

Abstract
Dicarboxylic fatty acids, taken as a nutritional supplement or produced endogenously via omega oxidation of monocarboxylic fatty acids, may have therapeutic potential for rare inborn errors of metabolism as well as common metabolic diseases such as type 2 diabetes. Breakdown of dicarboxylic acids yields acetyl-CoA and succinyl-CoA as products, the latter of which is anaplerotic for the TCA cycle. However, little is known about the metabolic pathways responsible for degradation of dicarboxylic acids. Here, we demonstrated with whole-cell fatty acid oxidation assays that both mitochondria and peroxisomes contribute to dicarboxylic acid degradation. Several mitochondrial acyl-CoA dehydrogenases were tested for activity against dicarboxylyl-CoAs. Medium-chain acyl-CoA dehydrogenase (MCAD) exhibited activity with both six and 12 carbon dicarboxylyl-CoAs, and the capacity for dehydrogenation of these substrates was significantly reduced in MCAD knockout mouse liver. However, when dicarboxylic acids were fed to normal mice, the expression of MCAD did not change, while expression of peroxisomal fatty acid oxidation enzymes was greatly upregulated. In conclusion, mitochondrial fatty acid oxidation, and in particular MCAD, contributes to dicarboxylic acid degradation, but feeding dicarboxylic acids induces only the peroxisomal pathway.
Keywords: fatty acid oxidation, dicarboxylic fatty acids, mitochondria, peroxisomes, acyl-CoA dehydrogenase, acyl-CoA oxidase
1. Introduction
Fatty acid omega oxidation is a poorly understood metabolic pathway characterized by the conversion of monocarboxylic fatty acids into fatty acids bearing a carboxyl group on both ends, dubbed dicarboxylic fatty acids (DCAs). Omega oxidation begins with hydroxylation of fatty acids in the ER by cytochrome P450 enzymes [1]. Hydroxylated fatty acids are processed into DCAs in the cytosol by alcohol and aldehyde dehydrogenases, and then converted to dicarboxylyl-CoAs (DC-CoAs) by an as-yet-unidentified acyl-CoA synthase enzyme [2,3]. DC-CoAs are catabolized through the usual fatty acid β-oxidation (FAO) pathways, although it is not currently clear whether peroxisomal β-FAO, mitochondrial β-FAO, or both pathways participate in elimination of DC-CoAs. It is known, however, that accumulation of hepatic DCAs is toxic and leads to liver failure and death in animal models [4,5].
DCAs are more water-soluble than their monocarboxylic counterparts and are well-tolerated in humans when administered either enterally or parenterally [6,7]. While normally thought of as a liver-specific substrate, exogenous DCAs can be used for energy by several organs including muscle and heart [8]. Dodecanedioc acid, a 12-carbon DCA (DC12), may have anti-diabetic properties [7,9]. Infusing DC12 into type 2 diabetic patients decreases plasma glucose levels to normal without affecting insulin levels. Gluconeogenesis may also be suppressed by DC12 [7,10]. Further, DC12 may be beneficial for mitochondrial function via anaplerotic effects on the TCA cycle [11]. There is mounting evidence to support the concept that DC12 can be chain-shortened in both peroxisomes and mitochondria to succinate, a four carbon DCA that can directly feed into the TCA cycle [10,11,12].
While normally considered a minor pathway of fatty acid metabolism, hepatic omega oxidation is known to increase under conditions that cause fatty acids to accumulate in the liver including diabetes, starvation, genetic fatty acid oxidation disorders, chronic alcohol consumption, and consumption of medium-chain triglycerides (MCTs) [4,13,14,15,16,17,18]. It is not presently known what molecular factors regulate the partitioning of fatty acids into the omega oxidation pathway. Nor is it understood what role is played by peroxisomes versus mitochondria in the disposal of DCAs. Some studies have supported a dominant role for mitochondria, others for peroxisomes [19,20]. Patients with either mitochondrial or peroxisomal FAO deficiencies display increased urinary excretion of DCAs [21,22]. Stimulating omega oxidation and endogenous DCA formation may be an attractive strategy for preventing fatty acid overload in the liver, and exogenous DCAs may be useful for diabetes and the metabolic syndrome, but before these mechanisms can be leveraged it needs to be determined which organelles and enzymes are required for DCA catabolism. For example, in mitochondria there are multiple acyl-CoA dehydrogenase (ACAD) enzymes with overlapping substrate specificities. There are human diseases associated with each of these enzymes; knowing which ACAD(s) are involved in DCA elimination is of importance. The benefits of DCAs may be overshadowed by their severe liver toxicity in the event that disposal is impeded [4,5]. Here, we used whole-cell models, knockout mouse strains, and purified recombinant ACAD enzymes to study DCA catabolism.
2. Materials and Methods
2.1. Animal studies.
All breeding and experimental protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC). Long-chain acyl-CoA dehydrogenase (LCAD) and medium-chain acyl-CoA dehydrogenase (MCAD) knockout mice were obtained from the Mutant Mouse Regional Resource Center and bred in-house. Male LCAD−/−and MCAD−/− mice age 10–12 weeks were used for experiments, and they were not fasted or stressed prior to tissue collection for enzyme activity assays. For DC12 feeding, wild-type mice were maintained on either powdered standard diet (control) or powdered diet mixed with 10% w/w DC12. Euthanasia was conducted using inhaled CO2 gas according to IACUC recommendations.
2.2. Cell isolation and culture.
Wild-type mouse embryonic fibroblasts (MEFs) were a kind gift of Dr. Eric Verdin (Buck Institute). Primary cardiomyocytes were isolated from 3-month old male Sprague-Dawley rats as described previously described [23]. Briefly, rats were i.p. injected with heparin at 50 units/100 g body weight and sacrificed. The hearts were removed from animals and perfused with buffer containing 10 mM HEPES, 113 mM NaCl, 4.7 mM KCl, 0.7 mM NaH2PO4, 0.6 mM KH2PO4, 12 mM 10 mM KHCO3, NaHCO3, 1.2 mM MgSO4, 30 mM taurine, 10 mM 2,3-butanedione monoxime and 5.5 mM glucose for 10 min followed by enzyme solution (collagenase 1.4 mg/mL, trypsin 8 unit/mL, neutral protease 0.2 unit/mL and CaCl2 50 μM) for 45–60 min. The hearts were then minced and agitated to get a single cell suspension. Cells were pelleted by centrifugation, resuspended with DMEM containing 20% FBS and grown on laminin-coated dishes. Day 3 cardiomyocytes were used for the experiments.
2.3. Radiolabeled fatty acid oxidation assays.
14C-labeled palmitic acid (C16) was from PerkinElmer while 14C-DC12 was from Moravek, Inc. Both were bound to bovine serum albumin. Cultured cells were harvested and resuspended in DMEM containing 5 mM glucose, 200 μM carnitine, and 125 μM of labeled fatty acid. Cells were rotated in a 37°C water bath for 1 hr. Peroxisomal FAO was measured by inhibiting mitochondria with either 2 mM freshly prepared KCN or with 100 μM etomoxir. Reactions were stopped with perchloric acid, centrifuged to clear precipitated material, and then the water-soluble 14C-labeled FAO products were isolated by extraction with chloroform/methanol and counted. Concurrent reactions with no cells were used as blank controls.
2.4. Enzyme activity assays.
Acyl-CoA dehydrogenase (ACAD) activity assays were performed on either 150 ng of purified recombinant ACAD protein or 200 μg of total mouse liver protein, with 25 μM of acyl-CoA substrate and 2 μM recombinant porcine ETF as the electron acceptor, as described [24]. C8, C12, and C16 acyl-CoAs were from Sigma (St. Louis, MO); dodecanedioyl-CoA (DC12-CoA) and 2,6-dimethylheptanoyl-CoA (2,6-C7-CoA) were from Toronto Research Chemicals, (Toronto, ON); adipoyl-CoA (DC6-CoA) was from CoALA Biosciences (Austin, TX). Reduction of ETF fluorescence was followed for 1 minute and used to calculate specific activity normalized to protein concentration. Acyl-CoA oxidase-1 (ACOX1) activity was assayed with 25 μM of acyl-CoA substrate and following H2O2 production with Amplex Red as described [25]. All recombinant enzymes were expressed and purified from E. coli as described [24,25].
2.5. Immunoblotting.
Liver homogenates were prepared from mice fed control diet or diet plus 10% w/w DC12 for seven days. Antibodies used for immunoblotting were: anti-MCAD and anti-LCAD (1:1000, gifts of Dr. Jerry Vockley); anti-ACOX1 (1:1000, Invitrogen); anti-L-peroxisomal bifunctional protein (gene name EHHADH, 1:3000, Abcam); anti-D-peroxisomal bifunctional protein (gene name HSD17B4, 1:500, Proteintech); anti-Cyp4a10 (1:1000, Invitrogen); anti-MTCO1 subunit of respiratory chain Complex IV (1:1000, Mitosciences); anti-succinate dehydrogenase subunit B of Complex II (1:1000, Mitosciences); and anti-β-actin (1:10,000, Proteintech). After incubation with HRP-conjugated secondary antibodies blots were visualized with chemiluminescence.
3. Results
3.1. Mitochondria contribute to the metabolism of dicarboxylic fatty acids (DCAs).
The capacity of mitochondria for metabolizing long-chain DCAs was interrogated by following the catabolism of 14C-labeled dodecanedioic acid (DC12) in the presence/absence of the irreversible mitochondrial FAO inhibitor etomoxir. In initial experiments 14C-DC12 was incubated with freshly prepared mouse liver homogenates, but no catabolism was observed (data not shown). We reasoned that breaking the cells may have compromised the as-yet-unidentified acyl-CoA synthase responsible for activating DC12 to DC12-CoA, which is thought to be localized to the endoplasmic reticulum and is known to be labile [3]. When 14C-DC12 was applied to intact wild-type mouse embryonic fibroblasts (MEFs), degradation to 14C-labeled FAO products was observed, and the rate of degradation was inhibited by more than 50% in the presence of etomoxir (Fig 1A). This suggested that mitochondrial FAO contributes to DC12 oxidation. Similar results were seen in primary mouse cardiomyocytes, where etomoxir inhibited 14C-DC12 oxidation by one-third (Fig 1B). As a control experiment, we tested the effects of etomoxir on the long-chain monocarboxylic fatty acid palmitate. Etomoxir inhibited palmitate oxidation by more than 90% as expected, confirming specificity of the inhibitor (Fig 1C). Finally, a second mitochondrial inhibitor, potassium cyanide (KCN), also inhibited cardiomyocyte 14C-DC12 oxidation by about 25% (Fig 1D). Together, these data indicate that mitochondria contribute to the cellular capacity for DC12 oxidation.
Figure 1. Mitochondria contribute to the metabolism of dicarboxylic fatty acids (DCAs).

Cultured wild-type mouse embryonic fibroblasts (MEFs, panel A) or primary mouse cardiomyocytes (B-D) were subjected to whole-cell fatty acid oxidation (FAO) assays. A, B) The mitochondrial FAO inhibitor etomoxir partially inhibits degradation of 14C-labeled dodecanedioc acid (DC12) in both MEFs and cardiomyocytes, indicating both peroxisomal and mitochondrial contribution. C) By comparison, etomoxir almost completely inhibits FAO of 14C-palmitate. D) Another mitochondrial inhibitor, KCN, gives similar results to etomoxir with 14C-DC12. *P<0.01 versus control.
3.2. Medium-chain acyl-CoA dehydrogenase (MCAD) uses DC12-CoA as substrate.
The first reaction in mitochondrial β-oxidation is catalyzed by the acyl-CoA dehydrogenase (ACAD) enzyme family. There are three ACAD enzymes with partially overlapping substrate specificities that were previously show to exhibit at least some activity against 12-carbon monocarboxylyl-CoA—medium-chain acyl-CoA dehydrogenase (MCAD), long-chain acyl-CoA dehydrogenase (LCAD), and very long-chain acyl-CoA dehydrogenase (VLCAD) [26]. To determine which of these three enzymes is active against 12-carbon dicarboxylyl-CoA, we tested the enzyme activities of human recombinant MCAD, LCAD, and VLCAD with DC12-CoA. We compared activity measured with DC12-CoA to that measured with each enzyme’s preferred substrate using the electron transferring-flavoprotein (ETF) fluorescence reduction assay [24]. MCAD was 28% active with DC12-CoA compared to its preferred substrate C8-CoA (Fig 2A), while LCAD and VLCAD were 72% and 4% active with DC12-CoA, respectively, compared to their preferred substrates (Fig 2B,C). To determine whether MCAD, LCAD, or both enzymes contribute to DC12 metabolism in vivo, we tested liver homogenates from wild-type, MCAD−/−, and LCAD−/− mice for ACAD activity using DC12-CoA. In MCAD−/− liver homogenates, but not LCAD−/− liver homogenates, the activity with DC12-CoA was significantly reduced (Fig 2D), indicating that MCAD contributes to the metabolism of DC12-CoA in liver mitochondria.
Figure 2. Medium-chain acyl-CoA dehydrogenase (MCAD) uses DC12-CoA as substrate.

Recombinant human MCAD (panel A), long-chain acyl-CoA dehydrogenase (LCAD, panel B), and very long-chain acyl-CoA dehydrogenase (VLCAD, panel C) were tested for enzymatic activity with dodecanedioyl-CoA (DC12-CoA) versus their preferred substrates (C8-CoA, 2,6-C7-CoA, and C16-CoA, respectively. Activities with DC12-CoA are expressed as a percentage of the preferred substrate. D) Liver homogenates from N=3 wild-type, MCAD−/−, and LCAD−/− mice were tested for acyl-CoA dehydrogenase activity with DC12-CoA. *P<0.01 versus wild-type.
3.3. Catabolism of DC6-CoA to succinyl-CoA.
Adipic acid, a 6-carbon DCA (DC6), is the hallmark species of dicarboxylic aciduria. The prevalence of DC6 in urine suggests that chain-shortening does not proceed from DC6-CoA to DC4-CoA (succinyl-CoA). Other studies indicate that succinyl-CoA is indeed formed in both peroxisomes and mitochondria during DCA catabolism [11,12]. Here, we investigated the ability of the rate-limiting enzymes ACOX1 (peroxisomes) and the ACADs (mitochondria) to dehydrogenate DC6-CoA, which is the first step in chain-shortening DC6-CoA to succinyl-CoA. ACOX1 is active against a broad range of acyl-CoA substrates but has highest activity with monocarboxylyl C12-CoA. Recombinant human ACOX1 was about half as active with DC12-CoA as it was with C12-CoA, but only 2% as active with DC6-CoA compared to C12-CoA (Fig 3A). These data suggest that peroxisomal formation of anaplerotic succinyl-CoA from DC6-CoA can occur, but most likely at a slow rate.
Figure 3. Catabolism of DC6-CoA to succinyl-CoA.

A) Recombinant human acyl-CoA oxidase-1 (ACOX1) was tested for enzymatic activity with its preferred substrate (C12-CoA) and the dicarboxylyl-CoAs dodecanedioyl-CoA (DC12-CoA) and adipoyl-CoA (DC6-CoA). B) Wild type mouse liver homogenates (N=3) assessed for mitochondrial acyl-CoA dehydrogenase activity with DC6-CoA versus DC12-CoA. C) Recombinant MCAD activity with DC6-CoA versus its preferred substrate C8-CoA. D) Liver homogenates from N=3 wild-type and MCAD−/− mice were tested for acyl-CoA dehydrogenase activity with DC6-CoA. *P<0.01 for indicated pairwise comparisons.
Multiple mitochondrial ACADs may be active against DC6-CoA. We began by interrogating total ACAD activity in wild-type mouse liver homogenates with DC6-CoA as compared to DC12-CoA. Activity against DC6-CoA was about 40% of that measured with DC12- CoA (Fig 3B). We next tested several recombinant ACADs for activity with DC6-CoA. LCAD has no measurable activity with DC6-CoA while short-chain acyl-CoA dehydrogenase (SCAD) and isovaleryl-CoA dehydrogenase (IVD) exhibited measurable, but near-zero, activities (data not shown). MCAD activity with DC6-CoA was 50-fold higher than that exhibited by SCAD or IVD, but this activity was still only 2.5% of the activity measured with MCAD’s preferred substrate C8-CoA (Fig 3C). Total ACAD activity against DC6-CoA was reduced by half in MCAD−/− mouse liver homogenates compared to wild-type homogenates (Fig 3D). These data show that mitochondria can also chain-shorten DCAs to succinyl-CoA at a slow rate, and that, in addition to its role in DC12 metabolism, MCAD contributes to DC6-CoA chain-shortening.
3.4. Feeding DC12 induces expression of peroxisomal, but not mitochondrial, FAO enzymes.
DC12 feeding has been reported to activate the transcription factor peroxisome proliferator activated receptor-α (PPARα), a master regulator of FAO gene expression, and increase mRNAs in mouse liver for multiple PPARα target genes including MCAD and LCAD [5]. To determine if this upregulation also occurs at the protein level, we fed wild-type mice a diet containing 10% w/w DC12 for one week. In contrast to what was reported at the mRNA level after 1-day of DC12 [5], after seven days there was no change in the protein levels of either MCAD or LCAD (Fig 4A). Two mitochondrial respiratory chain subunits, blotted as non-FAO mitochondrial control proteins, also showed no change. Western-blotting did, however, corroborate the previously reported induction of Cyp4a10, ACOX1, and the two peroxisomal bifunctional enzymes (L-PBE, D-PBE) downstream of ACOX1 (Fig 4B).
Figure 4. Feeding DC12 induces expression of peroxisomal, but not mitochondrial, FAO enzymes.
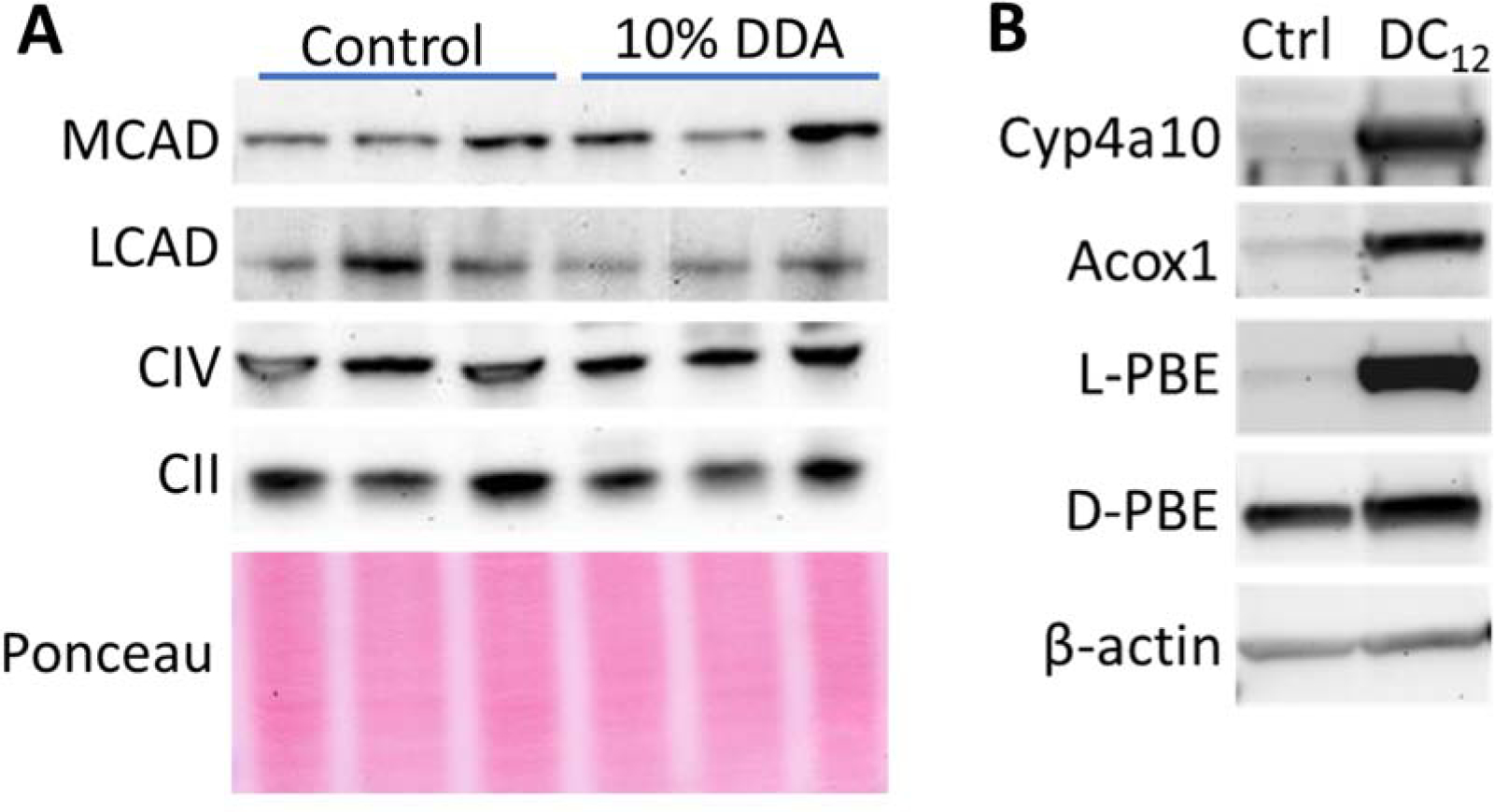
A) Immunoblotting of N=3 mouse liver lysates from mice fed normal diet versus normal diet plus 10% w/w dodecanedioic acid (DC12) for seven days. No change in expression was observed for the shown mitochondrial proteins. B) Representative blots confirm previously published finding of induced peroxisomal enzyme expression with DC12 feeding.
4. Discussion
Mitochondria have previously been implicated in the metabolism of DCAs [19]. Here, using whole cells we established that between 25–50% of DC12 can be metabolized through mitochondria, depending upon the cell type. Further, we have identified MCAD as a specific mitochondrial FAO enzyme involved in chain-shortening DCAs. Excretion of high levels of DC6 and DC8 in the urine has long been recognized as a feature of human MCAD deficiency [22]. It is possible that some portion of these accumulated medium-chain DCAs are due to reduced mitochondrial capacity for DCA catabolism in the absence of MCAD. However, given the high specificity of the omega oxidation system for C10 and C12 fatty acids [27], it is also possible that accumulating monocarboxylic C10 and C12 in the absence of MCAD drives omega oxidation and subsequent peroxisomal chain-shortening to DC6 and DC8, which are then excreted into the urine. Future work is needed to discriminate between these mechanisms of DCA excretion into the urine of MCAD patients.
Our observation that MCAD−/− mouse liver homogenates retain ~50% ACAD activity towards both DC12-CoA and DC6-CoA indicates that other ACAD enzymes are likely to play a role in DCA catabolism in vivo. For DC12-CoA, a portion of the non-MCAD activity is likely attributable to LCAD, which displayed activity with this substrate as a recombinant protein (Fig 2). For DC6-CoA, LCAD exhibited no activity. One ACAD which was not evaluated in the present studies, and which we speculate contributes to DCA metabolism, is glutaryl-CoA dehydrogenase (GCDH). GCDH decarboxylates the five-carbon DCA glutaryl-CoA, an intermediate in lysine degradation. While to our knowledge it has not been tested for activity with even-chain DCAs, it does have activity against even-chain monocarboxylic substrates 6 to 10 carbons in length [28]. Finally, GCDH patients excrete DC6 and DC8 in their urine [29]. Together these findings point to GCDH as the likely ACAD that supports MCAD in mitochondrial DCA metabolism. Future studies will express and purify recombinant GCDH in order to test this hypothesis.
Our work here supports previous studies that suggest that both mitochondria and peroxisomes can chain-shorten DCAs directly to succinyl-CoA [11,12,19]. Peroxisomal succinyl-CoA is released from peroxisomes as succinate, which can transfer into mitochondria [11]. Therefore, DCAs such as DC12 may prove to be useful anaplerotic therapies for mitochondrial long-chain FAO disorders and other inborn errors of metabolism. The current nutritional therapy for long-chain FAO disorders is triheptanoin, a synthetic triglyceride containing the odd-chain fatty acid C7. Two cycles of β-oxidation shortens C7 to C3, which is converted in two enzymatic steps to anaplerotic succinyl-CoA [30]. However, oral dosing with triheptanoin fails to increase serum C7 levels due to a strong first-pass liver effect, thereby limiting its effects in the periphery [31]. In contrast, oral DC12 rapidly increases DC12 levels in the blood [32]. This suggests that oral DC12 could potentially provide energy to the heart and muscles, which are often severely affected in mitochondrial long-chain FAO disorders. Further work is underway to evaluate the possible benefits of DCA therapy.
Highlights.
Both mitochondria and peroxisomes chain-shorten dicarboxylic fatty acids (DCAs).
Medium-chain acyl-CoA dehydrogenase is active against dicarboxylyl-CoAs.
Feeding DCAs induces liver expression of peroxisomal, but mitochondrial, enzymes.
Funding
This work was funded by the National Institute of Health grants DK090242 (ESG) and P30DK120531 (Pittsburgh Liver Research Center).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Literature Cited
- [1].Kim D, Cha GS, Nagy LD, Yun CH, Guengerich FP, Kinetic analysis of lauric acid hydroxylation by human cytochrome P450 4A11, Biochemistry 53 (2014) 6161–6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wanders RJ, Komen J, Kemp S, Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans, FEBS J 278 (2011) 182–194. [DOI] [PubMed] [Google Scholar]
- [3].Vamecq J, de Hoffmann E, Van Hoof F, The microsomal dicarboxylyl-CoA synthetase, Biochem J 230 (1985) 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mortensen PB, Gregersen N, The biological origin of ketotic dicarboxylic aciduria. II. In vivo and in vitro investigations of the beta-oxidation of C8-C16-dicarboxylic acids in unstarved, starved and diabetic rats, Biochim Biophys Acta 710 (1982) 477–484. [DOI] [PubMed] [Google Scholar]
- [5].Ding J, Loizides-Mangold U, Rando G, Zoete V, Michielin O, Reddy JK, Wahli W, Riezman H, Thorens B, The peroxisomal enzyme L-PBE is required to prevent the dietary toxicity of medium-chain fatty acids, Cell Rep 5 (2013) 248–258. [DOI] [PubMed] [Google Scholar]
- [6].Grego AV, Mingrone G, Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: an update, Clin Nutr 14 (1995) 143–148. [DOI] [PubMed] [Google Scholar]
- [7].Mingrone G, Castagneto-Gissey L, Mace K, Use of dicarboxylic acids in type 2 diabetes, Br J Clin Pharmacol 75 (2013) 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pourfarzam M, Bartlett K, Skeletal muscle mitochondrial beta-oxidation of dicarboxylates, Biochim Biophys Acta 1141 (1993) 81–89. [DOI] [PubMed] [Google Scholar]
- [9].Salinari S, Bertuzzi A, Gandolfi A, Greco AV, Scarfone A, Manco M, Mingrone G, Dodecanedioic acid overcomes metabolic inflexibility in type 2 diabetic subjects, Am J Physiol Endocrinol Metab 291 (2006) E1051–1058. [DOI] [PubMed] [Google Scholar]
- [10].Mortensen PB, The possible antiketogenic and gluconeogenic effect of the omega-oxidation of fatty acids in rats, Biochim Biophys Acta 620 (1980) 177–185. [DOI] [PubMed] [Google Scholar]
- [11].Jin Z, Bian F, Tomcik K, Kelleher JK, Zhang GF, Brunengraber H, Compartmentation of Metabolism of the C12-, C9-, and C5-n-dicarboxylates in Rat Liver, Investigated by Mass Isotopomer Analysis: ANAPLEROSIS FROM DODECANEDIOATE, J Biol Chem 290 (2015) 18671–18677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Westin MA, Hunt MC, Alexson SE, The identification of a succinyl-CoA thioesterase suggests a novel pathway for succinate production in peroxisomes, J Biol Chem 280 (2005) 38125–38132. [DOI] [PubMed] [Google Scholar]
- [13].Orellana M, Rodrigo R, Valdes E, Peroxisomal and microsomal fatty acid oxidation in liver of rats after chronic ethanol consumption, Gen Pharmacol 31 (1998) 817–820. [DOI] [PubMed] [Google Scholar]
- [14].Orellana M, Valdes E, Del Villar E, Microsomal and peroxisomal fatty acid oxidation in streptozotocin diabetic rat liver, Gen Pharmacol 28 (1997) 361–364. [DOI] [PubMed] [Google Scholar]
- [15].Orellana M, Fuentes O, Rosenbluth H, Lara M, Valdes E, Modulation of rat liver peroxisomal and microsomal fatty acid oxidation by starvation, FEBS Lett 310 (1992) 193–196. [DOI] [PubMed] [Google Scholar]
- [16].Gregersen N, Mortensen PB, Kolvraa S, On the biologic origin of C6-C10-dicarboxylic and C6-C10-omega-1-hydroxy monocarboxylic acids in human and rat with acyl-CoA dehydrogenation deficiencies: in vitro studies on the omega- and omega-1-oxidation of medium-chain (C6-C12) fatty acids in human and rat liver, Pediatr Res 17 (1983) 828–834. [DOI] [PubMed] [Google Scholar]
- [17].Gregersen N, Ingerslev J, The excretion of C6-C10-dicarboxylic acids in the urine of newborn infants during starvation. Evidence for omega-oxidation of fatty acids in the newborn, Acta Paediatr Scand 68 (1979) 677–681. [DOI] [PubMed] [Google Scholar]
- [18].Tserng KY, Griffin RL, Kerr DS, Distinction of dicarboxylic aciduria due to medium-chain triglyceride feeding from that due to abnormal fatty acid oxidation and fasting in children, Metabolism 45 (1996) 162–167. [DOI] [PubMed] [Google Scholar]
- [19].Kolvraa S, Gregersen N, In vitro studies on the oxidation of medium-chain dicarboxylic acids in rat liver, Biochim Biophys Acta 876 (1986) 515–525. [DOI] [PubMed] [Google Scholar]
- [20].Suzuki H, Yamada J, Watanabe T, Suga T, Compartmentation of dicarboxylic acid beta-oxidation in rat liver: importance of peroxisomes in the metabolism of dicarboxylic acids, Biochim Biophys Acta 990 (1989) 25–30. [DOI] [PubMed] [Google Scholar]
- [21].Rocchiccioli F, Aubourg P, Bougneres PF, Medium- and long-chain dicarboxylic aciduria in patients with Zellweger syndrome and neonatal adrenoleukodystrophy, Pediatr Res 20 (1986) 62–66. [DOI] [PubMed] [Google Scholar]
- [22].Kolvraa S, Gregersen N, Christensen E, Hobolth N, In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria: evidence for a defect in general acyl-CoA dehydrogenase, Clin Chim Acta 126 (1982) 53–67. [DOI] [PubMed] [Google Scholar]
- [23].Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, Kuliawat R, Liu H, Kuhn B, Cuervo AM, Muzumdar R, Humanin is an endogenous activator of chaperone-mediated autophagy, J Cell Biol 217 (2018) 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Mohsen AW, Kochersperger C, Solo K, Schmidt AV, Vockley J, Goetzman ES, An acyl-CoA dehydrogenase microplate activity assay using recombinant porcine electron transfer flavoprotein, Anal Biochem 581 (2019) 113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang Y, Bharathi SS, Beck ME, Goetzman ES, The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide, Redox Biol 26 (2019) 101253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wanders RJ, Ruiter JP, I.J. L, Waterham HR, Houten SM, The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results, J Inherit Metab Dis 33 (2010) 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mortensen PB, Gregersen N, The biological origin of ketotic dicarboxylic aciduria. In vivo and in vitro investigations of the omega-oxidation of C6-C16-monocarboxylic acids in unstarved, starved and diabetic rats, Biochim Biophys Acta 666 (1981) 394–404. [DOI] [PubMed] [Google Scholar]
- [28].Wu L, Qiao Y, Gao J, Deng G, Yu W, Chen G, Li D, Functional characterization of rat glutaryl-CoA dehydrogenase and its comparison with straight-chain acyl-CoA dehydrogenase, Bioorg Med Chem Lett 21 (2011) 6667–6673. [DOI] [PubMed] [Google Scholar]
- [29].Gregersen N, Brandt NJ, Ketotic episodes in glutaryl-CoA dehydrogenase deficiency (glutaric aciduria), Pediatr Res 13 (1979) 977–981. [DOI] [PubMed] [Google Scholar]
- [30].Gillingham MB, Heitner SB, Martin J, Rose S, Goldstein A, El-Gharbawy AH, Deward S, Lasarev MR, Pollaro J, DeLany JP, Burchill LJ, Goodpaster B, Shoemaker J, Matern D, Harding CO, Vockley J, Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial, J Inherit Metab Dis 40 (2017) 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kinman RP, Kasumov T, Jobbins KA, Thomas KR, Adams JE, Brunengraber LN, Kutz G, Brewer WU, Roe CR, Brunengraber H, Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats, Am J Physiol Endocrinol Metab 291 (2006) E860–866. [DOI] [PubMed] [Google Scholar]
- [32].Mingrone G, De Gaetano A, Greco AV, Capristo E, Benedetti G, Castagneto M, Gasbarrini G, Comparison between dodecanedioic acid and long-chain triglycerides as an energy source in liquid formula diets, JPEN J Parenter Enteral Nutr 23 (1999) 80–84. [DOI] [PubMed] [Google Scholar]