

Abstract
Despite extensive research in the pathogenesis, early detection, and therapeutic approaches of pancreatic ductal adenocarcinoma (PDAC), it remains a devastating and incurable disease. As the global incidence and prevalence of PDAC continue to rise, there is a pressing need to place strong emphasis on its prevention. Although it is widely recognized that cigarette smoking, a potentially modifiable risk factor, has been linked to PDAC development, its contribution to prognosis is still uncertain. Moreover, the mechanistic pathways of PDAC progression secondary to smoking are various and lack a summative narration. Herein, we update and summarize the direct and indirect roles cigarette smoking plays on PDAC development, review literature to conclude the impact cigarette smoking has on prognosis, and postulate a comprehensive mechanism for cigarette smoking–induced PDAC.
Key Words: pancreatic ductal adenocarcinoma, cigarette smoking, diabetes, obesity, genetics
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related mortality in the United States (US) and the seventh worldwide.1 Despite it not being among the most common cancers, PDAC causes almost 10% of all cancer-related deaths in both men and women.1 In addition, the approximately US $5 billion annual impact of the disease in the US presents a major economic concern.2 Frighteningly, the overall incidence of PDAC continues to increase at an alarming rate.3 There are more than 40,000 new PDAC diagnoses annually and nearly that number die of the disease each year in US.4 Being that PDAC leads to a near 100% mortality rate, in addition to the further investigation of the crucial countermeasures, there is a strong need to place an emphasis on disease prevention, namely, on its established risk factors.5
Chronic pancreatitis, diabetes, obesity, genetic makeup, intraductal pancreatic mucinous neoplasm (IPMN), and excessive alcohol consumption are all risk factors that have been documented to contribute to PDAC initiation with contributions of each ranging from 3% to 10% of the cases.6–9 Cigarette smoking has been determined, a major risk factor for PDAC, as it contributes to development of up to 25% of the cases.5,10,11 Moreover, elucidation of the underlying mechanistic action of smoking is paramount, as it can directly contribute to the prevention of PDAC, and perhaps mediate an important role in patient prognosis. Apart from disease stage, few prognostic factors have been identified. In particular, cigarette smoking and survival among patients with PDAC remain an unsettled and poorly characterized field.11 Hence, cigarette smoking deserves particular attention, and as an avoidable risk factor, the mechanistic unraveling would lead to a considerable reduction in the number of PDAC cases diagnosed annually.
Despite enormous research efforts at early diagnosis and treatment, PDAC continues to be a significant global health care burden. Lack of early detection in combination with its unresponsiveness to nearly all treatment modalities to date accounts for 1- and 5-year survival rates of merely 25% and 5%, respectively, in PDAC patients.12 For the past few years, multiple phase 3 trials of chemotherapeutic agents for advanced PDAC have failed, likely reflecting the refractory and complex nature of disease progression.3,13,14 Although the relationship between PDAC initiation and cigarette smoking has been extensively studied, the mechanism by which smoking acts to promote carcinogenesis, promote disease progression, and influence prognosis is yet to be revealed.5 Considering the above, knowledge and the methodology of PDAC prevention are still a major initiative given the highly malignant nature of this tumor. Herein, we summarize the current literature on the considerable involvement of cigarette smoking in PDAC development and review the relationship between cigarette smoking and the prognosis of patients with PDAC, as well as the mechanistic hypotheses presented for this association.
CIGARETTE SMOKING AS A RISK FACTOR FOR PDAC DEVELOPMENT
Over half a century ago, numerous studies determined a relationship between smoking and PDAC progression. These survey-based studies showed a 70% greater risk of developing PDAC in cigarette smokers compared with nonsmokers.5,15,16 More recently, a large meta-analysis found that current smokers have a 1.74-fold increased risk of developing PDAC in comparison with nonsmokers.17 In a large pooled analysis, cigarette smoking was associated with a staggering twofold increase for a risk of PDAC.18 Notably, even exposure to environmental tobacco smoke was found to be associated with an increased risk of PDAC.19 Current smokers were found to have a markedly elevated risk of PDAC development compared with nonsmokers with greater intensity, duration, and cumulative smoking dose.20 Smoking has also been associated with an earlier onset of PDAC development, and in certain scenarios, this can be as much as 20 years earlier than nonsmokers.5,21–23 Although various studies have established smoking as an undeniable risk factor for the initiation of PDAC, its role in patient prognosis has not been concluded. In addition, detailed studies on how smoking affects the survival of patients with PDAC are inconsistent and poorly represented throughout literature, hence demanding further examination.5
RECENT INSIGHTS INTO THE MECHANISMS OF SMOKING-INDUCED CARCINOGENESIS BY THE OTHER RISK FACTORS
Patients with chronic pancreatitis, largely due to smoking and alcohol abuse, carry up to a 10% risk of developing PDAC, with an eightfold increased risk 5 years after diagnosis.8,24,25 Nevertheless, chronic pancreatitis accounts for approximately only 3% of PDAC cases.6 The long-standing, unresolved inflammation of chronic pancreatitis is thought to underlie PDAC development. Disorders of the process of autophagy, or the process by which proteins and organelles are recycled via lysosomes, are common to both chronic pancreatitis and PDAC, arguing common features and interrelatedness of chronic pancreatitis and PDAC.26 In addition, the induction of pancreatic inflammation was found to accelerate oncogenic KRAS-induced fibrosis and PDAC development in mouse models.27 Importantly, pancreatitis has been shown to affect PDAC prognosis.28 The Nrdc gene may provide another connection between pancreatitis and PDAC development. The deletion of Nrdc resulted in spontaneous chronic pancreatitis concomitant with acinar-to-ductal conversion and pancreatic deletion of Nrdc dramatically accelerated KRAS-driven PDAC development in mice.29 The transforming growth factor-β1/Smad pathway has also been shown to connect both pancreatitis and PDAC.30 Galectin-1 expression, a common intermediate in the Smad pathway, in activated pancreatic satellite cells was found to promote fibrosis leading to both pancreatitis and PDAC development.20 Interestingly, infection with Enterococcus faecalis may be involved in chronic pancreatitis progression, ultimately leading to PDAC development.31
Patients with diabetes have an increased risk of developing PDAC by at least 2 times that of nondiabetic patients.32 Notably, a correlation between diabetes and prognosis of patients with PDAC has been established.33 Of recent, several mechanisms regarding the association between PDAC and type 2 diabetes were postulated. It was established that the phosphoinositide 3-kinase and nuclear factor kappa B pathway play an integral role in this process and the expression of polo-like kinase 1 enables insulin to promote the proliferation of pancreatic ductal epithelial cells, leading to PDAC development via both these pathways.34 Li et al35 postulated that the worse prognosis associated with diabetes-associated PDAC is due to the effect of hyperglycemia on the pancreatic microenvironment causing a transient hypoxia, ultimately responsible for promoting the metastatic ability of PDAC.
Not surprisingly, obesity was found to play a strong role in PDAC development. There even was a correlation between every unit increase in body mass index (BMI) to an increase in the risk of PDAC.36 The cellular defense mechanisms of inflammation and autophagy, thought to play an important role in PDAC, are dysregulated by visceral obesity. Therefore aside from a high-fat diet intake, the physiologic impact of being obese, even on a cellular level, has been shown to be as a risk factor for PDAC initiation.26 In addition, a connection between BMI and overall survival of PDAC patients was identified.37 However, other studies found conflicting evidence and could not prove an association between BMI and survival of patients with PDAC.38 In vivo studies revealed that adiponectin suppresses leptin-induced signal transducer and activator of transcription 3 (STAT3) signaling in PDAC.39 Inhibition of STAT3 signaling via activating the adiponectin receptor provides a potentially important approach to inhibit PDAC development.39 Mechanistically, a study revealed that obesity-driven PDAC exhibits accelerated growth and a striking transcriptional benefit for pathways regulating nitrogen metabolism—more specifically arginase 2.40 Importantly, a loss-of-function mutation, which acts as a silencing agent on arginase 2, has shown to reduce PDAC development.40 Furthermore, a number of obesity-sensitive pathways that manipulated by transcription activators such as YAP and TAZ reinforce KRAS signaling and mutation leading to prolonged KRAS activation and eventual PDAC progression.41
Although intraductal papillary mucinous neoplasms (IPMNs) are benign and make up only approximately 20% of the cystic lesions of the pancreas, they are the most extensively studied that this is secondary to their innate ability to transform into malignant lesions.42 In addition, the development and recent advances in endoscopic ultrasonography have largely influenced our understanding of IPMN and its subsequent risk for PDAC development. Intraductal papillary mucinous neoplasm with high-grade dysplasia has been shown to increase the risk of the subsequent development of PDAC.43 The presence of a mural nodule during endoscopic ultrasonography is the most reliable indicator for malignancy potential.44 Size greater than 3 cm, though once thought to be a major predictor of malignancy, has been shown to not be a significant factor leading to cancer development.45 The frequency of malignancy in main pancreatic duct lesions is close to 80%, whereas the frequency of malignancy in branch duct lesions is lower with an average of approximately 20%.46,47
Although mild drinking is not a risk factor for PDAC, excessive alcohol consumption has been found to be associated with PDAC development.48 However, as alcohol consumption is the leading cause of pancreatitis, which has been identified as a strong risk factor for PDAC development, it is debatable whether pancreatitis acts as the confounding factor. Further studies regarding mechanistic analyses of alcohol consumption and PDAC development would be required.
Hereditary factors contribute to somewhere between 5% and 10% of PDAC cases.8 Compared with individuals without a family history of PDAC, patients with a family history of PDAC have nearly a twofold increased risk for developing this lethal cancer.49 Although hereditary conditions and specific genes shown to induce PDAC are difficult to discern from each other, as they overlap greatly, the previously mentioned environmental factors play a central role in promoting these specific genes. Thus, aside from the hereditary nature patients with these genes possess, having an environmental factor increases the risk of PDAC by virtue of enhancing those genetic expression patterns. It also possible that genetics in its entirety is a risk factor outside the realm of the other environmental risk factors, although future studies are needed to establish its precise role. TP53 is one of the most frequently mutated genes in numerous cancers. Specifically, the role in PDAC is staggering, as it has been found mutated in 70% of PDAC cases.50 A study suggests that TP53 could be used as a biomarker for prognosis and perhaps even to guide therapy.51 Another major player identified to be involved with PDAC is the tumor suppressor protein SMAD4, which has been found to occur in approximately 50% of PDAC cases.50 It has also been suggested that SMAD4 inactivation could be used as a prognostic biomarker in PDAC.52 Numerous germ-line mutations including BRCA2, FANC-C, FANC-G, and PALB2 were found to be closely associated with PDAC development.53 Despite this, genetic disorders, which may involve some of these mutations, concur the highest risk for PDAC development. Oncogenic KRAS mutation is perhaps the most important trigger for PDAC initiation and is classically thought to play a vital role in disease progression as well. This signature genetic event has been found to be present in nearly 95% of PDAC cases.54 Importantly, the ability of KRAS to alter metabolic pathways, reroute glucose transportation, influence autophagy, and drive the cellular macropinocytosis (the uptake of extracellular nutrients) of materials has furthered our understanding of PDAC initiation and development.55 The elucidation of this connection has opened up new attempts at understanding the mechanistic driving force of PDAC and has orchestrated a new era of clinical investigation.54
THE RELATIONSHIP BETWEEN RISK FACTORS FOR PDAC AND CIGARETTE SMOKING
As illustrated in Figure 1, apart from its direct affects, cigarette smoke has indirect impacts on PDAC development, namely by activating and influencing its other risk factors. Exposure to tobacco smoking was found to be associated with an earlier diagnosis of chronic pancreatitis and predisposition to the development of pancreatic calcifications.56 It also has been identified as an independent risk factor for chronic pancreatitis.57 Numerous studies describe a strong relationship between cigarette smoking and the pathogenesis of diabetes. The development of complications, on both a microvascular and macrovascular level, has been proven to be modulated by cigarette smoke.58 In addition, smoking cessation limits microvascular disease and may also facilitate tighter glycemic control.59 A number of studies found that the odds of being obese increased progressively with smoking, such that male heavy smokers were more obese that male light smokers.60 Although smokers weigh less than nonsmokers, upon cessation their weight returns to that observed in nonsmokers, with some smokers, gaining even more weight.61 Furthermore, smoking plays an important indirect role in PDAC carcinogenesis by its ability to induce DNA methylation as well as create DNA adducts.5,62 These adducts collectively aggregate and can even activate KRAS mutations thus hastening PDAC development.63 Uniquely, having a smoking history is also one of the few valuable predictive factors for malignant IPMN that would thus warrant surgical resection.64 Cumulative pack-years were higher in patients who had PDAC concomitant with IPMN than in patients with just IPMN, arguing a strong role for smoking induced malignant IPMN.65 In a study by Zhang et al66 only patients who were both cigarette smokers and alcohol drinkers were associated with reduced survival from PDAC and the 2 risk factors interact with each other to promote the development of PDAC. Hence, taken collectively, aside from its direct effects, cigarette smoking yields indirect effects on PDAC development, namely, by modulating a number of its prominent risk factors.
FIGURE 1.
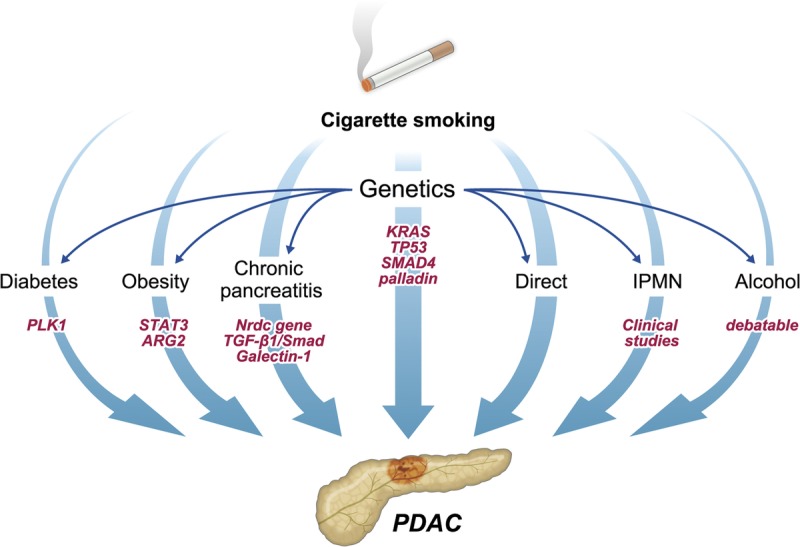
The diverse involvement of cigarette smoking in PDAC development and progression is illustrated in this diagram. Both the direct and indirect roles smoking plays in PDAC development are included. The direct affects are numerous, whereas the indirect affects occur through the modulation and/or activation of the various risk factors, namely, having a history of diabetes, IPMN, chronic pancreatitis, obesity, genetic mutations, and alcohol consumption. In addition, as illustrated, genetic factors (as effected by smoking) influence PDAC directly, as well as indirectly by modulating other risk factors.
THE IMPACT OF CIGARETTE SMOKING ON PDAC PROGNOSIS
To date, more than 7000 chemicals have been identified in tobacco smoke.4 Of these, 250 are known to be harmful, and 60 are classified as carcinogens containing extensively studied metabolites.4,67 Cigarette smoking might be the strongest environmental risk factor for PDAC development. In fact, cigarette smoking is associated with a reduced quality of life after diagnosis.68 A detrimental effect of smoking on prognosis in patients with malignancies that are etiologically associated with tobacco use, such as lung cancer, esophageal cancer, and squamous cell carcinoma of the head and neck has already been established.11,69 Furthermore, tobacco usage in other cancers, such as colon, gastric, cervical, prostate, and renal cell carcinoma has also been associated with worse survival outcomes in comparison to nonsmokers.70–74 However, evidence as to whether cigarette smoking accelerates disease progression, is associated with a harsher disease course, and thus affects prognosis in patients with PDAC is inconsistent and remains poorly concluded. Although limited, various study designs including retrospective analyses, prospective cohorts, case controls, and others, aimed at determining if in fact cigarette smoking affects PDAC prognosis (Table 1). The results were inconsistent and the topic is in desperate need of a conclusive edge. While some studies show no correlation, others show a statistically significant relationship between smoking and PDAC prognosis.
TABLE 1.
Clinical Studies Investigating the Relationship Between Cigarette Smoking and PDAC Prognosis
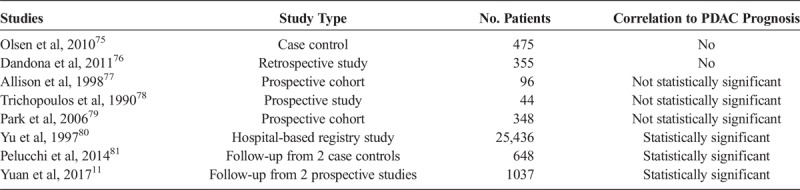
In a hospital-based case-control study by Olsen et al75 looking at 475 patients, no correlation between cigarette smoking and survival in patients with PDAC was found. However, the patient population identified had a large proportion (34%) of patients with resected disease; thus, overall survival may have been longer than typical scenarios.75 Similarly, a retrospective analysis including 355 patients spanning 14 years by Dandona et al76 concluded that cigarette smoking has no impact on PDAC patient survival after pancreaticoduodenectomy.
In a study looking at numerous prognostic factors for PDAC patients after resection, Allison et al77 found an association between prognosis and cigarette smoking. Although not statistically significant, the authors demonstrated that smoking does indeed affect patient survival. In a similar vein, Trichopoulos et al78 examining α1-antitrypsin concluded a nonstatistical association between PDAC survival and smoking. A well-crafted prospective cohort of 348 Korean men by Park et al79 demonstrated findings consistent with the above. Current smokers had a hazard ratio of 1.20 (95% confidence [CI], 0.84–1.72). In addition, they correlated smoking amount to prognosis as well.
Not surprisingly, cigarette smoking has also been shown to reduce survival in PDAC patients whom are diagnosed in advanced disease stages, namely, those whom have liver metastasis.82 A hospital-based registry study by Yu et al80 was one of the first large studies showing a significant correlation between cigarette smoking and PDAC prognosis. In a follow-up study containing data of PDAC patients enrolled in 2 Italian case-control studies, Pelucchi et al81 established a strong correlation between smoking and PDAC prognosis. For persons who ever smoked, the hazard ratio was 1.37 (95% CI, 1.14–1.65), for ex-smokers, the hazard ratio was 1.30 (95% CI, 1.03–1.65), and for current smokers, the hazard ratio was 1.42 (95% CI, 1.16–1.73). Even more importantly, increasing amount of smoking was associated with reduced survival. The hazard ratios of death were 1.26 (95% CI, 1.02–1.55) for smoking up to 19 cigarettes per day and 1.57 (95% CI, 1.26–1.96) for 20 or more cigarettes smoked per day.81 Moreover, increasing duration of smoking was associated with reduced survival with hazard ratios of 1.24 (95% CI, 0.98–1.57) for duration of smoking less than 30 years and 1.49 (95% CI, 1.22–1.83) of 30 or more years, as compared with never smokers. In addition, short-term ex-smokers had a higher mortality than did long-term ex-smokers.81 These results give further support to the detrimental role of smoking on patient's survival after PDAC diagnosis. A more recent study by Yuan et al11 examined more than 1000 patients from 2 large US prospective cohorts. While accounting for comorbid factors that smokers generally have, such as obesity, diabetes, and heart disease, there results maintained statistical significance. The multivariable-adjusted hazard ratio for death in current smokers was 1.37 (95% CI, 1.11–1.69) to that of never smokers.11 A significant negative trend in survival was observed for increasing pack-years of smoking with a hazard ratio for death of 1.49 (95% CI, 1.05–2.10) for 60 pack-years, and the authors found that regardless of time since quitting, survival among former smokers was similar to that for never smokers.11
Altogether, it conclusively seems that cigarette smoking plays an imperative role, in not only the etiology of PDAC but also its prognosis as well. Hence, groups at high risk for PDAC need to be educated continually to quit smoking not only to prevent cancer but also to improve survival rates. However, understanding how smoking effects the pancreas on a cellular level is of great importance as it can lead to a better mechanistic understanding for further prevention strategies for PDAC.
CIGARETTE SMOKING–MEDIATED MORPHOLOGICAL DAMAGE
Autopsy specimens have allowed us to gain a great wealth of knowledge regarding the distinct histopathological alterations pancreatic cells undergo as a result of cigarette smoking–induced insult to the pancreas.8 Pancreatic acinar cells, being the principal cells affected, were found to undergo marked hyperplasia and even dysplasia as a result of cigarette smoking.83 Laboratory rats exposed to environmental tobacco smoke also developed acinar cell damage similar to what was found in humans.84 Thus, it suggested that this may be a precursor lesion to acinar cell carcinoma.8 Nuclear atypia of both pancreatic ductal and acinar cells were seen secondary to cigarette smoking.8 The atypical nuclei demonstrated were different in shape, size, and staining character than normal pancreatic cells.85 In a study examining the immunohistochemical effects of cigarette smoke on laboratory rats, the Langerhans islet cells showed a marked decrease in insulin expression, whereas the glucagon expression increased in the pancreas.86 Hence, a possible diabetogenic effect of smoking can be observed by the increased glucagon-secreting cells and decreased insulin-secreting cells.86 This is in line with the fact that chronic cigarette smokers when compared with nonsmokers are generally intolerant to glucose.87
UNDERLYING MECHANISMS POSTULATING THE DIRECT IMPACT OF SMOKING ON PDAC
It is well established that exposure to cigarette smoke stimulates inflammatory cell infiltration including macrophages and mast cells leading to long-lasting fibrotic changes.67 Hence, inflammation- and fibrosis-mediated damage can explain how smoking induces morphological changes within the pancreas. Thus, cigarette smoking mediates enough damage on a cellular level to induce PDAC and alter pathways encouraging a harsher disease course. However, identifying the underlying mechanisms explaining how this occurs, to intervene at the appropriate pathological step therapeutically, is of critical importance.
A causal link between chronic inflammation and cancer has been well known. However, the exact molecular mechanism linking inflammation to cancer is unclear. Liu et al88 posed that the Src family kinases are key players in this connection as they have been demonstrated to be important in the activation of many cell types and also been shown to control production of cytokine tumor necrosis factor α in normal cells.89,90 Thus, an elevation and/or a dysregulation of the Src activity may play an integral role in the initiation of the invasive cell phenotype seen in precancerous cells.88 The potent Src-mediated stimulatory effects on malignant cell proliferation as well as the ability to inhibit cellular death can potentially increase the total mass of the tumor leading to cancer progression.88 Similarly, Wittel et al91 illustrated that exposure to cigarette smoke stimulated fibrosis and inflammation in the pancreas of rats.
Inflammation facilitates some of the important tenants of cancer cell survival. The inflammatory response acts via the production of inflammatory mediators, such as interleukin (IL)-6, IL-11, tumor necrosis factor α, and IL-1α. These, as instructed by oncogenic KRAS, activate STAT3 and nuclear factor kappa B transcription factors to promote cell survival and proliferation, as well as invasion.26 In addition, the surveillance immune system is suppressed by high inflammatory states.
The imbalance in protumor and antitumor immune cells as well as the absence of antitumor CD8+ killer T cells has been shown to promote PDAC development.26 In addition, inflammation has been shown to inhibit oncogene-induced senescence which in mice was shown to accelerate carcinogenesis.26 Elevated levels of inflammatory cells are closely associated with a stronger expression of vascular endothelial growth factor and basic fibroblast growth factor in human PDAC tissues compared with that of normal pancreatic tissues.67–92 Collectively, these act as a key mediator in tumor angiogenesis. Moreover, inflammation promotes metastasis by stimulating the epithelial-mesenchymal transition that has been demonstrated to be a necessary precursor for tumor development.26 Thus mechanistically, how carcinogenesis can be a by-product of inflammation secondary to cigarette smoke are elucidated.
The strong relationship between KRAS mutations and PDAC development begs explanation from a purely genetic perspective and is essential to understanding how carcinogenesis can be mediated via cigarette smoking.93 Reactive oxygen species (ROS) is a potent detoxification molecule responsible for killing cells suspicious of harboring deficiencies or disease, and smokers have an increased propensity toward increased KRAS mutations and patients with KRAS mutations were found to have decreased levels of ROS.93 With low levels of ROS, a permissive, proliferative, intracellular environment is established. This environment is one that has no stop to self-renewal and tumor cell growth.93 Hence, based on the association by Kong et al,93 it is conceivable how KRAS mutations secondary to smoking can directly contribute to PDAC initiation. As mentioned earlier, the formation of DNA adducts, transversions, and mutations as a result of the active metabolites present in cigarette smoke play a direct role in PDAC carcinogenesis.5
Pandol et al4 took the previously mentioned one step further that smoking acts in cooperation with activating KRAS to promote PDAC, based on the fact that treatment with 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone in KRAS-mutated mice resulted in advanced lesions that did not develop in control treated mice. These results indicate that smoking acts synergistically with KRAS to promote pancreatic carcinogenesis. Of note, the advanced lesions contained fibrotic stellate cells similar to human PDAC.94
Cigarette smoke also plays a role in PDAC development by its ability to alter normal cellular signaling pathways. Cigarette smoke stimulates pancreatic cancer cell growth through adrenergic receptor activation of cyclooxygenase-2, intracellular cyclic adenosine monophosphate, as well as by stimulating extracellular signal-regulated kinase phosphorylation in pancreatic ductal cells.94–96 Moreover, smoking compounds interact with pancreatic cells through nicotinic acetylcholine receptors (AChRs) such as through α7AChR.4,97
Cigarette smoking causes the aberrant expression of microRNA (miRNA) encoding genes, which in result effects protein synthesis.98–101 Thus, their aberrant expression could lead to the initiation and progression of malignancies.101,102 Zhang et al101 identified a panel of aberrantly expressed miRNAs in pancreatic duct epithelial cells exposed to cigarette smoke. In particular, the miR-25-3p level was significantly higher in smokers than in nonsmokers and in PDAC than in nontumor tissues and elevated levels were correlated with shorter survival time of patients.101 Not surprisingly, this oncogenic miR-25-3p has been reported in various other types of human carcinoma.103,104
There is an association between cigarette smoke exposure and the enzymatic levels of pancreatic cells illustrating that the alteration of enzyme levels may contribute to PDAC development and there was a ratio disturbance of trypsin, chymotrypsin, basal serum amylase, and pancreatic elastase expression in smokers.8 In addition, there was a decreased activity of pancreatic cell lysates in rats exposed to cigarette smoke.91
More recently, the direct effect cigarette smoke has on the microenvironment of tumor cells has become clear.105 Pancreatic cancer cells have an environment characterized by an extremely dense desmoplasia that surrounds the cells of this tumor.45,105 The desmoplasia contains myofibroblastic pancreatic stellate cells, extracellular matrix proteins, numerous collagen subtypes, hyaluronic acid, and immune cells.45,105,106 These factors enable the growth of cancer by providing a scaffold of angiogenesis factors.107 Evidence is emerging that there is a strong relationship between pancreatic cancer cells and stellate cells that results in rapid tumor growth and possibly metastasis.107 Cigarette smoke activates the stellate cells leading to secretion of extracellular matrix proteins, growth factors, and cytokines.4 These then interact with integrins, which transactivate the insulin-like growth factor-1 receptor, which in turn mediates the intracellular events that promote cancer cell survival and growth.108 In addition, they promote the survival of cancer cells through the activation of intracellular ROS-generating systems, which mediate prosurvival and proproliferative effects in cancer cells through their regulation of phosphatases and kinases.109
Moreover, the immunomodulation of the microenvironment has shown to be a cause of PDAC as alluded to earlier. There is a restricted immune surveillance secondary to the high inflammatory levels created by the microenvironment.55 In this environment, tumorigenesis is supported via paracrine cross talk between these immune cells and tumor cells.109 This, by a process of immune cell recruitment, eventually leads to a block in T cell–mediated immunity.55 Thus, the field of tumor immunology may be a potential therapeutic target to combat PDAC.
Taken collectively, smoking-induced PDAC occurs through an array of mechanisms (Fig. 2). Besides, cigarette smoking affects differently on pancreatic cells and on signaling pathways in PDAC growth as shown in Table 2. The combination of interfering with physiologic pathways, stimulating inflammation and fibrosis, as well as the powerful interactions cigarette smoke has with DNA, enables cigarette smoke to disrupt all the major pathways a normal cell needs to transition into a cancerous one, namely, self-sufficiency, limitless replication, evasion of apoptosis, sustained angiogenesis, invasion, and metastasis. By altering enzymatic secretion, morphologically damaging ductal as well as acinar cells, distorting the microenvironment, disarming the natural immune system, inducing high inflammatory states, and causing epigenetic alterations, in combination to the DNA changes seen, cigarette smoke is directly responsible for PDAC initiation and progression.
FIGURE 2.

The summative mechanistic approach of cigarette smoking–induced PDAC is summarized in this diagram. It consists of the ability of cigarette smoke to change the microenvironment, cause long-standing inflammation, increase miRNA expression, induce KRAS mutations, affect signaling pathways, and alter enzyme secretion. Understanding the combined mechanistic pathway will yield positive outcomes in terms of drug discovery and reduced PDAC-related mortality.
TABLE 2.
The Different Effects of Smoking on Cells and Pathways

CONCLUSIONS
As PDAC continues to be a stealthy killer, because of it generally being diagnosed in its advanced stages, both primary and secondary methods of treatment are warranted. Although effective preventative screening methods are yet to be discovered, understanding the avoidable risk factors of its development will help lower the incidence of the disease. Hence, cigarette smoking, a major risk factor for PDAC, not only has both a direct and an indirect influence on PDACs etiology but also affects its prognosis. Understanding this will hopefully aid in the promotion of cessation of smoking in patients across the globe and elucidating the mechanism of the disease will provide opportunities for the development of therapeutic strategies for PDAC patients. Furthermore, identifying key steps in the mechanism of smoking-induced disease will lead to a comprehensive understanding and potential treatments of all stages of PDAC development. As there still is a limited amount of research on the mechanistic pathogenesis of smoking-induced PDAC progression, further studies are still needed. Finally, as we are certainly getting closer to the crucial drug discovery to help prolong survival in patients with PDAC, the need of identifying the key steps in the mechanism, as laid out in this article, is essential.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Siegel R, American Cancer Society Cancer Facts & Figures. June 11, 2019. Available at https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. Accessed June 11, 2019.
- 2.Wilson LS, Lightwood JM. Pancreatic cancer: total costs and utilization of health services. J Surg Oncol. 1999;71:171–181. [DOI] [PubMed] [Google Scholar]
- 3.Wu W, He X, Yang L, et al. Rising trends in pancreatic cancer incidence and mortality in 2000-2014. Clin Epidemiol. 2018;10:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandol SJ, Apte MV, Wilson JS, et al. The burning question: why is smoking a risk factor for pancreatic cancer? Pancreatology. 2012;12:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Momi N, Kaur S, Ponnusamy MP, et al. Interplay between smoking-induced genotoxicity and altered signaling in pancreatic carcinogenesis. Carcinogenesis. 2012;33:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:197–209. [DOI] [PubMed] [Google Scholar]
- 7.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–2638. [DOI] [PubMed] [Google Scholar]
- 8.Wittel UA, Momi N, Seifert G, et al. The pathobiological impact of cigarette smoke on pancreatic cancer development (review). Int J Oncol. 2012;41:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker AE, Hernandez YG, Frucht H, et al. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World J Gastroenterol. 2014;20:11182–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med. 1996;156:2255–2260. [PubMed] [Google Scholar]
- 11.Yuan C, Morales-Oyarvide V, Babic A, et al. Cigarette smoking and pancreatic cancer survival. J Clin Oncol. 2017;35:1822–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review. June 11, 2019. Available at: https://seer.cancer.gov/archive/csr/1975_2003/. Accessed June 11, 2019.
- 13.Philip PA. Targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2(4 Suppl 2):S16–S19. [PMC free article] [PubMed] [Google Scholar]
- 14.Bayraktar S, Bayraktar UD, Rocha-Lima CM. Recent developments in palliative chemotherapy for locally advanced and metastatic pancreas cancer. World J Gastroenterol. 2010;16:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mack TM, Yu MC, Hanisch R, et al. Pancreas cancer and smoking, beverage consumption, and past medical history. J Natl Cancer Inst. 1986;76:49–60. [PubMed] [Google Scholar]
- 16.Silverman DT, Dunn JA, Hoover RN, et al. Cigarette smoking and pancreas cancer: a case-control study based on direct interviews. J Natl Cancer Inst. 1994;86:1510–1516. [DOI] [PubMed] [Google Scholar]
- 17.Iodice S, Gandini S, Maisonneuve P, et al. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393:535–545. [DOI] [PubMed] [Google Scholar]
- 18.Bosetti C, Lucenteforte E, Silverman DT, et al. Cigarette smoking and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann Oncol. 2012;23:1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vrieling A, Bueno-de-Mesquita HB, Boshuizen HC, et al. Cigarette smoking, environmental tobacco smoke exposure and pancreatic cancer risk in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2010;126:2394–2403. [DOI] [PubMed] [Google Scholar]
- 20.Lynch SM, Vrieling A, Lubin JH, et al. Cigarette smoking and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. Am J Epidemiol. 2009;170:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyle P, Maisonneuve P, Bueno de Mesquita B, et al. Cigarette smoking and pancreas cancer: a case control study of the search programme of the IARC. Int J Cancer. 1996;67:63–71. [DOI] [PubMed] [Google Scholar]
- 22.Raimondi S, Maisonneuve P, Löhr JM, et al. Early onset pancreatic cancer: evidence of a major role for smoking and genetic factors. Cancer Epidemiol Biomarkers Prev. 2007;16:1894–1897. [DOI] [PubMed] [Google Scholar]
- 23.Stewart SL, Cardinez CJ, Richardson LC, et al. Surveillance for cancers associated with tobacco use—United States, 1999-2004. MMWR Surveill Summ. 2008;57:1–33. [PubMed] [Google Scholar]
- 24.Setiawan VW, Monroe K, Lugea A, et al. Uniting epidemiology and experimental disease models for alcohol-related pancreatic disease. Alcohol Res. 2017;38:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirkegård J, Mortensen FV, Cronin-Fenton D. Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am J Gastroenterol. 2017;112:1366–1372. [DOI] [PubMed] [Google Scholar]
- 26.Gukovsky I, Li N, Todoric J, et al. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199–1209.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuang L, Zhan X, Bi Y, et al. Induction of pancreatic inflammation accelerates pancreatic tumorigenesis in mice. Methods Mol Biol. 2019;1882:287–297. [DOI] [PubMed] [Google Scholar]
- 28.Dzeletovic I, Harrison ME, Crowell MD, et al. Pancreatitis before pancreatic cancer: clinical features and influence on outcome. J Clin Gastroenterol. 2014;48:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikuta K, Fukuda A, Ogawa S, et al. Nardilysin inhibits pancreatitis and suppresses pancreatic ductal adenocarcinoma initiation in mice. Gut. 2019;68:882–892. [DOI] [PubMed] [Google Scholar]
- 30.Tang D, Wu Q, Zhang J, et al. Galectin-1 expression in activated pancreatic satellite cells promotes fibrosis in chronic pancreatitis/pancreatic cancer via the TGF-β1/Smad pathway. Oncol Rep. 2018;39:1347–1355. [DOI] [PubMed] [Google Scholar]
- 31.Maekawa T, Fukaya R, Takamatsu S, et al. Possible involvement of Enterococcus infection in the pathogenesis of chronic pancreatitis and cancer. Biochem Biophys Res Commun. 2018;506:962–969. [DOI] [PubMed] [Google Scholar]
- 32.Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon CY, Li D, Cleary S, et al. The Association of Recently Diagnosed Diabetes and Long-term Diabetes with survival in pancreatic cancer patients: a pooled analysis. Pancreas. 2018;47:314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu K, Wang W, Chen H, et al. Insulin promotes proliferation of pancreatic ductal epithelial cells by increasing expression of PLK1 through PI3K/AKT and NF-κB pathway. Biochem Biophys Res Commun. 2019;19:925–930. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Liu H, Qian W, et al. Hyperglycemia aggravates microenvironment hypoxia and promotes the metastatic ability of pancreatic cancer. Comput Struct Biotechnol J. 2018;16:479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berrington de Gonzalez A, Sweetland S, Spencer E. A meta-analysis of obesity and the risk of pancreatic cancer. Br J Cancer. 2003;89:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim B, Chung MJ, Park SW. Visceral obesity is associated with poor prognosis in pancreatic adenocarcinoma. Nutr Cancer. 2016;68:201–207. [DOI] [PubMed] [Google Scholar]
- 38.Jiang QL, Wang CF, Tian YT, et al. Body mass index does not affect the survival of pancreatic cancer patients. World J Gastroenterol. 2017;23:6287–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messaggio F, Mendonsa AM, Castellanos J. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget. 2017;8:85378–85391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaytouni T, Tsai PY, Hitchcock DS. Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat Commun. 2017;14:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eibl G, Rozengurt E. KRAS, YAP, and obesity in pancreatic cancer: a signaling network with multiple loops. Semin Cancer Biol. 2019;54:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weissman S, Thaker R, Zeffren N, et al. Intraductal papillary mucinous neoplasm of the pancreas: understanding the basics and beyond. Cureus. 2019;11:e3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rezaee N, Barbon C, Zaki A, et al. Intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia is a risk factor for the subsequent development of pancreatic ductal adenocarcinoma. HPB (Oxford). 2016;18:236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akita H, Takeda Y, Hoshino H, et al. Mural nodule in branch duct-type intraductal papillary mucinous neoplasms of the pancreas is a marker of malignant transformation and indication for surgery. Am J Surg. 2011;202:214–219. [DOI] [PubMed] [Google Scholar]
- 45.Hirono S, Tani M, Kawai M, et al. The carcinoembryonic antigen level in pancreatic juice and mural nodule size are predictors of malignancy for branch duct type intraductal papillary mucinous neoplasms of the pancreas. Ann Surg. 2012;255:517–522. [DOI] [PubMed] [Google Scholar]
- 46.Salvia R, Fernández-del Castillo C, Bassi C, et al. Main-duct intraductal papillary mucinous neoplasms of the pancreas: clinical predictors of malignancy and long-term survival following resection. Ann Surg. 2014;239:678–685; discussion 685–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez JR, Salvia R, Crippa S, et al. Branch-duct intraductal papillary mucinous neoplasms: observations in 145 patients who underwent resection. Gastroenterology. 2007;133:72–79; quiz 309–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang YT, Gou YW, Jin WW, et al. Association between alcohol intake and the risk of pancreatic cancer: a dose–response meta-analysis of cohort studies. BMC Cancer. 2016;16:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer. 2009;8:109–117. [DOI] [PubMed] [Google Scholar]
- 50.Cicenas J, Kvederaviciute K, Meskinyte I, et al. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel). 2017;9 pii: E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grochola LF, Taubert H, Greither T, et al. Elevated transcript levels from the MDM2 P1 promoter and low p53 transcript levels are associated with poor prognosis in human pancreatic ductal adenocarcinoma. Pancreas. 2011;40:265–270. [DOI] [PubMed] [Google Scholar]
- 52.Blackford A, Serrano OK, Wolfgang CL, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Heijden MS, Yeo CJ, Hruban RH, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63:2585–2588. [PubMed] [Google Scholar]
- 54.Bryant KL, Mancias JD, Kimmelman AC, et al. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. [DOI] [PubMed] [Google Scholar]
- 56.Maisonneuve P, Lowenfels AB, Müllhaupt B, et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut. 2005;54:510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Law R, Parsi M, Lopez R, et al. Cigarette smoking is independently associated with chronic pancreatitis. Pancreatology. 2010;10:54–59. [DOI] [PubMed] [Google Scholar]
- 58.Śliwińska-Mossoń M, Milnerowicz H. The impact of smoking on the development of diabetes and its complications. Diab Vasc Dis Res. 2017;14:265–276. [DOI] [PubMed] [Google Scholar]
- 59.Will JC, Galuska DA, Ford ES, et al. Cigarette smoking and diabetes mellitus: evidence of a positive association from a large prospective cohort study. Int J Epidemiol. 2001;30:540–546. [DOI] [PubMed] [Google Scholar]
- 60.Chiolero A, Faeh D, Paccaud F, et al. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008;87:801–809. [DOI] [PubMed] [Google Scholar]
- 61.Audrain-McGovern J, Benowitz NL. Cigarette smoking, nicotine, and body weight. Clin Pharmacol Ther. 2011;90:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hecht SS, Hoffmann D. N-nitroso compounds and tobacco-induced cancers in man. IARC Sci Publ. 1991;54–61. [PubMed] [Google Scholar]
- 63.Li D, Firozi PF, Zhang W, et al. DNA adducts, genetic polymorphisms, and K-ras mutation in human pancreatic cancer. Mutat Res. 2002;513:37–48. [DOI] [PubMed] [Google Scholar]
- 64.Kamata K, Takenaka M, Nakai A, et al. Association between the risk factors for pancreatic ductal adenocarcinoma and those for malignant intraductal papillary mucinous neoplasm. Oncology. 2017;93(suppl 1):102–106. [DOI] [PubMed] [Google Scholar]
- 65.Nakagawa T, Masuda A, Toyama H, et al. Smoking status and the incidence of pancreatic cancer concomitant with intraductal papillary mucinous neoplasm. Pancreas. 2017;46:582–588. [DOI] [PubMed] [Google Scholar]
- 66.Zhang S, Wang C, Huang H, et al. Effects of alcohol drinking and smoking on pancreatic ductal adenocarcinoma mortality: a retrospective cohort study consisting of 1783 patients. Sci Rep. 2017;7:9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoffmann D, Rivenson A, Abbi R, et al. A study of tobacco carcinogenesis: effect of the fat content of the diet on the carcinogenic activity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1993;53:2758–2761. [PubMed] [Google Scholar]
- 68.Deng Y, Tu H, Pierzynski JA, et al. Determinants and prognostic value of quality of life in patients with pancreatic ductal adenocarcinoma. Eur J Cancer. 2018;92:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuang JJ, Jiang ZM, Chen YX, et al. Smoking exposure and survival of patients with esophagus cancer: a systematic review and meta-analysis. Gastroenterol Res Pract. 2016;2016:7682387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chao A, Thun MJ, Jacobs EJ, et al. Cigarette smoking and colorectal cancer mortality in the cancer prevention study II. J Natl Cancer Inst. 2000;92:1888–1896. [DOI] [PubMed] [Google Scholar]
- 71.Mayadev J, Lim J, Durbin-Johnson B, et al. Smoking decreases survival in locally advanced cervical cancer treated with radiation. Am J Clin Oncol. 2018;41:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Y, Qi Y, Zhang J, et al. The impact of smoking on survival in renal cell carcinoma: a systematic review and meta-analysis. Tumour Biol. 2014;35:6633–6640. [DOI] [PubMed] [Google Scholar]
- 73.Fujino Y, Mizoue T, Tokui N, et al. Cigarette smoking and mortality due to stomach cancer: findings from the JACC study. J Epidemiol. 2005;15 Suppl 2:S113–S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zu K, Giovannucci E. Smoking and aggressive prostate cancer: a review of the epidemiologic evidence. Cancer Causes Control. 2009;20:1799–1810. [DOI] [PubMed] [Google Scholar]
- 75.Olson SH, Chou JF, Ludwig E, et al. Allergies, obesity, other risk factors and survival from pancreatic cancer. Int J Cancer. 2010;127:2412–2419. [DOI] [PubMed] [Google Scholar]
- 76.Dandona M, Linehan D, Hawkins W, et al. Influence of obesity and other risk factors on survival outcomes in patients undergoing pancreaticoduodenectomy for pancreatic cancer. Pancreas. 2011;40:931–937. [DOI] [PubMed] [Google Scholar]
- 77.Allison DC, Piantadosi S, Hruban RH, et al. DNA content and other factors associated with ten-year survival after resection of pancreatic carcinoma. J Surg Oncol. 1998;67:151–159. [DOI] [PubMed] [Google Scholar]
- 78.Trichopoulos D, Tzonou A, Kalapothaki V, et al. Alpha 1-antitrypsin and survival in pancreatic cancer. Int J Cancer. 1990;45:685–686. [DOI] [PubMed] [Google Scholar]
- 79.Park SM, Lim MK, Shin SA, et al. Impact of prediagnosis smoking, alcohol, obesity, and insulin resistance on survival in male cancer patients: National Health Insurance Corporation Study. J Clin Oncol. 2006;24:5017–5024. [DOI] [PubMed] [Google Scholar]
- 80.Yu GP, Ostroff JS, Zhang ZF, et al. Smoking history and cancer patient survival: a hospital cancer registry study. Cancer Detect Prev. 1997;21:497–509. [PubMed] [Google Scholar]
- 81.Pelucchi C, Galeone C, Polesel J, et al. Smoking and body mass index and survival in pancreatic cancer patients. Pancreas. 2014;43:47–52. [DOI] [PubMed] [Google Scholar]
- 82.Ouyang H, Ma W, Liu F, et al. Factors influencing survival of patients with pancreatic adenocarcinoma and synchronous liver metastases receiving palliative care. Pancreatology. 2017;17:773–781. [DOI] [PubMed] [Google Scholar]
- 83.Longnecker DS, Shinozuka H, Dekker A. Focal acinar cell dysplasia in human pancreas. Cancer. 1980;45:534–540. [DOI] [PubMed] [Google Scholar]
- 84.Wittel UA, Singh AP, Henley BJ, et al. Cigarette smoke-induced differential expression of the genes involved in exocrine function of the rat pancreas. Pancreas. 2006;33:364–370. [DOI] [PubMed] [Google Scholar]
- 85.Auerbach O, Garfinkel L. Histologic changes in pancreas in relation to smoking and coffee-drinking habits. Dig Dis Sci. 1986;31:1014–1020. [DOI] [PubMed] [Google Scholar]
- 86.Topsakal S, Ozmen O, Aslankoc R, et al. Pancreatic damage induced by cigarette smoke: the specific pathological effects of cigarette smoke in the rat model. Toxicol Res (Camb). 2016;5:938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hirai N, Kawano H, Hirashima O, et al. Insulin resistance and endothelial dysfunction in smokers: effects of vitamin C. Am J Physiol Heart Circ Physiol. 2000;279:H1172–H1178. [DOI] [PubMed] [Google Scholar]
- 88.Liu ST, Pham H, Pandol SJ, et al. Src as the link between inflammation and cancer. Front Physiol. 2013;4:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abram CL, Lowell CA. The diverse functions of Src family kinases in macrophages. Front Biosci. 2008;13:4426–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarang Z, Köröskényi K, Pallai A, et al. Translutaminase 2 null macrophages respond to lipopolysaccharide stimulation by elevated proinflammatory cytokine production due to an enhanced ανβ3 integrin-induced Src tyrosine kinase signaling. Immunol Lett. 2011;138:71–78. [DOI] [PubMed] [Google Scholar]
- 91.Wittel UA, Pandey KK, Andrianifahanana M, et al. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am J Gastroenterol. 2006;101:148–159. [DOI] [PubMed] [Google Scholar]
- 92.Foster JR, Idle JR, Hardwick JP, et al. Induction of drug-metabolizing enzymes in human pancreatic cancer and chronic pancreatitis. J Pathol. 1993;169:457–463. [DOI] [PubMed] [Google Scholar]
- 93.Kong B, Qia C, Erkan M, et al. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Edderkaoui M, Park C, Lee I, et al. Novel model of pancreatic neoplastic lesions induced by smoking compound NNK. In: Abstracts of Papers Submitted to the 42nd Annual Meeting of the American Pancreatic Association, November 2-5, 2011, Chicago, Illinois. Pancreas. 2011;40:1320.abstract. [Google Scholar]
- 95.Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol. 2005;131:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weddle DL, Tithoff P, Williams M, et al. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis. 2001;22:473–479. [DOI] [PubMed] [Google Scholar]
- 97.Askari MD, Tsao MS, Cekanova M, et al. Ethanol and the tobacco-specific carcinogen, NNK, contribute to signaling in immortalized human pancreatic duct epithelial cells. Pancreas. 2006;33:53–62. [DOI] [PubMed] [Google Scholar]
- 98.Teschendorff AE, Yang Z, Wong A, et al. Correlation of smoking-associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol. 2015;1:476–485. [DOI] [PubMed] [Google Scholar]
- 99.Xi S, Xu H, Shan J, et al. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J Clin Invest. 2013;123:1241–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kugel S, Sebastián C, Fitamant J, et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell. 2016;165:1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Bai R, Li M, et al. Excessive miR-25-3p maturation via N6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10:1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. [DOI] [PubMed] [Google Scholar]
- 103.Wu T, Chen W, Kong D, et al. miR-25 targets the modulator of apoptosis 1 gene in lung cancer. Carcinogenesis. 2015;36:925–935. [DOI] [PubMed] [Google Scholar]
- 104.Razumilava N, Bronk SF, Smoot RL, et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson JS, Pirola RC, Apte MV. Stars and stripes in pancreatic cancer: role of stellate cells and stroma in cancer progression. Front Physiol. 2014;5:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mollenhauer J, Roether I, Kern HF. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas. 1987;2:14–24. [DOI] [PubMed] [Google Scholar]
- 107.Pandol S, Gukovskaya A, Edderkaoui M, et al. Epidemiology, risk factors, and the promotion of pancreatic cancer: role of the stellate cell. J Gastroenterol Hepatol. 2012;27 Suppl 2:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vaquero EC, Edderkaoui M, Nam KJ, et al. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology. 2003;125:1188–1202. [DOI] [PubMed] [Google Scholar]
- 109.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. 2013;25:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]