

Abstract
While there now exist effective treatments for type 2 high, eosinophilic asthma, there are no specific therapies for 40–50% of people with asthma with other phenotypes, which result from poorly understood underlying pathological mechanisms. One such pathology is neutrophilic inflammation, which has been associated with interleukin (IL)-17 family cytokines. Human genetic studies identified IL-17 polymorphisms associated with asthma; in murine models of allergic airways disease, IL-17A contributes to airway hyperresponsiveness, and in humans, elevated airway IL-17A levels are repeatedly observed in severe asthma. However, the directionality of this association is unknown, and the assumption that IL-17 cytokines drive disease pathology remains speculative. Here, we explore the evidence underlying the relationship between IL-17 and asthma, we review lessons learned from investigating IL-17 in other inflammatory diseases, and discuss the possibility that IL-17 may even be protective in asthma rather than pathogenic. We also critically examine the newly proposed paradigm of a reciprocal relationship between type 2 and type 17 airways inflammation. In summary, we suggest an association between IL-17 and asthma, but research is needed examining the diverse functions of these cytokines, their longitudinal stability, their response to clinical interventions, and for mechanistic studies determining whether they are protective or pathogenic.
Short abstract
IL-17 cytokines have been implicated in neutrophilic asthma by genetic, murine and human data. Here, previous studies are critiqued and the assumption their dominant role is pathogenic rather than protective of airway epithelial barrier integrity is challenged. http://bit.ly/3axB4Zs
Introduction
Airways diseases are increasingly important causes of morbidity and mortality globally. Asthma prevalence increased markedly in recent decades, now affecting more than 300 million people worldwide and resulting in 1000 deaths each day [1], a figure comparable to deaths from malaria [1]. Likewise, chronic obstructive pulmonary disease (COPD) prevalence increased by 44% between 1990 and 2015, to 170 million people [2]. Despite major advances in treatment, particularly through guideline-directed increase in inhaled corticosteroid (ICS) use in asthma, there remain a significant number of patients for whom current treatment strategies are ineffective [3].
Where previously all patients were treated as though there was one unifying pathological process driving disease, there is now an appreciation of multiple asthma phenotypes, and that each may require different treatments according to each individual patient's underlying pathology [4]. Nonhierarchical clustering of multiple clinical and biological measurements identifies different asthma phenotypes with differing inflammatory patterns [5]. Histological analysis of sputum cells reveals distinct inflammatory subtypes: eosinophilic, neutrophilic, mixed granulocytic, and paucigranulocytic [6]. The most studied group is eosinophilic inflammation, which is driven by type 2 (T2) inflammation. T2 inflammation is characterised by expression of the cytokines interleukin (IL)-4, IL-5 and IL-13, which drive differentiation, maturation and maintenance of eosinophils, and their recruitment to the airways. There are a number of highly effective treatments for this phenotype, ranging from ICSs and oral corticosteroids to novel biologic therapies targeting these cytokines. However, people with asthma with other inflammatory phenotypes, especially neutrophilic sputum, are resistant to these treatments [7, 8].
The pathological mechanisms driving neutrophilic inflammation are not fully elucidated. Neutrophilic asthma has been associated with increased bacterial airway colonisation [9], and with increases in CXCL8, neutrophil elastase, neutrophil extracellular trap components [10, 11], and of caspase 1 and IL-1β, related to NLRP3 inflammasome activation [10, 11]. Neutrophilic inflammation is often associated with IL-17 cytokines, which induce epithelial cells to release chemokines and cytokines that attract neutrophils to the site of inflammation [12]. Additionally, IL-17A plays an important pathogenic role in rheumatological conditions particularly psoriasis and ankylosing spondylitis [13]. There are numerous reports correlating an excess of IL-17A with more severe forms of asthma [14–17].
In this review, we provide a summary of IL-17 biology, including its critical, protective function against bacterial and fungal infections. We explore the diverse members of the IL-17 family, their different receptors, their cellular sources, and their regulation. We examine the role of IL-17 in other diseases, and how this knowledge might enhance our understanding of airways diseases. We provide a comprehensive review of the evidence associating IL-17 with asthma, including recent data from the U-BIOPRED collaborative, and we challenge the assumed directionality of this association. We critically appraise the new paradigm of a reciprocal relationship between T2 and type 17 (T17) biology, and discuss why T17-targeted interventions trialled to date have proved ineffective. We propose the hypothesis that IL-17 may play a predominantly protective role in asthma and conclude by summarising the evidence for the role of IL-17 in asthma, posing questions that must be answered before we can be confident that it has a dominant causal role in disease pathology.
IL-17
Since the early 1990s a dominant paradigm has been of an inappropriate activation of T2 T-helper (Th2) cells causing atopic asthma, with reciprocal inhibition of type 1 cytokine-secreting T-helper (Th1) cells [18]. However, more recently, other T-cell subsets and types of inflammation have been recognised. Among these T-helper 17 (Th17) cells, inflammation characterised by release of IL-17A appears important in defence against extracellular bacteria and fungi [12]. Originally named CTLA8, IL-17A was first cloned in 1993 with the gene for its archetypal receptor, IL-17RA, cloned 2 years later. In 2005, Th17 cells were identified in mice as a distinct lineage of cluster of differentiation 4+ (CD4+) T-cells [19, 20], characterised by expression of IL-17A under the influence of the master transcription factor retinoic acid receptor-related orphan receptor γt (RORγt) and downstream signalling via Act-1 [12] (figure 1).
FIGURE 1.

Signalling pathways following interleukin (IL)-17 binding its cognate receptor, IL-17R. Both stimulatory (blue arrows) and inhibitory (red arrows) regulatory pathways are shown. ASF: alternative splicing factor; MAPK: mitogen-activated protein kinase; GCSF: granulocyte colony-stimulating factor; AMP: antimicrobial peptide.
IL-17 family cytokines
In humans, IL-17A is a 155-amino acid secreted glycoprotein, representing a family of homologous proteins [21]. IL-17F is most similar in structure and function to IL-17A, sharing 50% sequence homology and consisting of 158 amino acids. Interestingly, these cytokines form both homodimers and a heterodimer, each with differing levels of potency. As measured by downstream gene activation, the most potent is IL-17AA followed by IL-17AF and finally IL-17FF [22]. The significance of this remains unclear. Th17 responses via IL-17A and IL-17F are particularly involved in antimicrobial defence against extracellular pathogens such as bacterial and fungal infections. Uptake, processing and antigen presentation of such pathogens induces naïve Th0 cells in lymph nodes to differentiate into Th17 cells. Th17 cells migrate to the subepithelial space where, on pathogen recognition, they release IL-17 cytokines, which trigger antimicrobial responses in nearby cells, for instance epithelial release of CXCL8, a neutrophil chemokine, and β-defensin, an antimicrobial peptide that disrupts the stability of the microbial membrane.
Other members of the IL-17 family include IL-17B, IL-17C, IL-17D and IL-17E (now known as IL-25), and though they are classified as members of the same family, they have very different functions.
IL-17C is most abundantly released by epithelia after stimulation by IL-1β, tumour necrosis factor (TNF), various pathogens or through cell damage via Toll-like receptors (TLRs) 2 and 5 [23]. Subsequent IL-17C release promotes IL-17A and IL-17F release from Th17 cells, and thus may be an early driver of T17 inflammation, and indeed its release precedes that of IL-17A and IL-17F [24].
IL-17E, now known as IL-25, promotes T2 rather than T17 inflammation. Instead, IL-25 regulates T17 inflammation through directly inhibiting macrophage IL-23 release, and promoting dendritic cell IL-13 production, which in turn inhibits dendritic cell release of IL-23, IL-1β and IL-6, important in T17-polarising cytokines [25].
As little is currently known about IL-17B and IL-17D in this review we will focus predominantly on IL-17A and IL-17F.
Cellular sources of IL-17 cytokines
There are multiple cellular sources of IL-17 cytokines; it is not yet clear which are dominant. IL-17A release defines the distinct CD4+ Th17 cell lineage [19, 20]. Their differentiation from naïve T-cells requires cognate antigen presentation on major histocompatibility complex (MHC) class II and a specific cytokine milieu [20]. These co-stimulatory cytokines released by the antigen-presenting cell include IL-6, IL-1β, transforming growth factor (TGF)-β, IL-21 and IL-23. IL-23 is essential for Th17 cell survival. IL-23 comprises two subunits, a unique p19 subunit and a p40 subunit shared with IL-12 [26]. As IL-12 causes differentiation of naïve T-cells into Th1 cells, therapeutic blockade of the IL-12/IL-23 p40 subunit causes inhibition of both T17 and type 1 inflammation. Transcription factors, stimulating and effector cytokines for each T-helper cell subtype are shown in table 1 [27].
TABLE 1.
Human T-helper (Th) cell subtypes
| Type | Co-stimulatory cytokine | Transcription factors | Effector cytokines | Target |
| Th1 | IL-12 | T-bet, STAT1 | IFN-γ, TNF, IL-2 | Intracellular bacteria, viruses and mycobacteria |
| Th2 | IL-4 | STAT6, GATA3 | IL-4, IL-5, IL-13 | Extracellular parasites |
| Th17 | IL-23 | STAT3, RORγt | IL-17A, IL-17F, IL-21, IL-22 | Extracellular bacteria and fungi |
IL: interleukin; STAT: signal transducer and activator of transcription; IFN: interferon; TNF; tumour necrosis factor; RORγt: retinoic acid receptor-related orphan receptor γt.
In addition to Th17 cells, other cell types release IL-17A, many sharing with Th17 cells a dependence on IL-23, the master transcription factor RORγt and expression of the chemokine receptor CCR6 [28–30].
MHC class I-restricted CD8+ T-cells releasing IL-17A (Tc17 cells) are found in the lungs of mice challenged with influenza A [31]. Moreover IL-17A is a dominant cytokine produced by a range of unconventional T-cells. These include γδ; T-cells, which comprise a functionally diverse group of cells restricted by a range of nonclassical MHC-like molecules [18]. The Vγ4+ subset particularly sequesters to the lung and produces IL-17A. In murine models of ovalbumin (OVA)-induced allergic inflammation γδ;17 are more prevalent than Th17 cells [32]. Adoptive transfer of γδ; T-cells is protective against allergen-induced airway hyperresponsiveness (AHR) and enhances resolution of eosinophilic T2 inflammation. Blocking γδ; T-cells exacerbates injury whilst transfer of Th17 cells or IL-17-deficient γδ; T-cells has no effect.
Natural killer T (NKT) cells, which recognise glycolipids presented on CD1d, also release IL-17A. IL-17A-producing NKT cells are elevated in a murine model of ozone-induced AHR, and are associated with increased airway neutrophils [33]. Blocking CD1d prevents ozone-induced AHR. Interestingly allergen-induced AHR does not result in an increase in NKT-17 cells, implicating IL-17A in some but not all mechanisms of AHR.
Mucosal-associated invariant T (MAIT) cells are a major unconventional T-cell subset defined by a semi-invariant T-cell receptor, like invariant NKT cells, but restricted by MHC-related protein-1 (MR1), and including CD8+, CD4+ and double negative subsets. MAIT cells highly express RORγt and are strong IL-17 producers [34, 35]. They can respond to a wide variety of bacterial and viral pathogens, so MAIT-derived IL-17 may protect against mucosal bacterial and Candida infections. Recent work in humans and mice has shown that activated MAIT cells promote epithelial wound repair [36, 37], which may be a dominant function in damaged airways. Indeed, while activated MAIT cells produce copious amounts of IL-17A, and to a lesser extent IL-17F, during an acute infection, after resolution of infection IL-17A expression is downregulated and IL-17F becomes predominant [36], which could reconcile discrepant observations from murine models in which IL-17 molecules appear to be either proinflammatory or proresolution in different contexts and at different time points.
Innate lymphoid cells are innate immune cells derived from a common lymphoid precursor, which express a range of natural cytotoxicity receptors, but do not express antigen-specific B-cell or T-cell receptors [38]. The ILC3 subset is defined by expression of RORγt, the capacity to produce IL-17A and responsiveness to IL-1β and IL-23. Preliminary data show ILC3 being detected in bronchoalveolar lavage (BAL) in humans [39], and an ILC3 gene signature being increased in nasal epithelium in asthma [40].
Bone-marrow-derived neutrophils both produce and respond to IL-17A [41]. IL-6 and IL-23 are required for this expression, and interestingly these cytokines also induce DECTIN2, a pattern recognition receptor, which augments IL-17 receptor expression, allowing neutrophil-derived IL-17A to act as in a paracrine manner, recruiting further neutrophils via epithelial CXCL8.
Likewise, B-cells produce IL-17A and IL-17F. IL-17A is released earlier (induced by TGF-β and IL-23 at 24–48 h) and IL-17F later, being more dependent on IL-23 and IL-6 at 72 h. Given the decreasing hierarchy of potency, from IL-17AA to IL-17AF to IL-17FF, IL-17F may act later as a negative regulator of the otherwise potently proinflammatory IL-17A [42].
Other cell types, including macrophages, also release IL-17A, but this may be related to their endocytic capacity rather than de novo IL-17A synthesis [12].
Overall diverse cell types produce both IL-17A and IL-17F, but they may be responding to different cytokines and may differ in the kinetics and quantities they release. How these differences contribute to normal biology and to immune pathology is unclear.
What are the main cellular targets of the IL-17 cytokines?
IL-17 cytokines act on numerous cells, with different IL-17 family members signalling through different receptor subtypes, potentially inducing differential effects in different tissues. IL-17RA is the obligate co-receptor to which other IL-17R subtypes bind. IL-17A and IL-17F signal through the IL-17RA–IL-17RC receptor; IL-17C through IL-17RA–IL-17RE; and IL-17B and IL-17E through IL-17RA–IL-17RB. The receptor for IL-17D is unknown [43, 44].
IL-17 receptors are widely expressed in lung tissues. Polarised lung epithelium responds to IL-17A and IL-17F but only on the basolateral surface due to the surface-specific expression of IL-17RA and IL-17RC [45, 46]. In children with severe treatment-refractory asthma IL-17RA-positive cells are enriched in the submucosa and epithelium, and stimulation of their primary bronchial epithelial cells with IL-17A enhances mRNA expression of IL-17RA and IL-17RC [47].
Airway smooth muscle cells express IL-17RA and IL-17RC. IL-17A drives proliferation, migration and contraction of airway smooth muscle [48–50]. Blocking IL-17RA or IL-17RC alone partially inhibits IL-17A/IL-17F-induced airway smooth muscle cell migration [50]. Blocking IL-17RA is more effective at inhibiting migration induced by IL-17A than IL-17F and blocking IL-17RC is more effective for IL-17F than IL-17A. Blocking both receptors results in full inhibition of smooth muscle migration induced by either cytokine, an effect mediated partly through the intracellular signalling kinase p38-MAPK.
Human lung endothelial cells also express IL-17RA and IL-17RC, and release the neutrophil chemoattractant CXCL1 in response to both IL-17A and IL-17F [51]. Likewise, human eosinophils express both IL-17RA and IL-17RC, releasing neutrophil chemokines in response to IL-17 stimulation [52]. In advanced COPD IL-17RA and IL-17RC expression is increased in mast cells. High concentrations of IL-17A induce proangiogenic factors from these cells, potentially contributing to vascular remodelling [53].
Although many lung cells express IL-17RA and IL-17RC receptors no comprehensive studies have explored the differential expression of these receptors in the lungs in airways disease.
IL-17 signalling pathways and regulation
IL-17RA binds the adapter Act-1, which then signals via two distinct pathways [54]. In its “canonical” response, Act-1 ubiquitylates the receptor TRAF6, resulting in NF-ĸB and MAPK activation, which subsequently activate cell signalling proteins ERK, p38-MAPK and JNK (figure 1). These directly activate IL-17 target genes. In its “noncanonical” pathway Act-1 activates TRAF2 and TRAF5, which sequester the RNA destabiliser SF2 [12], preventing degradation of unstable inflammatory chemokine mRNA transcripts [55]. Therefore IL-17 may have a complex role in driving inflammation, as it induces some inflammation on its own but has a more profound effect in synergy with other inflammatory cytokines by prolonging the half-life of their mRNAs.
Negative feedback is provided by TRAF3 and TRAF4 competitively binding IL-17RA in place of Act-1, and by A20 which is released downstream of IL-17R stimulation and deubiquitylates TRAF6 [56]. As IL-17A stimulation increases neutrophil apoptosis [57–59] IL-17A may also regulate neutrophilic inflammation, preventing excessive accumulation of neutrophils and associated bystander damage. This is supported by evidence from a sepsis model, where intravenous Staphylococcus aureus induced more IL-23 in IL-17A knockout mice than in wild types [60]. It is often assumed that IL-17 is proinflammatory, perhaps because it is known to be elevated during infections, but it may also have significant anti-inflammatory effects, which might even be their predominant action in some physiological contexts.
IL-17 functions
The predominant roles of IL-17 cytokines are considered to be in maintaining the integrity of the epithelial surface barrier, and in responding to extracellular bacteria and fungi through the recruitment of neutrophils and the release of antimicrobial peptides. Multiple genes are activated upon stimulation of the IL-17 receptor, including the neutrophil chemo-attractants CXCL1, CXCL5, CXCL8 (IL-8) and CCL2 [61–63]; granulocyte–macrophage colony-stimulating factor, which drives neutrophil production in the bone marrow [64]; CCL20, which recruits CCR6+ cells producing IL-17A [65]; claudin, important for epithelial tight junction integrity [66]; and antimicrobial peptides including β-defensins, S100 proteins and Lipocalin-2 [67].
IL-17 in other inflammatory diseases
Many diseases have been linked with dysregulated IL-17 signalling. Psoriasis and inflammatory bowel disease share underlying chronic inflammation and dysregulation of the epithelial barrier with airways diseases.
Psoriasis
Psoriasis was initially considered a Th1-driven disease. The type 1 cytokines interferon (IFN)-γ and TNF, are elevated in psoriatic lesions [68] and blocking IL-12-driven Th1 differentiation through IL-12/IL-23 p40 inhibitors proved clinically effective [69], although this was before the role of IL-23 in the development of Th17 cells was appreciated. However, more recently, data from genetic studies, mouse models, pathway analysis and therapeutic trials suggest that T17 inflammation is the dominant process.
In mice, IL-23 blockade is effective at blocking the IL-12/23 p40 subunit [70], implying that it is the IL-23 component that is pathogenic. Likewise, mice lacking IL-17RA are as resistant to psoriasis induced by the TLR7/TLR8 ligand imiquimod as those receiving IL-23 blockade [71].
In humans, IL-17A, IL-17F and IL-17C are elevated in active psoriatic lesions [23]. In genetic studies, risk alleles upstream and downstream of IL-17 expression are associated with psoriasis [72].
Biologics targeting the IL-17 pathway are highly effective in psoriasis. In addition, IL-12/IL-23 p40 blockade induces early reductions in IL-23 and IL-17A, with later reductions in Th1-associated genes [73]. IL-23 p19 blockade is also effective [74], as is IL-17A blockade by secukinumab and ixekizumab or IL-17RA blockade by brodalumab [75]. Furthermore, clinical response correlates with serum IL-17AA homodimer levels, with a decrease in serum IL-17AA levels of almost 60% in treatment responders at week 12, but only 16% in nonresponders [76]. IL-17A may also have a pathological action in atopic dermatitis, where it impairs the epidermal tight junction barrier [77].
Inflammatory bowel disease
In contrast to psoriasis, IL-17 may play a protective role in inflammatory bowel disease (IBD). In animal models, adoptive transfer of IL-17-deficient T-cells exacerbated disease and IFN-γ release [78]. Likewise, IL-23p19-deficient mice had worse disease, with low IL-17 levels and high IFN-γ levels, whilst antagonism of the IL-12/IL-23 p40 subunit (hence IL-12 and IL-23) reduced colitis [79]. An additional protective mechanism of IL-17A may be reciprocal inhibition of Th1-derived IFN-γ.
Consistently with this, naïve T-cells co-cultured with dendritic cells from Crohn's disease express high IFN-γ and only minimal IL-17A [80]. Therapeutic IL-17 blockade in psoriasis is associated with increased frequency of IBD exacerbations in patients with pre-existing IBD [81], and is potentially associated with de novo IBD onset [82]. Anti-IL-17A biologics provided no clinical benefit in IBD and may have increased exacerbations [83, 84]. Therefore, increased submucosal IL-17A observed in IBD [85] may represent not a pathological, but rather a protective response to epithelial damage and subsequent commensal invasion.
Thus IL-17A and IL-17F are important in maintaining the barrier function of epithelial cells, for epithelial repair and countering bacterial invasion. The marked differential efficacy of IL-17 blockade in psoriasis versus IBD derives from the differing underlying pathobiologies: psoriasis results from pathologically increased IL-17 signalling, inducing hyper-proliferation and aberrant differentiation of keratinocytes, whereas in IBD, IL-17 signalling is protective against commensal bacterial invasion.
IL-17 in asthma
Asthma has been linked with IL-17 dysregulation by genetic analyses, clinical associations, in vitro studies and murine models. Human IL-17 gene polymorphisms are associated with asthma in diverse populations [86–88]. IL-17A was originally proposed to contribute to airway neutrophilia, which characterises the neutrophilic phenotype [6, 89–91] based on in vitro studies demonstrating that human bronchial epithelial cells stimulated with IL-17A expressed more CXCL8 mRNA, which was chemotactic to neutrophils [92]. IL-17A also stimulates expression of the mucin MUC5B, potentially implicating it in airway remodelling [93].
Several groups reported preliminary associations between IL-17A and human asthma. Molet et al. [94] found more IL-17A+ cells in BAL and sputum of 11 people with asthma than seven healthy controls and showed that in vitro dexamethasone reduced IL-17A-induced IL-6 release from airway fibroblasts. Barczyk et al. [95] found a possible, modest negative correlation of sputum IL-17A and AHR, though no significant differences overall between asthma and health. Sun et al. [96] reported increased sputum IL-17A correlating with disease severity and with sputum neutrophils, CXCL8 and myeloperoxidase. Blood IL-17 levels also correlated with airway neutrophils. Another group reported a correlation between sputum IL-17A mRNA and sputum neutrophils [15].
Similarly, some bronchoscopy studies report increases in airway tissue IL-17 inflammation. An immunohistochemistry study of bronchial biopsies found a correlative increase in IL-17A and IL-17F in people with increasing asthma severity [14], with two studies reporting double-positive IL-4/IL-17-producing T-cells in patients with severe asthma [30, 97]. A further study showed increased numbers of IL-17-secreting ILC3s in the sputum of patients with severe asthma [39]. However, such weak clinical associations from these small studies have not been replicated consistently. In a large bronchoscopy study of 84 volunteers, we found no evidence of increased IL-17A protein in asthma in serum, sputum or BAL [98]. Furthermore using intracellular cytokine staining on activated T-cells we found no significant differences in frequencies of Th17 or γδT17 cells between patients with asthma and healthy people in peripheral blood, sputum, BAL or bronchial biopsies at stable state, nor did we observe significant differences in peripheral blood or sputum Th17 cell frequencies during an exacerbation [98].
Exacerbations are a major cause of morbidity and mortality in asthma [99]. Most are induced by viruses and characterised by neutrophilic inflammation [100, 101] implicating Th17 cells. Diesel fume exposure is associated with elevated serum IL-17 in childhood asthma, and cigarette smoke with elevated IL-17A and neutrophilic inflammation in asthma [102, 103]. One immunohistochemical study found bronchial IL-17F+ cells correlated with both airway neutrophils and asthma exacerbation rates, with IL-17F co-localising with Th and cytotoxic T-cells [16, 104]. Using flow cytometry in 19 children, the frequency of circulating MAIT-17 cells was threefold higher in asthma exacerbators than in nonexacerbators and there were modest correlations positively with the number of severe exacerbations and negatively with symptom scores [104].
Murine models of allergic airways disease link IL-17A with a cardinal physiological feature of asthma, AHR [105–107]. Mice sensitised to OVA by inhalation, but not intraperitoneally, developed strong airway Th17 cell responses, associated with AHR, which was less prominent in IL17ra−/− mice [106]. Likewise, Th17-dependent AHR could be induced by epicutaneous sensitisation to OVA, and again, not via intraperitoneal injection [107], underlining the importance of the barrier epithelium in the development of these responses. Using inhaled dust mite allergen, Chesne et al. [105] induced airway Th2 and Th17 cells, and found anti-IL-17A antibody slightly reduced AHR, whilst IL-17A had a direct contractile effect on airway smooth muscle. Mice deficient in Th17 cells were resistant to developing AHR, despite normal IL-17A-producing γδ; T-cells [48]. Conversely, in another study [32] γδ; T-cells were important in reducing AHR at the point of allergic inflammation, an effect independent of conventional Th17 cells, suggesting that Th17 cells may be associated more with pathology and γδ; T-cells with bronchial homeostasis, perhaps dependent on the timing of Th17 induction.
In humans, IL-17A (and not IL-17F) enhanced smooth muscle cell contractility with methacholine, suggesting that IL-17A may drive hyperresponsiveness [48]. Irvin et al. [97] reported a weak negative correlation (r= −0.35) between BAL IL-17A and forced expiratory volume in 1 s (FEV1), and a moderate negative correlation between frequencies of dual Th2/Th17 in BAL and AHR (r= −0.52). As IL-17F is produced later in an inflammatory response it may be important in modulating the proinflammatory function of IL-17A. IL-17A also stimulates human bronchial epithelial production of CCL28, which increases chemotaxis of IgE-containing B-cells, potentially important in allergic inflammation [108].
A recent large analysis from the U-BIOPRED cohort used whole-genome transcriptomics on epithelial brushing and bronchial biopsy specimens in 91 people with asthma and 46 healthy controls [109]. They found an IL-17 gene signature in 22 of 91 patients' bronchial biopsies, and an IL-13 gene signature in 9 patients. The IL-17-high group were characterised by frequent exacerbations, airway neutrophilia, and decreased lung microbial diversity. Differentially expressed pathways in IL-17-high asthma were shared with those reported as altered in psoriasis and included genes regulating epithelial barrier function and defence mechanisms, such as IL1B, IL6, CXCL8, and β-defensin.
Currently it remains unclear whether IL-17 is driving asthma pathology in a particular subset of patients, or whether it is present as a response to some other cause, such as epithelial damage related to the underlying asthma, or an increased bacterial airway colonisation present in those with asthma.
IL-17 and corticosteroids in airways disease
Corticosteroids are effective in reducing symptoms and exacerbations in asthma, and have multiple anti-inflammatory effects. However non-eosinophilic asthma is less responsive to corticosteroids [7, 110]. IL-17 could contribute to this corticosteroid insensitivity through several processes (figure 2).
FIGURE 2.

Flow diagram exploring the putative links between elevated interleukin (IL)-17A levels and severe asthma. As is commonly supposed, IL-17A may drive a particularly severe form of the disease (1), or the reciprocal may be true that severe asthma results in elevated IL-17A levels, through for instance, the disruption and damage to the epithelial cell layer that is commonly seen in asthmatic airways (2). Given that, by definition, all people with severe asthma are on high-dose inhaled corticosteroids (ICS), it is possible that through ICS-induced suppression of local inflammation there is a predisposition to infection that causes an appropriate increase in IL-17A to combat that infection (3). Alternatively, IL-17A may induce corticosteroid resistance in the epithelium, requiring higher doses of ICS for the same effect (4). IL-17R: interleukin-17 receptor.
In human cell lines, IL-17A reduced the sensitivity of epithelial cell TNF-induced CXCL8 production to budesonide, mediated by PI3K activation [111]. Th2 cells polarised in vitro transferred to OVA-sensitised mice caused increased eosinophilic inflammation, sensitive to dexamethasone [112]. Conversely in mice, transferred Th17 cells caused neutrophilic inflammation resistant to dexamethasone. Both cell types, however, induced AHR. Dexamethasone can also promote and maintain Th17 differentiation in mice in vivo [113] and Th cells from patients with steroid-resistant asthma produce higher levels of IL-17 than from patients with steroid-sensitive asthma or healthy controls [114].
We recently examined gene signatures in human bronchial epithelium and flow-sorted T-cells from sputum and BAL of 19 healthy control participants and 46 participants with asthma [115]. In mild, steroid-naïve asthma, the IL-13 response genes POSTN, SERPINB2 and CLCA1 were upregulated. This signal was suppressed in those with moderate asthma, which we attributed to their use of maintenance ICSs. In contrast, in severe, predominantly neutrophilic asthma a different gene profile was observed. TCN1 and MMP9 were upregulated, consistent with neutrophilia, as were IL-17-inducible chemokines (CXCL1, CXCL2, CXCL3, CXCL8, and CSF3) in sputum T-cells. A functional pathway analysis showed strong upregulation of innate defence-response genes, including TLR2, CD14 and JUN, suggestive of a response to airway bacteria in these individuals driving both the neutrophilia, and the steroid-insensitive IL-17 responses.
IL-17A is also associated with steroid unresponsiveness in COPD, a disease more commonly associated with chronic bacterial airway infection. Analysis of airway epithelial cell brushings from patients with COPD from the GLUCOLD and SPIROMICS studies found upregulation of an 11-gene IL-17-induced gene signature in approximately one-third of patients with COPD, associated with increased airway obstruction and decreased response to corticosteroids [116]. Thus, IL-17 may either promote steroid-unresponsive disease, or constitute a protective response to bacterial infection. In particular, nontypeable Haemophilus influenzae (NTHi) is associated with COPD and with neutrophilic asthma [9, 117, 118] and induces IL-17-mediated airways inflammation in murine models [118–120]. In mice, Th17 cells may be protective against this bacterium [121]. Furthermore, in human asthma we have directly observed an in vivo BAL Th17 cell response to H. influenzae infection in the same BAL sample (figure 3). We performed whole-genome metagenomic sequencing of BALF and paired measurement of BAL Th17 frequencies using intracellular cytokine staining in a cohort of 60 patients with asthma and 24 healthy subjects [98]. H. influenzae was identified as highly abundant in one individual with severe neutrophilic asthma, characterised by marked neutrophilia in sputum (71%) and BALF (68%) and a strikingly elevated Th17 frequency (11.3% of T-cells) in the same BALF sample, consistent with a direct, local Th17 cell response to the pathogen in the airways.
FIGURE 3.
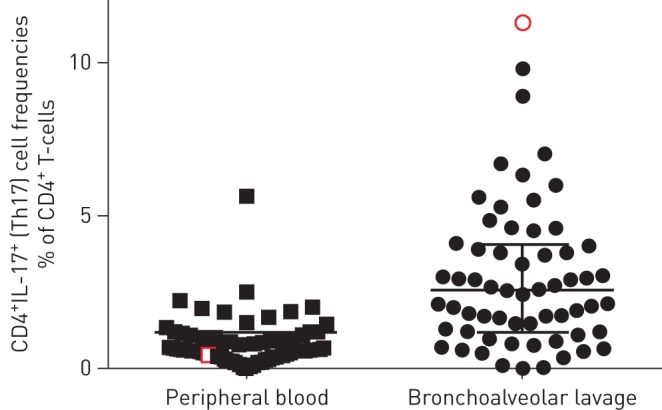
Human airway bronchoalveolar lavage T-helper (Th)17 cell frequencies in health and asthma. Frequencies of live CD3+CD4+ interleukin (IL)-17-secreting (Th17) cells were measured using intracellular cytokine staining after ex vivo stimulation in peripheral blood mononuclear cells and bronchoalveolar lavage fluid (BALF) obtained from 60 patients with asthma and 24 healthy subjects [99]. In one individual in this cohort, Haemophilus influenzae was identified as a highly abundant pathogen by deep sequencing of BALF. This finding was also confirmed on routine culture of the same sample. This 63-year-old man exhibited marked neutrophilia in sputum (71%) and BALF (68%) and his Th17 frequency in the same BALF sample (red) of 11.3% of CD4+ T-cells was strikingly elevated above the group median (2.6%), representing the highest frequency observed in the study, consistent with a direct, local Th17 cell response to the pathogen in the airways. In the 12 months prior to the study, the subject had experienced 20 exacerbations requiring oral steroids. After initiation of long-term antibiotics following the culture result, they experienced a 1.1-point fall in asthma control score and suffered only one steroid-treated exacerbation in the next 18 months.
IL-17A has been investigated in several murine models. In an OVA-induced allergic airways disease model, NTHi infection increased airway neutrophilia and IL-17-expressing pulmonary macrophages, neutrophils and T-cells [9, 118]. In a lipopolysaccharide (LPS) model of bacterial infection, LPS induced neutrophilia, BAL IL-17A and lymphatic Th17 cells, whereas IL-17A knockout mice exhibited worsened eosinophilic inflammation [122]. Similar IL-17-dependent airway neutrophilia was found in another allergen LPS model [123] and in a model using simultaneous challenge with LPS, elastase and respiratory syncytial virus, associated with IL-17-dependent mucus cell hyperplasia [124]. Because NTHi is an obligate human pathogen, an important limitation of these models is that they require short-term, high-dose infection, or chronic recurrent bacterial inoculations [125], but without the development of the chronic, persistent airway infection characteristic of NTHi in the human airways. Thus, further mechanistic studies require development of a model in which NTHi can persist spontaneously in the lower airways. Several limitations are frequently highlighted for murine models: asthma does not occur in animals (except perhaps cats and horses) [126], there are known differences between strains and immunisation protocols, and these protocols depend on unphysiological sensitisation procedures, generating inflammation more reminiscent of allergic alveolitis than asthma [127]. Thus, research in airways disease should avoid an overreliance on animal models, which can often be difficult to extrapolate to the complex and uniquely human disease of asthma.
Is there a reciprocal relationship between T2 and T17 inflammation?
A recent human transcriptomic study postulated the hypothesis that IL-17-driven inflammation may be induced by corticosteroids [128]. The authors defined T2 and T17 epithelial gene signatures through quantitative PCR on epithelial cells stimulated in vitro with IL-13 or with IL-17A and TNF, and used this to analyse microarray data from endobronchial biopsies from 51 patients with asthma. T2 and T17 gene signature scores were negatively correlated and were mutually exclusive with no patient high in both signatures. These subgroups did not differ phenotypically in spirometry, airway hyperresponsiveness or exacerbation frequency. It was suggested that the T17 signature was associated with inhaled or oral corticosteroid use, whereas the T2 signature was only found in those patients with eosinophilic inflammation. The study did not report on bacterial airway colonisation.
In a murine model of allergic inflammation in the same report, selective blockers of the T2 pathway induced increase in tissue IL-17 expression in the lungs and lymph nodes and a proportional increase in pulmonary neutrophil frequencies, concomitant with a decrease in eosinophil frequencies in the lungs. This relative neutrophilia was prevented by simultaneous inhibition of both T2 and T17 pathways. The authors propose that in asthma, a predisposition to T2 inflammation is driven towards T17-mediated inflammation by corticosteroid therapy [128]. This hypothesis would have important implications for patients receiving type 2-targeting biologics, although the study has provoked some controversy [129, 130]. The three-group classification may be a simplification and lacks a validation cohort, measures of neutrophilic inflammation were not significantly different between groups, nor were ICS or oral corticosteroid use [128], whereas tissue eosinophils and other markers of T2 inflammation were prevalent in many patients classified as Th17-high [129].
Given this potential relationship between T2 and T17 inflammation, combined administration of anti-IL-13 and anti-IL-17A is being considered for patients with asthma. A mouse model of T2/T17-high asthma [131] suggests that at individually subtherapeutic doses, anti-IL-13 and anti-IL-17A, given in combination, limits asthma-like symptoms without induction of compensatory T17 responses. However, whether this will extend to humans is yet to be investigated. Even in mice, another report found that IL-13 blockade was effective without inducing the development of neutrophilic inflammation [132], and the neutrophilia observed in mice by Choy et al. [128] may be partly due to the well-known direct effect of corticosteroids in promoting neutrophil mobilisation and survival. Moreover, the mutually exclusive T2/T17 findings of Choy et al. are yet to be replicated in an independent validation cohort [133] or shown at protein level [129]. There are as yet no prospective clinical data showing development of T17 inflammation in people receiving blockade of T2 pathways. Whether such an increase T17 inflammation would be harmful is also unknown [130], and will be important to investigate.
Targeting IL-17 in airways disease
To date there have been three studies targeting the IL-17 pathway in airways disease. Brodalumab, a biologic targeting IL-17 receptor, did not improve asthma symptoms or FEV1 in 302 patients with partially controlled moderate-to-severe asthma [134]. The study was negative for the primary outcome (p=0.37) of change in Asthma Control Questionnaire at 12 weeks, and for all secondary outcomes. The authors report an improvement in Asthma Control Questionnaire in the subgroup with high bronchodilator reversibility, but this was of small magnitude, without a dose–response effect across groups, and was based on a p-value of 0.02 in one of nine subgroup analyses, the other eight of which were negative. Importantly no statistical correction was made for these multiple comparisons. Moreover, this finding was also not confirmed in a subsequent, unpublished phase 3 study [135]. However, there was no stratification of patients in this study according to asthma inflammatory phenotype so any meaningful signal in a subpopulation may therefore have been missed.
In the second study, inhibition of CXCR2, the receptor for the neutrophil chemokine CXCL8, was trialled without success. Although an attempt was made at patient stratification, the criteria (blood eosinophils <0.5×109 cells per L, blood neutrophils >2.7×109 cells per L) would not have reliably predicted neutrophilic airway inflammation, again suggesting that a potentially important signal may have been lost [136]. This study is noteworthy in that it also reported exacerbation rates over a 12-month period. Indeed, the authors questioned the contribution that neutrophils make to the pathobiology of asthma severity or exacerbations, except in the context of infective exacerbations. Arguably as a primary outcome for a trial, changes in FEV1 are of much less clinical relevance to patients than changes in frequencies of exacerbations, which are responsible for the majority of morbidity in asthma, which are more likely to be linked to changes in airway inflammation induced by IL-17 modulation, and which have proved to be highly effective outcomes in trials of ICSs and T2 biologics.
The third study was a placebo-controlled trial of an anti-IL-17 (CNTO 6785) in COPD [137], randomising 93 patients to treatment and 94 to placebo, with the primary end-point of FEV1 change at week 16 and secondary end-points of rescue medication use, quality of life and exacerbations. No end-points showed statistically significant differences. A small improvement in FEV1 % predicted in patients with frequent exacerbations was neither statistically nor clinically meaningful; however, this study is limited by the short duration of treatment and a low baseline exacerbation rate.
Of relevance to the suggested reciprocal relationship between T2 and T17 pathologies, a recent phase 1 trial of a humanised bispecific IgG4 antibody that binds and neutralises IL-13 and IL-17AA, IL-17AF and IL-17FF recently reported results [138]. Although the drug showed satisfactory tolerability, the study was terminated early due to frequent antidrug antibodies.
Conclusions
Several questions related to IL-17 in asthma remain outstanding. Firstly, can a group of patients with severe asthma driven by IL-17 be identified, and is their pathology stable over time? Secondly, what are the dominant cellular sources of IL-17 in asthma? Thirdly, does the biology of patients with T2-associated eosinophilic inflammation change to T17-associated neutrophilic inflammation in response to treatments targeted against T2 pathways? Fourthly, will targeting IL-17 in asthma with elevated IL-17 levels result in reduced inflammation and symptoms, or be ineffective, or even predispose to further infections and worse symptoms. Clearly, further studies addressing these questions are mandated, given the central role of the IL-17 family in mucosal barrier immunity.
Footnotes
Author contributions: G.M. Hynes and T.S.C. Hinks jointly conceived the article, conducted the literature review and drafted the manuscript. Both authors approved the final manuscript.
Conflict of interest: G.M. Hynes reports grant support from UCB during the writing of this review.
Conflict of interest: T.S.C. Hinks reports GBP 1 648 000 in fellowships from the Wellcome Trust (grants 088365/z/09/z, 104553/Z/14/Z and 211050/Z/18/Z) and a GBP 25 000 Wellcome–Beit fellowship during the writing of this review; and a GBP 750 lecture fee from AstraZeneca, a GBP 765 lecture fee from TEVA and a GBP 2100 fee for an educational video from Peer Voice, outside the submitted work.
Support statement: This work was supported by grants from the Wellcome Trust (104553/z/14/z and 211050/Z/18/z) and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (to T.S.C. Hinks) and a University of Oxford–UCB fellowship (G.M. Hynes). The views expressed are those of the authors and not those of the National Health Service or NIHR.
References
- 1.The Global Asthma Report 2018. Global Asthma Network, Auckland, NZ, 2018. [Google Scholar]
- 2.Soriano JB, Abajobir AA, Abate KH, et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med 2017; 5: 691–706. doi: 10.1016/S2213-2600(17)30293-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomson NC. Novel approaches to the management of noneosinophilic asthma. Ther Adv Respir Dis 2016; 10: 211–234. doi: 10.1177/1753465816632638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavord ID, Beasley R, Agusti A, et al. After asthma: redefining airways diseases. Lancet 2018; 391: 350–400. doi: 10.1016/S0140-6736(17)30879-6 [DOI] [PubMed] [Google Scholar]
- 5.Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 2008; 178: 218–224. doi: 10.1164/rccm.200711-1754OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson JL, Scott R, Boyle MJ, et al. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology 2006; 11: 54–61. doi: 10.1111/j.1440-1843.2006.00784.x [DOI] [PubMed] [Google Scholar]
- 7.Pavord ID, Brightling CE, Woltmann G, et al. Non-eosinophilic corticosteroid unresponsive asthma. Lancet 1999; 353: 2213–2214. doi: 10.1016/S0140-6736(99)01813-9 [DOI] [PubMed] [Google Scholar]
- 8.Chung KF. Asthma phenotyping: a necessity for improved therapeutic precision and new targeted therapies. J Intern Med 2016; 279: 192–204. doi: 10.1111/joim.12382 [DOI] [PubMed] [Google Scholar]
- 9.Green BJ, Wiriyachaiporn S, Grainge C, et al. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS ONE 2014; 9: e100645. doi: 10.1371/journal.pone.0100645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright TK, Gibson PG, Simpson JL, et al. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 2016; 21: 467–475. doi: 10.1111/resp.12730 [DOI] [PubMed] [Google Scholar]
- 11.Lachowicz-Scroggins ME, Dunican EM, Charbit AR, et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. Am J Respir Crit Care Med 2019; 199: 1076–1085. doi: 10.1164/rccm.201810-1869OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol 2018; 10: a028522. doi: 10.1101/cshperspect.a028522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 2018; 55: 379–390. doi: 10.1007/s12016-018-8702-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Ramli W, Prefontaine D, Chouiali F, et al. Th17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 2009; 123: 1185–1187. doi: 10.1016/j.jaci.2009.02.024 [DOI] [PubMed] [Google Scholar]
- 15.Bullens DM, Truyen E, Coteur L, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell-driven inflammation and granulocytic influx? Respir Res 2006; 7: 135. doi: 10.1186/1465-9921-7-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricciardolo FLM, Sorbello V, Folino A, et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol 2017; 140: 395–406. doi: 10.1016/j.jaci.2016.10.034 [DOI] [PubMed] [Google Scholar]
- 17.Sorbello V, Ciprandi G, Di Stefano A, et al. Nasal IL-17F is related to bronchial IL-17F/neutrophilia and exacerbations in stable atopic severe asthma. Allergy 2015; 70: 236–240. doi: 10.1111/all.12547 [DOI] [PubMed] [Google Scholar]
- 18.Godfrey DI, Uldrich AP, McCluskey J, et al. The burgeoning family of unconventional T cells. Nat Immunol 2015; 16: 1114–1123. doi: 10.1038/ni.3298 [DOI] [PubMed] [Google Scholar]
- 19.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6: 1123–1132. doi: 10.1038/ni1254 [DOI] [PubMed] [Google Scholar]
- 20.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6: 1133–1141. doi: 10.1038/ni1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity 2019; 50: 892–906. doi: 10.1016/j.immuni.2019.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright JF, Guo Y, Quazi A, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem 2007; 282: 13447–13455. doi: 10.1074/jbc.M700499200 [DOI] [PubMed] [Google Scholar]
- 23.Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 2013; 190: 2252–2262. doi: 10.4049/jimmunol.1201505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramirez-Carrozzi V, Sambandam A, Luis E, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 2011; 12: 1159–1166. doi: 10.1038/ni.2156 [DOI] [PubMed] [Google Scholar]
- 25.Kleinschek MA, Owyang AM, Joyce-Shaikh B, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med 2007; 204: 161–170. doi: 10.1084/jem.20061738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000; 13: 715–725. doi: 10.1016/S1074-7613(00)00070-4 [DOI] [PubMed] [Google Scholar]
- 27.Kaiko GE, Horvat JC, Beagley KW, et al. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology 2008; 123: 326–338. doi: 10.1111/j.1365-2567.2007.02719.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castro G, Liu X, Ngo K, et al. RORγt and RORα signature genes in human Th17 cells. PLoS ONE 2017; 12: e0181868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willing A, Jager J, Reinhardt S, et al. Production of IL-17 by MAIT cells is increased in multiple sclerosis and is associated with IL-7 receptor expression. J Immunol 2018; 200: 974–982. doi: 10.4049/jimmunol.1701213 [DOI] [PubMed] [Google Scholar]
- 30.Cosmi L, Maggi L, Santarlasci V, et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol 2010; 125: 222–230. doi: 10.1016/j.jaci.2009.10.012 [DOI] [PubMed] [Google Scholar]
- 31.Hamada H, de la Luz Garcia-Hernandez M, Reome JB, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol 2009; 182: 3469–3481. doi: 10.4049/jimmunol.0801814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murdoch JR, Lloyd CM. Resolution of allergic airway inflammation and airway hyperreactivity is mediated by IL-17-producing γδ T cells. Am J Respir Crit Care Med 2010; 182: 464–476. doi: 10.1164/rccm.200911-1775OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pichavant M, Goya S, Meyer EH, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 2008; 205: 385–393. doi: 10.1084/jem.20071507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinks TS. Mucosal-associated invariant T cells in autoimmunity, immune-mediated diseases and airways disease. Immunology 2016; 148: 1–12. doi: 10.1111/imm.12582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosgrove C, Ussher JE, Rauch A, et al. Early and nonreversible decrease of CD161++ /MAIT cells in HIV infection. Blood 2013; 121: 951–961. doi: 10.1182/blood-2012-06-436436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinks TSC, Marchi E, Jabeen M, et al. Activation and in vivo evolution of the MAIT cell transcriptome in mice and humans reveals tissue repair functionality. Cell Rep 2019; 28: 3249–3262. doi: 10.1016/j.celrep.2019.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leng T, Akther HD, Hackstein CP, et al. TCR and inflammatory signals tune human MAIT cells to exert specific tissue repair and effector functions. Cell Rep 2019; 28: 3077–3091. doi: 10.1016/j.celrep.2019.08.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells: how did we miss them? Nat Rev Immunol 2013; 13: 75–87. doi: 10.1038/nri3349 [DOI] [PubMed] [Google Scholar]
- 39.Kim HY, Lee HJ, Chang YJ, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 2014; 20: 54–61. doi: 10.1038/nm.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hekking PP, Loza MJ, Pavlidis S, et al. Pathway discovery using transcriptomic profiles in adult-onset severe asthma. J Allergy Clin Immunol 2018; 141: 1280–1290. doi: 10.1016/j.jaci.2017.06.037 [DOI] [PubMed] [Google Scholar]
- 41.Taylor PR, Roy S, Leal SM Jr, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat Immunol 2014; 15: 143–151. doi: 10.1038/ni.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez-Tello A, Halwani R, Li R, et al. IL-17A and IL-17F expression in B lymphocytes. Int Arch Allergy Immunol 2012; 157: 406–416. doi: 10.1159/000329527 [DOI] [PubMed] [Google Scholar]
- 43.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 2009; 9: 556–567. doi: 10.1038/nri2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brembilla NC, Senra L, Boehncke W-H. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol 2018; 9: 1682–1682. doi: 10.3389/fimmu.2018.01682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McAllister F, Henry A, Kreindler JL, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-α and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol 2005; 175: 404–412. doi: 10.4049/jimmunol.175.1.404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuestner RE, Taft DW, Haran A, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol 2007; 179: 5462–5473. doi: 10.4049/jimmunol.179.8.5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andersson CK, Adams A, Nagakumar P, et al. Intraepithelial neutrophils in pediatric severe asthma are associated with better lung function. J Allergy Clin Immunol 2017; 139: 1819–1829. doi: 10.1016/j.jaci.2016.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudo M, Melton AC, Chen C, et al. IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med 2012; 18: 547–554. doi: 10.1038/nm.2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang Y, Al-Alwan L, Risse PA, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J 2012; 26: 5152–5160. doi: 10.1096/fj.12-208033 [DOI] [PubMed] [Google Scholar]
- 50.Chang Y, Al-Alwan L, Risse PA, et al. TH17 cytokines induce human airway smooth muscle cell migration. J Allergy Clin Immunol 2011; 127: 1046–1053. doi: 10.1016/j.jaci.2010.12.1117 [DOI] [PubMed] [Google Scholar]
- 51.Fujie H, Niu K, Ohba M, et al. A distinct regulatory role of Th17 cytokines IL-17A and IL-17F in chemokine secretion from lung microvascular endothelial cells. Inflammation 2012; 35: 1119–1131. doi: 10.1007/s10753-011-9419-0 [DOI] [PubMed] [Google Scholar]
- 52.Cheung PFY, Wong CK, Lam CWK. Molecular mechanisms of cytokine and chemokine release from eosinophils activated by IL-17A, IL-17F, and IL-23: implication for Th17 lymphocytes-mediated allergic inflammation. J Immunol 2008; 180: 5625–5635. doi: 10.4049/jimmunol.180.8.5625 [DOI] [PubMed] [Google Scholar]
- 53.Roos AB, Mori M, Gura HK, et al. Increased IL-17RA and IL-17RC in end-stage COPD and the contribution to mast cell secretion of FGF-2 and VEGF. Respir Res 2017; 18: 48. doi: 10.1186/s12931-017-0534-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swaidani S, Liu C, Zhao J, et al. TRAF regulation of IL-17 cytokine signaling. Front Immunol 2019; 10: 1293. doi: 10.3389/fimmu.2019.01293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun D, Novotny M, Bulek K, et al. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol 2011; 12: 853–860. doi: 10.1038/ni.2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garg AV, Ahmed M, Vallejo AN, et al. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci Signal 2013; 6: ra44–ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang L, Yao C, Jin Q, et al. [Effect of interleukin-17 on neutrophil apoptosis in patients with tuberculosis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2014; 30: 833–836. [PubMed] [Google Scholar]
- 58.Wang W, Zhou A, Zhang X, et al. Interleukin 17A promotes pneumococcal clearance by recruiting neutrophils and inducing apoptosis through a p38 mitogen-activated protein kinase-dependent mechanism in acute otitis media. Infect Immun 2014; 82: 2368–2377. doi: 10.1128/IAI.00006-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silverpil E, Glader P, Hansson M, et al. Impact of interleukin-17 on macrophage phagocytosis of apoptotic neutrophils and particles. Inflammation 2011; 34: 1–9. doi: 10.1007/s10753-010-9201-8 [DOI] [PubMed] [Google Scholar]
- 60.Silverpil E, Wright AK, Hansson M, et al. Negative feedback on IL-23 exerted by IL-17A during pulmonary inflammation. Innate Immun 2013; 19: 479–492. doi: 10.1177/1753425912470470 [DOI] [PubMed] [Google Scholar]
- 61.Chen K, Pociask DA, McAleer JP, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One 2011; 6: e20333. doi: 10.1371/journal.pone.0020333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo J, An X, Yao Y, et al. Epigenetic regulation of IL-17-induced chemokines in lung epithelial cells. Mediat Inflamm 2019; 2019: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.You Z, Ge D, Liu S, et al. Interleukin-17 induces expression of chemokines and cytokines in prostatic epithelial cells but does not stimulate cell growth in vitro. Int J Med Biol Front 2012; 18: 629–644. [PMC free article] [PubMed] [Google Scholar]
- 64.Brown RL, Sequeira RP, Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun 2017; 8: 1512. doi: 10.1038/s41467-017-01803-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 2007; 204: 2803–2812. doi: 10.1084/jem.20071397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 2015; 43: 727–738. doi: 10.1016/j.immuni.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Archer NK, Adappa ND, Palmer JN, et al. Interleukin-17A (IL-17A) and IL-17F are critical for antimicrobial peptide production and clearance of Staphylococcus aureus nasal colonization. Infect Immun 2016; 84: 3575–3583. doi: 10.1128/IAI.00596-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-γ, interleukin-2, and tumor necrosis factor-α, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol 1999; 113: 752–759. doi: 10.1046/j.1523-1747.1999.00749.x [DOI] [PubMed] [Google Scholar]
- 69.Toichi E, Torres G, McCormick TS, et al. An anti-IL-12p40 antibody down-regulates type 1 cytokines, chemokines, and IL-12/IL-23 in psoriasis. J Immunol 2006; 177: 4917–4926. doi: 10.4049/jimmunol.177.7.4917 [DOI] [PubMed] [Google Scholar]
- 70.Nakajima K, Kanda T, Takaishi M, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol 2011; 186: 4481–4489. doi: 10.4049/jimmunol.1000148 [DOI] [PubMed] [Google Scholar]
- 71.van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009; 182: 5836–5845. doi: 10.4049/jimmunol.0802999 [DOI] [PubMed] [Google Scholar]
- 72.Ray-Jones H, Eyre S, Barton A, et al. One SNP at a time: moving beyond GWAS in psoriasis. J Invest Dermatol 2016; 136: 567–573. doi: 10.1016/j.jid.2015.11.025 [DOI] [PubMed] [Google Scholar]
- 73.Krueger JG, Fretzin S, Suarez-Farinas M, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol 2012; 130: 145–154. doi: 10.1016/j.jaci.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reich K, Gooderham M, Thaci D, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet 2019; 394: 576–586. doi: 10.1016/S0140-6736(19)30952-3 [DOI] [PubMed] [Google Scholar]
- 75.von Csiky-Sessoms S, Lebwohl M. What's new in psoriasis? Dermatol Clin 2019; 37: 129–136. doi: 10.1016/j.det.2018.11.001 [DOI] [PubMed] [Google Scholar]
- 76.Fitz L, Zhang W, Soderstrom C, et al. Association between serum interleukin-17A and clinical response to tofacitinib and etanercept in moderate to severe psoriasis. Clin Exp Dermatol 2018; 43: 790–797. doi: 10.1111/ced.13561 [DOI] [PubMed] [Google Scholar]
- 77.Yuki T, Tobiishi M, Kusaka-Kikushima A, et al. Impaired tight junctions in atopic dermatitis skin and in a skin-equivalent model treated with interleukin-17. PLoS One 2016; 11: e0161759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O'Connor W J, Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol 2009; 10: 603–609. doi: 10.1038/ni.1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 2006; 116: 1310–1316. doi: 10.1172/JCI21404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sakuraba A, Sato T, Kamada N, et al. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn's disease. Gastroenterology 2009; 137: 1736–1745. doi: 10.1053/j.gastro.2009.07.049 [DOI] [PubMed] [Google Scholar]
- 81.Hohenberger M, Cardwell LA, Oussedik E, et al. Interleukin-17 inhibition: role in psoriasis and inflammatory bowel disease. J Dermatolog Treat 2018; 29: 13–18. doi: 10.1080/09546634.2017.1329511 [DOI] [PubMed] [Google Scholar]
- 82.Wang J, Bhatia A, Krugliak Cleveland N, et al. Rapid onset of inflammatory bowel disease after receiving secukinumab infusion. ACG Case Rep J 2018; 5: e56–e56. doi: 10.14309/crj.2018.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012; 61: 1693–1700. doi: 10.1136/gutjnl-2011-301668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Targan SR, Feagan B, Vermeire S, et al. A randomized, double-blind, placebo-controlled phase 2 study of brodalumab in patients with moderate-to-severe Crohn's disease. Am J Gastroenterol 2016; 111: 1599–1607. doi: 10.1038/ajg.2016.298 [DOI] [PubMed] [Google Scholar]
- 85.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003; 52: 65–70. doi: 10.1136/gut.52.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Du J, Han J-C, Zhang Y-J, et al. Single-nucleotide polymorphisms of IL-17 gene are associated with asthma susceptibility in an Asian population. Med Sci Monit 2016; 22: 780–787. doi: 10.12659/MSM.895494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silva MJ, de Santana MBR, Tosta BR, et al. Variants in the IL17 pathway genes are associated with atopic asthma and atopy makers in a South American population. Allergy Asthma Clin Immunol 2019; 15: 28. doi: 10.1186/s13223-019-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawaguchi M, Takahashi D, Hizawa N, et al. IL-17F sequence variant (His161Arg) is associated with protection against asthma and antagonizes wild-type IL-17F activity. J Allergy Clin Immunol 2006; 117: 795–801. doi: 10.1016/j.jaci.2005.12.1346 [DOI] [PubMed] [Google Scholar]
- 89.Fahy JV, Kim KW, Liu J, et al. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol 1995; 95: 843–852. doi: 10.1016/S0091-6749(95)70128-1 [DOI] [PubMed] [Google Scholar]
- 90.Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 1999; 160: 1001–1008. doi: 10.1164/ajrccm.160.3.9812110 [DOI] [PubMed] [Google Scholar]
- 91.Fahy JV. Eosinophilic and neutrophilic inflammation in asthma. Proc Am Thorac Soc 2009; 6: 256–259. doi: 10.1513/pats.200808-087RM [DOI] [PubMed] [Google Scholar]
- 92.Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol 1999; 162: 2347–2352. [PubMed] [Google Scholar]
- 93.Fujisawa T, Chang MM, Velichko S, et al. NF-κB mediates IL-1β- and IL-17A-induced MUC5B expression in airway epithelial cells. Am J Respir Cell Mol Biol 2011; 45: 246–252. doi: 10.1165/rcmb.2009-0313OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Molet S, Hamid Q, Davoine F, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 2001; 108: 430–438. doi: 10.1067/mai.2001.117929 [DOI] [PubMed] [Google Scholar]
- 95.Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med 2003; 97: 726–733. doi: 10.1053/rmed.2003.1507 [DOI] [PubMed] [Google Scholar]
- 96.Sun YC, Zhou QT, Yao WZ. Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chin Med J 2005; 118: 953–956. [PubMed] [Google Scholar]
- 97.Irvin C, Zafar I, Good J, et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol 2014; 134: 1175–1186. doi: 10.1016/j.jaci.2014.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hinks TS, Zhou X, Staples KJ, et al. Innate and adaptive T cells in asthmatic patients: relationship to severity and disease mechanisms. J Allergy Clin Immunol 2015; 136: 323–333. doi: 10.1016/j.jaci.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008; 372: 1107–1119. doi: 10.1016/S0140-6736(08)61452-X [DOI] [PubMed] [Google Scholar]
- 100.Johnston SL, Pattemore PK, Sanderson G, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995; 310: 1225–1229. doi: 10.1136/bmj.310.6989.1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wimalasundera SS, Katz DR, Chain BM. Characterization of the T cell response to human rhinovirus in children: implications for understanding the immunopathology of the common cold. J Infect Dis 1997; 176: 755–759. doi: 10.1086/514101 [DOI] [PubMed] [Google Scholar]
- 102.Brandt EB, Kovacic MB, Lee GB, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol 2013; 132: 1194–1204. doi: 10.1016/j.jaci.2013.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siew LQC, Wu SY, Ying S, et al. Cigarette smoking increases bronchial mucosal IL-17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clin Exp Allergy 2017; 47: 740–750. doi: 10.1111/cea.12907 [DOI] [PubMed] [Google Scholar]
- 104.Lezmi G, Abou Taam R, Dietrich C, et al. Circulating IL-17-producing mucosal-associated invariant T cells (MAIT) are associated with symptoms in children with asthma. Clin Immunol 2018; 188: 7–11. doi: 10.1016/j.clim.2017.11.009 [DOI] [PubMed] [Google Scholar]
- 105.Chesne J, Braza F, Chadeuf G, et al. Prime role of IL-17A in neutrophilia and airway smooth muscle contraction in a house dust mite-induced allergic asthma model. J Allergy Clin Immunol 2015; 135: 1643–1643. doi: 10.1016/j.jaci.2014.12.1872 [DOI] [PubMed] [Google Scholar]
- 106.Wilson RH, Whitehead GS, Nakano H, et al. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med 2009; 180: 720–730. doi: 10.1164/rccm.200904-0573OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He R, Oyoshi MK, Jin H, et al. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci USA 2007; 104: 15817–15822. doi: 10.1073/pnas.0706942104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scanlon KM, Hawksworth RJ, Lane SJ, et al. IL-17A induces CCL28, supporting the chemotaxis of IgE-secreting B cells. Int Arch Allergy Immunol 2011; 156: 51–61. doi: 10.1159/000322178 [DOI] [PubMed] [Google Scholar]
- 109.Ostling J, van Geest M, Schofield JPR, et al. IL-17-high asthma with features of a psoriasis immunophenotype. J Allergy Clin Immunol 2019; 144: 1198–1213. doi: 10.1016/j.jaci.2019.03.027 [DOI] [PubMed] [Google Scholar]
- 110.Cowan DC, Cowan JO, Palmay R, et al. Effects of steroid therapy on inflammatory cell subtypes in asthma. Thorax 2010; 65: 384–390. doi: 10.1136/thx.2009.126722 [DOI] [PubMed] [Google Scholar]
- 111.Zijlstra GJ, Ten Hacken NH, Hoffmann RF, et al. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur Respir J 2012; 39: 439–445. doi: 10.1183/09031936.00017911 [DOI] [PubMed] [Google Scholar]
- 112.McKinley L, Alcorn JF, Peterson A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 2008; 181: 4089–4097. doi: 10.4049/jimmunol.181.6.4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol 2013; 6: 335–346. doi: 10.1038/mi.2012.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nanzer AM, Chambers ES, Ryanna K, et al. Enhanced production of IL-17A in patients with severe asthma is inhibited by 1alpha,25-dihydroxyvitamin D3 in a glucocorticoid-independent fashion. J Allergy Clin Immunol 2013; 132: 297–304. doi: 10.1016/j.jaci.2013.03.037 [DOI] [PubMed] [Google Scholar]
- 115.Singhania A, Wallington JC, Smith CG, et al. Multi-tissue transcriptomics delineates the diversity of airway T cell functions in asthma. Am J Respir Cell Mol Biol 2018; 58: 261–270. doi: 10.1165/rcmb.2017-0162OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Christenson SA, van den Berge M, Faiz A, et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J Clin Invest 2019; 129: 169–181. doi: 10.1172/JCI121087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Simpson JL, Daly J, Baines KJ, et al. Airway dysbiosis: Haemophilus influenzae and tropheryma in poorly controlled asthma. Eur Respir J 2016; 47: 792–800. doi: 10.1183/13993003.00405-2015 [DOI] [PubMed] [Google Scholar]
- 118.Essilfie A-T, Simpson JL, Dunkley ML, et al. Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma. Thorax 2012; 67: 588–599. doi: 10.1136/thoraxjnl-2011-200160 [DOI] [PubMed] [Google Scholar]
- 119.Essilfie AT, Simpson JL, Horvat JC, et al. Haemophilus influenzae infection drives IL-17-mediated neutrophilic allergic airways disease. PLoS Pathog 2011; 7: e1002244. doi: 10.1371/journal.ppat.1002244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Essilfie AT, Horvat JC, Kim RY, et al. Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax 2015; 70: 458–467. doi: 10.1136/thoraxjnl-2014-206067 [DOI] [PubMed] [Google Scholar]
- 121.Li W, Zhang X, Yang Y, et al. Recognition of conserved antigens by Th17 cells provides broad protection against pulmonary Haemophilus influenzae infection. Proc Natl Acad Sci 2018; 115: E7149–E7157. doi: 10.1073/pnas.1802261115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao S, Jiang Y, Yang X, et al. Lipopolysaccharides promote a shift from Th2-derived airway eosinophilic inflammation to Th17-derived neutrophilic inflammation in an ovalbumin-sensitized murine asthma model. J Asthma 2017; 54: 447–455. doi: 10.1080/02770903.2016.1223687 [DOI] [PubMed] [Google Scholar]
- 123.Barboza R, Camara NO, Gomes E, et al. Endotoxin exposure during sensitization to Blomia tropicalis allergens shifts Th2 immunity towards a TH17-mediated airway neutrophilic inflammation: role of TLR4 and TLR2. PLoS ONE 2013; 8: e67115. doi: 10.1371/journal.pone.0067115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mebratu YA, Tesfaigzi Y. IL-17 plays a role in respiratory syncytial virus-induced lung inflammation and emphysema in elastase and LPS-injured mice. Am J Respir Cell Mol Biol 2018; 58: 717–726. doi: 10.1165/rcmb.2017-0265OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang X, Wang Y, Zhao S, et al. Long-term exposure to low-dose Haemophilus influenzae during allergic airway disease drives a steroid-resistant neutrophilic inflammation and promotes airway remodeling. Oncotarget 2018; 9: 24898–24913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Holmes AM, Solari R, Holgate ST. Animal models of asthma: value, limitations and opportunities for alternative approaches. Drug Discov Today 2011; 16: 659–670. doi: 10.1016/j.drudis.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 127.Zosky GR, Sly PD. Animal models of asthma. Clin Exp Allergy 2007; 37: 973–988. doi: 10.1111/j.1365-2222.2007.02740.x [DOI] [PubMed] [Google Scholar]
- 128.Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 2015; 7: 301ra129. doi: 10.1126/scitranslmed.aab3142 [DOI] [PubMed] [Google Scholar]
- 129.Lynch JP, Ferreira MA, Phipps S. Th2/Th17 reciprocal regulation: twists and turns in the complexity of asthma phenotypes. Ann Transl Med 2016; 4: Suppl. 1, S59. doi: 10.21037/atm.2016.10.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Poynter ME. Do insights from mice imply that combined Th2 and Th17 therapies would benefit select severe asthma patients? Ann Transl Med 2016; 4: 505. doi: 10.21037/atm.2016.11.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim D, McAlees JW, Bischoff LJ, et al. Combined administration of anti-IL-13 and anti-IL-17A at individually sub-therapeutic doses limits asthma-like symptoms in a mouse model of Th2/Th17 high asthma. Clin Exp Allergy 2019; 49: 317–330. doi: 10.1111/cea.13301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Manni ML, Mandalapu S, McHugh KJ, et al. Molecular mechanisms of airway hyperresponsiveness in a murine model of steroid-resistant airway inflammation. J Immunol 2016; 196: 963–977. doi: 10.4049/jimmunol.1501531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shum AK. T cell types that take your breath away. Sci Transl Med 2015; 7: 301fs333. [DOI] [PubMed] [Google Scholar]
- 134.Busse WW, Holgate S, Kerwin E, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med 2013; 188: 1294–1302. doi: 10.1164/rccm.201212-2318OC [DOI] [PubMed] [Google Scholar]
- 135.Pavord ID, Beasley R, Agusti A, et al. After asthma: redefining airways diseases. Lancet 2017; 391: 350–400. doi: 10.1016/S0140-6736(17)30879-6 [DOI] [PubMed] [Google Scholar]
- 136.O'Byrne PM, Metev H, Puu M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2016; 4: 797–806. doi: 10.1016/S2213-2600(16)30227-2 [DOI] [PubMed] [Google Scholar]
- 137.Eich A, Urban V, Jutel M, et al. A randomized, placebo-controlled phase 2 trial of CNTO 6785 in chronic obstructive pulmonary disease. COPD 2017; 14: 476–483. doi: 10.1080/15412555.2017.1335697 [DOI] [PubMed] [Google Scholar]
- 138.Staton TL, Peng K, Owen R, et al. A phase I, randomized, observer-blinded, single and multiple ascending-dose study to investigate the safety, pharmacokinetics, and immunogenicity of BITS7201A, a bispecific antibody targeting IL-13 and IL-17, in healthy volunteers. BMC Pulm Med 2019; 19: 5. doi: 10.1186/s12890-018-0763-9 [DOI] [PMC free article] [PubMed] [Google Scholar]