

Abstract
Leigh syndrome, or infantile necrotizing subacute encephalopathy (OMIM #256000), is one of the most common manifestations of mitochondrial dysfunction, due to mutations in more than 75 genes, with mutations in respiratory complex I subunits being the most common cause. In the present study, we used the recently described PHP.B serotype, characterized by efficient capacity to cross the blood-brain barrier, to express the hNDUFS4 gene in the Ndufs4−/− mouse model of Leigh disease. A single intravenous injection of PHP.B-hNDUFS4 in adult Ndufs4−/− mice led to a normalization of the body weight, marked amelioration of the rotarod performance, delayed onset of neurodegeneration, and prolongation of the lifespan up to 1 year of age. hNDUFS4 protein was expressed in virtually all brain regions, leading to a partial recovery of complex I activity. Our findings strongly support the feasibility and effectiveness of adeno-associated viral vector (AAV)-mediated gene therapy for mitochondrial disease, particularly with new serotypes showing increased permeability to the blood-brain barrier in order to achieve widespread expression in the central nervous system.
Keywords: mitochondrial diseases, gene therapy, AAV, Ndufs4, complex I, OXPHOS, PHP.B, Leigh syndrome
Graphical Abstract
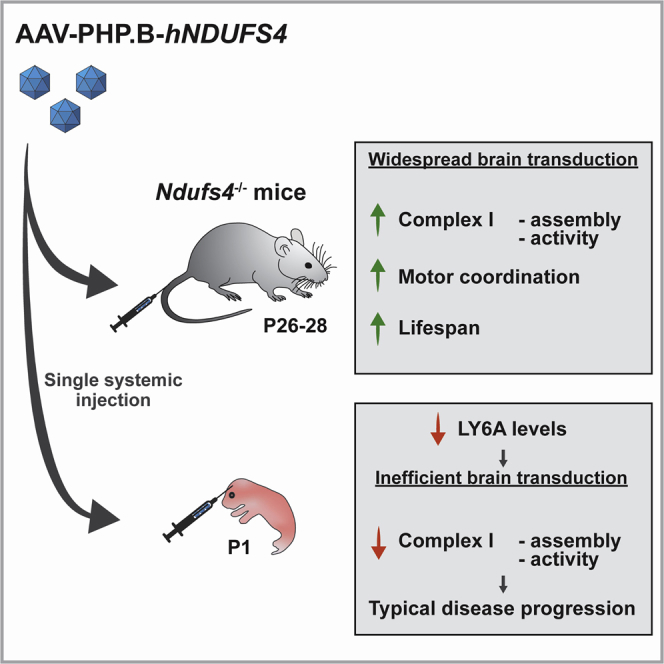
Systemically injected AAV-PHP.B efficiently crosses the blood-brain barrier in mice. In this study, a PHP.B-hNDUFS4 vector was used in both adult and newborn Ndufs4−/− mice, a model of mitochondrial neurodegeneration. Amelioration of the phenotype was observed in adults, but not newborns, due to low expression of the viral receptor in pups. Keywords:mitochondrial diseases, gene therapy, AAV, Ndufs4, complex I, OXPHOS, PHP.B, Leigh syndrome
Introduction
Adeno-associated viral vector (AAV)-based gene therapy has become, in recent years, a reality for the treatment of several human conditions, and in particular for genetic diseases. Several studies used AAVs in preclinical models of mitochondrial disorders either to re-express the missing/mutated gene1, 2, 3, 4, 5, 6, 7, 8, 9 or to express nucleases able to selectively shift mtDNA heteroplasmy.10, 11, 12, 13, 14, 15 These studies highlight the potential of AAV-based gene targeting to achieve radical, curative therapy of mitochondrial disorders, which are usually highly disabling, complex conditions characterized by multisystem dysfunction, with a prevalence of neurological impairment. Targeting the brain, however, is still very challenging because of the presence of the blood-brain barrier (BBB). The BBB is formed by continuous non-fenestrated endothelial cells, the presence of astrocyte endfeet ensheathing the capillary, and pericytes embedded in the capillary basement membrane. This system vascularizes the central nervous system (CNS) and tightly regulates the movement of ions, molecules, and cells between the blood and the brain. The BBB allows the passage of some molecules by passive diffusion, as well as the selective transport of molecules such as glucose, water, and amino acids that are crucial to neural function. However, the BBB restricts the passage of pathogens, but also the diffusion of many solutes in the blood, and large or hydrophilic molecules into the cerebrospinal fluid (CSF), while it allows the diffusion of hydrophobic molecules (O2, CO2, hormones) and small polar molecules. Cells of the BBB actively transport metabolic products such as glucose across the barrier using specific transport proteins.33 Several routes have been exploited in order to bypass the BBB and deliver AAVs to the brain in both preclinical models and humans.16 Local administration has been used to deliver therapeutic genes to peripheral sensory organs, while the intrathecal route can target specific areas of the brain parenchyma. These approaches have the advantage of reducing the amount of virus required, as well as the risk of ectopic expression, but they have limited applicability in the case of cell-autonomous defects, where extensive correction in a large number of cells is needed. In these cases, delivery via the CSF by intracerebroventricular or intracisternal injection has been exploited. Finally, a nasal route has been shown to deliver therapeutic genes to different areas of the brain, and intramuscular injections have shown efficacy to target spinal motor neurons via retrograde axonal transport. Although the above approaches have been used in several preclinical, and now also clinical, settings, the development of AAV serotypes with improved permeability to the BBB is essential to deliver the therapeutic gene by systemic injection, thus overcoming the need for invasive surgical techniques and increased target diffusion, one major limitation of the current approaches.
Recently, an engineered AAV9, called AAV-PHP.B, has demonstrated a remarkable capacity to cross the BBB when intravenously (i.v.) injected in adult C57BL/6 mice, and to extensively target both neurons and glial cells.17 This serotype was obtained by screening a library of AAV variants subject to selective pressure for increased capacity to cross the BBB. This exceptional property was subsequently found to be mediated by a membrane glycoprotein of the brain endothelia (LY6A-GPI), which is spontaneously mutated in some mouse strains and is not present in primates.18, 19, 20, 21 As a consequence, AAV-PHP.B does not cross the BBB when i.v. injected in these “resistant” mouse strains and in primates as well (including, presumably, humans). However, AAV-PHP.B seems to be as efficient as AAV9, the current serotype of choice for therapy of neurological disorders, to effectively target cortical and spinal neurons by the intrathecal route.22
In spite of the detailed mechanistic information that elucidates the unique BBB permeability of AAV-PHP.B, there is a limited number of reports exploring the use of AAV-PHP.B in disease models.21,23,24
Leigh syndrome (LS), or infantile necrotizing subacute encephalomyopathy (OMIM #256000), is one of the most common manifestations of mitochondrial dysfunction. More than 75 loci have been linked to LS, with respiratory complex I defects being the most common cause.25 No treatment in addition to supportive care to relieve symptoms is available for LS. In this study, we explored the applicability of widespread targeting of the brain via i.v. recombinant AAV-PHP.B for the therapy of brain impairment in LS by taking advantage of the Ndufs4−/− mouse model, a widely acknowledged model of LS.
Results
A Single i.v. Injection of AAV-PHP.B-hNDUFS4 Ameliorates the Clinical Phenotype of Ndufs4−/− Mice
We injected two cohorts of Ndufs4−/− mice between postnatal day 26 (P26) and P28 with 1012 or 2 × 1012 viral genomes (vg). Since no difference was observed in any of the analyzed parameters (not shown), we pooled the data from the two concentrations. We culled one group of animals (n = 7) 2 months after the injection and kept the rest (n = 22) alive to build a survival curve. At the age of injection, the Ndufs4−/− mice were slightly but significantly smaller than the wild-type (WT) littermates (Figure 1A), with hardly any neurological symptom, although they already showed the reversible fur shedding typical of the disease.26 Ten days after injection of AAV-PHP.B-hNDUFS4, treated Ndufs4−/− mice started to gain weight and became virtually indistinguishable from the WT littermates, while the untreated Ndufs4−/− littermates started to lose weight and eventually died between 45 and 60 days after birth. The motor coordination of Ndufs4−/− mice, measured by a standard accelerating rotarod test, was improved in AAV-PHP.B-hNDUFS4-treated versus untreated mice Ndufs4−/− mice (Figure 1B). At seven weeks of age, untreated mice could barely stand on the rotating bar, while treated animals could stay on the bar for up to 150 s. Although this value was still significantly lower than that of the WT animals (220 ± 15 s, p < 0.01), the difference with untreated affected littermates was highly significant (p < 0.0001). The rotarod performance of treated animals was similar at 12 versus 7 weeks. No untreated animal survived beyond 7–8 weeks of age.
Figure 1.

AAV-PHP.B-hNDUFS4 Improves the Clinical Phenotype of Ndufs4−/− Mice
(A) Body weights during the first 90 days of animals of the three genotypes. Note the recovery up to normal levels of Ndufs4−/− mice about 10 days after the injection. (B) Rotarod performance during an accelerated protocol. At 12 weeks no Ndufs4−/− mice were alive. The bars indicate mean ± SEM (n = 6/group). ∗∗p < 0.01, ∗∗∗∗p < 0.0001, calculated by two-way ANOVA. 7w (7 weeks) and 12w (12 weeks) represent the ages of the animals. (C) Determination of the AAV-PHP.B viral genomes in Ndufs4−/− tissues. (D) Western blot analysis of hNDUFS4 in the brain.
AAV-PHP.B-Mediated Expression of Therapeutic hNDUFS4 Increases Complex I Activity in the Tissues of Ndufs4−/− Mice
We sacrificed the first cohort of AAV-PHP.B-hNDUFS4-treated Ndufs4−/− mice at 3 months of age (2 months post-injection) and analyzed the viral vector distribution in different tissues. Approximately 10 vg/diploid genome (dg) were detected in brain and liver, 2–3 vg/dg in heart, and 0.3 vg/dg in skeletal muscle (Figure 1C). Since the phenotype of Ndufs4−/− mice is mainly related to the absence of the protein in the brain,27 we next investigated in greater detail the effects of hNDUFS4 expression in the brain of AAV-PHP.B-injected animals. These results are in line with published data for the AAV-PHP.B serotype.17 Anti-hNDUFS4 cross-reacting material was detected by western blot immunovisualization in brain samples (Figure 1D). However, the amount of NDUFS4 protein was lower than in the WT untreated brains.
Immunohistochemistry using an anti-NDUFS4 antibody showed positive areas and cells in all of the brain regions, including areas particularly relevant for the Ndufs4−/− pathology such as the olfactory bulb (OB) (Figure 2A) and vestibular nuclei (VN) (Figure 3A).27 Importantly, no anti-NDUFS4-positive staining was present in Ndufs4−/− mice. Astrogliosis and microgliosis were observed in the same areas by anti-GFAP and anti-CD68 immunostaining, respectively (Figures 2B, 2C, 3B, and 3C). Scattered CD68-positive cells were also detected in the cerebellar posterior lobule (Figure S1). H&E staining showed extensive areas of spongiform neurodegeneration in the OB (Figure 2D) and VN (Figure 3D) of untreated Ndufs4−/− mice. Massive neurodegeneration was detected in the OB, but not VN, by PathoGreen, which selectively stains degenerating neurons (Figures 2E and 3E). These alterations were partially prevented by, but still obviously present in, AAV-PHP.B-hNDUFS4 OB (Figure 2), whereas a trend to a decrease of PathoGreen-positive cells was observed in the VN (where the PathoGreen staining was altogether much milder than in the OB also in untreated animals). Evident astrogliosis was present in both the OB and VN of treated animals. In addition, the hippocampus, which did not show obvious alterations in untreated Ndufs4−/− mice, showed a number of PathoGreen-positive neurons in some treated animals (Figure S2). It should be noted, however, that untreated animals were sacrificed at 7 weeks of age, while treated animals were culled at 12 weeks. Therefore, progression of neurodegeneration may explain the presence of PathoGreen-positive hippocampal neurons in treated versus untreated Ndufs4−/− individuals.
Figure 2.

Histological and Immunohistochemical Analysis of the Olfactory Bulb Sacrificed at 100 Days
(A) Immunohistochemical analysis using an anti-NDUFS4 antibody. Scale bars, 10 μm. (B) Analysis of astrogliosis by immunodecoration with an anti-GFAP antibody. Scale bars, 10 μm. (C) Analysis of microgliosis by immunodecoration with an anti CD68 antibody. Scale bars, 10 μm. (D) H&E staining. Black arrows indicate the areas of vacuolization; red arrows indicate degenerating neurons. Scale bars, 10 μm. (E) PathoGreen staining. Note the massive staining in untreated Ndufs4−/− animals, which was partially rescued by AAV-PHP.B-hNDUFS4 treatment. Scale bars, 20 μm.
Figure 3.

Histological and Immunohistochemical Analysis of the Vestibular Nucleus Sacrificed at 100 Days
(A) Immunohistochemical analysis using an anti-NDUFS4 antibody. Scale bars, 50 μm. (B) Analysis of astrogliosis by immunodecoration with an anti-GFAP antibody. Scale bars, 10 μm. (C) Analysis of microgliosis by immunodecoration with an anti-CD68 antibody. Scale bars, 10 μm. (D) H&E staining. Note the presence of spongiform vacuolization in VN of untreated Ndufs4−/− animals, which is absent in AAV-PHP.B-hNDUFS4-treated animals. Scale bars, 50 μm. (E) PathoGreen staining. No positive cells are detected in the VN of either treated or untreated mice at this stage. Scale bars, 20 μm.
We then measured complex I activity in brain homogenates of untreated and AAV-PHP.B-injected mice (Figure 4A). Rotenone-sensitive NADH (reduced nicotinamide adenine dinucleotide)-CoQ (coenzyme Q) reductase activity was profoundly decreased in Ndufs4−/− mice (16.3% ± 3.86% compared to WT; p < 0.0001). In contrast, complex I activity was 63.5% ± 11.26% of the WT (p < 0.05) in AAV-PHP.B-injected animals.
Figure 4.

Analysis of Complex I
(A) Spectrophotometric activity of complex I (CI) normalized to that of citrate synthase (CS) in brain homogenates of the different genotypes (n = 4 for WT and Ndufs4−/− mice; n = 7 for AAV-PHP.B-hNDUFS4-treated mice). ∗p < 0.05, ∗∗∗p < 0.001, by one-way ANOVA. (B) In-gel activity for complex I. Note the complete absence of the complex I band in Ndufs4−/− samples. (C) Western blot analysis of first-dimension BNGE with an antibody specific for complex I (NDUFA9). Note that only the 830-kDa subassembly band is present in Ndufs4−/− samples, while both fully assembled and subassembly bands are present after treatment with AAV-PHP.B-hNDUFS4. An antibody specific for complex II was used as control (SDHB). SC, supercomplexes.
Finally, blue native gel electrophoresis and in-gel activity on isolated brain mitochondria revealed that the activity associated with fully assembled complex I, which was completely absent in naive Ndufs4−/− mice, was restored in Ndufs4−/− mice, although it was lower than in WT mitochondria (Figure 4B). Accordingly, western blot immunovisualization revealed robust recovery of fully assembled complex I, although there was persistency of the N-module-less 830-kDa subassembly, typically observed in Ndufs4-lacking complex I (Figure 4C).
AAV-PHP.B-hNDUFS4 Prolongs the Lifespan of Ndufs4−/− Mice
We then analyzed the survival probability of AAV-PHP.B-hNDUFS4-treated versus untreated Ndufs4−/− mice by Kaplan-Meier distribution and log rank analysis in the second cohort of animals. The median lifespan of the treated animals was significantly higher than that of untreated littermates (100 versus 55 days, log rank test; p < 0.0001) (Figure 5A). Interestingly, approximatively 30% of the treated animals survived up to 1 year of age and were culled in apparently good health. Unexpectedly, four animals injected in different days died suddenly between 90 and 100 days after birth. Post-mortem analysis showed a massively enlarged stomach (Figure S3). H&E analysis of the stomach, small intestine, and large intestine did not reveal any obvious histological alteration (not shown). Other AAV-PHP.B-treated animals developed the typical signs of the disease, although at a later stage than untreated littermates, and were sacrificed when body weight dropped by 20% and ataxia became evident. All of the mice that were sacrificed because of poor health conditions showed clear signs of neurodegeneration by histological and PathoGreen staining. (Figure S4).
Figure 5.

Survival Curve in Adult and Newborn Ndufs4−/− Mice
(A) Kaplan-Meier survival probability in Ndufs4−/− adult mice. Significance was calculated with a log rank test (median lifespan 55 days for Ndufs4−/− vs 100 for AAV-PHP.B-hNDUFS4; p<0.0001). (B) Kaplan-Meier survival probability in Ndufs4−/− newborn mice. Significance was calculated with a log rank test. (C) Determination of the AAV-PHP.B viral genomes in Ndufs4−/− tissues of animals injected at birth (P1). (D) Western blot analysis of hNDUFS4 in the brain of animals injected at birth.
Overall, these data suggest that the treatment with AAV-PHP.B-hNDUFS4 at P26–P28 is highly effective in delaying, if not blocking, the disease progression.
AAV-PHP.B-Mediated Gene Therapy Is Ineffective in Newborn Ndufs4−/− Mice
Next, we wondered whether anticipating the treatment could improve the clinical outcome. We thus treated a group of Ndufs4−/− mice at P1 by i.v. injection (1012 vg/mouse) through the temporal vein. Surprisingly, the survival curve overlapped that of untreated littermates (Figure 5B). We sacrificed the animals around day 40, when they started to lose weight and developed severe ataxia. AAV-PHP.B copy number in the tissues of injected pups was similar to that of the injected adults, with even higher AAV-PHP.B copy numbers in skeletal muscle (Figure 5C). Importantly, the copy number in the brain was lower than in adults and showed much higher variability, suggesting an immature import system of the AAV vector. Accordingly, the hNDUFS4 protein levels were barely detectable in the brains of AAV-PHP.B-treated animals (Figure 5D). These data suggest that the AAV-PHP.B crossed the BBB much less effectively in pups than in adults. We hypothesized that this could be due to lower expression of LY6A receptor in P1 pups. To test this, we used an anti-LY6A antibody on brain homogenates from P1 and P30 mice. Western blot immunovisualization showed a marked difference, with P30 animals expressing much higher levels of LY6A than newborn mice (Figure S5).
Discussion
We showed that a single i.v. injection of AAV-PHP.B-hNDUFS4 in pre-symptomatic, young adult Ndufs4−/− mice could significantly prolong their lifespan up to 1 year and improve their clinical phenotype. These results are by far more encouraging than those we presented previously by using AAV9, likely because the i.v. delivery facilitates the distribution of the vector throughout the brain, and for the intrinsic tropism of AAV-PHP.B for both neuronal and glial cell types.
Several animals died during the observation time, in part because of the disease progression, and in part as a consequence of a sudden and so far unexplained gastroparesis. Note, however, that gastroparesis has been reported as a complication in several mitochondrial diseases, including MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes), POLG1 mutations, and MNGIE (mitochondrial neurogastrointestinal encephalopathy).28 Although we did not find any obvious reason for the variable outcome of the treatment, several hypotheses can be proposed. One possibility is that, although no obvious signs of neurodegeneration were detected at the age of injection, we cannot exclude that the disease was already ongoing. This is also suggested by recent data showing that mutations in SURF1, a common cause of Leigh disease due to defective complex IV, impair neurogenesis.29 A second hypothesis stems from the observation that complex I activity is only partially restored, although hNDUFS4 is extensively expressed in the mouse brain upon AAV-PHP.B delivery. Accordingly, the amount of hNDUFS4 is consistently lower than the endogenous protein. In addition, substantial amounts of the 830-kDa assembly intermediate typically detected in Ndufs4−/− mitochondria26 are still present. These data suggest that the expression levels are not sufficient to ensure full rescue of the phenotype, despite the high titers used in this study. One possibility to be explored is that the hNDUFS4 is not efficiently incorporated into the mouse fully assembled complex I. This would also explain our previous observation that only partial rescue of the Ndufs4−/− phenotype was obtained with AAV9 double systemic and intracerebroventricular injections.2
The development of the AAV-PHP.B serotype raised great expectations for the gene therapy of neurological and neurodegenerative diseases. Unfortunately, these expectations have been challenged by the discovery of a critical role for the membrane glycoprotein LY6A, which is mutated in some mouse strains and is absent in humans, in enabling this serotype to cross the BBB.18,19,30 We also showed that an additional limitation of AAV-PHP.B is related to low expression of LY6A in newborns. Accordingly, barely detectable levels of hNDUFS4 were present in the brains of animals injected at P1.
Despite the limitations of AAV-PHP.B serotype, our data support the idea that AAV-mediated gene therapy can be life-changing for patients with severe, neurological mitochondrial disease. Future efforts are needed to identify additional candidate vectors with the same BBB permeability of AAV-PHP.B, but improved applicability in humans.
Materials and Methods
Reagents
All chemicals were from Sigma. Antibodies used included the following: anti-NDUFS4 (1:100 for immunohistochemistry [IHC] and 1:1,000 for western blot [WB]) was from Novus Biologicals (catalog no. NBP1-31465); anti-CD68 (1:100) was from Abcam (catalog no. ab125212); anti-GFAP (1:1,000) was from Sigma-Aldrich (catalog no. G3893); anti-NDUFA9, and anti-SDHB (1:1,000) were from MitoSciences (catalog no. ab14713). Secondary antibodies were from Promega (catalog nos. W4011 [rabbit] and W4021 [mouse]). PathoGreen histofluorescent stain (1,000X in water) was from Biotium (catalog no. 80027-5mL). The Novolink Polymer Detection System was from Leica Biosystems (catalog no. RE7140-K). The anti-LY6A antibody (dilution 1:1,000) was from Abcam (catalog no. ab109211).
Animals
All animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 (PPL: P6C20975A) and EU Directive 2010/63/EU. The mice were kept on a C57BL/6 background, and WT littermates were used as controls. The animals were maintained in a temperature- and humidity-controlled animal care facility with a 12-h light/12-h dark cycle and free access to water and food, and they were sacrificed by cervical dislocation. Body weight was monitored daily up to 200 days after injection.
Vector Construction, Production, and Injection
AAV-PHP.B was kindly provided by Dr. B.E. Deverman and used for the packaging of CMV (cytomegalovirus)-hNDUFS4 as described.2 In adult mice, AAV particles were administered systemically by tail vein injection. Pups were injected via the temporal vein, using a 30G, 30° bevelled needle syringe.
Rotarod Analysis
A rotarod apparatus (Ugo Basile) was used to assess coordination skills. After two acclimation sessions, the mice underwent three trial sessions at least 20 min apart, using a standard acceleration protocol pre-set by the constructor.
Immunoblotting
Mouse tissues were homogenized in 10 vol of 10 mM potassium phosphate buffer (pH 7.4). Mitochondrial-enriched fractions were collected after centrifugation at 800 × g for 10 min in the presence of protease inhibitors, and frozen and thawed three times in liquid nitrogen. Protein concentration was determined by the Lowry method. Aliquots, 30 μg each, were run through a 12% SDS-PAGE and electroblotted onto a polyvinylidene fluoride (PVDF) membrane, which was then immunodecorated with different antibodies.
For blue native gel electrophoresis (BNGE) analysis, brain mitochondria isolated as previously described31 were resuspended in 1.5 M aminocaproic acid, 50 mM Bis-Tris/HCl (pH 7) and 4 mg of dodecyl maltoside/mg of protein, and incubated for 5 min on ice before centrifuging at 20,000 × g at 4°C. 5% Coomassie G250 was added to the supernatant. 100 μg was separated by 4%–12% gradient BNGE and electroblotted on nitrocellulose membranes for immunodetection.
Genome DNA Extraction and Quantitative PCR
Total DNA was extracted and analyzed as previously described.2
Biochemical Analysis
Tissues were snap-frozen in liquid nitrogen and homogenized in 10 mM phosphate buffer (pH 7.4). The spectrophotometric activity of complex I and citrate synthase was measured as previously described.32
Morphological Analysis
For histological and immunohistochemical analyses, brains from AAV-PHP.B-hNDUFS4-treated and untreated animals were fixed in 10% neutral buffered formalin (NBF) for a few days at room temperature and then included in paraffin wax. 4-μm-thick sections were used for analysis. Hematoxylin and eosin staining was performed by the standard method. PathoGreen stain was performed by following the manufacturer’s instructions. Immunohistochemistry was performed using a Novolink Polymer Detection System and specific antibodies against the indicated proteins.
Statistical Analysis
All numerical data are expressed as mean ± SEM. A one- or two-way ANOVA test with Tukey’s correction was used for multiple comparisons (see figure legends for details); Kaplan-Meier distribution and a log rank test were used for survival analysis. Differences were considered statistically significant for p <0.05.
Author Contributions
M.Z. and C.V. designed the study, C.V. and P.S.-P. performed the in vivo experiments, P.S.-P. performed the biochemical and molecular analysis, M.L.-S. performed the BNGE experiments, and R.C. performed the histological and histochemical analyses.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We are grateful to Dr. Ivano Di Meo for helpful suggestions and discussions during the study. This work was supported by the Telethon Foundation (grant GGP19007 to M.Z.); an NRJ-Institute de France grant (to M.Z.); the Fondazione Onlus Luigi Comini (to M.Z. and C.V.); and by a core grant from the Medical Research Council (MC_UU_00015/5). P.P. is supported by Marie Sklodowska-Curie ITN-REMIX grant 721757.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.026.
Contributor Information
Massimo Zeviani, Email: massimo.zeviani@unipd.it.
Carlo Viscomi, Email: carlo.viscomi@unipd.it.
Supplemental Information
References
- 1.Bottani E., Giordano C., Civiletto G., Di Meo I., Auricchio A., Ciusani E., Marchet S., Lamperti C., d’Amati G., Viscomi C., Zeviani M. AAV-mediated liver-specific MPV17 expression restores mtDNA levels and prevents diet-induced liver failure. Mol. Ther. 2014;22:10–17. doi: 10.1038/mt.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Meo I., Marchet S., Lamperti C., Zeviani M., Viscomi C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017;24:661–667. doi: 10.1038/gt.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira C.V., Peralta S., Arguello T., Bacman S.R., Diaz F., Moraes C.T. Myopathy reversion in mice after restauration of mitochondrial complex I. EMBO Mol. Med. 2020;12:e10674. doi: 10.15252/emmm.201910674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piguet F., de Montigny C., Vaucamps N., Reutenauer L., Eisenmann A., Puccio H. Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich ataxia. Mol. Ther. 2018;26:1940–1952. doi: 10.1016/j.ymthe.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pirozzi M., Quattrini A., Andolfi G., Dina G., Malaguti M.C., Auricchio A., Rugarli E.I. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Invest. 2006;116:202–208. doi: 10.1172/JCI26210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki-Hatano S., Saha M., Rizzo S.A., Witko R.L., Gosiker B.J., Ramanathan M., Soustek M.S., Jones M.D., Kang P.B., Byrne B.J. AAV-mediated TAZ gene replacement restores mitochondrial and cardioskeletal function in Barth syndrome. Hum. Gene Ther. 2019;30:139–154. doi: 10.1089/hum.2018.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres-Torronteras J., Viscomi C., Cabrera-Pérez R., Cámara Y., Di Meo I., Barquinero J., Auricchio A., Pizzorno G., Hirano M., Zeviani M., Martí R. Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Mol. Ther. 2014;22:901–907. doi: 10.1038/mt.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres-Torronteras J., Cabrera-Pérez R., Vila-Julià F., Viscomi C., Cámara Y., Hirano M., Zeviani M., Martí R. Long-term sustained effect of liver-targeted adeno-associated virus gene therapy for mitochondrial neurogastrointestinal encephalomyopathy. Hum. Gene Ther. 2018;29:708–718. doi: 10.1089/hum.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L., Slone J., Li Z., Lou X., Hu Y.-C., Queme L.F., Jankowski M.P., Huang T. Systemic administration of AAV-Slc25a46 mitigates mitochondrial neuropathy in Slc25a46−/− mice. Hum. Mol. Genet. 2020;29:649–661. doi: 10.1093/hmg/ddz277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bacman S.R., Williams S.L., Hernandez D., Moraes C.T. Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a “differential multiple cleavage-site” model. Gene Ther. 2007;14:1309–1318. doi: 10.1038/sj.gt.3302981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacman S.R., Williams S.L., Garcia S., Moraes C.T. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010;17:713–720. doi: 10.1038/gt.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacman S.R., Williams S.L., Duan D., Moraes C.T. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012;19:1101–1106. doi: 10.1038/gt.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacman S.R., Kauppila J.H.K., Pereira C.V., Nissanka N., Miranda M., Pinto M., Williams S.L., Larsson N.G., Stewart J.B., Moraes C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018;24:1696–1700. doi: 10.1038/s41591-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gammage P.A., Viscomi C., Simard M.-L., Costa A.S.H., Gaude E., Powell C.A., Van Haute L., McCann B.J., Rebelo-Guiomar P., Cerutti R. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018;24:1691–1695. doi: 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira C.V., Bacman S.R., Arguello T., Zekonyte U., Williams S.L., Edgell D.R., Moraes C.T. mitoTev-TALE: a monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 2018;10:e8084. doi: 10.15252/emmm.201708084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudry E., Vandenberghe L.H. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron. 2019;102:263. doi: 10.1016/j.neuron.2019.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hordeaux J., Yuan Y., Clark P.M., Wang Q., Martino R.A., Sims J.J., Bell P., Raymond A., Stanford W.L., Wilson J.M. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood-brain barrier. Mol. Ther. 2019;27:912–921. doi: 10.1016/j.ymthe.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Q., Chan K.Y., Tobey I.G., Chan Y.A., Poterba T., Boutros C.L., Balazs A.B., Daneman R., Bloom J.M., Seed C., Deverman B.E. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE. 2019;14:e0225206. doi: 10.1371/journal.pone.0225206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuzaki Y., Tanaka M., Hakoda S., Masuda T., Miyata R., Konno A., Hirai H. Neurotropic properties of AAV-PHP.B are shared among diverse inbred strains of mice. Mol. Ther. 2019;27:700–704. doi: 10.1016/j.ymthe.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morabito G., Giannelli S.G., Ordazzo G., Bido S., Castoldi V., Indrigo M., Cabassi T., Cattaneo S., Luoni M., Cancellieri C. AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol. Ther. 2017;25:2727–2742. doi: 10.1016/j.ymthe.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liguore W.A., Domire J.S., Button D., Wang Y., Dufour B.D., Srinivasan S., McBride J.L. AAV-PHP.B administration results in a differential pattern of CNS biodistribution in non-human primates compared with mice. Mol. Ther. 2019;27:2018–2037. doi: 10.1016/j.ymthe.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.György B., Meijer E.J., Ivanchenko M.V., Tenneson K., Emond F., Hanlon K.S., Indzhykulian A.A., Volak A., Karavitaki K.D., Tamvakologos P.I. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of Usher syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 2018;13:1–13. doi: 10.1016/j.omtm.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim J.-A., Yi H., Gao F., Raben N., Kishnani P.S., Sun B. Intravenous injection of an AAV-PHP.B vector encoding human acid α-glucosidase rescues both muscle and CNS defects in murine pompe disease. Mol. Ther. Methods Clin. Dev. 2019;12:233–245. doi: 10.1016/j.omtm.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. doi: 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- 26.Kruse S.E., Watt W.C., Marcinek D.J., Kapur R.P., Schenkman K.A., Palmiter R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quintana A., Kruse S.E., Kapur R.P., Sanz E., Palmiter R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finsterer J., Frank M. Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therap. Adv. Gastroenterol. 2017;10:142–154. doi: 10.1177/1756283X16666806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inak G., Rybak-Wolf A., Lisowski P., Jüttner R., Zink A., Mlody B., Glažar P., Secker C., Ciptasari U.H., Stenzel W. SURF1 mutations causative of Leigh syndrome impair human neurogenesis. bioRxiv. 2019 doi: 10.1101/551390. [DOI] [Google Scholar]
- 30.Batista A.R., King O.D., Reardon C.P., Davis C., Shankaracharya, Philip V., Gray-Edwards H., Aronin N., Lutz C., Landers J., Sena-Esteves M. Ly6a differential expression in blood-brain barrier is responsible for strain specific central nervous system transduction profile of AAV-PHP.B. Hum. Gene Ther. 2020;31:90–102. doi: 10.1089/hum.2019.186. [DOI] [PubMed] [Google Scholar]
- 31.Fernández-Vizarra E., López-Pérez M.J., Enriquez J.A. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- 32.Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Daneman R., Prat A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.