

Abstract
Phosphoinositides are lipid signaling molecules that act as master regulators of cellular signaling. Recent studies have revealed novel roles of phosphoinositides in myriad cellular processes, and multiple human diseases mediated by misregulation of phosphoinositide signaling. This review will present a timely summary of recent discoveries in phosphoinositide biology, specifically their role in regulating unexpected signaling pathways, modification of signaling outcomes downstream of integral membrane proteins, and novel roles in lipid transport. This has revealed new roles of phosphoinositides in regulating membrane trafficking, immunity, cell polarity, and response to extracellular signals. A specific focus will be on novel opportunities to target phosphoinositide metabolism for treatment of human diseases, including cancer, pathogen infection, developmental disorders, and immune disorders.
Introduction
Phosphoinositides (PPIns) are lipid signaling molecules that coordinate numerous aspects of membrane trafficking and cell signaling in eukaryotic cells. Their action is essential in cell growth, metabolism, and cell death [1]. The enzymes that modify phosphoinositides (kinases, phosphatases, and lipases) are critically linked to multiple human diseases, with mutations leading to cancer, immune disorders, developmental disorders, and inflammatory diseases [2–4]. There are 7 different PPIn species (Fig. 1A), with all of them acting as regulators of temporal and spatial signaling events (Fig. 1B). The canonical view of phosphoinositide signaling has focused on their role in both the recruitment (Fig. 2A) and allosteric activation of proteins at specific intracellular locations (Fig. 2B). Phosphoinositide mediated recruitment of proteins to specific organelles can mark their final destination in membrane trafficking [5], modify biophysical properties of membranes [6], and provide spatial regulation of signaling.
Figure 1: Identity and cellular localization of phosphoinositides in cells.

A. Phosphoinositides are composed of two acyl chains attached to a glycerol backbone, with a myo-inositol headgroup. There are a total of seven different phosphoinositide species that can be generated downstream of the precursor phosphatidylinositol through phosphorylation of the hydroxyls on the inositol headgroup. These include phosphatidylinositol 3-phosphate (PI3P), phosphatidylinositol 4-phosphate (PI4P), phosphatidylinositol 5-phosphate (PI5P), phosphatidylinositol 3,4 bis-phosphate (PI(3,4)P2, phosphatidylinositol 3,5 bis-phosphate (PI(3,4)P2, phosphatidylinositol 3,4 bis-phosphate (PI(4,5)P2, and phosphatidylinositol 3,4,5 tris-phosphate (PI(3,4,5)P2 (referred to as PIP3). The different human lipid kinases and phosphatases that generate them are indicated in the legend according to the numbers. Phosphoinositide conversion reactions that are not fully established are marked with a ? sign. *The conversion of PI4,5P2 to PI5P has been implicated to involve the PIP4P1/PIP4P2 genes (TMEM55A/TMTM55B proteins), however, recent work indicates the biological activity of these proteins is not driven through PI4,5P2 phosphatase activity [88]. The & sign indicates reactions that have only recently been identified [89].
B. The generation of phosphoinositides are master regulators of temporal and spatial localization of cellular signaling and membrane trafficking events, with their location tightly restricted through the action of the lipid kinases and phosphatases that generate them. They play key roles in secretion from the Golgi, endocytosis, and endo-lysosomal trafficking of membranes [90].
Figure 2: Phosphoinositides roles in protein recruitment/allosteric activation, modulation of integral membrane proteins, and lipid transport.

A. Role of Phosphoinositides in the recruitment of proteins to specific intracellular locations. Many PPIn binding domains have been identified, although many of these domains have varying levels of specificity, and also frequently require coincidence detection of other signals (including both additional lipid and protein binding partners). Phosphoinositides can also regulate protein recruitment outside of lipid binding domains, including polybasic stretches, and non-canonical lipid binding sites.
B. Roles of phosphoinositides in allosteric activation of signaling enzymes. Example of the allosteric activation of the pro-growth kinase Akt (PKB) downstream of PIP3, where PIP3 binding to the PH domain disrupts an inhibitory PH-kinase interface, followed by PIP3 activated phosphorylation of Akt by phosphoinositide dependent kinase 1 (PDK1).
C. Phosphoinositides are key regulators of integral membrane proteins, including ion channels, G-protein coupled receptors, and lipid scramblases, flippases and floppases. Phosphoinositides can regulate integral membrane function through allosteric conformational changes and/or through modulating their coupling to protein binding partners.
D. Phosphoinositides can mediate the transport of lipids against their concentration gradient through the coordinated action of lipid kinases, phosphatases, and lipid transport proteins (LTPs).
However, recent years has seen an explosion in our understanding of other key roles of phosphoinositides, including their role in modulating integral membrane protein signaling (Fig. 2C), and regulating lipid transport (Fig. 2D). This review will highlight recent discoveries in phosphoinositide biology, focusing on four specific areas: novel roles of phosphoinositides in signaling, phosphoinositide regulation of integral membrane proteins, how phosphoinositides and lipid transport proteins mediate lipid exchange against thermodynamic gradients, and finally novel discoveries towards treating human disease by targeting phosphoinositide metabolizing enzymes. For more comprehensive coverage of PPIn function in e.g. cytoskeletal dynamics and nuclear function, we refer the reader to several recent, comprehensive reviews [7,8].
Non-canonical roles of phosphoinositides in recruitment of cellular signaling machinery
The canonical view of phosphoinositide signaling is the recruitment of specific lipid binding domains to intracellular membranes, with multiple domains putatively identified as specific PPIn binders (PX, PH, FYVE, etc., for an in-depth review readers are advised to consult [9,10]).The recruitment of these domains to specific intracellular organelles can mediate signaling not only through localization, but also through lipid binding mediated conformational changes [11–13]. Exhaustive analysis of these specific domains is revealing a more complicated picture. Detailed biochemical and biophysical studies of 39 of the 49 PX domains found in the human genome [14] revealed striking differences in PPIn binding. Four different clusters of PX domains were identified based on their lipid specificity: those that do not specifically bind lipid (i), specific PI3P binders (ii), those that bind other PPIn species (iii), and those that bind PI3P along with other PPIn species (iv). These distinct groups are not coupled to their evolutionary history. This highlights the extreme importance in the vigorous biochemical, biophysical and cellular analysis of the lipid binding specificity of putative PPIn binding domains. Intriguingly, phosphorylation of the lipid binding loop of the PX domain of sorting Nexin 3 (SNX3) disrupts membrane recruitment, and reveals another level of how membrane recruitment can be regulated by PTMs [15].
Many proteins are known to be recruited to phosphoinositides independent of PPin binding domains. This can be mediated through multiple different molecular mechanisms, including disordered polybasic motifs. Our focus is on recent discoveries of signaling complexes that are regulated by phosphoinositide recruitment/activation in the absence of PPIn binding domains. This includes the spatially localized PI(3,4)P2 plasma membrane recruitment of RasGAP proteins to mediate cell polarity [16], the PI4P-mediated dispersed Trans-Golgi recruitment of the NLRP3 inflammasome to promote inflammatory signaling [17], the PI(4,5)P2 mediated plasma membrane recruitment of the inflammatory DNA sensing cGAS protein to govern self-nonself discrimination [18], and the PI3P mediated pre-autophagasome (PAS) recruitment of the core autophagy protein ATG16L1 to mediate autophagy [19]. This highlights a nuanced picture of PPIn recruitment, where recruitment will be dependent not only on specific PPIn binding domains, but also through non-canonical PPIn binding sites in proteins. Important to consider is that these sites will likely exhibit less strict dependency on any PPIn species, and will require extensive testing of other coincidence signals that may mediate recruitment.
Phosphoinositides as regulators of integral membrane protein signaling
The binding of phosphoinositides to integral membrane proteins can modulate their activity, and can allow for activation of integral membrane proteins to only occur when they are in specific membrane organelles. Phosphoinositides can regulate integral membrane proteins through multiple mechanisms: they can induce allosteric conformational changes and/or mediate coupling to protein binding partners. Phosphoinositides have long been known to be key regulators of ion channels, with PI(4,5)P2 identified as a key regulator of channel opening [20,21], with the first molecular insight on the role of PI(4,5)P2 being shown in the structure of the Kir2.2 ion channel [22]. For the Ca2+ ion channel TRPML1 the binding of PI(4,5)P2 inhibits channel opening, while binding of PI(3,5)P2 leads to channel activation [23], providing a lipid mediated switch making sure the channel is only active in PI(3,5)P2 containing endolysosomal compartments. This reveals the potential multifaceted roles of PPIn in both positively and negatively regulating integral membrane protein signaling. For example, this could play an important role in coordinating how long-range signals can be propagated physiologically, with modification of PI(4,5)P2 levels discovered to play an important role in blood flow in the neural vasculature: this could be controlled by the ability of PI(4,5)P2 to activate Kir2.1 ion channels and inhibit Transient receptor potential cation channel subfamily V member 4 (TRPV4) channels [24,25], which can be further tuned by GqPCR receptors activating PLC and depleting PI(4,5)P2 levels.
Advances in native mass spectrometry [26–28], Cryo electron microscopy [23,29-36], and molecular dynamic simulations [37–40] have been particularly powerful in providing insight into the molecular mechanisms that mediate how PPIn can bind and regulate integral membrane protein function [41]. A survey of a selection of recent structures of phosphoinositides bound to different membrane proteins (ion channels, flippases) and the molecular basis for how they regulate protein function is shown in Fig. 3. An important note is that for almost all interactions of PPIn with integral membrane protein there is no distinct polypeptide that mediates binding, which is instead mediated by the complex tertiary and quaternary protein architecture; as such probes for specific PPIns derived from integral membrane proteins should be approached with caution [42].
Figure 3: Structural basis for the regulation of integral membrane proteins by PPIns.
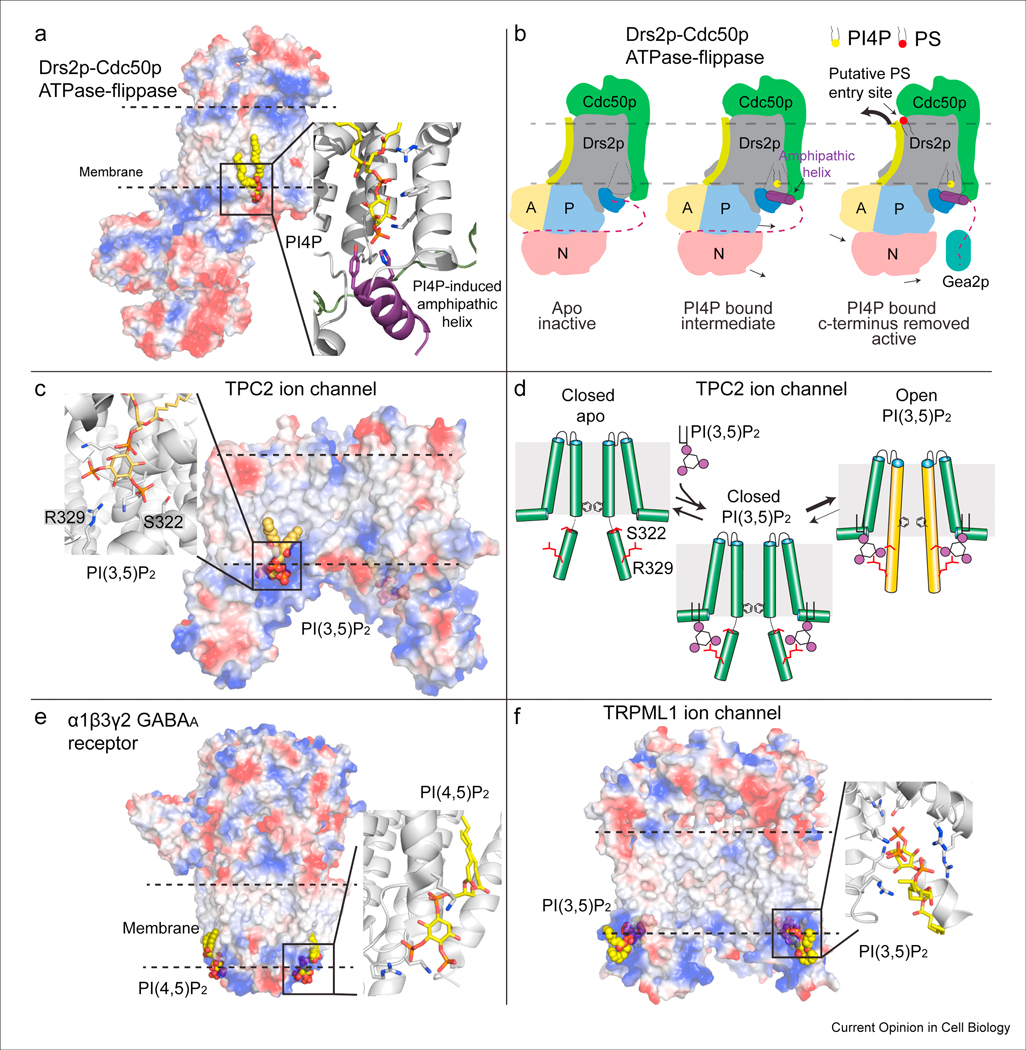
A. Cryo-EM structure of the yeast dimeric complex of the ATPase flippase Drs2pCdc50p [29]. The protein complex is shown as a surface representation, highlighting charged pockets that mediate lipid binding. The amphipathic helix that binds PI4P is colored purple, with the residues that bind specifically to PI4P shown as sticks.
B. A cartoon schematic of the conformational changes that occur during PI4P binding in the flippase catalytic cycle [29,91]. The Cdc50p protein is shown in green, with the Drs2p protein colored according to its domains, with the A, P, and N domains colored in yellow, blue and red respectively. The inhibitory c-terminus of Drs2p is shown as a dotted line, which is attached to the amphipathic helix that forms upon PI4P binding. The coordinated binding of PI4P and disruption of the c-terminal inhibitory interaction through binding to the Arf-GEF Gea2p leads to an allosteric conformational change in TM2 (colored lime) that opens a putative PS lipid binding pocket, allowing for lipid transfer.
C. Cryo-EM structure of the open active form of the human Na2+ selective two pore channel (TPC2) bound to PI3,5P2 [35]. The protein complex is shown as a surface representation, highlighting charged pockets that mediate lipid binding. The specific residues that mediate phosphoinositide binding are shown as sticks. The Arg and Ser residues that interact with PI3,5P2 are labeled.
D. Cartoon schematic of the molecular mechanism of how PI3,5P2 mediates ion channel opening (only the 6 TM-I domain is shown for simplicity). Binding of PI3,5P2 biases the equilibrium to the open conformation through allosteric conformational changes in the IS6 helix (colored in orange in the open conformation). This helix contains Ser-322 and Arg-329 which interact with PI3,5P2, and putatively bias the channel towards an open conformation.
E. Cryo-EM structure of the type A γ-aminobutyric acid (GABAA) pentameric ligand gated ion channel bound to PI(4,5)P2 [30]. The protein complex is shown as a surface representation, highlighting charged pockets that mediate lipid binding. The specific residues that mediate phosphoinositide binding are shown as sticks.
F. Cryo-EM structure of the Transient receptor potential mucolipin 1 (TRPML1) ion channel bound to PI(3,5)P2 [23]. The protein complex is shown as a surface representation, highlighting charged pockets that mediate lipid binding. The specific residues that mediate phosphoinositide binding are shown as sticks.
The activation of G-protein coupled receptors (GPCRs) can be allosterically modulated by surrounding phospholipids [43], and potentially this may play a role in controlling their oligomeric state [44]. The adenosine A2A GPCR can bind PI(4,5)P2, with this interaction potentiating G-protein activation [27]. GPCRs are able to integrate multiple signals, and generate specific downstream outputs, including G-proteins and beta-arrestin signals [45]. The formation of the GPCR beta arrestin complex requires GPCR phosphorylation downstream of GPCR kinases (GRKs). The presence of PI(4,5)P2 promotes formation of a complex between the beta-2 adrenergic GPCR with the GPCR kinase GRK5 [46]. Beta-arrestin mediated endocytosis of GPCRs is proposed to require arrestin binding to phosphoinositides [47]. Recent work suggests the possibility of an active beta-arrestin conformation on membranes that is catalytically activated by GPCRs, but independent of a stable GPCR-arrestin complex. The dissociation of signaling competent active beta-arrestin is putatively mediated by phosphoinositides [48]. Biased agonists can specifically favor one pathway over another, with the exact molecular details of this process being unknown. GPCRs primarily generate signals at the plasma membrane, but can also be active in endosomal compartments.
Together these studies bring up an intriguing possibility. They suggest that phosphoinositides may spatially organize multiple aspects of GPCR signaling, both affecting the coupling of GPCRs to G proteins, GPCR kinases and arrestin, and affecting the activation of signaling proteins downstream of GPCRs. This suggests different PPIn species may in some cases act as spatially localized lipid switches to bifurcate and modulate GPCR signaling.
The disruption of PPIn regulation of integral membrane signaling can lead to human disease through surprising ways. In Niemann-Pick Type C Disease the disruption of lysosomal transport of cholesterol leads to upregulation of the ATP-binding cassette transporter ABCA1 (ABC1A), which through its PI(4,5)P2 floppase activity decreases PM PI(4,5)P2 levels, leading to inactivation of the KCNQ2/3 potassium channel and aberrantly modified neuronal signaling patterns [49].
One of the key implications for the role of phosphoinositides in regulating integral membrane signaling is that unlike in peripheral membrane recruitment this interaction will almost certainly be driven by both interactions between the PPIn headgroup, and the acyl chains. The majority of PPIn species contain an 18:0 sn-1 chain, and a 20:4 sn2 acyl chain [50], however, there is extensive variability within different cells and tissues. This has long been known to be critical in the activation of the Kir2.1 channel, with PIP2 containing saturated acyl chains only weakly activating channel opening [51]. This has also been shown for the ER protein SEIPIN which can bind PA and PI3P and exhibits a strong binding preference towards unsaturated fatty acids [31]. Further biophysical and biochemical analysis will be required to define the full complement of mechanisms by which specific PPIn species (head group and acyl chain variants) may activate specific integral membrane signaling processes.
Phosphoinositides and Lipid Transfer Proteins (LTPs).
Evidence is emerging that lipid transport proteins (LTPs) can utilize phosphoinositides in a novel way (Figure 4): as a means to couple the energy of ATP hydrolysis to the transport of lipid cargoes against a concentration gradient at membrane contact sites (MCS). So far, evidence implicates this mechanism for enrichment of TGN cholesterol and PM PS [52,53]. In both cases, members of the oxysterol binding protein (OSBP) related protein (ORP) family and the PPIn PI4P are involved.
Figure 4: PPIn synthesis powers lipid transfer.

Principles of counter ion (A) and counter-lipid (B) transport. In both cases, transport of a cargo (green) is powered by flow of the counter-molecule (red) down its concentration gradient. Ultimately, the chemical gradient of counter-molecule is established via ATP hydrolysis. This concept mirrors the textbook example of exchangers in ion homeostasis. For example, sodium and calcium ions are both maintained at low cytosolic concentrations relative to the extracellular milieu. The sodium calcium exchanger (NCX) helps maintain low cytosolic calcium concentrations in the absence of ATP hydrolysis, by exchanging cytosolic calcium ions for extracellular sodium ions. Thus calcium moves against its electrochemical gradient (out of cells), but powered by the flow of sodium down its own gradient (into cells). Ultimately, ATP powers this cycle through the sodium-potassium ATPases that actively pump sodium out of cells and maintains the gradient. The same has been proposed for lipid transfer proteins of the ORP family: These can exchange a sterol or phospholipid that is synthesized in the ER for PI4P in another membrane [10]. This membrane is anchored to the ER by the ORP protein itself, usually by PI4P through a PH domain at one end and through an ER anchor (or interaction with the ER receptor VAPa/b) at the other. The ORD domain then exchanges PI4P and cargo lipid. Hydrolysis of the PI4P in the ER by the SAC1 lipid phosphatase ensures vectoral transfer of PI4P, which is unavailable for the return step. Thus the cargo lipid is moved instead, against its own concentration gradient. Ultimately, ATP hydrolysis powers this cycle through the PI 4-kinases that maintains high PI4P levels in the non-ER membrane.
Although this is an apparently novel concept relative to the protein recruitment and activation mechanisms traditionally associated with PPIn signaling, there is a unifying theme: In both cases, PPIn synthesis couples the energy of ATP hydrolysis to the nonequilibrium acquisition of lipid or protein molecules (or their activity) in restricted membrane compartments. This is an essential function for multi-organelle eukaryotic cells.
For ORP function, PI4P has two roles: firstly, as the counter lipid for the exchange cycle; and secondly, in its canonical capacity, to anchor the ORP to the acceptor membrane (See figure 2). Thus, lipid transfer can only be accomplished when PI4P levels are sufficient to localize the ORP to the MCS. This creates a putative feedback mechanism: PI4P synthesis at the TGN by PI4KB was reported to be inhibited by high sterol concentrations, depleting PI4P and thus de-localizing OSBP and stopping further sterol traffic [54]. The cycle can be re-initiated at distal TGN sites in response to another, cholesterol-activated PI4K, PI4K2A [54].
A similar feedback mechanism regulates ORP5 and −8 proteins at the PM [55]. The ORP5 and ORP8 PH domains have an adjacent polybasic domain that together facilitate concurrent binding of both PI4P and PI(4,5)P2, which co-operate to localize the LTP to ER-PM contact sites. This forms the basis of a homeostatic mechanism: when the cardinal regulatory PM lipid PI(4,5)P2 is depleted (e.g. by phospholipase C), ORP5/8 is not localized to the PM and cannot transfer PI4P to the ER; in this way, all newly synthesized PM PI4P can be directed towards PI(4,5)P2 synthesis until levels of the latter recover sufficiently [55]. Intriguingly, such a feedback mechanism must be balanced by a component that stops PI4P to PI(4,5)P2 conversion once levels are restored. Whilst these data imply that PI(4,5)P2 plays an exclusive regulatory role, PI(4,5)P2 transfer by ORP5 and 8 has also been reported, with this lipid putatively being transported instead of PI4P [56,57]. However, these data are not consistent with other reports that ORP5 and ORP8 over-expression can deplete PM PI4P, but not PI(4,5)P2 [52,55]. For now, the exact roles of PI4P and PI(4,5)P2 as localizing factors versus exchange currency are still subject to debate.
Another lipid that is greatly enriched at the PM is cholesterol, so it has been tempting to speculate this may be fueled by a PPIn-mediated exchange cycle. The first empirical evidence for such a cycle has just been reported, with the observation that ORP2 can facilitate cholesterol enrichment and PI(4,5)P2 depletion from the PM [57]. PM cholesterol homeostasis also requires negative feedback, for instance when highdensity lipoproteins (HDLs) deliver exogenous cholesterol to the PM of steroidogenic tissues. This is removed by a novel family of ER proteins, GRAMD1-A, -B and -C (a.k.a. “aster” proteins). Asters localize to PM contact sites in response to elevated cholesterol levels via their GRAM domains, facilitating removal of cholesterol via the VASt/aster lipid transfer domain [58]. In this case, cholesterol flows down a concentration gradient, so counter-transport is not required, and PPIn are not implicated in their function or localization [58,59]. On the other hand, PPIns are needed to localize the related GRAMD2a and -b proteins, which lack the transfer domain but do form ER-PM contact sites that house other lipid and ion exchange reactions [59].
Elegant and fulfilling as these lipid exchange mechanisms are, they are not quite established enough to enter the textbook. Although ORPs and other LTPs can certainly facilitate lipid exchange reactions in the test tube (e.g. [53,56,60]), whether this reflects their true function in cells, or is simply a test tube manifestation of a more subtle activity, is actively debated [61]. It has been proposed instead that several LTPs, including ORPs, can utilize lipid transfer domains to present PPIn to kinases and phosphatases in membranes replete with their lipid counter-ligand. Thus, rather than exchangers, they are sensors of local lipid composition that charge PPIn synthesis (or turnover) as a function of membrane composition – a so called “instructive synthesis” model in the context of kinase presentation [61] or “instructive metabolism” more generally. Indeed, recent work has shown that ORP4 can function to present PI(4,5)P2 to PLCβ3 for hydrolysis at the PM [62]. Other work has proposed a hybrid of the models, whereby ORP-mediated lipid transfer produces a nanoscale environment enriched with sterol and unsaturated PS that favors PIP5K activation and PI(4,5)P2 synthesis [63]. However, another group has reported that mammalian PIP5K are in fact most potently activated by PI(4,5)P2 itself [64]. This produces a feed-forward scenario, with local physical constraints on lipid enrichment and diffusion together with local phosphatase activity constraining PI(4,5)P2 synthesis [64].
A key problem for the field is that most evidence in intact cells for lipid exchange relies on PPIn depletion in response to LTP enrichment, or vice versa (e.g. [52,54–56]): yet precisely the same result can be expected from the instructive synthesis model. How can we differentiate the two models? Certainly, lipid exchange cycles demand segregation of PI kinase and phosphatase activity to distinct membrane compartments, whereas instructive synthesis does not. This has recently been tested for the PI4P phosphatase SAC1, which predominantly localizes to the ER. Treatment of cells with oxidants that block SAC1 activity lead to accumulation of PI4P in the ER, consistent with failure to degrade ORP-transported molecules in this compartment [65]. Likewise, molecular engineering shows that an additional ~6 nm of length is needed between the ER anchor and catalytic domain before the enzyme becomes capable of “reaching” substrate in the plasma membrane, implying substrate must be transferred to it in the ER [65]. That said, it has recently been reported that the versatile TGN protein, FAPP1, is able to complex with SAC1 and VAP proteins to stimulate activity of ER-localized SAC1 at TGN MCSs [66], consistent with an instructive metabolism model.
ORP2-mediated PI(4,5)P2 exchange to drive sterol enrichment is supported by the fact that sterol enrichment (and PI(4,5)P2 depletion) is inhibited by knock-down of endosomal PI(4,5)P2 phosphatases [57]. Likewise, PI(4,5)P2 exchange at ER-PM contact sites [56] requires the localization of an ER phosphatase to degrade this lipid; INPP5K seems like an enzyme to fit this role, though its catalytic activity is not yet explicitly linked to PM PS and PI(4,5)P2 homeostasis; instead, it is required for ER tubule morphogenesis at the cellular level [67], with loss of function in humans causing a form of muscular dystrophy [68,69]. Clearly, delineating the functional distribution of PPIn phosphatase activities at the subcellular level, and how this couple to LTP function, requires further work. This will be a key goal to resolve lipid transfer and instructive synthesis.
Finally, although both instructive synthesis and lipid exchange predict the same change in PPIn levels in target compartments, the fate of the lipid differs. Exchange specifically predicts that the degraded PPIn molecules will be delivered to a recipient compartment for degradation. It is this specific transfer step that has yet to be demonstrated in living cells. The failure to observe it can be explained if lipid exchange reactions simply don’t happen in living cells; but they are also explained by the necessarily close coupling of PPIn degradation to transport. It therefore seems that if lipid transfer to a destination membrane can be demonstrated in intact cells, the case for lipid exchange cycles will be greatly strengthened. This will require acute control of LTP activity, and/or careful, precise and acute decoupling from the degradative process. These experiments will prove tricky, though with increasingly sophisticated molecular, optical and chemical genetic approaches (e.g. [54,65,66]) the requisite experimental tool kit may be available. Whichever model ends up favored by the evidence and enters the realm of textbook knowledge (and it could be both, depending on the specific molecular circumstance), it is clear that PPIn signaling is central to lipid as well as protein organization.
Novel insight into targeting pathological phosphoinositide metabolism in disease
The most clinically advanced strategy targeting pathologies in phosphoinositide signaling has been the targeting the PI3K pathway in cancer, immunodeficiencies, and growth disorders. Currently there are four clinically approved PI3K inhibitors [2,70–73], however, there has been little success in targeting solid tumors with this approach, with various deleterious side effects. Recent discoveries suggest there may be unique additional opportunities to target this pathway in disease. Inhibitors specific to the PIK3CA PI3K isoform lead to a large increase in insulin signaling, which can limit their efficacy. Suppressing this insulin feedback through either dietary or pharmacological approaches shows promise for increased efficacy of PI3K inhibitors as cancer therapeutics [74]. It has also been found that in breast cancers there are frequent occurrences of two mutations within the same allele of PIK3CA, leading to increased PI3K activity and tumor growth [75]. This is consistent with unique mutations in PIK3CA leading to activation by different molecular mechanisms [76]. Patients expressing the doubly mutated PIK3CA showed a more positive clinical response to PI3K inhibitors, which suggests that cancers driven by these mutations are excellent targets for antiPI3K therapeutics.
In addition to cancer, mutation of PIK3CA is a driver of overgrowth disorders (PIK3CA-related overgrowth syndromes [PROS]), with treatment using PIK3CA specific inhibitors dramatically decreasing symptoms in patients [77]. Activating mutations in the PIK3CD isoform of PI3K is a causative agent of primary immunodeficiencies [78], and PIK3CD specific inhibitors are showing promise as a clinical treatment for these patients [79]. The lipid phosphatase PTEN that antagonizes the PI3K pathway has been discovered to be inactivated by poly-ubiquitination mediated by the E3 ligase WWP1, with small molecule WWP1 inhibitors representing a novel mechanism to reactivate anti-tumorigenic PTEN activity [80].
While PI3K inhibitors are the most clinically advanced molecules for modulation of pathological PPIn metabolism, exciting pre-clinical experiments are revealing potential therapeutic strategies. Myotubular myopathy (MTM) is a childhood muscle disease mediated by inactivating mutations in the MTM lipid phosphatases, which dephosphorylate PPIn at the 3’ position [81]. The development of PIK3C2B inhibitors may be useful as MTM therapeutics [82], but surprisingly it has been found in mouse models that the clinically approved estrogen receptor inhibitor tamoxifen can ameliorate some of the symptoms of MTM [83]. The full mechanistic details of how tamoxifen improves the MTM phenotype is unknown, however, it is proposed to function mainly through the estrogen receptor pathway by extra-nuclear estrogen receptor alpha signaling. Finally, inhibitors towards the parasitic variants of the lipid kinase PI4KB show promise as single-dose anti-malarial [84,85] and anti-cryptosporidium [86] agents, with clinical trials currently ongoing in malaria [87].
Conclusions and Future Directions
Phosphoinositides are master regulators of signaling in almost every intracellular membrane compartment. The development of novel tools to interrogate PPIn metabolism have revealed exciting new insight into their roles in controlling membrane trafficking, metabolism, autophagy, and signaling. The classical understanding of phosphoinositide signaling was as messengers that could recruit and/or activate soluble effector proteins. As we have seen, our knowledge of the repertoire of such proteins and their associated functions is still expanding. Moreover, driven especially by the revolution in structural biology, we now know that these lipids are also crucial to the regulation of integral membrane proteins. We have also discussed evidence that PPIn may in fact fulfil a novel role as an energy currency to drive non-vesicular lipid transport. In the meantime, knowledge of the classical PPIn-driven pathways is now maturing into a state where pharmacologic inactivation of their metabolism can alleviate a variety of malignant, infectious and inflammatory diseases. As our basic mechanistic knowledge of PPIn signaling expands, we expect therapeutic opportunities derived from this knowledge to expand even further.
Acknowledgements
Work in the Burke laboratory is supported by research grants from CIHR (CRN142393), NSERC (2014–05218), and the Cancer Research Society (CRS 24368) along with salary awards from CIHR (New investigator award), and the Michael Smith Foundation for Health Research (Scholar 17686). Work in the Hammond laboratory is supported by National Institutes of Health grant 1R35GM119412–01.
Footnotes
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflict of interest
‘The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balla T: Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93:1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goncalves MD, Cantley LC: Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379:2052–2062.* An excellent review on the involvement of PI3Ks in pathological conditions, and novel therapeutic strategies to target them in disease,
- 3.Burke JE: Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71:653–673. [DOI] [PubMed] [Google Scholar]
- 4.Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke SL, Pillay N, et al. : Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46:376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ketel K, Krauss M, Nicot A-S, Puchkov D, Wieffer M, Müller R, Subramanian D, Schultz C, Laporte J, Haucke V: A phosphoinositide conversion mechanism for exit from endosomes. Nature 2016, 529:408–412. [DOI] [PubMed] [Google Scholar]
- 6.Rahajeng J, Kuna RS, Makowski SL, Tran TTT, Buschman MD, Li S, Cheng N, Ng MM, Field SJ: Efficient Golgi Forward Trafficking Requires GOLPH3-Driven, PI4PDependent Membrane Curvature. Developmental Cell 2019, 50:573–585.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickson EJ, Hille B: Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 2019, 476:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiume R, Faenza I, Sheth B, Poli A, Vidalle MC, Mazzetti C, Abdul SH, Campagnoli F, Fabbrini M, Kimber ST, et al. : Nuclear Phosphoinositides: Their Regulation and Roles in Nuclear Functions. Int J Mol Sci 2019, 20:2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pemberton JG, Balla T: Polyphosphoinositide-Binding Domains: Insights from Peripheral Membrane and Lipid-Transfer Proteins. Adv. Exp. Med. Biol. 2018, 287:733–61. [Google Scholar]
- 10.Lemmon M: Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9:99–111. [DOI] [PubMed] [Google Scholar]
- 11.Amatya N, Wales TE, Kwon A, Yeung W, Joseph RE, Fulton DB, Kannan N, Engen JR, Andreotti AH: Lipid-targeting pleckstrin homology domain turns its autoinhibitory face toward the TEC kinases. Proc. Natl. Acad. Sci. U.S.A. 2019, 116:21539–21544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joseph RE, Wales TE, Fulton DB, Engen JR, Andreotti AH: Achieving a Graded Immune Response: BTK Adopts a Range of Active/Inactive Conformations Dictated by Multiple Interdomain Contacts. Structure 2017, 25:1481–1494.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lučić I, Rathinaswamy MK, Truebestein L, Hamelin DJ, Burke JE, Leonard TA: Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad. Sci. U.S.A. 2018, 115:E3940–E3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandra M, Chin YK-Y, Mas C, Feathers JR, Paul B, Datta S, Chen K-E, Jia X, Yang Z, Norwood SJ, et al. : Classification of the human phox homology (PX) domains based on their phosphoinositide binding specificities. Nat Commun 2019, 10:1528.** This manuscript provides an excellent blueprint to understand how putative lipid binding domains interact with PPIn containing membranes. Using a combined biochemical, biophysical, and cellular approach they classified the interaction of 39 of the 49 PX domains in the human genome, revealing a diversity of membrane binding patterns.
- 15.Lenoir M, Ustunel C, Rajesh S, Kaur J, Moreau D, Gruenberg J, Overduin M: Phosphorylation of conserved phosphoinositide binding pocket regulates sorting nexin membrane targeting. Nat Commun 2018, 9:993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Edwards M, Swaney KF, Singh N, Bhattacharya S, Borleis J, Long Y, Iglesias PA, Chen J, Devreotes PN: Mutually inhibitory Ras-PI(3,4)P2 feedback loops mediate cell migration. Proc. Natl. Acad. Sci. U.S.A. 2018, 115:E9125–E9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Chen ZJ: PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564:71–76. * This study intriguingly finds a novel role of PI4P in mediating NLRP3 inflammasome activation. Importantly this study not only showed binding to PI4P in vitro, but showed that decreasing Golgi PI4P using a Rapamycin inducible system decreases inflammasome localization and activation" xlink:type="simple">* This study intriguingly finds a novel role of PI4P in mediating NLRP3 inflammasome activation. Importantly this study not only showed binding to PI4P in vitro, but showed that decreasing Golgi PI4P using a Rapamycin inducible system decreases inflammasome localization and activation.
- 18.Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, Kagan JC: Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176:1432–1446.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudley LJ, Cabodevilla AG, Makar AN, Sztacho M, Michelberger T, Marsh JA, Houston DR, Martens S, Jiang X, Gammoh N: Intrinsic lipid binding activity of ATG16L1 supports efficient membrane anchoring and autophagy. EMBO J. 2019, 38:e100554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilgemann DW, Ball R: Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 1996, 273:956–959. [DOI] [PubMed] [Google Scholar]
- 21.Huang CL, Feng S, Hilgemann DW: Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998, 391:803–806. [DOI] [PubMed] [Google Scholar]
- 22.Hansen SB, Tao X, MacKinnon R: Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011, 477:495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fine M, Schmiege P, Li X: Structural basis for PtdInsP2-mediated human TRPML1 regulation. Nat Commun 2018, 9:4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT: Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. U.S.A. 2018, 115:E3569–E3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT: PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 2018, 7:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, LoCaste CE, Liu W, Poltash ML, Russell DH, Laganowsky A: Selective binding of a toxin and phosphatidylinositides to a mammalian potassium channel. Nat Commun 2019, 10:1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yen H-Y, Hoi KK, Liko I, Hedger G, Horrell MR, Song W, Wu D, Heine P, Warne T, Lee Y, et al. : PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559:423–427.** This study shows the critical tole of PI(4,5)P2 in regulating the active conformational state of GPCRs, and promotes coupling to G-proteins. Importantly this study demonstrated that binding of PS did not promote G-protein coupling, and highlights the importance of specific PPIn binding in GPCR regulation.
- 28.Garcia-Alai MM, Heidemann J, Skruzny M, Gieras A, Mertens HDT, Svergun DI, Kaksonen M, Uetrecht C, Meijers R: Epsin and Sla2 form assemblies through phospholipid interfaces. Nat Commun 2018, 9:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timcenko M, Lyons JA, Januliene D, Ulstrup JJ, Dieudonné T, Montigny C, Ash M-R, Karlsen JL, Boesen T, Kühlbrandt W, et al. : Structure and autoregulation of a P4ATPase lipid flippase. Nature 2019, 571:366–370.** This study provides the first molecular insight into how PI4P binding can regulate the activity of a lipid flippase. By comparing inactive apo states, and a fully activated form where the inhibitory c-terminus is removed with PI4P present, the authors can map the full molecular details of how the flippase can be activated.
- 30.Laverty D, Desai R, Uchański T, Masiulis S, Stec WJ, Malinauskas T, Zivanov J, Pardon E, Steyaert J, Miller KW, et al. : Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 2019, 565:516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan R, Qian H, Lukmantara I, Gao M, Du X, Yan N, Yang H: Human SEIPIN Binds Anionic Phospholipids. Developmental Cell 2018, 47:248–256.e4. [DOI] [PubMed] [Google Scholar]
- 32.She J, Guo J, Chen Q, Zeng W, Jiang Y, Bai X-C: Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556:130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirschi M, Herzik MA, Wie J, Suo Y, Borschel WF, Ren D, Lander GC, Lee S-Y: Cryoelectron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 2017, 550:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, She J, Zeng W, Guo J, Xu H, Bai X-C, Jiang Y: Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 2017, 550:415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.She J, Zeng W, Guo J, Chen Q, Bai X-C, Jiang Y: Structural mechanisms of phospholipid activation of the human TPC2 channel. Elife 2019, 8:814.* This study reveals the molecular basis for how TPC2 ion channels can be allosterically modulated by PI(3,5)P2 binding to mediate channel opening. Comparison to the apo state reveals the specific reorientation of amino acids by PI3,5P2 binding to mediate channel opening.
- 36.Hughes TET, Pumroy RA, Yazici AT, Kasimova MA, Fluck EC, Huynh KW, Samanta A, Molugu SK, Zhou ZH, Carnevale V, et al. : Structural insights on TRPV5 gating by endogenous modulators. Nat Commun 2018, 9:4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chavent M, Karia D, Kalli AC, Domański J, Duncan AL, Hedger G, Stansfeld PJ, Seiradake E, Jones EY, Sansom MSP: Interactions of the EphA2 Kinase Domain with PIPs in Membranes: Implications for Receptor Function. 2018, 26:1025–1034.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu K, Jiang T, Cui Y, Tajkhorshid E, Hartzell HC: A network of phosphatidylinositol 4,5-bisphosphate binding sites regulates gating of the Ca2+-activated Cl- channel ANO1 (TMEM16A). Proc. Natl. Acad. Sci. U.S.A. 2019, 116:19952–19962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le SC, Jia Z, Chen J, Yang H: Molecular basis of PIP2-dependent regulation of the Ca2+-activated chloride channel TMEM16A. Nat Commun 2019, 10:3769–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song W, Yen H-Y, Robinson CV, Sansom MSP: State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 2019, 27:392–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson CV, Rohacs T, Hansen SB: Tools for Understanding Nanoscale Lipid Regulation of Ion Channels. Trends in Biochemical Sciences 2019, doi: 10.1016/j.tibs.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammond GRV, Takasuga S, Sasaki T, Balla T: The ML1Nx2 Phosphatidylinositol 3,5-Bisphosphate Probe Shows Poor Selectivity in Cells. PLoS ONE 2015, 10:e0139957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, Kobilka BK, Govaerts C: Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nature Chemical Biology 2016, 12:35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta K, Donlan JAC, Hopper JTS, Uzdavinys P, Landreh M, Struwe WB, Drew D, Baldwin AJ, Stansfeld PJ, Robinson CV: The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 2017, 541:421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weis WI, Kobilka BK: The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87:897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues JPGLM, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, et al. : Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell 2017, 169:407–421.e16.* This study using an integrative structural biology approach provides molecular insight into how GRKs interact with and phosphorylate GPCRs. Intriguingly, there is a very strong dependence for GRK interaction driven by PI(4,5)P2, which together with ref. 25 reveals important insight into how PPin may play a key role in regulating multiple aspects of GPCR regulation.
- 47.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH: Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 1999, 18:871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita J-B, Dror RO, Zastrow von M: Catalytic activation of β-arrestin by GPCRs. Nature 2018, 557:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vivas O, Tiscione SA, Dixon RE, Ory DS, Dickson EJ: Niemann-Pick Type C Disease Reveals a Link between Lysosomal Cholesterol and PtdIns(4,5)P2 That Regulates Neuronal Excitability. Cell Rep 2019, 27:2636–2648.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traynor-Kaplan A, Kruse M, Dickson EJ, Dai G, Vivas O, Yu H, Whittington D, Hille B: Fatty-acyl chain profiles of cellular phosphoinositides. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohács T, Chen J, Prestwich GD, Logothetis DE: Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J. Biol. Chem. 1999, 274:36065–36072. [DOI] [PubMed] [Google Scholar]
- 52.Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P: INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ERplasma membrane contacts. Science 2015, 349:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B: A fourstep cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ERGolgi tether OSBP. Cell 2013, 155:830–843. [DOI] [PubMed] [Google Scholar]
- 54.Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas-Gervais S, Antonny B: Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J. 2017, 36:3156–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sohn M, Korzeniowski M, Zewe JP, Wills RC, Hammond GRV, Humpolickova J, Vrzal L, Chalupská D, Veverka V, Fairn GD, et al. : PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J. Cell Biol. 2018, 217:1797–1813.** This paper demonstrates a homeostatic mechanism to prioritize essential PI(4,5)P2 functions above PI4P-driven lipid transport. Essentially, PI4P-driven PS transfer to the PM only occurs when sufficient PI(4,5)P2 is present to localize the transfer proteins.
- 56.Ghai R, Du X, Wang H, Dong J, Ferguson C, Brown AJ, Parton RG, Wu J-W, Yang H: ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and regulate its level at the plasma membrane. Nat Commun 2017, 8:757–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Ma Q, Qi Y, Dong J, Du X, Rae J, Wang J, Wu W-F, Brown AJ, Parton RG, et al. : ORP2 Delivers Cholesterol to the Plasma Membrane in Exchange for Phosphatidylinositol 4, 5-Bisphosphate (PI(4,5)P2). Mol. Cell 2019, 73:458–473.e7. [DOI] [PubMed] [Google Scholar]
- 58.Sandhu J, Li S, Fairall L, Pfisterer SG, Gurnett JE, Xiao X, Weston TA, Vashi D, Ferrari A, Orozco JL, et al. : Aster Proteins Facilitate Nonvesicular Plasma Membrane to ER Cholesterol Transport in Mammalian Cells. Cell 2018, 175:514–529.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Besprozvannaya M, Dickson E, Li H, Ginburg KS, Bers DM, Auwerx J, Nunnari J: GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. Elife 2018, 7:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G: Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol. 2011, 195:965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Mousley CJ, Lete MG, Bankaitis VA: An equal opportunity collaboration between lipid metabolism and proteins in the control of membrane trafficking in the trans-Golgi and endosomal systems. Curr. Opin. Cell Biol. 2019, 59:58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong W, Xu M, Li C, Zhu B, Cao X, Li D, Chen H, Hu C, Li R, Luo C, et al. : ORP4L Extracts and Presents PIP2 from Plasma Membrane for PLCβ3 Catalysis: Targeting It Eradicates Leukemia Stem Cells. Cell Rep 2019, 26:2166–2177.e9.** An evocative paper suggesting that rather than transporting lipids, this lipid transfer protein in cells presents its PPIn ligand to phospholipases C, thus facilitating signaling as opposed to non-vesciular lipid transport.
- 63.Nishimura T, Gecht M, Covino R, Hummer G, Surma MA, Klose C, Arai H, Kono N, Stefan CJ: Osh Proteins Control Nanoscale Lipid Organization Necessary for PI(4,5)P2 Synthesis. Mol. Cell 2019, 75:1043–1057.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hansen SD, Huang WYC, Lee YK, Bieling P, Christensen SM, Groves JT: Stochastic geometry sensing and polarization in a lipid kinase-phosphatase competitive reaction. Proc. Natl. Acad. Sci. U.S.A. 2019, 116:15013–15022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zewe JP, Wills RC, Sangappa S, Goulden BD, Hammond GR: SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. Elife 2018, 7:1019.* This paper presents evidence that PI4P dephosphorylation occurs mainly in the ER where the major PI4P phosphatase, SAC1, is present. Furthermore, it is demonstrated that in cells, the enzyme cannot “reach” across the gap between ER and PM at contact sites to be active. This implies PI4P breakdown occurs exclusively in the realm of lipid transport.
- 66.Venditti R, Masone MC, Rega LR, Di Tullio G, Santoro M, Polishchuk E, Serrano IC, Olkkonen VM, Harada A, Medina DL, et al. : The activity of Sac1 across ER-TGN contact sites requires the four-phosphate-adaptor-protein-1. J. Cell Biol. 2019, 218:783–797.* However, this paper suggests that at ER-TGN contact sites, the ER protein VAPA/B and TGN protein FAPP1 may orientate ER-localized SAC1 to degrade PI4P in TGN membranes. This reveals that specific membrane contact sites may allow for trans activity of lipid phosphatases.
- 67.Dong R, Zhu T, Benedetti L, Gowrishankar S, Deng H, Cai Y, Wang X, Shen K, De Camilli P: The inositol 5-phosphatase INPP5K participates in the fine control of ER organization. J. Cell Biol. 2018, 217:3577–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Osborn DPS, Pond HL, Mazaheri N, Dejardin J, Munn CJ, Mushref K, Cauley ES, Moroni I, Pasanisi MB, Sellars EA, et al. : Mutations in INPP5K Cause a Form of Congenital Muscular Dystrophy Overlapping Marinesco-Sjögren Syndrome and Dystroglycanopathy. Am. J. Hum. Genet. 2017, 100:537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiessner M, Roos A, Munn CJ, Viswanathan R, Whyte T, Cox D, Schoser B, Sewry C, Roper H, Phadke R, et al. : Mutations in INPP5K, Encoding a Phosphoinositide 5Phosphatase, Cause Congenital Muscular Dystrophy with Cataracts and Mild Cognitive Impairment. Am. J. Hum. Genet. 2017, 100:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al. : Alpelisib for PIK3CA-Mutated, Hormone ReceptorPositive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380:1929–1940.* This study describes the first successful clinical application of a PI3K inhibitor towards a solid tumor.
- 71.Flinn IW, O’Brien S, Kahl B, Patel M, Oki Y, Foss FF, Porcu P, Jones J, Burger JA, Jain N, et al. : Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood 2018, 131:877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, et al. : PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370:1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dreyling M, Santoro A, Mollica L, Leppä S, Follows GA, Lenz G, Kim WS, Nagler A, Panayiotidis P, Demeter J, et al. : Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35:3898–3905. [DOI] [PubMed] [Google Scholar]
- 74.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. : Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560:499–503.** Despite the critical role of PI3Ks in cancer formation and progression, there has been limited success in targeting them in solid tumors. This study provides a possible mechanistic reason for this lack of success, through PI3K inhibitor mediated insulin feedback Suppression of this feedback by either diet or pharmacological intervention greatly improved PI3K inhibitor efficacy in mouse models, and provides exciting possibilities of increasing PI3K inhibitor efficacy in cancer treatment.
- 75.Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, Ladewig E, Gorelick A, Lin T-Y, Toska E, et al. : Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 2019, 366:714–723.** This manuscript describes the occurrence of double mutations on a single allele of PIK3CA in cancer, leading to increased kinase activity and PI3K inhibitor sensitivity. This supports the hypothesis of ref. 76, which showed that different mutations in PIK3CA lead to activation by unique mechanisms, suggesting that they could act synergistically.
- 76.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL: Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc. Natl. Acad. Sci. U.S.A. 2012, 109:15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong J-P, Blanc E, Johnson SC, Hoguin C, Boccara O, et al. : Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 13:195.* Along with their role in cancer progression, PI3K mutations also mediate PI3K related overgrowth syndromes (PROS), with this study showing clear efficacy of using of PIK3CA specific inhibitor in patients.
- 78.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K: PI3Kδ and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16:702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rao VK, Webster S, Dalm VASH, Sediva A, van Hagen, Holland S, Rosenzweig SD, Christ AD, Sloth B, Cabanski M, et al. : Effective “Activated PI3Kδ Syndrome”targeted therapy with the PI3Kδ inhibitor leniolisib. Blood 2017, doi: 10.1182/blood2017-08-801191.* Mutation of PIK3CD is a driver of primary immunodeficiencies. This paper shows that targeted therapy using PIK3CD specific inhibitors are showing excellent clinical response in APDS patients.
- 80.Lee Y-R, Chen M, Lee JD, Lin S-Y, Fu T-M, Chen H, Ishikawa T, Chiang S-Y, Katon J, Zhang Y, et al. : Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364:eaau0159.* This study identified a novel regulatory mechanism controlling the activity of the lipid phosphatase PTEN, where inhibiting a WWP1 E3 ligase that downregulates PTEN, it is possible to increase the tumor suppressor activity of PTEN. Small molecules found in cruciferous vegetables inhibit WWP1 activity, and provide a new basis for drug design.
- 81.Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, Coy JF, Klauck SM, Poustka A, Dahl N: A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13:175–182. [DOI] [PubMed] [Google Scholar]
- 82.Sabha N, Volpatti JR, Gonorazky H, Reifler A, Davidson AE, Li X, Eltayeb NM, Dall’Armi C, Di Paolo G, Brooks SV, et al. : PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Invest. 2016, 126:3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maani N, Sabha N, Rezai K, Ramani A, Groom L, Eltayeb N, Mavandadnejad F, Pang A, Russo G, Brudno M, et al. : Tamoxifen therapy in a murine model of myotubular myopathy. Nat Commun 2018, 9:4849–16.* This paper describes the exciting and unexpected discovery that the clinically approved estrogen receptor modulator tamoxifen improves symptoms of myotubular myopathy in a mouse model of disease, and reveals a novel therapeutic strategy for MTM patients.
- 84.Kato N, Comer E, Sakata-Kato T, Sharma A, Sharma M, Maetani M, Bastien J, Brancucci NM, Bittker JA, Corey V, et al. : Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McNamara CW, Lee MCS, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BKS, Chatterjee AK, McCormack SL, et al. : Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manjunatha UH, Vinayak S, Zambriski JA, Chao AT, Sy T, Noble CG, Bonamy GMC, Kondreddi RR, Zou B, Gedeck P, et al. : A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 2017, 546:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paquet T, Le Manach C, Cabrera DG, Younis Y, Henrich PP, Abraham TS, Lee MCS, Basak R, Ghidelli-Disse S, Lafuente-Monasterio MJ, et al. : Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med 2017, 9:eaad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Willett R, Martina JA, Zewe JP, Wills R, Hammond GRV, Puertollano R: TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat Commun 2017, 8:1580–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P, Nakanishi H, Eguchi S, Koizumi A, Sasaki J, et al. : PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K. Mol. Cell 2017, 68:566–580.e10.** This study is an important conceptual advance defining the role of PTEN in degrading PI3,4P2, and reveal how initial in vitro results on substrate specificity can be misleading of true biological activity. The careful cellular and organism level analysis of PTEN’s role in regulating PI3,4P2 levels in this study reveals a new aspect of PTEN function, and may play an important aspect in its ability to act as a tumor suppressor.
- 90.Wang H, Lo W-T, Haucke V: Phosphoinositide switches in endocytosis and in the endolysosomal system. Curr. Opin. Cell Biol. 2019, 59:50–57. [DOI] [PubMed] [Google Scholar]
- 91.Bai L, Kovach A, You Q, Hsu H-C, Zhao G, Li H: Autoinhibition and activation mechanisms of the eukaryotic lipid flippase Drs2p-Cdc50p. Nat Commun 2019, 10:4142–10. [DOI] [PMC free article] [PubMed] [Google Scholar]