

Abstract
Mild traumatic brain injury results in aberrant free radical generation, which is associated with oxidative stress, secondary injury signaling cascades, mitochondrial dysfunction, and poor functional outcome. Pharmacological targeting of free radicals with antioxidants has been examined as an approach to treatment, but has met with limited success in clinical trials. Conventional antioxidants that are currently available scavenge a single free radical before they are destroyed in the process. Here, we report for the first time that a novel regenerative cerium oxide nanoparticle antioxidant reduces neuronal death and calcium dysregulation after in vitro trauma. Further, using an in vivo model of mild lateral fluid percussion brain injury in the rat, we report that cerium oxide nanoparticles also preserve endogenous antioxidant systems, decrease macromolecular free radical damage, and improve cognitive function. Taken together, our results demonstrate that cerium oxide nanoparticles are a novel nanopharmaceutical with potential for mitigating neuropathological effects of mild traumatic brain injury and modifying the course of recovery.
Keywords: cerium oxide, nanoparticles, oxidative stress, traumatic brain injury
Introduction
Mild traumatic brain injury (mTBI) affects millions each year, absorbing substantial annual healthcare costs. Despite extensive research, limited treatments exist because of the complex primary and secondary injury mechanisms underlying the pathophysiology. In mTBI, the primary injury often results in little or no cell death in the brain.1 The secondary injury phase is characterized by biochemical mechanisms that produce cellular dysfunction into the post-injury period, resulting in a sequelae of pathologies. One of the mechanisms of secondary injury is aberrant free radical production and increased oxidative stress.2–6 Despite endogenous brain antioxidants such as superoxide dismutase (SOD), catalase, and glutathione (GSH); excessive free radical production after TBI overwhelms endogenous defenses, leaving the brain in an abnormal state of high oxidative stress, persisting into the chronic phase after injury.7,8 This increase in oxidative stress has been coupled to alterations in cognitive performance.9–11 Traditional antioxidants have been utilized in attempts to mitigate the pathological effects of TBI,12–14 but have met with limited success in clinical trials.15 A single molecule of these antioxidants scavenges only a single free radical, before loss of activity. Because of the chronic, high levels of oxidative stress induced by TBI, more efficacious antioxidants need to be active for extended periods of time.
Cerium is a rare earth element of the lanthanide series, with multiple valence states (+3, fully reduced; or +4, fully oxidized) imparting redox activity. The crystal lattice structure of the oxide form affords even more redox capacity because of electron “holes” or oxygen defects in the nanoparticle matrix.16 Creation and annihilation of oxygen vacancies and alterations in cerium valence impart exceptional redox activity to cerium oxide nanoparticles (CeONPs), which is further enhanced by the increased surface area and quantum lattice alterations that occur at the nanoscale.17,18 In contrast to traditional antioxidants, the radical scavenging activity of CeONPs is regenerative under biological conditions,19–21 permitting sustained activity.
Research has shown that CeONPs preserved the life span of mixed organotypic cultures of brain cells and pure neurons, while preserving normal calcium signaling during the extended life span.19,22 CeONPs also protected neurons and other cell types from free radical challenge and decreased inflammatory functions.23–27 In Drosophila models, CeONPs extended the life span and protected flies from challenge with free radical generating agents.23 In a rat model of Parkinson's disease, we have shown that CeONPs restored dopamine levels in the striatum, and dramatically improved neuronal survival in the substantia nigra.28 CeONPs have also been used to enhance vascularization of bone grafts29 and preserve retinal function in models of macular degeneration and retinal dysfunction.30
Here, we test the ability of CeONPs to improve survival and signaling function in an in vitro model of mTBI, and decrease oxidative stress and improve functional outcome after in vivo mTBI in the rat. We demonstrate for the first time that CeONPs increase neuronal survival and preserve intracellular free signaling in a tissue culture model of TBI, reduce oxidative stress, preserve endogenous calcium antioxidant activity, and ultimately improve cognitive function when administered after mTBI in the rat.
Methods
CeONPs
CeONPs were obtained as a 1.2% solution in water, pharmaceutical grade, from Nanophase, Inc. (Romeoville, IL). Stocks were probe sonicated and resuspended in saline-citrate buffer at the desired concentrations. This buffer maintains a uniform particle suspension and prevents agglomeration.22,31 The average particle size in suspension was 10 nm as determined by transmission electron microscopy. Surface area (Brunauer–Emmett–Teller [BET]) was 88.6 m2/g with a BET diameter of 9.7. The amount of Ce3+ in the particles as delivered was 33.6%, and CeONPs were endotoxin-free.
To confirm nanoparticle distribution to the brain, we administered rats (n = 6) a single tail vein injection. As direct measurement of the nanoparticle is not possible, we measured tissue cerium as an indicator of nanoparticle accumulation, using inductively coupled plasma mass spectrometry and inductively coupled optical emission spectroscopy (analyzed by Cerium Labs Inc., Austin, TX).
In vitro TBI
Mixed organotypic neuronal cultures were prepared from 1–2 day-old neonatal rats and grown in silastic-bottomed tissue culture wells (Flex Plate, Flex Cell, Hillsborough, NC) as previously described.32–35 Cells were injured using a model 94A Cell Injury Controller (Custom Design & Fabrication, Virginia Commonwealth University, Richmond, VA).36 Briefly, the injury controller delivers a 50 msec pulse of compressed gas, which transiently displaces the silastic membrane, along with its adherent cells, to varying degrees controlled by the pulse pressure. We have arbitrarily defined a membrane displacement of 5.5 mm as a mild stretch, 6.5 mm as a moderate stretch, and 7.5 mm as a severe stretch.33,34 These degrees of membrane displacement correspond to a biaxial strain (or stretch) of 0.31 (31%), 0.38 (38%), and 0.54 (54%), respectively. This range of strain has been shown to be relevant to what occurs in humans subject to rotational acceleration-deceleration injuries.37
On days 12–14 in vitro, cultures were delivered a mild or moderate injury. CeONPs (1 nM–1 μM) were delivered to the cultures 1 h later. Controls received either vehicle or CeONPs, at the same time points, without injury. Neuronal damage was assessed at 24 h after injury by measuring uptake of propidium iodide (PrI). Cultures were stained with PrI as previously described33 and injured cells were counted and expressed as injured cells/mg protein. Protein was determined by the method of Lowry.38 To assure that cell loss or detachment had not occurred during the 24 h post-injury period, cells were permeabilized and total PrI stained nuclei were counted and were consistent with those of uninjured cultures. Total protein in the supernatant media was also measured, which remained unchanged between injured and control cultures.
For measurement of [Ca2+]i, neurons were loaded with Fura-2 and [Ca2+]i was determined with microspectrophotometric imaging, as previously described by our group.32,34,35 Results represent ∼50 neurons examined per culture field, in triplicate, in three separate experiments.
In vivo TBI
The University Institutional Animal Care and Use Committee at Virginia Tech approved the experimental protocols described herein. Adult male Sprague–Dawley rats (Harlan Inc., Livermore, CA) weighing 300–350 g were acclimated for at least 3 days with food and water provided ad libitum. After acclimation, animals were delivered a mild lateral fluid percussion brain injury (mLFPI) as previously described.39 Briefly, rats were anesthetized with an induction of 4% isoflurane with oxygen and maintained at 2.5% isoflurane with oxygen during the procedure. A standard craniectomy was performed under aseptic conditions by trephining a 4.8 mm hole between bregma and lambda above the right parietal cortex. A modified 20G needle hubcap (2.6 mm inside diameter) was placed over the exposed dura and bonded in place with cyanoacrylate adhesive. Dental cement was poured around the hub for support during injury. mLFPI was delivered directly to the intact dura, using a fluid percussion device (Custom Design & Fabrication, Virginia Commonwealth University, Richmond, VA), consisting of a Plexiglas cylindrical reservoir 60 cm long and 4.5 cm in diameter and filled with sterile distilled water. Fitted at the end of the metal housing is a 5 mm tube that terminates with a male Luer-Loc fitting. This fitting was connected to the female fitting implanted in the rat. Injury was produced by the metal pendulum striking the piston of the injury device, creating a pressure pulse and injecting a volume of water into the closed cranial cavity, producing a brief displacement and deformation of brain tissue. Animals received an impact with an average peak pressure of 1.47 atm, which is consistent with mild injury.40,41 No significant differences in peak pressures were observed between treatment groups.
The biological actions of CeONPs are known to have a bell-shaped dose-response curve and accumulate in brain.19,23 Therefore, large, repetitive daily doses over lengthy time periods were not used initially. Dosing used in these studies was based on that determined from our prior studies in a Parkinson's disease model,28 and from studies in which animals were pretreated with CeONPs prior to injury.42 The dosing paradigms were as follows: three injections over 3 h post-injury (3 × ) or five injections over 48 h post-injury (5 × ). The 3 × groups received injections at 1 min, 15 min, and 3 h following injury, whereas the 5 × groups received additional injections at 24 and 48 h. A low dose of 0.05 μg/g (LD) and a high dose 0.5 μg/g (HD) was used within each injection paradigm. Therefore, four CeONP treatment groups were included in this study: 3 × LD, 3 × HD, 5 × LD, and 5 × HD. The total cumulative dose for the 3 × dosing regimen was 0.15 μg/g (LD) and 1.5 μg/g (HD). For the 5 × dosing, the total cumulative LD and HD were 0.25 μg/g and 2.5 μg/g, respectively. Sham controls received the craniectomy surgery without any injury or drug administration. Injury-only animals received the craniectomy surgery and injury, but were not administered CeONPs.
Assessment of oxidative stress in the brain
For biochemical assessment of oxidative stress, animals were euthanized 2h after the last injection of CeONPs. Brains were rapidly collected on ice in a cold room. Upon collection, brains were inspected for signs of bleeding or hematoma at the injury site, but none were found. Brains were placed into an ice-cold tissue slicing matrix (Electron Microscopy Sciences) and divided into three sections: a rostral section, a middle section containing the injury site, and a caudal section. Assays were conducted on the middle sections, containing the site of injury. Sections were then divided into ipsilateral and contralateral and placed in ice-cold phosphate-buffered saline (pH 7.4) containing butylated hydroxytoluene. Sections were lysed by sonication for 30 sec (Fisher Model 100 Sonic Dismembrator), followed by storage at −80°C. Total protein for each section was measured using the method of Lowry.38
Oxidative stress was measured by two main parameters. The first was activity of endogenous antioxidants and the second was by measurement of oxidative damage to cellular components. For measurement of endogenous antioxidants, the activity of the enzymes SOD and catalase, and the amount of reduced and oxidized GSH were determined. Catalase activity was measured by the decomposition of H2O2 in a methanol medium, producing formaldehyde. Formaldehyde was measured colorimetrically with 4-amino-3-hydrzino-5-mercapto-1,2,4-triazole.43 Results are expressed as H2O2 consumed/min/mg protein. Total SOD (Cu/Zn, Mn, and Fe) was measured using the xanthine/xanthine oxidase and hypoxanthine system to generate superoxide radicals, which were then measured by conversion of a tetrazolium salt to formazan dye, using a kit from Cayman Chemical.44 One unit of SOD was defined as the amount of enzyme that produced 50% dismutation of the superoxide radical. GSH/GSH disulfide (GSSG) ratios were also measured with a kit from Cayman Biochemical, using GSH reductase for the quantification of GSH.45
For measurement of oxidative damage to cellular macromolecules, we assessed lipid peroxidation via formation of lipid hydroperoxides (LOOH) and 3-nitrotyrosine (NT). LOOH are a widely acknowledged byproduct of lipid peroxidation and a marker for oxidative stress damage.46 LOOH were measured using an assay kit from Cayman Chemical. First, LOOH were extracted into chloroform/methanol as per kit directions, using aliquots of brain homogenate containing 1.0 mg of protein. LOOH were measured directly by virtue of the redox reaction of the LOOHs with ferrous ions, to form a stable chromogen with thiocyanate, allowing for measurement of absorption at 500 nm. Each assay was performed in triplicate. Results were measured as the total amount of LOOH per sample, and are expressed at a percentage of control. NT was measured by competitive enzyme-linked immunosorbent assay (ELISA) (Abcam, Cambridge, MA).47 NT is a product of NO-dependent damage to proteins, which marks the protein for subsequent degradation.48 Briefly, NT standard or brain homogenate (50 mL) was added to each well of a 96 well plate followed by addition of horseradish peroxidase conjugated anti-NT antibody. Plates were incubated for 1 h after which they were washed four times. Trimethylbenzidine was added and plates were incubated for 30 min in the dark, producing a colored product that was read in an ELISA plate-reader at 450 nm.
Behavioral assessment following injury
To determine the effect of CeONPs on cognitive function after mTBI, we conducted a behavioral study to evaluate short-term memory deficits utilizing the standardized novel object recognition test (NOR).49–51 Animals were divided into eight groups: sham, injury-only, vehicle, 3 × LD, 3 × HD, 5 × LD, and 5 × HD. The same injury and treatment conditions were used as described previously. An additional vehicle group was included that received injury and isovolumetric injections of vehicle.
NOR testing was conducted 14 days following injury as previously described by our group.52 Briefly, animals were acclimated to the training environment 2 days prior to injury. The task then occurred in two phases 14 days following injury: T1 (familiarization phase) and T2 (testing phase). During T1, a rat was placed in the 80 × 80 cm testing arena with two identical objects and allowed to explore for 5 min. Twenty minutes later, the rat was returned to the testing arena, where one of the objects had been swapped with a new object. Novel object location was randomly assigned to each T2 trial. Animals were allowed 5 min to explore. All trials were recorded and tracked using EthoVision XT™ (Noldus Information Technology, Leesburg, VA). Three-point tracking was performed during each trial and included tracking of the nose-point (blue arrows), center of the body (red squares), and base of the tail (purple squares) (Fig. 6) . Exploration was measured when the nose-point of the animal entered a 4 cm zone outside of the object. Independent assessors blind to treatments verified accurate tracking of the nose-point.
FIG. 6. .

Mild traumatic brain injury (mTBI) alters object exploration in the Novel Object Recognition Test. This figure shows a representative EthoVision tracing of the Novel Object Recognition test quantified in Figure 7. The yellow arrows represent the novel object. Blue arrows in the tracking represent nose-point tracking, whereas red squares indicate body center tracking and purple squares indicate tail base tracking. The injured animal, treated with vehicle (left) spent similar time at both objects, hence an inability to discriminate between novel and familiar objects. The animal receiving a chronic high dose of cerium oxide nanoparticles (CeONPs) showed preferential investigation of the novel object, indicative of improved memory.
For analysis, a discrimination index was calculated for each trial (time spent exploring the novel object divided by the total time at either object) at the 1 min time point in the T1 phase.49,53 A ratio of 0.5 indicates equal exploration of both objects during the trial. Consistent with our published protocols,52 intact memory was interpreted as exploring the novel object significantly more than the familiar object (score >0.5). A memory deficit was interpreted as null object preference (≤0.5); the animal did not discriminate the novel object. Poor discrimination of the novel objects and less time spent in exploration reflects damage to memory formation.49,51
Statistical analysis
For molecular and cellular assessments following in vivo and in vitro injury models, statistical comparisons were made among treatment groups using a two way ANOVA with Tukey's post-hoc tests. For behavioral assessments, average NOR preference for each treatment group was compared with a NOR preference of 0.5 using a Students t test. P values <0.05 were considered significant.
Results
Cerium oxide nanoparticles
Figure 1 shows a transmission electron microscopy (TEM) of the “as-delivered” CeONP suspension, with an average particle size of 10 nm and a uniform, non-agglomerated mixture. CeONPs suspended in saline-citrate reached the brain, as seen in Figure 2, which reports accumulation of cerium within the brain and/or cerebral vasculature 24 h after administration of a single injection to uninjured animals. As shown, a single 0.05 μg/g dose of CeONPs administered to the rat via the tail vein increased levels of cerium by 3.6-fold, and a 0.5 μg/g dose increased them by 5.8-fold in the brain and/or cerebral vasculature. We would note that in our prior studies19,23 we found that CeONPs are not metabolized in the brain and accumulate after dosing, remaining in tissues for up to 6 months after administration.54 No adverse pathological effects were produced by the doses of CeONPs used in these studies, or doses 10-fold higher.54
FIG. 1.
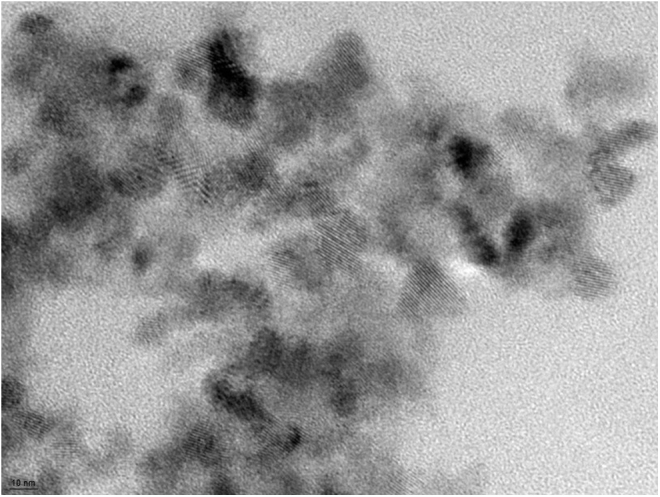
Cerium oxide nanoparticles. Representative transmission electron microscopy (TEM) image of the nanoparticle solution as delivered, in saline-citrate buffer (average particle size 10 nm). Note lack of agglomeration and uniform particle size. Scale bar = 10 nm.
FIG. 2.

Brain distribution of cerium oxide nanoparticles (CeONPs). As a measurement of CeONP content, brain cerium concentrations were measured by inductively coupled plasma mass spectrometry (ICP-MS) and inductively coupled plasma optical emission spectrometry (ICP-OES) after a single injection of CeONP at the concentrations shown (Cerium Labs, Austin, TX). Even animals unexposed to CeONP have a basal level of cerium in the brain, as observed in saline-citrate treated controls. Note the dose-dependent increase in brain cerium with increasing concentration of CeONP. *Significant from saline, p < 0.01.
CeONPs decrease neuronal damage after in vitro TBI
Our previous work demonstrated that CeONPs protect brain and other cell types from oxidative stress.19 Because TBI increases oxidative stress, we tested the hypothesis that CeONPs improve neuronal survival and maintain function after in vitro TBI. These studies utilized a well-characterized in vitro model of TBI that demonstrates delayed neuronal death, calcium dysregulation, and many other similarities with mammalian TBI.33–35 We have previously shown that in vitro TBI induces neuronal damage and death by 24 h post-injury.35 We reasoned that if CeONPs are potent antioxidants, then post-injury treatment should decrease neuronal death and damage observed 24 h post-injury.
Uninjured cultures showed trace amounts of PrI uptake (Fig. 3A). As expected, mild or moderate injury increased PrI uptake at 24 h. However, in cells treated with CeONPs 1 h post-injury, PrI uptake was reduced as compared with untreated cultures, over the concentration range tested, with 10 nM CeONP being the most effective dose. At the mild injury level, 10 nM CeONPs decreased PrI uptake to control (uninjured) levels, whereas at the moderate injury level, 10 nM CeONP reduced PrI uptake by 78%. The highest dose examined, 1 μM CeONPs, reduced PrI uptake at 24 h post-injury, but was not as efficacious as the lower doses, similar to responses we have seen in other studies.19 We hypothesize that because CeONPs are particulate, higher doses may be unable to enter the cell because of limited uptake mechanisms, or may induce damage by a high concentration of extracellular particulates. These data demonstrate that CeONPs are highly efficacious at blocking neuronal damage 24 h after in vitro injury.
FIG. 3.

Cerium oxide nanoparticles (CeONPs) increase neuronal survival and improve glutamate signaling after in vitro traumatic injury. In panel a, mixed organotypic brain cell cultures underwent in vitro traumatic brain injury (TBI) at mild (5.5 mm stretch) and moderate (6.5 mm stretch) injury levels. CeONPs were delivered 1 h post-injury, at the indicated concentrations. Controls received saline-citrate. Neuronal death or damage was quantified by propidium iodide (PrI) uptake 24 h after injury. Results are expressed as injured cells/mg protein and are representative of three separate experiments performed in triplicate. #Significant from uninjured cultures, p < 0.01; *Significant from injury alone, p < 0.01. For panel b, cultures underwent in vitro TBI, followed by treatment with 10 nM CeONP 1 h later (right hand set of bars) or saline-citrate (left hand set of bars). The glutamate-stimulated change in [Ca2+]i was measured with Fura-2 microspectrophotometery and imaging. Results represent the μ ± SE of the change in [Ca2+]i produced by glutamate stimulation, from three separate experiments each with at least 50 neurons in the field, performed in duplicate. CeONP treatment blocked the perturbations in glutamate-stimulated calcium signaling after injury. *Significant from no injury, p < 0.01.
CeONPs improve glutamate-mediated calcium signaling after in vitro TBI
Next, we tested the hypothesis that CeONPs could improve neuronal function after in vitro injury. In prior work, we demonstrated alterations in glutamate-stimulated [Ca2+]i signaling after in vitro injury.32–35 Here, we determined whether CeONP treatment could abrogate these effects.
Uninjured neurons stimulated with 100 μM glutamate responded with an average change in [Ca2+]i of 203 ± 12 nM, which was similar in uninjured neurons treated with CeONPs (Fig. 3B). Twenty-four and 48 h after mild injury (Fig. 3B), the response to glutamate was decreased, indicating perturbation of signaling, as we have previously reported.33 However, in cells treated with CeONPs 1 h after mild injury, the response to glutamate was the same as that in uninjured cells. Twenty-four hours after moderate injury, the neuronal response to glutamate was dramatically increased, suggestive of excitotoxicity at this increased level of injury, as we have previously shown.33 Then, at 48 h after injury, the response to glutamate was depressed, below normal levels, consistent with our prior reports. In contrast, cells treated with CeONPs showed no aberrations in glutamate signaling at 24 and 48 h after mild or moderate in vitro injury. These results indicate that CeONPs, delivered 1 h after injury, can block injury-induced aberrant glutamate signaling, possibly preserving neuronal function after traumatic injury.
CeONPs preserve endogenous antioxidants in a mammalian model of mTBI
Because CeONPs preserved neuronal survival and function after in vitro injury, we tested the efficacy of CeONPs using an in vivo model of mTBI. For these studies, male Sprague–Dawley rats were delivered an mLFPI of 1.47 atm, followed by delivery of CeONP at the 3 × LD, 3 × HD, 5 × LD, and 5 × HD injection schemes, as described in the Methods section. Animals were euthanized and the brain removed 2 h after the last dose of CeONPs, at a total of 24 or 48 h after injury.
To determine whether CeONPs preserve endogenous antioxidant function following TBI, we investigated the activity of SOD, catalase, and the ratio of reduced GSH/oxidized GSH in the ipsilateral and contralateral brains of rats delivered mTBI or shams, at 24 and 48 h after injury, with or without 3 × or 5 × CeONP treatment. As shown in Figure 4A, catalase activity significantly decreased in the ipsilateral and contralateral sides of injured animals as compared with shams, at both 24 and 48 h after injury, which is consistent with TBI-induced oxidative stress damage.11 CeONP at the 3 × LD significantly improved catalase activity compared with the injury-only animals, whereas the 3 × HD administration returned catalase activity to sham levels. Similar results were observed with the 5 × dosing paradigm (Fig. 4A). Injured animals displayed a continued decrease in catalase activity in ipsilateral and contralateral hemispheres at 48 h after injury. CeONP treatment significantly improved catalase levels in both the ipsi- and contralateral sides at the LD. The 5 × HD administration of CeONPs increased catalase activity above that seen in shams. This is not that surprising, because CeONPs are known to exhibit catalase-like activity,18 and this increase could be caused by the accumulation of CeONPs in the brain.
FIG. 4.

Cerium oxide nanoparticles (CeONPs) restored endogenous antioxidants after mild traumatic brain injury (mTBI) in vivo. Panel a shows that CeONP administration significantly increased catalase activity, restoring normal levels at higher doses (p < 0.01). In panel b, similar improvement in function is noted for superoxide dismutase (SOD), with chronic high doses being equivalent to shams. In panel c, we observe that CeONP improves glutathione (GSH)/ GSH disulfide (GSSG) ratios, retuning them to near normal levels with the chronic high dose paradigm. *Significant from sham, p < 0.01; #Significant from injury, p < 0.01.
SOD plays a critical role in degradation of toxic superoxide radicals. Because SOD activity is decreased by TBI, we determined whether CeONPs could block this effect. Total (Cu/Zn, Mn, and Fe) SOD activity was significantly decreased in both ipsilateral and contralateral hemispheres after injury, as compared with shams (Fig. 4B), with the greatest decrease in the ipsilateral hemisphere as expected. Once again, these deficits were mitigated through CeONP administration. Both the 3 × LD and 3 × HD treatments significantly improved total SOD activity levels, with the HD returning SOD activity to near that of shams. At 48 h post-injury, SOD levels remained significantly depressed in both ipsi- and contralateral sections (Fig. 4B). Animals receiving the 5 × LD of CeONPs again showed significantly improved SOD levels, compared with injury-only animals. However in the 5 × HD group, total SOD activity was returned to levels consistent with shams.
Reduced GSH is oxidized to GSSG upon the conversion of peroxide radicals to water, and is subsequently regenerated by GSH reductase. The ratio of the two (GSH/GSSG) is used as an indicator of the GSH distribution between redox states, and the antioxidant capacity of a tissue. Prior studies have demonstrated that this critical endogenous antioxidant ratio declines after TBI. Consistent with these observations, Figure 4C shows that the GSH/GSSG ratio substantially declined 24 h after injury. The 3 × LD and 3 × HD injection schemes significantly improved the GSH/GSSG ratio, with the HD being more effective, particularly on the contralateral side of the brain where GSH/GSSG was returned to the level observed in shams. At 48 h post-injury, the GSH/GSSG ratio remained depressed in injury-only animals. Five injections of CeONPs significantly increased GSH/GSSG ratio on both ipsi- and contralateral sides. The contralateral side GSH/GSSG ratio was returned to normal following the 5 × LD, whereas both sides achieved normal GSH/GSSG ratios with the 5 × HD dose of CeONPs. Taken together, these results demonstrate that CeONPs significantly improve endogenous antioxidant activity after mLFPI.
CeONPs reduce macromolecular oxidative damage in vivo
Next, we investigated whether CeONPs could mitigate macromolecular damage caused by excessive free radical production in brain tissue. For these studies, we measured two byproducts of radical damage: LOOH and NT. LOOH are a widely acknowledged byproduct of lipid peroxidation and marker for oxidative damage to the cellular membrane.46 As shown in Figure 5A, mLFPI increased LOOH as compared with sham at 24 and 48 h after injury, indicating oxidative damage to cellular lipids. Three drug injections of CeONPs mitigated this increase in LOOH. The HD was most efficacious, in which CeONPs returned levels of LOOH to those consistent with shams in both ipsi- and contralateral sections. When delivered in the five injection dosing paradigm, both LD and HD of CeONPs significantly decreased LOOH, again to levels observed in shams.
FIG. 5.

Cerium oxide nanoparticles (CeONPs) decreased oxidative macromolecular damage after mild traumatic brain injury (mTBI) in vivo. Panel a demonstrates the decrease in lipid hydroperoxides formed after mTBI in CeONP-treated rats, with the chronic high dose group showing lipid hydroperoxides equal to those of sham animals. Panel b demonstrates a decrease in 3-nitrosotyrosine levels in animals treated with CeONP. Again, the chronic high dose reduced levels to that of sham animals.*Significant from sham, p < 0.01; #Significant from injury, p < 0.01.
NT is a product of free radical damage to proteins, which alters function and marks the protein for subsequent degradation.48 NT was significantly increased in the ipsilateral hemisphere after mLFPI (Fig. 5B) by 48% and 41% compared with shams, at both 24 and 48 h post-injury, respectively. Interestingly, no significant increase in NT occurred in the injured contralateral cortices. Administration of the LD of CeONPs, in either injection scheme (3 × or 5 × ), did not significantly affect NT. However administration of the HD of CeONPs decreased NT levels after 3 × administration, by 9% (Fig. 5b). When delivered in the 5 × scheme (Fig. 5B) the HD of CeONPs returned NT levels to those observed in shams. Taken together, these results demonstrate the ability of CeONPs to reduce oxidative damage to cellular macromolecules following mTBI.
CeONPs improve cognitive function in a mammalian model of mTBI
Figure 6 shows EthoVision tracking results of the NOR tests of an injured rat treated with vehicle (left panel), compared with an injured rat receiving a 5 × HD treatment (right panel). Note the similar exploration of each object for the injury+vehicle animal in the left panel, indicating memory deficit. In contrast, the CeONP-treated animal showed significantly increased exploration of the novel object, indicating intact memory. Figure 7 shows the calculated novel object preferences for the different treatment groups. As expected, the sham group discriminated the novel object (p = 0.0383 compared with the 0.5 null index). The injury-only group showed a null preference of ∼0.5 and there was a similar result for the injury+vehicle group, indicative of memory deficit. Neither the 3 × LD nor the 3 × HD schemes appeared to have a real effect on novel object preference. Although this was not statistically significant, animals receiving 5 × LD showed trending improvements compared with the 0.5 null NOR index (p = 0.0794). However, animals in the 5 × HD treatment group showed similar behavior to the sham group and showed a novel object preference significantly higher than 0.5 (p = 0.0041), demonstrating improved memory. The results suggest that CeONP administration can improve cognitive deficits after mLFPI when delivered in the 5 × HD paradigm.
FIG. 7.

Chronic cerium oxide nanoparticle (CeONP) administration after mild traumatic brain injury (mTBI) improves performance in the Novel Object Recognition (NOR) Test. This graph depicts the novel object preferences for each treatment group. The dotted line represents the novel object preference score of an animal unable to discern between novel and familiar objects (indicative of memory deficit). Intact memory was observed in the sham and chronic high dose group, as the novel object recognition value was significantly higher than 0.5 (p = 0.0383 and p = 0.0041, respectively). Injury-induced memory deficits resulted in novel object preferences not significantly different from 0.5. The chronic low dose treatment group showed trending differences in memory improvement (score >0.5) with p = 0.0794. The chronic high dose animals showed significant improvement in NOR scores, which were similar to those of controls. Data are expressed as μ+SE. *Significant from 0.5, p < 0.5.
Discussion
We report here, for the first time, that CeONPs decrease the biochemical and functional sequelae of mTBI in vitro and in vivo. CeONPs are potent, regenerative free radical scavengers and mitochondrial protectants. The antioxidant capacity of CeONPs derive from three properties of these nanoparticles as shown in Figure 8. First, cerium is present in either the +3 or +4 state, and interconversions between these valences imparts high antioxidant potential. Second, CeONPs have oxygen vacancies or “electron holes” that further participate in radical scavenging and regeneration of the free radical scavenging capacity. Finally, the decrease in particle size at the nanoscale imparts high surface area for free radical reactions and their interactions with ionic species in water. All three properties contribute to the superior and regenerative activity of CeONPs.20,23 The hypothesized mechanism of action of CeONPs is shown in Figure 8. CeONPs are known to have SOD-like and catalase-like actions. For example, if a superoxide radical is generated, it will interact with Ce3+ ions and oxygen vacancies as shown. In so doing, it will be converted to H2O2, while cerium is converted to the +4 state, and an oxygen vacancy is filled.18 Additional final electron acceptors come from the CeONP hydration shell.20 Via a catalase-like activity, CeONPs can then convert H2O2 to O2 + 4H+, by again interacting with ionic species of water in the hydration shell, conversion of Ce4+ back to the 3+ valence state, and formation of an oxygen vacancy. The entire cycle regenerates the CeONP. However, in the biological environment, this action is ongoing in a continuous cycle. Although we use superoxide and H2O2 as examples, any radical species can enter into the cycle for detoxification.
FIG. 8.

Hypothesized mechanism of action of cerium oxide nanoparticles (CeONPs). In a CeONP nanoparticle, the cerium atom exists in the 3+ and 4+ valence states, bound to oxygen and containing oxygen vacancies (O2-x). When exposed to a superoxide radical for example, it exhibits superoxide dismutase (SOD)-like activity, and Ce3+ is converted to Ce4+, with a corresponding change in oxygen vacancies. There is also likely a contribution to this reaction from the hydration shell around the CeONP. Superoxide is converted to H2O2. Via catalase-like activity involving Ce4+ and the hydration shell, H2O2 is converted to O2 + 4H+, and cerium valence to 3+ (with corresponding changes in oxygen vacancies), regenerating the original CeONP state. In the biological milieu, this action exists in a continuous cycle, depending on the ionic species exposed to the CeONPs, the hydration shell, and any surrounding ionic species. Although we utilized superoxide and H2O2 as examples, radicals scavenged could be any number of biologically relevant free radicals, with the ionic species entering at any point in the cycle.
We and others have previously demonstrated the in vitro radical scavenging properties of the CeONPs used in these studies,19,23,30 showing that they do not generate free radicals under biological conditions, but scavenge superoxide, hydroxyl, and nitroxyl radicals, and radical species produced during aggregation of amyloid β (1–42) peptide. Further, we demonstrated that post-insult delivery of CeONPs could protect Drosophila from paraquat toxicity,23 and act as a disease-modifying agent in a mouse model of Parkinson's disease.27 Estevez and coworkers55 demonstrated the efficacy of CeONPs in a rat brain slice model of stroke. Because increases in oxidative stress and mitochondrial dysfunction have been implicated as causes for the neuropathological sequelae of TBI,1–3 we reasoned that CeONPs may be effective in reducing pathological effects of TBI.
Using an in vitro model for TBI, we found that a single post-injury dose of CeONPs (10 nM) reduced cell damage and death after mild and moderate TBI. After mild injury, CeONPs decreased damage to the levels of uninjured controls, and to near-control levels after moderate injury. Other groups have also demonstrated the efficacy of CeONPs in reducing free-radical mediated oxidative stress to neurons in culture.26,27
To assess the efficacy of CeONPs at preserving normal neuronal function after in vitro TBI, we examined [Ca2+]i dysregulation. In prior work,32,33 we demonstrated extensive [Ca2+]i dysregulation in neurons after in vitro TBI, showing that 24 h after mild injury, the [Ca2+]i elevation produced by glutamate was depressed through 24 and 48 h post-injury. After moderate injury, we found that the glutamate-stimulated [Ca2+]i elevation was decreased 24 h after injury, followed by a hyper-responsive phase. Our results shown here are consistent with these reports. However, a single 10 nM dose of CeONPs, delivered 1 h post-injury, completely blocked these effects, and injured neurons maintained normal [Ca2+]i responses to glutamate. These observations suggest that mitigating the pro-oxidative environment with CeONPs following injury may aide in the preservation of neuronal signaling and function.
To further determine the efficacy of CeONPs in mTBI, we investigated their ability to mitigate the pathological depletion of endogenous antioxidant systems7,8,11 using an in vivo model. Both injection paradigms utilized showed significant improvement of catalase, SOD, and GSH/GSSG ratios compared with the injury-only group (Fig. 4). Sham levels of these endogenous antioxidant systems were reached following the 5 × HD paradigm. The ratio of redox states of GSH decreased following TBI, indicating the relative abundance of nonreactive oxidized GSH. However, following treatment of CeONP, the balance between redox states of GSH is recovered. With a relative increase in GSH, the antioxidant ability of the tissue was improved. SOD levels were also restored, and catalase activity was increased above sham levels. As CeONPs are known to have catalase and SOD-like properties in vitro, it is presently unclear as to whether the restored SOD and catalase levels were directly caused by CeONPs, or caused by preservation of endogenous SOD and catalase by virtue of CeONP radical scavenging. Given the low doses of CeONPs used in this study, it is likely that both potential mechanisms come into play. Restoration of SOD, catalase, and GSH enzymes correlated with mitigation of oxidatively induced macromolecular damage. Levels of LOOH and NT were returned to sham levels following the 4 × HD injection scheme (Fig. 5). These results suggest that administration of CeONPs following mTBI provides support for the endogenous antioxidant defenses and avoids depletion of such systems. By doing so, damage induced from the pro-oxidative environment is mitigated.
A key question is whether these biochemical improvements can translate to improved function. In the injury-only group, we observed signs of antioxidant depletion, oxidative damage, and impaired memory. When treated with CeONPs in the 5 × HD injection scheme, we observed improved antioxidant function and reduced oxidative damage, which correlated with intact recognition memory following mTBI. Improved cognition of the animals receiving CeONPs may be attributable to potential increased neuronal survivability by ameliorating the oxidative environment. In vitro experimentation has shown CeONPs' ability to improve neuronal functionality through calcium signaling (Fig. 3). Thus, the ability of CeONP to relieve oxidative stress and preserve neuron function may manifest as improvements to memory following mTBI, as observed here.
A question arises as to the toxicity of CeONPs. Several reports suggest toxicity of CeONPs and other nanoparticles.56 However, the doses used in these previous studies are ∼1000-fold higher than those utilized here. In a recent study conducted by our group, we found no evidence of pathological or neurological consequences at doses up to 5 μg/g delivered intravenously to mice and rats54 for up to 6 months post-administration of a single dose. However, because a given dose persists in the brain and is not metabolized, longer duration studies would be appropriate.
Conclusion
In summary, our results demonstrated for the first time that the actions of CeONPs in the brain may improve the pathological outcomes of mTBI. Although more biochemical and behavioral studies are needed, this study demonstrates a new prospect for treatment of mTBI: a CeONP nanopharmaceutical.
Acknowledgments
This work was supported by NIH-NINDS – NS072873 to B.A.R. and P.J.V. and DOD-CDRMP W81XWH11-2-0057 to B.A.R.
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Hylin M.J., Orsi S.A., Zhao J., Bockhorst K., Perez A., Moore A.N., and Dash P.K. (2013). Behavioral and histopathological alterations resulting from mild fluid percussion injury. J. Neurotrauma 30, 702–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith S.L., Andrus P.K., Zhang J.R., and Hall E.D. (1994). Direct measurement of hydroxyl radicals, lipid peroxidation, and blood–brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 11, 393–404 [DOI] [PubMed] [Google Scholar]
- 3. Kontos H.A., and Wei E.P. (1986). Superoxide production in experimental brain injury. J. Neurosurg. 64, 803–807 [DOI] [PubMed] [Google Scholar]
- 4. Lafon–Cazal M., Pietri S., Culcasi M., and Bockaert J. (1993). NMDA-dependent superoxide production and neurotoxicity. Nature 364, 535–537 [DOI] [PubMed] [Google Scholar]
- 5. Globus M.Y., Alonso O., Dietrich W.D., Busto R., and Ginsberg M.D. (1995). Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J. Neurochem. 65, 1704–1711 [DOI] [PubMed] [Google Scholar]
- 6. Chan P.H. (2001). Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 21, 2–14 [DOI] [PubMed] [Google Scholar]
- 7. Ansari M.A., Roberts K.N., and Scheff S.W. (2008). Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 45, 443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez–Rodriguez A., Egea–Guerrero J.J., Murillo–Cabezas F., and Carrillo–Vico A. (2014). Oxidative stress in traumatic brain injury. Curr. Med. Chem. 21, 1201–1211 [DOI] [PubMed] [Google Scholar]
- 9. Praticò D., Clark C.M., Liun F., Lee V.M., and Trojanowski J.Q. (2002). Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of alzheimer disease. Arch. Neurol. 59, 972–976 [DOI] [PubMed] [Google Scholar]
- 10. Kannan K., and Jain S.K. (2000). Oxidative stress and apoptosis. Pathophysiology 7, 153–163 [DOI] [PubMed] [Google Scholar]
- 11. Tyurin V.A., Tyurina Y.Y., Borisenko G.G., Sokolova T.V., Ritov V.B., Quinn P.J., Rose M., Kochanek P., Graham S.H., and Kagan V.E. (2000). Oxidative stress following traumatic brain injury in rats. J. Neurochem. 75, 2178–2189 [DOI] [PubMed] [Google Scholar]
- 12. Saito I., Asano T., Sano K., Takakura K., Abe H., Yoshimoto T., Kikuchi H., Ohta T., and Ishibashi S. (1998). Neuroprotective effect of an antioxidant, ebselen, in patients with delayed neurological deficits after aneurysmal subarachnoid hemorrhage. Neurosurg. 42, 269–277 [DOI] [PubMed] [Google Scholar]
- 13. Hu G., Lyeth B.G., Zhao X., Mitchell J.B., and Watson J.C. (2003). Neuroprotection by the stable nitroxide 3-carbamoyl-proxyl during reperfusion in a rat model of transient focal ischemia. J. Neurosurg. 98, 393–396 [DOI] [PubMed] [Google Scholar]
- 14. Cuzzocrea S., McDonald M.C., Mazzon E., Siriwardena D., Costantino G., Fulia F., Cucinotta G., Gitto E., Cordaro S., Barberi I., De Sarro A., Caputi A.P., and Thiemermann C. (2000). Effects of tempol, a membrane-permeable radical scavenger, in a gerbil model of brain injury. Brain Res. 875, 96–106 [DOI] [PubMed] [Google Scholar]
- 15. Narayan R.K., Michel M.E., Ansell B., Baethmann A., Biegon A., Bracken M.B., Bullock M.R., Choi S.C., Clifton G.L., Contant C.F., Coplin W.M., Dietrich W.D., Ghajar J., Grady S.M., Grossman R.G., Hall E.D., Heetderks W., Hovda D.A., Jallo J., Katz R.L., Knoller N., Kochanek P.M., Maas A.I., Majde J., Marion D.W., Marmarou A., Marshall L.F., McIntosh T.K., Miller E., Mohberg N., Muizelaar J.P., Pitts L.H., Quinn P., Riesenfeld G., Robertson C.S., Strauss K.I., Teasdale G., Temkin N., Tuma R., Wade C., Walker M.D., Weinrich M., Whyte J., Wilberger J., Young A.B., and Yurkewicz L. (2002). Clinical trials in head injury. J. Neurotrauma 19, 503–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis V.T., and Thompson J.S. (2002). Measurement of the electron affinity of cerium. Phys. Rev. Lett. 88, 073003. [DOI] [PubMed] [Google Scholar]
- 17. Celardo I., De Nicola M., Mandoli C., Pedersen J.Z., Traversa E., and Ghibelli L. (2011). Ce3+ ions determine redox-dependent anti-apoptotic effect of cerium oxide nanoparticles. A.C.S. Nano 5, 4537–4549 [DOI] [PubMed] [Google Scholar]
- 18. Celardo I., Pedersen J.Z., Traversa E., and Ghibelli L. (2011). Pharmacological potential of cerium oxide nanoparticles. Nanoscale 3, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 19. Rzigalinski B.A., Meehan K., Davis R.M., Xu Y., Miles W.C., and Cohen C.A. (2006). Radical nanomedicine. Nanomedicine (Lond.) 1, 399–412 [DOI] [PubMed] [Google Scholar]
- 20. Andrievsky G.V., Bruskov V.I., Tykhomyrov A.A., and Gudkov S.V. (2009). Peculiarities of the antioxidant and radioprotective effects of hydrated C60 fullerene nanostuctures in vitro and in vivo. Free Radic. Biol. Med. 47, 786–793 [DOI] [PubMed] [Google Scholar]
- 21. Aneggi E., Boaro M., Leitenburg C.d., Dolcetti G., and Trovarelli A. (2006). Insights into the redox properties of ceria-based oxides and their implications in catalysis. J. Alloys Compd. 408–412, 1096–1102 [Google Scholar]
- 22. Singh N., Cohen C.A., and Rzigalinski B.A. (2007). Treatment of neurodegenerative disorders with radical nanomedicine. Ann. N. Y. Acad. Sci. 1122, 219–230 [DOI] [PubMed] [Google Scholar]
- 23. Rzigalinski B.A., Meehan K., Whiting M.D., Dillon C.E., and Hockey K. (2011). Antioxidant Nanoparticles, in: Nanomedicine in Health and Disease. R. J. Hunger, and V.R. Preedy (eds.). CRC Press: New York, pps. 100–122 [Google Scholar]
- 24. Ciofani G., Genchi G.G., Liakos I., Cappello V., Gemmi M., Athanassiou A., Mazzolai B., and Mattoli V. (2013). Effects of cerium oxide nanoparticles on PC12 neuronal-like cells: proliferation, differentiation, and dopamine secretion. Pharm. Res. 30, 2133–2145 [DOI] [PubMed] [Google Scholar]
- 25. Tsai Y.Y., Oca–Cossio J., Agering K., Simpson N.E., Atkinson M.A., Wasserfall C.H., Constantinidis I., and Sigmund W. (2007). Novel synthesis of cerium oxide nanoparticles for free radical scavenging. Nanomedicine (Lond.) 2, 325–332 [DOI] [PubMed] [Google Scholar]
- 26. Schubert D., Dargusch R., Raitano J., and Chan S. W. (2006). Cerium and yttrium oxide nanoparticles are neuroprotective. Biochem. Biophys. Res. Commun. 342, 86–91 [DOI] [PubMed] [Google Scholar]
- 27. Das M., Patil S., Bhargava N., Kang J. F., Riedel L.M., Seal S., and Hickman J.J. (2007). Auto-catalytic ceria nanoparticles offer neuroprotection to adult rat spinal cord neurons. Biomaterials 28, 1918–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frey A., Bates J.A., Sholar C.A., Hockey K.S., and Rzigalinski B.A. (2014). Cerium Oxide Nanoparticles as disease-modifying therapy for Parkinson's disease. Society of Neuroscience Abstract No. 199(01) [Google Scholar]
- 29. Xiang J., Li J., He J., Tang X., Dou C., Cao Z., Yu B., Zhao C., Kang F., Yang L., Dong S., and Yang X. (2016). Cerium oxide nanoparticle modified scaffold interface enhances vascularization of bone grafts by activating calcium channel of mesenchymal stem cells. ACS Appl. Mater. Interfaces 8, 4489–4499 [DOI] [PubMed] [Google Scholar]
- 30. Walkey C., Das S., Seal S., Erlichman J., Heckman K., Ghibelli L., Traversa E., McGinnis J.F., and Self W.T. (2015). Catalytic properties and biomedical applications of cerium oxide nanoparticles. Environ. Sci. Nano 2, 33–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polak P., and Shefi O. (2015). Nanometric agents in the service of neuroscience: manipulation of neuronal growth and activity using nanoparticles. Nanomedicine 11, 1467–1479 [DOI] [PubMed] [Google Scholar]
- 32. Weber J.T., Rzigalinski B.A., Willoughby K.A., Moore S.F., and Ellis E.F. (1999). Alterations in calcium-mediated signal transduction and intracellular calcium stores after in vitro injury of pure embryonic neurons. Cell Calcium 26, 289–299 [DOI] [PubMed] [Google Scholar]
- 33. Weber J.T., Rzigalinski B.A., and Ellis E.F. (2001). Traumatic injury of cortical neurons causes changes in intracellular calcium stores and capacitative calcium influx. J. Biol. Chem. 276, 1800–1807 [DOI] [PubMed] [Google Scholar]
- 34. Zhang L., Rzigalinski B.A., Ellis E.F., and Satin L.S. (1996). Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274, 1921–1923 [DOI] [PubMed] [Google Scholar]
- 35. Ahmed S.M., Rzigalinski B.A., and Ellis E.F. (2000). Alterations in mitochondrial energetics after in vitro traumatic injury of astrocytes and neurons. J. Neurochem. 74, 1951–1960 [PubMed] [Google Scholar]
- 36. Rzigalinski B.A., Weber J.T., Willoughby K.A., and Ellis E.F. (1998). Intracellular free calcium dynamics in stretch-injured astrocytes. J. Neurochem. 70, 2377–2385 [DOI] [PubMed] [Google Scholar]
- 37. Schreiber D., Gennarelli T.A., and Meaney D.F. (1995). Proceedings of the 1995 International Research Conference on Biomechanics of Impact, in: International Research Council of Biokinetics of Impact. A. Charpenne (ed.). Lyon, pps. 233–244 [Google Scholar]
- 38. Lowry O.H., Rosebrough N.J., Farr A.L., and Randall R.J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 39. Whiting M.D., and Hamm R.J. (2006). Traumatic brain injury produces delay-dependent memory impairment in rats. J. Neurotrauma 23, 1529–1534 [DOI] [PubMed] [Google Scholar]
- 40. Titus D.J., Furones C., Atkins C.M., and Dietrich W.D. (2015). Emergence of cognitive deficits after mild traumatic brain injury due to hyperthermia. Exp. Neurol. 263, 254–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Griesbach G.S., Tio D.L., Nair S., and Hovda D.A. (2014). Recovery of stress response coincides with responsiveness to voluntary exercise after traumatic brain injury. J. Neurotrauma 31, 674–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Whiting M.D., Rzigalinski B.A., and Ross J.S. (2009). Cerium oxide nanoparticles improve neuropathological and functional outcome following traumatic brain injury. J. Neurotrauma 26, 101 [Google Scholar]
- 43. Johansson L.H., and Borg L.A. (1988). A spectrophotometric method for determination of catalase activity in small tissue samples. Anal. Biochem. 174, 331–336 [DOI] [PubMed] [Google Scholar]
- 44. Marklund S. (1980). Distribution of CuZn superoxide dismutase and Mn superoxide dismutase in human tissues and extracellular fluids. Acta. Physiol. Scand Suppl 492, 19–23 [PubMed] [Google Scholar]
- 45. Baker M.A., Cerniglia G.J., and Zaman A. (1990). Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal. Biochem. 190, 360–365 [DOI] [PubMed] [Google Scholar]
- 46. Ayala A., Munoz M.G., and Arguelles S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brahmajothi M.V., Tinch B.T., Wempe M.F., Endou H., and Auten R.L. (2014). Hyperoxia inhibits nitric oxide treatment effects in alveolar epithelial cells via effects on L-type amino acid transporter-1. Antioxid. Redox Signal. 21, 1823–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ischiropoulos H. (1998). Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch. Biochem. Biophys. 356, 1–11 [DOI] [PubMed] [Google Scholar]
- 49. Ennaceur A. (2010). One-trial object recognition in rats and mice: methodological and theoretical issues. Behav. Brain Res. 215, 244–254 [DOI] [PubMed] [Google Scholar]
- 50. Aggleton J.P., Albasser M.M., Aggleton D.J., Poirier G.L., and Pearce J.M. (2010). Lesions of the rat perirhinal cortex spare the acquisition of a complex configural visual discrimination yet impair object recognition. Behav. Neurosci. 124, 55–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reger M.L., Hovda D.A., and Giza C.C. (2009). Ontogeny of rat recognition memory measured by the novel object recognition task. Dev. Psychobiol. 51, 672–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sajja V.S., Perrine S.A., Ghoddoussi F., Hall C.S., Galloway M.P., and VandeVord P.J. (2014). Blast neurotrauma impairs working memory and disrupts prefrontal myo-inositol levels in rats. Mol. Cell Neurosci. 59, 119–126 [DOI] [PubMed] [Google Scholar]
- 53. Gaskin S., Tardif M., Cole E., Piterkin P., Kayello L., and Mumby D.G. (2010). Object familiarization and novel-object preference in rats. Behav. Processes 83, 61–71 [DOI] [PubMed] [Google Scholar]
- 54. Jortner B.S., Ehrich M., Hinckley J., and Rzigalinski B.A. (2015). Safety studies on a cerium containing nanomaterial. Society For Toxicology Annual Meeting, Abstract [Google Scholar]
- 55. Estevez A.Y., Pritchard S., Harper K., Aston J.W., Lynch A., Lucky J.J., Ludington J.S., Chatani P., Mosenthal W.P., Leiter J.C., Andreescu S., and Erlichman J.S. (2011). Neuroprotective mechanisms of cerium oxide nanoparticles in a mouse hippocampal brain slice model of ischemia. Free Radic. Biol. Med. 51, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 56. Li Y., Li P., Yu H., and Bian Y. (2016). Recent advances (2010–2015) in studies of cerium oxide nanoparticles' health effects. Environ. Toxicol. Pharmacol. 44, 25–29 [DOI] [PubMed] [Google Scholar]