

Abstract
Tumors arise through waves of genetic alterations and clonal expansion that allow tumor cells to acquire cancer hallmarks, such as genome instability and immune evasion. Recent genomic analyses showed that the vast majority of cancer driver genes are mutated in a tissue-dependent manner, that is, are altered in some cancers but not others. Often the tumor type also affects the likelihood of therapy response. What is the origin of tissue specificity in cancer? Recent studies suggest that both cell-intrinsic and cell-extrinsic factors play a role. On one hand, cell type–specific wiring of the cell signaling network determines the outcome of cancer driver gene mutations. On the other hand, the tumor cells’ exposure to tissue-specific microenvironments (e.g. immune cells) also contributes to shape the tissue specificity of driver genes and of therapy response. In the future, a more complete understanding of tissue specificity in cancer may inform methods to better predict and improve therapeutic outcomes.
Keywords: Cancer driver genes, Tissue specificity, DNA damage response, Cancer immune evasion, Mutations, Aneuploidy
Introduction
It has long been known that cancer driver genes can have profoundly different effects in different tissues. For example, germline mutations in BRCA1/BRCA2 increase the risk of breast and ovarian cancers much more than other types of cancer, whereas mutations in the mismatch repair (MMR) pathway contribute to colorectal cancer. At the somatic level, KRAS mutations are frequent in lung, colon, and pancreatic tumors but not in other cancers. Recent genomic analyses of human cancers have confirmed and expanded this notion, revealing that the vast majority of cancer driver genes, with a few exceptions (e.g. MYC), are mutated or amplified/deleted in a tissue-specific way (Figure 1) [1,2]. These data also indicate that tumor cells from different tissues often acquire similar cancer hallmarks, such as genome instability and proliferation, in a tissue type–dependent manner.
Figure 1. Tissue specificityspecificity of cancer driver genes.
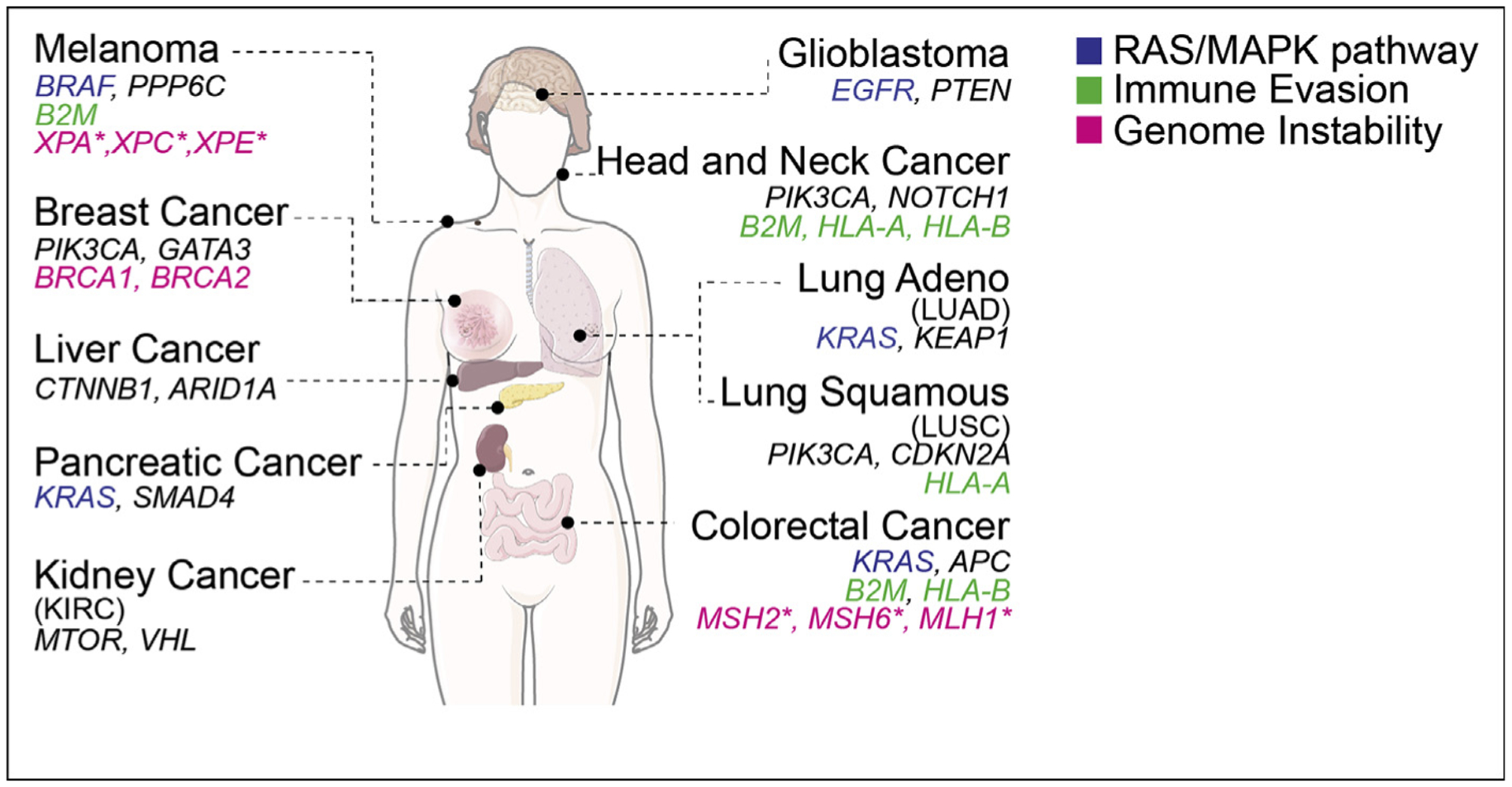
For each tumor type representative mutated cancer driver genes are shown including genes acting in the RAS/MAPK pathway, immune evasion and genome instability. Lung cancers refer to NSCLC (non–small-cell lung cancer) either adeno-carcinoma (LUAD) or squamous cell carcinoma (LUSC) as indicated. *: germline only.
One of the cancer hallmarks that have recently received increased attention is cancer immune evasion, owing to novel outstanding therapeutic opportunities. The explosive growth of the field of immune oncology in the past few years has revealed high tissue specificity of both (1) the frequency of mutations in genes and pathways that control cancer immune evasion (e.g., B2M and HLA-A/B) and (2) the efficacy of cancer immunotherapy – current immunotherapy strategies such as checkpoint inhibitors can achieve enduring clinical benefit in a fraction of patients affected by certain tumor types (e.g. melanoma or lung cancer) but are less effective for other cancers (e.g. pancreatic cancers) [3,4].
What are the causes of tissue specificity of cancer driver genes and of therapy response? Tissue specificity of cancer genes may be explained by tissue-specific expression level (the fact that a gene is expressed at different levels across tissues), but computational analyses suggest that this is generally not the case [5]. Instead, recent studies indicate a different scenario [6–9]. On one hand, each cell in the body has a specific signaling network that depends on its developmental origin. This circuitry (cell-intrinsic) plays a role in determining the type of outcome of mutations in a certain gene (e.g. increased proliferation or genome instability) [6,7,9]. On the other hand, tumor cells interact with tissue-specific microenvironments (cell-extrinsic; e.g. immune cell composition or exposure to hormones) that can influence the way mutations in specific cancer genes and pathways are selected for during tumorigenesis [8]. Here, we will cover recent advances in the field, focusing on aspects of the DNA damage response (DDR) and cancer immune evasion. We will provide specific examples of how both tumor cell-intrinsic and cell-extrinsic factors cooperate to determine the tissue specificity of cancer driver genes and pathways.
Tissue specificity of DDR genes and genome instability
DDR genes are among the most mutated genes in cancer [10]. Although some of them are mutated also at the somatic level (such as BRCA1/2), most DDR genes are mutated mainly at the germline level and promote the development of tissue-specific cancers. For example, germline mutations in the nucleotide excision repair (NER) pathway (e.g. XPA, XPC, XPE) induce xeroderma pigmentosum (XP), a hereditary skin syndrome that promotes cutaneous cancers. Patients with germline mutations in the homologous recombination pathway (e.g. BRCA1 and BRCA2) have an increased risk of developing breast and ovarian cancers, whereas germline mutations in the MMR pathway (e.g. MSH2, MSH6, and MLH1) contribute to hereditary nonpolyposis colorectal cancer (also known as Lynch syndrome) [11]. Despite the tissue specificity of these cancers, genes involved in DNA repair pathways are expressed at a similar level across tissues including those from which the cancer originates [5,12]. Distinct mechanisms involving cell-extrinsic and cell-intrinsic factors have been explored to explain the tissue-specific effects of DDR mutations.
First, cell-extrinsic factors such as exposure to environmental mutagens and hormones can impact the tissue specificity of mutations in DDR genes (Figure 2). Different organs and tissues are exposed to specific mutagens which give rise to specific type of DNA damage and related mutation signatures in cancer genomes [13,14]. For instance, UV light induces several types of DNA lesions including pyrimidine dimers, which can be repaired by NER. Accordingly, NER pathway genes are highly mutated in skin cancers [5]. Organs such as the mammary gland are not directly exposed to environmental mutagens but can be affected by endogenous genotoxic metabolites. For instance, excess estrogen can be metabolized into quinone radicals that induce oxidative damage in DNA; BRCA1 acts in a tissue-specific manner to regulate these estrogen-metabolizing enzymes [15]. Thus, it is possible that in the case of BRCA1, as in NER-deficient skin cancers, the tissue specificity of cancer driver genes arises from the interaction between the exposure to tissue-specific mutagens and DNA repair pathways.
Figure 2. Model for the possible cellular and molecular interactions driving the tumor specificity of DDR genes.
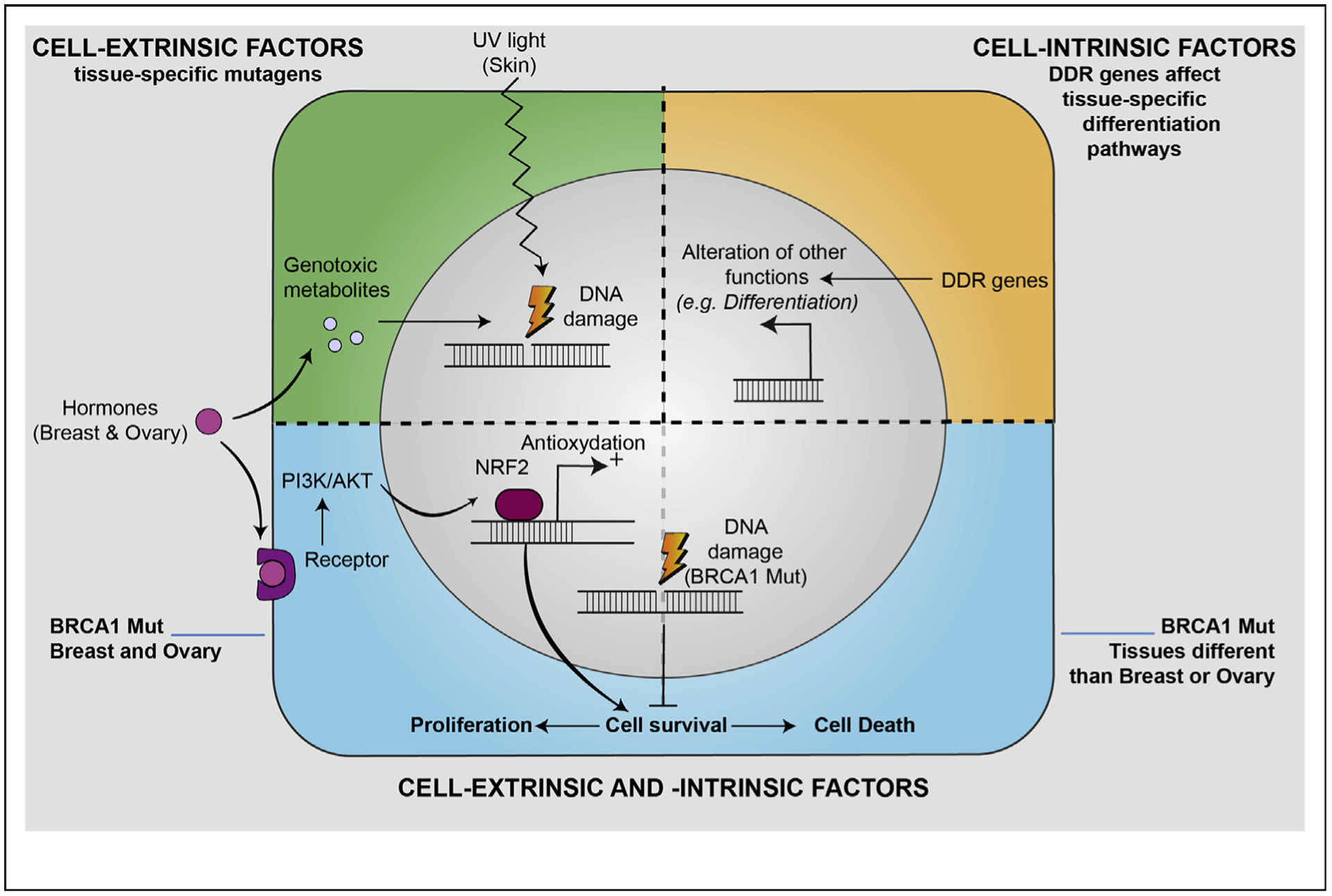
Upper left: tissue-specific cell-extrinsic factors, such as UV light or hormones, can determine DNA damage and promote cell transformation. Upper right: DDR genes can affect other cellular signaling pathways, such as cellular differentiation as in the case of BRCA1 inhibition that can promote the maintenance of a stem cell–like behavior in mammary epithelial cells. Lower: cell-extrinsic and -intrinsic pathways can interact and affect cell autonomous pathways. For instance, estrogen can promote survival of BRCA1-mutated cells, thus protecting them from oxidative stress. DDR , DNA damage response.
A second possibility is that the outcome of DDR gene mutations depends on the interaction between cell-extrinsic factors (e.g. hormones) and cell type–specific circuitry of signaling pathways. For BRCA1, it has been proposed that the tissues where the tumors manifest may be the only ones in which complete loss of the DNA repair gene is tolerated through hormone-dependent proliferative signals [16]. Supporting this hypothesis, it has been suggested that estrogen plays a fundamental role by specifically protecting mammary (and possibly ovarian) epithelial cells (MECs) from the oxidative stress-induced cell death occurring in the absence of BRCA1 [9] (Figure 2). This effect is mediated by the binding of estrogen to its receptor (ER) that can subsequently regulate NRF2 via the PI3K-AKT pathway. Recent studies also suggest another mechanism in which mature BRCA1-haplodeficient MECs, expressing ER and progesterone receptor, can stimulate the progesterone receptor–dependent secretion of RANKL. Through a paracrine signaling, RANKL then activates its receptor RANK, localized on luminal progenitor cells (LPCs) that lack ER and triggers subsequent LPC proliferation through NF-κB pathway [17,18]. Interestingly, LPCs have been proposed to be the cell-of-origin of the basal-like subtype of breast cancer and BRCA1-deficient LPCs display a greater sensitivity to ionizing radiation and replication stress than wild-type LPCs. Thus, activation of NF-κB may increase the genomic instability of RLPCs by promoting their proliferation, which in turn induces additional replication-associated DNA damage and constantly activates intrinsic NF-κB activity, thereby freeing the proliferation of LPCs from hormonal influences [18].
Third, tissue specificity of DDR mutations in cancer could be explained by cell-intrinsic factors, that is, the fact that DNA repair genes can modulate cell type–specific cellular signaling pathways that are critical for tumorigenesis, such as differentiation of tissue stem cells (Figure 2). For example, apart from its involvement in DNA repair, several in vitro and in vivo studies have proposed a direct role of BRCA1 in cell fate determination. Indeed, depletion of BRCA1 impairs differentiation of MECs and maintains a stem cell–like behavior [19,20]. BRCA1 activates the NOTCH pathway by transcriptional upregulation of NOTCH ligands and receptors, in breast cells. This regulation is important for normal breast differentiation, as knockdown of NOTCH signaling components results in loss of ER (estrogen receptor) and luminal marker expression [21]. In addition, BRCA1 negatively regulates SLUG protein stability, which promotes breast differentiation, as SLUG acts by functionally suppressing human breast progenitor cell lineage commitment and differentiation [22]. BRCA1 has also been shown, specifically in MECs, to positively regulate the transcription of SIRT1, a deacetylating enzyme involved in many functions including the regulation of telomere length and the induction of apoptosis. Accordingly, BRCA1-haplodeficient human MECs exhibit significant telomere shortening and chromosomal instability [23]. Hence, BRCA1 can contribute to tumorigenesis in hormone-sensitive and ER-negative populations through synergic interactions and a tumor-promoting environment specific to breast tissue.
Finally, other factors must be considered. Colorectal cancers arise from rapidly dividing stem cells and are also associated with mutations in MMR genes [5]. Indeed, DNA mismatches occur frequently at the replication fork of dividing cells, and thus MMR is especially crucial to prevent the fast accumulation of DNA replication errors and extensive genomic microsatellite instability in intestinal stem cells [1]. Accordingly, context-specific mutation signatures associated with replication timing were retrieved at point mutations affecting various colorectal cancer driver genes in adult stem cells of the colon, the cells of origin of this cancer [24]. Moreover, cell-of-origin chromatin and epigenomic features are the best predictors of cancer mutation rates, suggesting that variations of the epigenomic landscape across tissues may also contribute to cell type–specific mutagenesis [25]. Therefore, context-specific differences in features such as replication timing or chromatin structure can also shape the tumor-specific effects of DDR mutations and the acquisition of genome instability in human cancer.
Tissue specificity of cancer immune evasion
Evading recognition and killing by the immune system represents a crucial hallmark of cancer and is targeted by immunotherapy strategies [3,4]. Although some tumor-agnostic markers of response have been identified, such as the extent of cytotoxic immune infiltrate, there is an increasing recognition of the role of tissue- and context-specific determinants of cancer immune evasion and therapy response [8].
As with other cancer hallmarks such as genome instability (see to the aforementioned information), the mechanisms of cancer immune evasion and the likelihood of responding to immunotherapy vary across tumor types. Similarly to other cancer driver genes, the spectrum and frequency of point mutations or deletion/amplification in cancer genes that drive immune escape is highly tissue-specific. For example, genes that are crucial for antigen presentation such as B2M, HLA-A, and HLA-B act as tumor suppressors in a fraction of skin melanoma, colorectal, head and neck, or lung cancers [26,27] but not in breast, ovarian, or pancreatic cancers (Figure 3). Because antigen presentation is necessary for Tcell–emediated recognition and killing of tumor cells, a high (vs low) frequency in the inactivation of this pathway suggests a high (vs low) degree of selection to escape T cell recognition [28]. Perhaps not surprisingly, the tumor types showing the highest rate of clinical response after anti-PD1 and/or anti-CTLA-4 immunotherapies (which reactivate mainly T cell–mediated immunity) are the same tumor types that display significant inactivation of genes involved in antigen presentation. In fact, current immunotherapy strategies have demonstrated clinical benefit mainly in melanoma, lung, colorectal, head and neck cancers but much less in other tumor types such as pancreatic or ovarian cancers [3,4,29–32].
Figure 3. Tissue-specific features affecting immune response across cancer types.
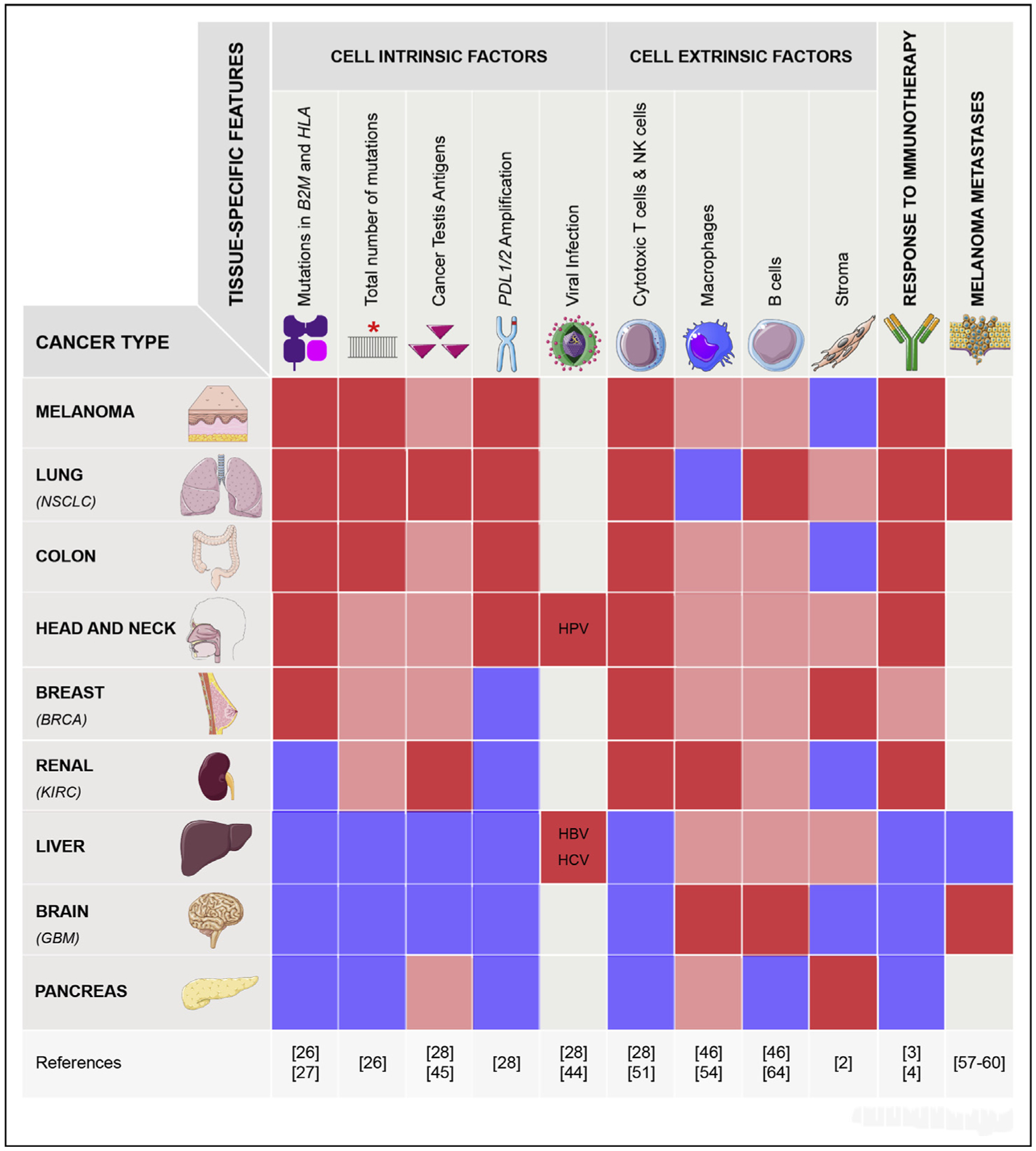
The different levels of each feature are depicted as a heatmap-like system (dark red: high, light red: medium and blue: low or absent. Gray refers to information not available). For the response to immune checkpoint inhibitors, the dark red indicates that a response has been achieved in a fraction of patients and that it has been FDA approved for this tumor type. The light red for the BRCA type refers to only one immunotherapy approved by the FDA in 2019 and performed in combination with chemotherapy (Schmid et al., NEJM, 2019). For the cancer testis antigens, we show its average expression in the tumors, not its correlation with cytotoxicity or immunotherapy (see text for details). NSCLC, non–small-cell lung cancer; BRCA, breast invasive carcinoma; KIRC, kidney renal cell carcinoma; GBM, glioblastoma multiforme.
Why do distinct tumor types display different patterns of mutations in cancer genes driving immune evasion and different response to immunotherapy? Among the possible causes are (1) how cellular pathways implicated in immune recognition are regulated in tumor cells from different tissues (cell-intrinsic factors) and (2) how tumor cells interact with tissue-specific immune microenvironments (cell-extrinsic factors) (Figure 3) [8,33].
First, among the tumor cell–intrinsic properties, different tumor types display different loads of point mutations and aneuploidy that can alter tumor immune properties. For example, owing to UV- and smoke-related DNA damage, melanomas and lung cancers have among the highest number of point mutations (and thus neoantigens) that can promote tumor immune recognition [34–36]. Although this hypothesis can in part explain why highly mutated tumor types are more likely to respond to immunotherapy, it does not explain why tumors with, on average, lower amount of mutations such as kidney tumors (and in part head and neck tumors), can still show a good response rate. Even though the total number of mutations is positively associated with T cell infiltrates in some tumor types, neoantigen and mutation burden are not always good predictors of cytotoxic infiltrate and immunotherapy response [28,37,38]. The level of aneuploidy is associated with lower amount of cytotoxic markers both within and across many cancer types and could explain in part why tumor types mainly driven by copy number alterations, such as ovarian and pancreatic cancers, tend to respond poorly to immunotherapy [39–42].
Second, in addition to the total load of point mutations or aneuploidy, alterations in specific cancer genes primarily involved in other cellular pathways can affect the cancer immune phenotype [43]. Although mutations in some cancer drivers are associated with high (e.g. CASP8 and EP300) or low (e.g. IDH1 and APC) level of cytotoxic markers across most tumor types, many associations between mutations and immune markers are tumor type–specific [42,44]. For example, TP53-mutant tumors tend to display a lower level of cytotoxic markers than TP53-wild-type tumors in pancreatic, colorectal, and head, and neck cancers, whereas in breast and lung cancers and in sarcomas TP53 mutations are linked to higher immune infiltrate [43–45].
Third, re-expression within the tumor cells of cancer testis antigens or endogenous retroviruses, which are normally restricted to the germline, is associated with different cancer immune properties in a tumor type–specific manner. For example, expression of cancer testis antigens positively correlates with cytotoxic immune infiltrates in breast, lung, cervix, bladder, uterine, and stomach cancer [28,45]. In contrast, in melanoma, MAGEA antigen expression predicts resistance to CTLA4 blockade [37]. Expression of viral antigens by tumor cells positively correlates with the IFNγ level and other cytotoxic markers in cervix, kidney, and head and neck cancers (positive for human papilloma virus) and stomach cancers (positive for Epstein–Barr virus) [28,44].
Tumor-extrinsic features that characterize the tissue-and organ-specific immune microenvironment also shape the strategies used by cancer cells to evolve during tumor development. The immune system has evolved and diversified across different parts of the body, depending on the extent and spectrum of pathogens’ exposure. Thus, the composition, differentiation, and activation status of immune cells vary across tissues. Lung and gut mucosae, which are directly or indirectly exposed to exogenous viruses, have developed a strong local army of tissue-resident immune cells — especially T cells and B cells — to fight the infections locally as compared with internal organs such as ovary, breast, or pancreas, which are not as directly exposed to exogenous pathogens [33]. In addition, the level of markers of activated T and NK (Natural Killer) cells is higher in normal lung, colon, and ileum tissues than in other tissues such as brain, ovary, and kidney [28,46–48].
Overall, across tissues, the extent and spectrum of the immune infiltrate in tumors follow a trend similar to normal tissues, suggesting that the pre-existing tissue-specific microenvironment is often retained in the tumors. For example, colon or stomach cancers and brain or liver tumors display among the highest or lowest level of T and NK cell cytolytic markers, respectively [28,44,49–51], similar to their normal counterpart. However, there are notable exceptions. For example, melanoma, head and neck, and kidney cancers are among the tumor types with high proportion of IFNγ-dominant subtype, characterized by high abundance of cytotoxic markers cells (e.g. CD8 T and NK cells) [28,44,49,52], much higher than their adjacent normal tissues. Other tumor types, such as a large proportion of pancreatic adenocarcinoma, display myeloid-inflamed stroma that may contribute to a weak response to immunotherapy in these patients [53,54].
Trafficking and homing of immune cells to different organs can also vary widely and play different roles in tumorigenesis and response to immunotherapy. For example, a recent study using a syngeneic model of melanoma showed that, in the skin, melanoma cells recruit blood-derived inflammatory monocytes that contribute to antibody-dependent tumor cell killing, whereas in the lung, shrinking of melanoma metastases depends on tissue-resident macrophage populations [55]. Organ-specific microbiomes can regulate, in different ways, systemic immune responses, as in the case of the intestinal microbiome, which can not only promote systemic antitumor immune responses [56] but also the local immune microenvironment, as in the case of the lung microbiome, which promotes tumorigenesis through recruitment of γ/δ T cells [57,58].
Finally, if the local microenvironment plays a significant role in shaping the anticancer immune response, then metastatic lesions derived from the same tumors but located in different organs should undergo tissue-dependent immune response and regulation. Recent data from patients with metastatic cancer suggest that this may be the case [59,60]. Compared with metastases (from melanoma, colorectal, or lung cancer) to other sites, metastases to the liver tend to show a lower cytotoxic T cell/Treg ratio and a weaker response to immunotherapy [60–62]. This suggests that the strong immune tolerance of the liver, whose local immune system evolved to limit harmful immune response against food and microbiota, promotes immune tolerance against cancer cells and limits the efficacy of immunotherapy. Interestingly, compared with the liver, melanoma metastases to the brain, an immune privileged organ also characterized by low cytotoxic molecules, respond well to immunotherapies, similar to nonbrain lesions [63]. Although other explanations are possible, it is plausible that in brain metastases the physical disruption of the blood–brain barrier profoundly alters the local microenvironment, promoting leukocyte infiltration in this otherwise immune privileged organ [28,50,64].
Final remarks and open questions
Recent studies highlight how the crosstalk between cell type–specific rewiring of signaling pathways [7,65] and tissue type–specific microenvironment [8] shapes the tumor type–specific mutational frequency of cancer driver genes, as well as response to therapy. Although, we have covered some aspects of tumor type–specific response to immunotherapy, tumor type often also dictates response to targeted therapies. For example, inhibition of BRAF is highly effective in BRAF-mutated melanoma but not in BRAF-mutated colon cancers [66] and recent data from a phase I clinical trial suggest that a new KRASG12C inhibitor may be more effective in lung tumors than in colorectal tumors harboring the same mutation [67].
Several outstanding questions remain to be addressed: (1) How do cancer driver genes (especially those that are ubiquitously expressed and perform core cellular functions such as DDR) interact with cell type–specific genetic networks to promote cancer initiation and progression? Our understanding, as described previously, is quite limited to a few examples and tissues. (2) How do oncogenic mutations in specific cancer drivers or aneuploidy events interact with the tumor microenvironment to promote cancer hallmarks such as cancer immune evasion and therapy response? A better understanding of tumor type specificity will allow us to decipher context-dependent roles of cancer driver genes and context-dependent therapeutic vulnerabilities.
Acknowledgments
This work was supported by a postdoctoral fellowship from the National Cancer Center (J.J. Bianchi), by grant from the Cancer Research UK Grand Challenge and the Mark Foundation for Cancer Research (C5470/A27144 to T. Davoli as a member of the SPECIFICANCER Team), R00 CA212621 to T. Davoli and by the Melanoma Research Alliance (Young Investigator Award).
Footnotes
Conflict of interest statement
Nothing declared.
References
Papers of particular interest, published within the period of review, have been highlighted as:
** of outstanding interest
- 1.Schneider G, Schmidt-Supprian M, Rad R, Saur D: Tissue-specific tumorigenesis: context matters. Nat Rev Cancer 2017, 10.1038/nrc.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.**.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. : Cell-of-Origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study described a comprehensive analysis of human cancers using different molecular features, showing that the cell-of-origin is a critical determinant of the molecular classification of tumors.
- 3.Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J: Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015, 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Wolchok JD: Cancer immunotherapy using checkpoint blockade. Science 2018, 10.1126/science.aar4060.80-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer MH, Serrano L: Cell type-specific properties and environment shape tissue specificity of cancer genes. Sci Rep 2016, 10.1038/srep20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.**.Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. : Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 2018, 10.1016/j.cell.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; By performing genome-wide functional screenings in human cells from different tissues, this study showed that there is a high degree of tissue-specificity in genes that drive proliferation, which recapitulates the tissue-specificty in cancer driver genes.
- 7.**.Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. : Tissue-specific oncogenic activity of KRASA146T. Cancer Discov 2019, 9: 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated how expression of different KRAS oncogenic mutations (which show tissue-specific frequencies in human cancers) drives tissue-specific effects on cell proliferation and differentiation in mouse tissues, recapitulating the tissue-specific frequencies of these KRAS mutations in human tumors.
- 8.**.Salmon H, Remark R, Gnjatic S, Merad M: Host tissue determinants of tumour immunity. Nat Rev Cancer 2019, 10.1038/s41568-019-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article represents a comprehensive review of tissue- and organ-specific immune microenvironments of human tumors.
- 9.Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li YT, Molyneux S, et al. : Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A 2014, 111: 4472–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dietlein F, Thelen L, Reinhardt HC: Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet 2014, 30:326–339. [DOI] [PubMed] [Google Scholar]
- 11.Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E: DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8:16–29. [DOI] [PubMed] [Google Scholar]
- 12.Lage K, Hansen NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK, Szallasi Z, Jensen TS, Brunak S: A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci U S A 2008, 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. : Signatures of mutational processes in human cancer. Nature 2013, 10.1038/nature12477. [DOI] [Google Scholar]
- 14.**.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. : The repertoire of mutational signatures in human cancer. bioRxiv 2019, 10.1101/322859. [DOI] [Google Scholar]; This article and the one above represent landmark papers on the analysis of mutational signatures in human tumors.
- 15.Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, Orr KS, Vohhodina J, Kavanagh JN, Madden AF, et al. : Brca1 deficiency exacerbates estrogen-induced dna damage and genomic instability. Cancer Res 2014, 74:2773–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elledge SJ, Amon A: The BRCA1 suppressor hypothesis: an explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell 2002, 10.1016/S1535-6108(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 17.Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Rohrbach K, Huang LY, et al. : RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 2016, 22:933–939. [DOI] [PubMed] [Google Scholar]
- 18.Sau A, Lau R, Cabrita MA, Nolan E, Crooks PA, Visvader JE, Pratt MAC: Persistent activation of NF-kB in BRCA1-deficient mammary progenitors drives aberrant proliferation and accumulation of DNA damage. Cell Stem Cell 2016, 19:52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH: Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A 2005, 102:9176–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD, Dontu G, Wicha MS: BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A 2008, 105:1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley NE, Nic An, Tsaoir CB, Blayney JK, Oram LC, Crawford NT, D’Costa ZC, Quinn JE, Kennedy RD, Harkin DP, Mullan PB: BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res 2013, 41:8601–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S, et al. : Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P, Keller PJ, Glover E, Richardson AL, Cowan J, et al. : Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat Commun 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blokzijl F, De Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. : Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538:260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, et al. : Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ: Cumulative haploinsufficiency and trip-losensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.**.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. : Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes one of the most comprehensive analyses of point mutated cancer driver genes across tumor types.
- 28.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N: Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH: Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol 2018, 10.1038/s41575-018-0005-x. [DOI] [PubMed] [Google Scholar]
- 30.Fan C (Ava), Reader J, Roque DM: Review of immune therapies targeting ovarian cancer. Curr Treat Options Oncol 2018, 10.1007/s11864-018-0584-3. [DOI] [PubMed] [Google Scholar]
- 31.Getz G, Gabriel SB, Cibulskis K, Lander E, Sivachenko A, Sougnez C, Lawrence M, Kandoth C, Dooling D, Fulton R, et al. : Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, et al. : Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018, 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 33.Pao W, Ooi CH, Birzele F, Ruefli-Brasse A, Cannarile MA, Reis B, Scharf SH, Schubert DA, Hatje K, Pelletier N, et al. : Tissue-specific immunoregulation: a call for better understanding of the “immunostat” in the context of cancer. Cancer Discov 2018, 10.1158/2159-8290.CD-17-1320. [DOI] [PubMed] [Google Scholar]
- 34.Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, Duyvesteyn K, Haidari S, van Hoeck A, Onstenk W, et al. : Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher TN, Schreiber RD: Neoantigens in cancer immunotherapy. Science 2015, 348:69–74. [DOI] [PubMed] [Google Scholar]
- 36.Schumacher TN, Scheper W, Kvistborg P: Cancer Neoantigens. Annu Rev Immunol 2019, 10.1146/annurev-immunol-042617-053402. [DOI] [PubMed] [Google Scholar]
- 37.Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, Langer R, Lee PC, Gusenleitner D, Keskin DB, et al. : Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 2018, 10.1016/j.cell.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, Gajewski TF: Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A 2016, 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, Yi S, Lin SY, Sahni N: Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat Commun 2018, 10.1038/s41467-018-03730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davoli T, Uno H, Wooten EC, Elledge SJ: Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399 80-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.**.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. : Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 2018, 33:676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes an analysis of aneuploidy across tumor types showing that aneuploidy patterns recapitulate cell and tissue of origin. Together with the paper above, it also shows that aneuploidy level is negatively correlated with cytotoxic immune markers.
- 42.Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, et al. : Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep 2013, 3:2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.**.Wellenstein MD, de Visser KE: Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]; This article represents a review of how many cell-intrinsic oncogenic pathways, which are not primarily involved in immunological processes, contribute to shape the interaction between tumor cells and the immune system.
- 44.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. : The immune landscape of cancer. Immunity 2018, 10.1016/j.immuni.2018.03.023. [DOI] [Google Scholar]
- 45.**.Tamborero D, Rubio-Perez C, Muiños F, Sabarinathan R, Piulats JM, Muntasell A, Dienstmann R, Lopez-Bigas N, Gonzalez-Perez A: A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin Cancer Res 2018, 10.1158/1078-0432.CCR-17-3509. [DOI] [PubMed] [Google Scholar]; This paper and the one above shows a comprehensive computational analyses of tumors based on immune-related features, including genetic features predicting immunological subtypes and therapy response.
- 46.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. : Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016, 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJC, Bickham KL, Lerner H, Goldstein M, Sykes M, et al. : Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong MT, Ong DEH, Lim FSH, Teng KWW, McGovern N, Narayanan S, Ho WQ, Cerny D, Tan HKK, Anicete R, et al. : A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity 2016, 10.1016/j.immuni.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z: Pan-cancer immunogenomic analyses reveal genotypeimmunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 2017, 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 50.Li B, Li T, Pignon JC, Wang B, Wang J, Shukla SA, Dou R, Chen Q, Hodi FS, Choueiri TK, et al. : Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet 2016, 10.1038/ng.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. : The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015, 21:938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. : Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, et al. : Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep 2017, 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C: Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res 2017, 10.1158/0008-5472.CAN-16-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.**.Lehmann B, Biburger M, Brückner C, Ipsen-Escobedo A, Gordan S, Lehmann C, Voehringer D, Winkler T, Schaft N, Dudziak D, et al. : Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci Immunol 2017, 10.1126/sciimmunol.aah6413. [DOI] [PubMed] [Google Scholar]; This study uses syngeneic mouse models to show that depending on the tissue type where the tumor is located, different myeloid cells are recruited to the tumor site and exert distinct functions in the cancer microenvironment.
- 56.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF: The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 10.1126/science.aao3290.80-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, et al. : Commensal microbiota promote lung cancer development via gd T cells. Cell 2019, 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, Trinchieri G: Microbes and cancer. Annu Rev Immunol 2017, 35:199–228. [DOI] [PubMed] [Google Scholar]
- 59.**.Jiménez-Sánchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, Gill MB, Park KJ, Zivanovic O, Konner J, et al. : Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017, 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that metastatic lesions from the same patient located in different sites display different immune microenvironments and therapy responses.
- 60.Bilen MA, Shabto JM, Martini DJ, Liu Y, Lewis C, Collins H, Akce M, Kissick H, Carthon BC, Shaib WL, et al. : Sites of metastasis and association with clinical outcome in advanced stage cancer patients treated with immunotherapy. BMC Canc 2019, 10.1186/s12885-019-6073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, Rosenblum M, Harview CL, Taube JM, Handley N, et al. : Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res 2017, 10.1158/2326-6066.CIR-16-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee J, Mehdizadeh S, Tsai K, Algazi A, Rosenblum M, Daud A, Bluestone JA: Immunological insights into liver metastasis associated resistance to checkpoint blockade cancer immunotherapy. J Immunol May 1, 2018, 200(1 Supplement). 122.26. [Google Scholar]; This paper and the two above show how liver metastases display a weaker response to immunotherapy compared to metastases in other sites for patients affected by advanced melanoma, colorectal, or lung cancer.
- 63.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. : Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med 2018, 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu X, Zhang J, Wang J, Fu J, Li T, Zheng X, Wang B, Gu S, Jiang P, Fan J, et al. : Landscape of B cell immunity and related immune evasion in human cancers. Nat Genet 2019, 10.1038/s41588-018-0339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haigis KM, Cichowski K, Elledge SJ: Tissue-specificity in cancer: the rule, not the exception. Science 2019, 363: 1150–1152. 80-. [DOI] [PubMed] [Google Scholar]
- 66.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R: Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 67.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. : The KRASG12C inhibitor, MRTX849, provides insight toward therapeutic susceptibility of KRAS mutant cancers in mouse models and patients. Cancer Discov 2019, 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]