoA-GDP (Table 1). The Yersinia spp. T3SS effector YopE has a single GAP domain that targets RhoA and triggers activation of pyrin in BMDMs [21], [25], [26]. Pseudomonas aeruginosa ExoS and ExoT are bifunctional effectors with N-terminal GAP domains and C-terminal ADPRT domains (Table 1). The GAP domains of ExoS and ExoT target RhoA and are predicted to activate pyrin (Table 1). Yersinia YopT is a cysteine protease that cleaves upstream of the C-terminal Cys in RhoA (either -GDP or -GTP bound forms) (Table 1). Detachment of RhoA from membranes by YopT triggers the pyrin inflammasome in BMDMs [21], [26]. Defects in geranylgeranylation, such as that seen in the autoinflammatory disease hyperimmunoglobulinemia D syndrome (HIDS), can trigger the pyrin inflammasome in part due to decreased RhoA localization to membranes [18], [33].
Taken together, the above data suggest that pyrin activation is triggered when RhoA molecules in a host cell are prevented from interacting with transducers by one of the above mechanisms. This concept is in line with data showing that the RhoA transducer PRK is critical for negative regulation of pyrin [18], [21]. Given that RhoA molecules normally cycle between active and inactive conformations and on and off membranes in the host cell, it is possible that there is a threshold number of active RhoA molecules in a cell needed for negative regulation of pyrin. Bacterial toxins and effectors may trigger activation of pyrin when active RhoA drops below this threshold. This model is consistent with recent data suggesting that FMF mutations are hypermorphic and increase the threshold for negative regulation of pyrin [34].
PRK kinases are activated by RhoA and negatively regulate pyrin by phosphorylation of 14–3-3 binding sites
The PRKs are members of the protein kinase C family and are among the best characterized RhoA transducers. Several studies show that PRKs negatively regulate pyrin by phosphorylation of two serines that form a binding site for a dimer of the 14–3-3ε protein [18]–[20]. Park et al. used siRNA knockdown and inhibitors or activators to show that PRK isoforms 1 and 2 act redundantly to negatively regulate pyrin [18]. The 14–4-3 interaction was disrupted following treatment of BMDMs with C3, suggesting that pyrin is dephosphorylated upon activation. PRK1 was shown to bind to pyrin via its C-terminal kinase domain [18]. Recombinant PRK1 or PRK2 phosphorylated serines 208 and 242 located between the PYD and B-box in human pyrin (Fig. 1) [18]. These phosphorylated residues were previously shown to mediate binding of 14–3-3 to pyrin [35]. Codon changes at positions 208 or 242 resulted in constitutively active pyrin and loss of 14–3-3 binding [18]. Masters et al. discovered that a human autoinflammatory disease associated with neutrophilic dermatosis (and clinically distinct from FMF) is caused by an S242R codon change and a dominant gain of function mutation in MEFV [20]. Gao et al. significantly extended these results by showing that murine pyrin is negatively regulated by 14–3-3 binding to the corresponding S205 and S241 (Fig. 1) [19]. Phospho-specific monoclonal antibodies were developed and used to show that the regulatory serines in mouse and human pyrin were dephosphorylated upon treatment with TcdB and other RhoA-targeting toxins [19].
Together, these data suggest that PRKs activated by RhoA negatively regulate pyrin by phosphorylation of two serines, creating a binding site for a 14–3-3 dimer. This may keep pyrin in an inactive conformation. When RhoA is modified by bacterial toxins or effectors, this negative regulation pathway is interrupted. To counter negative regulation by PRKs, a role for a phosphatase in positive regulation of pyrin has been suggested [18]–[20] but additional work is required to address this possibility.
Regulation of pyrin by microfilaments
Pyrin can associate with actin or tubulin microfilaments [4], [36] and there is evidence that these cytoskeletal structures can regulate pyrin at different levels.
Inhibition of actin polymerization with cytochalasin D does not trigger the pyrin inflammasome [17]. However, variants of the actin-depolymerizing protein Wdr1, which increase actin filaments, trigger activation of pyrin [37], [38]. Mice homozygous for the Wdr1rd allele have an autoinflammatory disease that is partially dependent on pyrin-dependent IL-18 release from monocytes [37]. The Wdr1rd allele has a mRNA splicing mutation that results in reduced production of the wild-type protein as well as synthesis of a shorter mutant protein with reduced half-life and incorrect folding. Monocytes from Wdr1rd/rd mice cultured ex vivo do not spontaneously release IL-18 at higher levels than wild-type cells, but do so in response to TcdB or LPS [37]. Inhibition of actin polymerization using latrunculin-B reduces release of IL-18 from Wdr1rd/rd monocytes, suggesting that aberrant actin polymerization triggers the pyrin inflammasome [37]. An autoinflammatory disease in two patients was linked to the presence of a homozygous missense mutation in WDR1 [38]. The patients had increased serum IL-18. Monocyte-derived dendritic cells and neutrophils from the patients released higher IL-18 than controls in response to LPS. When the mutant Wrd1 protein and pyrin were overexpressed in HEK-293 cells, the Wrd1 protein formed abnormal aggregates that co-localized with pyrin. The authors suggest that these aggregates trigger spontaneous ASC oligomerization and pyrin inflammasome activation [38].
Colchicine is a microtubule polymerization inhibitor and an effective treatment for FMF [4]. Colchicine inhibits activation of the pyrin inflammasome [7], [39] and recent experiments suggest that microtubule dynamics can regulate pyrin at different levels [18], [19], [22].
Park et al. used relatively low concentrations of colchicine (0.25 μM or 100 ng/ml) and obtained evidence that colchicine inhibits pyrin activation in C3-treated BMDMs, due to increased RhoA-GTP levels [18]. This was attributed to the known ability of colchicine to activate RhoA by stimulating the release of GEF-H1 by microtubule depolymerization [18].
Gao et al. showed that colchicine and other more potent tubulin inhibitors prevent activation of inflammasomes downstream of pyrin dephosphorylation in BMDMs treated with TcdA and other RhoA toxins [19]. Even in the presence of the relatively higher concentrations of colchicine (10 μM) pyrin was dephosphorylated and released from 14–3-3, but failed to oligomerize ASC [19]. Similar results were obtained by Van Gorp et al. using BMDMs and PBMCs treated with TcdA [22]. The microtubule-stabilizing drug paclitaxel (taxol) had an intermediate [19] to no effect [22] on pyrin inflammasome activation; this discrepancy may be due to the use of different cells (BMDMs vs. PBMCs, respectively) or other experimental conditions. Different conclusions have also been reached on the ability of colchicine to inhibit the pyrin inflammasome in FMF PBMCs or monocytes [18], [22], likely due to the use of different stimuli and concentrations of the drug.
Mechanistically these results suggest that microtubules can serve as a platform for pyrin to assume a conformation that is conducive to ASC oligomerization [19], [22]. In transfected HEK-293 cells endogenous β-tubulin co-immunoprecipitated with full length human pyrin, as well as the individual PYD or a C-terminal region (amino acids 375–781) containing the B-box, coiled-coil, and B30.2 domains [22]. Additional studies are needed to understand how the interaction of pyrin with tubulin via these two regions contributes to inflammasome assembly.
Role of the pyrin inflammasome in host-microbe interactions
Burkholderia cenocepacia
B. cenocepacia is a Gram-negative bacterium that infects the lungs of immunocompromised patients. B. cenocepacia infections are most frequent in cystic fibrosis (CF) and chronic granulomatous disease (CGD) patient populations. While B. cenocepacia often causes chronic infection, severe cases of these infections can become bacteremic, leading to sepsis resulting in high mortality rates [40]. B. cenocepacia infections are difficult to treat due to this bacterium’s high antibiotic resistance [40].
It has been known for some time that B. cenocepacia disrupts the cellular actin cytoskeleton by inactivating Rac1 and Cdc42 using its type VI secretion system (T6SS) [41], [42]. B. cenocepacia’s ability to activate the pyrin inflammasome was first demonstrated by Gavrilin et al. 2012 [11]. In this study, B. cenocepacia K56–2 infection of human monocytes and THP-1 cells resulted in caspase-1 cleavage and IL-1β secretion dependent on pyrin expression and the B. cenocepacia T6SS [11].
The mechanistic basis for the activation of the pyrin inflammasome by the B. cenocepacia T6SS was later revealed in 2014 by Xu et al in studies that demonstrated that pyrin becomes activated in response to bacterial modifications that inactivate RhoA [17]. B. cenocepacia infection was shown to result in modification of RhoA at N41, a residue adjacent to the Switch I region. When this residue is deamidated it becomes an aspartic acid (Table 1). B. cenocepacia J2315 infection of WT BMDMs resulted in caspase-1 cleavage but this was not observed in WT BMDMs infected with B. cenocepacia Δhcp (lacking the T6SS) or in Mefv−/− BMDMs [17]. Intranasal (i.n.) infections of WT, Asc−/− and Mefv−/− mice with B. cenocepacia J2315 show that the ASC adaptor protein and pyrin are required for the recruitment of inflammatory cells seen by H&E at 12 hours post infection [17].
The T6SS effector, designated TecA was discovered by Aubert et al. 2016 [24]. In vitro experiments confirmed that TecA was the T6SS effector protein responsible for the cytoskeletal disruption in ANA-1 macrophages and that TecA deamidates RhoA at N41 [24]. Analysis of TecA’s amino acid sequence showed high conservation of 3 residues, cysteine-41, histidine-105 and aspartic acid-148, that were predicted to form a catalytic triad by in silico structure modeling [24]. Point mutations of cysteine-41 and histidine-105 to alanine resulted in the loss of TecA’s ability to deamidate RhoA and Rac1, cleave caspase-1 and cause cell death in primary murine BMDMs. Finally, in intranasal and intraperitoneal mouse infection models, pyrin inflammasome activation by B. cenocepacia J2315 TecA lead to increased immune cell infiltration into the lung and decreased mortality in WT mice compared to Mefv−/− mice [24]. The decreased mortality was associated with reduction in bacterial loads in spleen and liver. The authors concluded that pyrin inflammasome activation by TecA is host protective.
Future studies of B. cenocepacia TecA may focus on TecA induced pyrin inflammasome activation in the context of CF and CGD as these populations of individuals are at higher risk and may have intrinsic susceptibility to B. cenocepacia. It is assumed that deamidation of N41 of RhoA by TecA results in inactivation by the same mechanism as ADP ribosylation of this residue by C3 (Table 1), but this remains to be established. Structural and biochemical studies of TecA modifications of RhoA may elucidate this mechanism.
Bordetella pertussis Ptx
Pertussis toxin (Ptx) is one of the major virulence factors of B. pertussis, the agent of pertussis or whooping cough [43]. Ptx is a hexameric (AB5) toxin, which is taken up by host cell endocytosis, travels by retrograde transport through the ER, and the active A or S1 subunit enters the cytosol. The A monomer ADP ribosylates the α subunits of the heterotrimeric Gi and Go proteins. In the case of Gi, the modified G protein is unable to inhibit adenylyl cyclase, resulting in increased production of cyclic AMP (cAMP).
Evidence indicates that Ptx contributes to respiratory pathology in pertussis, although the underlying mechanism remains unclear. In a non-infection setting, Ptx is used to increase experimental autoimmune encephalomyelitis (EAE), the most widely used animal model of multiple sclerosis (MS). Dumas et al. obtained evidence that pyrin plays a role in Ptx-induced EAE [23]. The authors used a model in which transgenic mice with lymphocytes specific for myelin antigen are given intraperitoneal injection of Ptx to induce EAE, and in this context pyrin deficient mice had reduced EAE. Ptx was shown previously in this model to trigger systemic production of IL-6 and recruitment and adhesion of leukocytes and lymphocytes to blood vessels in the brain. The authors found that Ptx induced TLR4-dependent synthesis of pro-IL-1β and pyrin in CD11b+ leukocytes in the peritoneal cavity. Mature IL-1β was released into the peritoneal cavity and in turn stimulated systemic production of IL-6 from stromal cells in response to Ptx. This required the catalytic activity of the A subunit. Mice lacking pyrin, Asc or caspase-1 had diminished peritoneal IL-1β, and systemic IL-6 in response to injection of Ptx. Ex vivo administration of Ptx induced synthesis of pro-IL-1β and pyrin in CD11b+ leukocytes, but not release of IL-1β, suggesting that triggering of the inflammasome requires an additional unknown signal in vivo. Pyrin was important for adhesion of leukocytes but not lymphocytes to the blood vessels in the brain. Thus, Ptx induces lymphocyte recruitment to the brain independent of pyrin.
Overall, the data suggest that pyrin stimulates neutrophil adhesion to blood vessels, but is not required for the effector phase of EAE, when myelin-specific lymphocytes are activated and recruited to the brain. An association between FMF mutations and MS has been suggested, but remains controversial, and requires additional study. Additionally, it is unclear if Ptx-triggered activation of pyrin plays a role in B. pertussis pathogenesis. IL-1 signaling is important for clearance of B. pertussis in a murine lung infection model, however, this was independent of caspase-1 [44]. Furthermore, the adenylate cyclase toxin, not Ptx, may be more important for activating IL-1 signaling during pulmonary infection [45].
Clostridium difficile
C. difficile is a Gram-positive, anaerobic, spore-forming bacterium and the most frequent cause of nosocomial colitis in the developed world [46], [47]. Broad-spectrum antibiotics alter the gut microbiota, disrupting the body’s natural defense against C. difficile. Disruption of the microbiota allows C. difficile to overgrow in the colon. Virulent strains typically produce one or both of the large glucosylating toxins TcdA and TcdB. TcdA and TcdB both preferentially glucosylate RhoA,B,C over other Rho GTPases [48] although TcdB seems to be more active and more important for virulence than TcdA.
Purified TcdA and TcdB trigger the pyrin inflammasome in BMDMs and human monocytes [17], [19], [22], [49]. During the initial stage of infection it is likely that TcdA and TcdB initially intoxicate intestinal epithelial cells (IECs), causing IEC cell death and breakdown of the mucosal barrier [27], [50]. Breakdown of the barrier elicits inflammation, recruitment of neutrophils and monocytes to the mucosa, and permits translocation not of C. difficile but of commensal bacteria. Although IECs do not express pyrin [27], [29], pyrin is expressed in neutrophils and monocytes, and if these cells become intoxicated with TcdA or TcdB, it could amplify inflammation. Consistently, injection of purified TcdA or TcdB into cecal or ileal loops in mice induces IL-1β release and immunopathology [16], [49]. This response was dependent on the caspase-1 adaptor ASC [16]. Ex vivo infection of BMDMs or PBMCs with live TcdA+ TcdB+ C. difficile elicits caspase-1 cleavage and IL-1β release [22]. This response was reduced when the C. difficile strain was TcdA- TcdB- or the macrophages lacked pyrin [22].
In mice pretreated with antibiotics, ASC deficiency results in increased susceptibility to C. difficile infection, due to reduced chemokine production and reduced neutrophil recruitment, which leads to increased translocation of commensals [51], indicating that an ASC-dependent inflammasome(s) is important for protection against C. difficile. However, challenge of pyrin deficient mice with C. difficile showed no difference in weight loss, stool, or bacteria in stool as compared to wild-type [27]. The effect of pyrin deficiency on release of IL-1β or mucosal inflammation was not determined in this study. TcdA and TcdB triggered IEC apoptosis via intrinsic caspase-3/7 pathway, restricting C. difficile [27]. Thus, pyrin does not appear to be important for host protection in this model, and the function of ASC in this context remains unclear.
Yersinia spp
In the genus Yersinia, three species are well-described human pathogens: Y. pestis, the causative agent of the Plague, and Y. pseudotuberculosis and Y. enterocolitica, which cause gastroenteritis [32]. These species are pathogenic due to a variety of virulence factors they share, and a plasmid-encoded type III secretion system (T3SS) is among the most critical for virulence [32]. Through the assembly of this apparatus, Yersinia can secrete its Yop effector proteins, which modulate normal host physiology and lead to immune evasion [32]. The Yersinia T3SS engages several different inflammasomes in infected host cells [52]. Seven Yops are secreted into the host cell and among them four interact with the pyrin inflammasome pathway: YopE and YopT modify RhoA, YopM inhibits pyrin and YopJ cooperates with YopM in this process (Table 1, Fig. 2).
Figure 2.

Yersinia spp. use a T3SS to inject Yop effectors into the infected host cell. YopE and YopT target RhoA. YopE acts as a Rho GAP, which inactivates the pool of RhoA in the host cell by shifting the equilibrium from the GTP to the GDP bound state. YopT acts by cleaving RhoA upstream of the terminal cysteine residue. This removes RhoA from the membrane where it can no longer effectively interact with transducers. YopM and YopJ prevent activation of the pyrin inflammasome YopM hijacks kinases RSK and PRK, interacts with pyrin and maintains pyrin in its phosphorylated and inactive state. Finally, YopJ acetylates MAPKKs, which may directly inhibit pyrin activation and or reduce expression of genes encoding inflammasome components such as pro-IL-1β.
Abbreviations: Yersinia outer protein (Yop); GTPase activating proteins (GAP); guanosine triphosphate (GTP); guanosine diphosphate (GDP); ribosomal s6 kinase (RSK); mitogen-activated protein kinase kinase (MAPKK)
LaRock et al. discovered that YopM inhibits a caspase-1 inflammasome to promote Yersinia virulence [53]. It was later shown that YopM specifically inhibits pyrin to counteract YopE- and YopT-triggered activation of this inflammasome pathway [21], [25], [54] (Fig. 2). In addition, YopJ was shown to cooperate with YopM to inhibit the pyrin inflammasome pathway [25], [54], [55]. Thus, Yersinia uses YopE and YopT to intentionally inactivate RhoA, and employs YopM and YopJ to counteract the compensatory protective immune mechanism of the pyrin inflammasome (Fig. 2).
YopE and YopT target RhoA, Rac1 and Cdc42 and promote virulence by inhibiting phagocytic signaling pathways. The underlying mechanisms by which the YopE GAP or the YopT protease trigger the pyrin inflammasome have been studied by infecting BMDMs with Yersinia yopM mutants [21], [25], [26]. YopE activity is dominant in this regard: YopT-triggered pyrin activation is detected only in the absence of YopE in Y. pseudotuberculosis and not at all in Y. pestis [21], [25]. The Y. pestis YopT isoform has a D298G substitution relative to Y. pseudotuberculosis, which could explain its lower pyrin-activating activity. YopE also dominates over YopT in terms of virulence activity, and some strains of Y. pseudotuberculosis have inactivating deletions in yopT [26]. Similar to the toxins and effectors that covalently modify switch I in RhoA (Table 1), Y. pseudotuberculosis YopE and YopT were shown to trigger dephosphorylation of pyrin [26]. Interestingly, although they triggered the same conserved response, YopT caused slower dephosphorylation of pyrin in comparison to YopE [26]. In addition, protein stability and RhoA specificity were critical for YopE to trigger pyrin dephosphorylation. Together, these results demonstrate that all RhoA-modifying toxins and effectors trigger activation of pyrin by a conserved dephosphorylation mechanism, and that different features of the bacterial proteins affect the efficiency of this process [26].
The YopM effector has several proposed roles in Yersinia virulence [52]. Key to YopM’s role in inhibiting pyrin is the central leucine rich repeat (LRR) domain, and a conserved C-terminal 20 amino acid tail. Each Yersinia strain encodes one YopM isoform, but these isoforms can vary in numbers of LRRs from 13–21. All YopM isoforms tested to date inhibit pyrin equally [21]. The LRR and C-terminal regions lack apparent enzymatic activity and instead act to bind host protein kinases [21], [56]–[58]. The C-terminal tail binds to and activates isoforms of the ribosomal S6 kinases (RSKs), is essential to inhibit pyrin, and appears to mediate multimerization of YopM [25], [56]–[60]. The LRR region binds to and activates PRKs [56], [58], [60], and hijacks these kinases to interact with and keep pyrin phosphorylated. A better understanding of how YopM hijacks PRKs and RSKs to inhibit pyrin, and why the LRR variability exists among isoforms, remain important goals in the field.
The acetyltransferase activity of YopJ inactivates multiple MAPKKs and the MAPKKK TAK1 (Table 1). In naïve BMDMs, YopJ counteracts pyrin by inhibiting synthesis of inflammasome components such as IL-1β [25], [54]. However, in BMDMs primed with LPS for 5 hours or overnight, YopJ activity reduces the amount of IL-1β released upon infection with a Yersinia yopM mutant by 50% [25], [54], [55]. In this context YopJ may inhibit infection-induced expression of inflammasome components [25], [54], or a pathway important for post-translational regulation of pyrin [55]. YopJ does not appear to prevent dephosphorylation of pyrin [26], but may reduce pyrin-dependent cleavage of caspase-1 [55].
Mouse infection models demonstrate the importance of the pyrin inflammasome for host protection against Yersinia, and the relative importance of YopM and YopJ for counteracting this response. In a Y. pestis subcutaneous infection model representing bubonic plague, YopM and YopJ act redundantly to counteract the pyrin inflammasome [25], [54]. The virulence defect of a Y. pestis yopM yopJ mutant is restored in mice lacking pyrin, IL-1β, IL-18, indicating that both cytokines are important for protection [25], [54]. In a Y. pseudotuberculosis intragastric infection model a yopM is significantly attenuated and a role for YopJ in promoting virulence was evident only in the absence of YopM [55]. The virulence defect of a yopM mutant was fully rescued in Mefv−/− mice infected with Y. pseudotuberculosis by intravenous route [21]. This was shown by restored lethality and bacterial growth in spleen and liver [21]. Taken together, these results suggest that the pyrin inflammasome protects against systemic infection by reducing bacterial loads, and that the importance of YopM and YopJ for counteracting this host response depends on the species of Yersinia and the route of infection.
Conclusions and perspectives
Bacterial effector proteins that disarm RhoA or disrupt RhoA signaling are the most well understood triggers of the pyrin inflammasome. Pyrin may play a role in autophagy, tumor suppression and detecting microbial metabolites but the pathway of activation is poorly understood [28]–[30]. Pyrin is regulated on multiple levels including phosphorylation and by cellular microfilaments however, the mechanism behind regulation by the latter is not fully known [19], [37], [38]. Elucidating the role of the pyrin inflammasome in vivo requires additional work for most pathogens with RhoA modifying toxins and effectors covered in this review. Yersinia spp. have evolved effectors to prevent inflammasome formation which greatly increases its virulence but no other pathogens with similar effectors are known [21], [25], [54], [55]. Future ex vivo and in vivo host-microbe interaction studies on the role of toxin- and effector-triggered pyrin inflammasome activation will shed light on the role of pyrin in the innate immune response in opportunistic versus professional human pathogens and in different bodily sites.
Highlights.
The pyrin inflammasome is triggered in response to RhoA inactivation.
Bacterial effectors and toxins that inactivate RhoA vary in mechanism.
Pyrin induced inflammation protects against systemic bacterial infection.
Familial Mediterranean Fever is caused by mutations in the pyrin gene MEFV.
Selective pressure by plague may have driven gain of function MEFV mutations.
Acknowledgments
We thank the following individuals for reading and providing feedback on this review: to be determined
Funding
Financial support for the authors of this review came from the Department of Microbiology and Immunology and the Geisel School of Medicine at Dartmouth, Dartmouth College, the NIAID under award R01AI099222. N.A.L was supported by the Dartmouth Cystic Fibrosis Training Program (T32HL134598). N.P.M. was supported by Science Without Borders/CAPES – Brazil.
Abbreviations
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- PRK
protein kinase C-related kinase
- T3SS
type 3 secretion system
- T6SS
type 6 secretion system
- ECVs
extracellular vesicles
- GAP
GTPase activating proteins
- ASC
apoptosis-associated speck-like protein
- IL
interleukin
- GSDMD
gasdermin-D
- PSP
protein serine/threonine phosphatase
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
• Of special interest
•• Of outstanding interest
- [1].Brewer SM, Brubaker SW, and Monack DM, “Host inflammasome defense mechanisms and bacterial pathogen evasion strategies,” Curr. Opin. Immunol., vol. 60, pp. 63–70, 2019. [DOI] [PubMed] [Google Scholar]
- [2].Zhao Y and Shao F, “Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence,” Curr. Opin. Microbiol, vol. 29, pp. 37–42, 2016. [DOI] [PubMed] [Google Scholar]
- [3].Manthiram K, Zhou Q, Aksentijevich I, and Kastner DL, “The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation,” Nat. Immunol, vol. 18, no. 8, pp. 832–843, 2017. [DOI] [PubMed] [Google Scholar]
- [4].Schnappauf O, Chae JJ, Kastner DL, and Aksentijevich I, “The Pyrin Inflammasome in Health and Disease,” Front. Immunol, vol. 10, no. 1745, pp. 1–15, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chae JJ et al. , “Gain-of-Function Pyrin Mutations Induce NLRP3 Protein - Independent Interleukin-1β Activation and Severe Autoinflammation in Mice,” Immunity, vol. 34, no. 5, pp. 755–768, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yu J et al. , “Cryopyrin and pyrin activate caspase-1, but not NF-kB, via ASC oligomerization,” Cell Death Differ, vol. 13, pp. 236–249, 2006. [DOI] [PubMed] [Google Scholar]
- [7].Yu J, Farias A, Hwang I, Fernandes-Alnemri T, and Alnemri ES, “Ribotoxic Stress through p38 Mitogen-activated Protein Kinase Activates in Vitro the Human Pyrin Inflammasome,” J. Biol. Chem, vol. 288, no. 16, pp. 11378–11383, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seshadri S, Duncan MD, Hart JM, Gavrilin MA, and Wewers MD, “Pyrin Levels in Human Monocytes and Monocyte-Derived Macrophages Regulate IL-1β Processing and Release,” J. Immunol, vol. 179, no. 2, pp. 1274–1281, 2007. [DOI] [PubMed] [Google Scholar]
- [9].Gaidt MM and Hornung V, “Alternative inflammasome activation enables IL-1β release from living cells,” Curr. Opin. Immunol, vol. 44, pp. 7–13, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gavrilin MA et al. , “Pyrin Critical to Macrophage IL-1 Response to Francisella Challenge,” J. Immunol, vol. 182, no. 12, pp. 7982–7989, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gavrilin MA et al. , “Activation of the Pyrin Inflammasome by Intracellular Burkholderia cenocepacia,” J. Immunol, vol. 188, no. 7, pp. 3469–3477, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rajamuthiah R, Mylonakis E, Rajamuthiah R, and Mylonakis E, “Effector triggered immunity,” Virulence, vol. 5, no. 7, pp. 697–702, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jones JD, Vance RE, and Dangl JL, “Intracellular innate immune surveillance devices in plants and animals,” Science (80-. )., vol. 354, no. 6316, pp. aaf6395–1–8, 2016. [DOI] [PubMed] [Google Scholar]
- [14].Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, and Dong J, “Non-canonical inflammasome activation targets caspase-11,” Nature, vol. 479, no. 7371, pp. 117–121, 2011. [DOI] [PubMed] [Google Scholar]
- [15].V Jafari N et al. , “Clostridium difficile Modulates Host Innate Immunity via Toxin-Independent and Dependent Mechanism(s),” PLoS One, vol. 8, no. 7, pp. 1–10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ng J et al. , “Clostridium difficile Toxin–Induced Inflammation and Intestinal Injury Are Mediated by the Inflammasome,” Gastroenterology, vol. 139, no. 2, pp. 542–552.e3, 2010. [DOI] [PubMed] [Google Scholar]
- [17].Xu H et al. , “Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome,” Nature, vol. 513, no. 7517, pp. 237–241, 2014. [DOI] [PubMed] [Google Scholar]
- [18].Park YH, Wood G, Kastner DL, and Chae JJ, “Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS,” Nat. Immunol, vol. 17, no. 8, pp. 914–921, 2016.•• This study is the first to demonstrate a role for the RhoA-activated serine-threonine kinase PKN (also known as PRK) in negative regulation of pyrin. PKN was shown to interact with pyrin and phosphorylate two serine residues that are bound by the 14–3-3 protein. This negative regulation mechanism was interupted by FMF mutations in pyrin, or when RhoA was inactivated by bacterial toxins or loss of geranylgeranylation that occurs in the autoinflammatory disease HIDS.
- [19].Gao W, Yang J, Liu W, Wang Y, and Shao F, “Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation,” Proc. Natl. Acad. Sci, vol. 113, no. 33, pp. E4857–E4866, 2016.•• This study demonstates that human and mouse pyrin is negatively regulated by phosphorylation of two serine residuces and 14–3-3 binding. By developing phospho-specific pyrin monoclonal antibodies, the authors were able to show for the first time that bacterial toxins that inactivate RhoA trigger pyrin dephosphorylation. In addition, this is the first study to show that microtubule inhibitors such as colchicine can prevent inflammasome assembly downstream of pyrin dephosphorylation.
- [20].Masters SL et al. , “Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation,” Sci. Transl. Med, vol. 8, no. 332, pp. 1–10, 2016.•• Masters et al. discovered that a human disease termed pyrin-associated autoinflammation with neutrophilic dermatosis is caused by a dominant gain of function mutation in MEFV, corresponding to a S242R codon change. The S242R change prevents normal negative regulation of pyrin by phosphorylation and 14–3-3 binding.
- [21].Chung LK et al. , “The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome,” Cell Host Microbe, vol. 20, no. 3, pp. 296–306, 2016.•• This study is the first to show that targeting of RhoA by the Y. pseudotuberculosis effectors YopE or YopT can trigger the pyrin inflammasome. Additionally, it was shown that a third effector, YopM, binds to pyrin and hijacks host kinases to keep pyrin phosphorylated and inactivated. The specific inhibition of pyrin by YopM was shown to be essential for Y. pseudotuberculosis virulence in a mouse systemic infection model.
- [22].Van Gorp H, V Saavedra PH, De Vasconcelos NM, and Van Opdenbosch N, “Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation,” Proc. Natl. Acad. Sci, vol. 113, no. 50, pp. 5–10, 2016.• This study demonstrates that microtubule inhibitors can prevent inflammasome assembly downstream of dephosphorylation of the wild-type pyrin protein. Surprisingly, microtubule inhibitors did not prevent pyrin inflammasome assembly in PBMCs of FMF patients infected with C. difficile or intoxicated with TcdA. These results suggest that gain of function mutations in the B30.2 domain bypass the requirment for microtubules in pyrin inflammasome assembly.
- [23].Dumas A et al. , “The Inflammasome Pyrin Contributes to Pertussis Toxin-Induced IL-1β Synthesis, Neutrophil Intravascular Crawling and Autoimmune Encephalomyelitis,” PLoS Pathog, vol. 10, no. 5, pp. 1–14, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Aubert DF et al. , “A Burkholderia Type VI Effector Deamidates Rho GTPases to Activate the Pyrin Inflammasome and Trigger Inflammation,” Cell Host Microbe, vol. 19, no. 5, pp. 664–674, 2016.•• Aubert et al. identified TecA as the T6SS effector in B. cenocepacia that is responsible for triggering the pyrin inflammasome. TecA is shown to deamindate N41 of RhoA and N39 of Rac1. TecA and pyrin were required for lung immunopathology in response to lung infection of mice with B. cenocepacia. TecA and pyrin decreased bacterial loads and virulence of B. cenocepacia during mouse systemic infection, demonstrating the host protective role of this response against invasive pathogens.
- [25].Ratner D et al. , “The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation,” PLoS Pathog, vol. 12, no. 12, pp. 1–17, 2016.• This study demonstrates that the Y. pestis effector YopM inhibits the pyrin inflammasome, which is activated in response to RhoA targeting by YopE. Mice lacking either pyrin or IL-18 were equally and highly susceptible to lethal bubonic plague infection by a yopM yopJ mutant, demonstrating the importance of this innate response for host protection against systemic Y. pestis infection.
- [26].Medici NP, Rashid M, and Bliska JB, “ Characterization of Pyrin Dephosphorylation and Inflammasome Activation in Macrophages as Triggered by the Yersinia Effectors YopE and YopT,” Infect. Immun, vol. 87, no. 3, pp. 1–13, 2019.• This study demonstrates that pyrin dephosphorylation is triggered by Yersinia effectors that target RhoA by distinct mechanisms. Non-covalent inactivation of RhoA by the GAP YopE and detachment of RhoA from membranes by the protease YopT triggered pyrin dephosphorylation in BMDMs infected with Yersinia. This study also demonstrated that distinct features of different effectors such as RhoA specificity or stability determine the efficiency of pyrin dephosphorylation and inflammasome assembly.
- [27].V Saavedra PH et al. , “Apoptosis of intestinal epithelial cells restricts Clostridium difficile infection in a model of pseudomembranous colitis,” Nat. Commun, vol. 9, no. 4846, pp. 1–10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, and Johansen T, “TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity,” J. Cell Biol, vol. 210, no. 6, pp. 973–989, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sharma D, Malik A, Guy CS, Karki R, Vogel P, and Kanneganti TD, “Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis,” Gastroenterology, vol. 154, no. 4, pp. 948–964, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alimov I et al. , “Bile acid analogues are activators of pyrin inflammasome,” J. Biol. Chem, vol. 294, no. 10, pp. 3359–3366, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim JG et al. , “Regulation of RhoA GTPase and various transcription factors in the RhoA pathway,” J. Cell. Physiol, vol. 233, no. 9, pp. 6381–6392, 2018. [DOI] [PubMed] [Google Scholar]
- [32].Chung LK and Bliska JB, “Yersinia versus host immunity: how a pathogen evades or triggers a protective response,” Curr. Opin. Microbiol, vol. 29, pp. 56–62, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Akula MK et al. , “Control of the innate immune response by the mevalonate pathway,” Nat. Immunol, vol. 17, no. 8, pp. 922–930, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jamilloux Y et al. , “The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes,” Pathog. Dis, vol. 76, no. 3, pp. 1–9, 2018. [DOI] [PubMed] [Google Scholar]
- [35].A. S. Jéru I, Papin S, L’hoste S, Duquesnoy P, Cazeneuve C, Camonis J, “Interaction of Pyrin With 14.3.3 in an Isoform-Specific and Phosphorylation-Dependent Manner Regulates Its Translocation to the Nucleus,” ARTHRITIS Rheum, vol. 52, no. 6, pp. 1848–1857, 2005. [DOI] [PubMed] [Google Scholar]
- [36].Yang J, Xu H, and Shao F, “Immunological function of familial Mediterranean fever disease protein Pyrin,” Sci. China Life Sci, vol. 57, no. 12, pp. 1156–1161, 2014. [DOI] [PubMed] [Google Scholar]
- [37].Kim ML et al. , “Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β,” J. Exp. Med, vol. 212, no. 6, pp. 927–938, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Standing ASI et al. , “Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin- regulatory gene WDR1,” J. Exp. Med, vol. 214, no. 1, pp. 59–71, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu J et al. , “Pyrin Activates the ASC Pyroptosome in Response to Engagement by Autoinflammatory PSTPIP1 Mutants,” Mol. Cell, vol. 28, no. 2, pp. 214–227, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mahenthiralingam E, Urban TA, and Goldberg JB, “The multifarious, multireplicon Burkholderia cepacia complex,” Nat. Rev. Microbiol, vol. 3, no. 2, pp. 144–156, 2005. [DOI] [PubMed] [Google Scholar]
- [41].Flannagan RS et al. , “Burkholderia cenocepacia disrupts host cell actin cytoskeleton by inactivating Rac and Cdc42,” Cell. Microbiol, vol. 14, no. 2, pp. 239–254, 2012. [DOI] [PubMed] [Google Scholar]
- [42].Rosales-Reyes R, Skeldon AM, Aubert DF, and Valvano MA, “The Type VI secretion system of Burkholderia cenocepacia affects multiple Rho family GTPases disrupting the actin cytoskeleton and the assembly of NADPH oxidase complex in macrophages,” Cell. Microbiol, vol. 14, no. 2, pp. 255–273, 2012. [DOI] [PubMed] [Google Scholar]
- [43].Carbonetti NH, “Contribution of pertussis toxin to the pathogenesis of pertussis disease,” FEMS Pathog. Dis, vol. 73, no. 8, pp. 1–8, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Place DE, Muse SJ, Kirimanjeswara GS, and Harvill ET, “Caspase-1-Independent Interleukin-1β Is Required for Clearance of Bordetella pertussis Infections and Whole- Cell Vaccine-Mediated Immunity,” PLoS One, vol. 9, no. 9, pp. 1–9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dunne A et al. , “Inflammasome Activation by Adenylate Cyclase Toxin Directs Th17 Responses and Protection against Bordetella pertussis,” J. Immunol, vol. 185, no. 3, pp. 1711–1719, 2010. [DOI] [PubMed] [Google Scholar]
- [46].Abt MC, Mckenney PT, and Pamer EG, “Clostridium difficile colitis: pathogenesis and host defence,” Nat. Publ. Gr, vol. 14, no. 10, pp. 609–620, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Awad MM et al. , “Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen,” Gut Microbes, vol. 5, no. 5, pp. 579–593, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Genth H et al. , “Difference in Mono-O-Glucosylation of Ras Subtype GTPases Between Toxin A and Toxin B From Clostridioides difficile Strain 10463 and Lethal Toxin From Clostridium sordellii Strain 6018,” Front. Microbiol, vol. 9, no. 3078, pp. 1–10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cowardin CA, Jackman BM, Noor Z, Burgess SL, Feig AL, and Petri WA, “Glucosylation Drives the Innate Inflammatory Response to Clostridium difficile Toxin A,” Infect. Immun, vol. 84, no. 8, pp. 2317–2323, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Leslie JL et al. , “Persistence and Toxin Production by Clostridium difficile within Human Intestinal Organoids Result in Disruption of Epithelial Paracellular Barrier Function,” Infect. Immun, vol. 83, no. 1, pp. 138–145, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hasegawa M, Kamada N, and Jiao Y, “Protective Role of Commensals against Clostridium difficile Infection via an IL-1β−Mediated Positive-Feedback Loop,” J. Immunol, vol. 189, no. 6, pp. 3085–3091, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Philip NH, Zwack EE, and Brodsky IE, “Activation and Evasion of Inflammasomes by Yersinia,” in Current Topics in Microbiology and Immunology, vol. 397, 2016, pp. 69–90. [DOI] [PubMed] [Google Scholar]
- [53].Stewart MK and Cookson BT, “Evasion and interference: intracellular pathogens modulate caspase-dependent inflammatory responses,” Nat. Publ. Gr, vol. 14, no. 6, pp. 346–359, 2016. [DOI] [PubMed] [Google Scholar]
- [54].Ratner D et al. , “Manipulation of Interleukin-1β and Interleukin-18 Production by Yersinia pestis Effectors YopJ and YopM and Redundant Impact on Virulence,” J. Biol. Chem, vol. 291, no. 19, pp. 9894–9905, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schoberle TJ, Chung LK, Mcphee JB, Bogin B, and Bliska JB, “Uncovering an Important Role for YopJ in the Inhibition of Caspase-1 in Activated Macrophages and Promoting Yersinia pseudotuberculosis Virulence,” Infect. Immun, vol. 84, no. 4, pp. 1062–1072, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mcdonald C, Vacratsis PO, Bliska JB, and Dixon JE, “The Yersinia Virulence Factor YopM Forms a Novel Protein Complex with Two Cellular Kinases,” J. Biol. Chem, vol. 278, no. 20, pp. 18514–18523, 2003. [DOI] [PubMed] [Google Scholar]
- [57].Mccoy MW, Marre ML, Lesser CF, and Mecsas J, “The C-Terminal Tail of Yersinia pseudotuberculosis YopM Is Critical for Interacting with RSK1 and for Virulence,” Infect. Immun, vol. 78, no. 6, pp. 2584–2598, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McPhee JB, Mena P, and Bliska JB, “Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence,” Infect. Immun, vol. 78, no. 8, pp. 3529–3539, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chung LK et al. , “IQGAP1 Is Important for Activation of Caspase-1 in Macrophages and Is Targeted by Yersinia pestis Type III Effector YopM,” MBio, vol. 5, no. 4, pp. 1–11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hentschke M, Berneking L, Campos CB, Buck F, Ruckdeschel K, and Aepfelbacher M, “Yersinia Virulence Factor YopM Induces Sustained RSK Activation by Interfering with Dephosphorylation,” PLoS One, vol. 5, no. 10, p. e13165, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]