

Abstract
Invariant natural killer T (iNKT) cells recognize activating self and microbial lipids presented by CD1d. CD1d can also bind non-activating lipids, such as sphingomyelin. We hypothesized that these serve as endogenous regulators and investigated humans and mice deficient in acid sphingomyelinase (ASM), an enzyme that degrades sphingomyelin. We show that ASM absence in mice leads to diminished CD1d-restricted antigen presentation and iNKT cell selection in the thymus, resulting in decreased iNKT cell levels and resistance to iNKT cell-mediated inflammatory conditions. Defective antigen presentation and decreased iNKT cells are also observed in ASM-deficient humans with Niemann-Pick disease, and ASM activity in healthy humans correlates with iNKT cell phenotype. Pharmacological ASM administration facilitates antigen presentation and restores the levels of iNKT cells in ASM-deficient mice. Together, these results demonstrate that control of non-agonistic CD1d-associated lipids is critical for iNKT cell development and function in vivo and represents a tight link between cellular sphingolipid metabolism and immunity.
Invariant natural killer T (iNKT) cells are an important lymphocyte population that sense self- and microbial lipids presented by the major histocompatibility complex (MHC) class I-like glycoprotein CD1d1. In response to these antigens, iNKT cells rapidly release large arrays of mediators, making them powerful and early modulators of immune pathways2. The self-reactivity of iNKT cells is also critical for their development in the thymus3, where iNKT cells are positively selected by CD1d-bearing thymocytes4. While there have been great efforts to identify CD1d-binding, iNKT cell-activating lipids (that is, lipid antigens5), iNKT cell activation is also amenable to negative regulation by CD1d-associated lipids that do not stimulate the iNKT cell antigen receptor (TCR). As such, iNKT cell activation is anticipated to be influenced by the balance of CD1d-associated antigenic and non-antigenic lipids. However, little is known about the functional relevance of non-antigenic lipids that potentially impede CD1d-restricted iNKT cell activation. Sphingolipids, which are abundantly present in the cell membrane6, are a major class of CD1d-associated lipids7,8. Sphingomyelin, a dominant sphingolipid in mammals, has been reported to be a non-stimulatory CD1d-associated lipid in vitro9, leading us to hypothesize that it may regulate CD1d access to potentially agonistic lipids.
Sphingomyelin is degraded by sphingomyelinases into ceramide and phosphorylcholine10. In lysosomes, one of the sites where the exchange and loading of lipids onto CD1d takes place11, ASM is the primary enzyme responsible for sphingomyelin degradation12,13. In light of the non-stimulatory nature of sphingomyelin in vitro9, we sought to understand the consequences of sphingomyelin accumulation on iNKT cell function. To do so, we used mice with homozygous deficiency in the gene encoding ASM (Smpd1−/−; called ‘Asm−/−’ here)14 that develop age-dependent accumulation of sphingomyelin and a clinical phenotype resembling ASM deficiency in humans, known as Niemann-Pick disease (NPD) types A and B, where NPD-A is the more severe form of disease first manifesting during infancy. We demonstrate that control of the cellular abundance of sphingomyelin by ASM regulates the presentation of endogenous and exogenous lipid antigens by CD1d in thymocytes and dendritic cells (DCs) and thus the abundance and activation of iNKT cells. In the absence of ASM in mice, sphingomyelin levels increased in hematopoietic cells, resulting in decreased CD1d-restricted antigen presentation, impaired iNKT cell development in the thymus and reduced abundance and activation of iNKT cells, defects that were reversed by the transfer of wild-type bone marrow or administration of recombinant human ASM (rhASM). These observations can be extended to humans with or without NPD and establish ASM, through its control of sphingomyelin levels, as an important regulator of iNKT cells with potential therapeutic implications.
Results
ASM is active in the hematopoietic system and required for iNKT cell development.
Although it is well established that ASM is expressed in tissues where the clinical phenotypes of NPD-A and NPD-B are most prominent, such as the liver and brain as well as in some hematopoietic cells, such as macrophages15, little is known about its function in the immune system. Therefore, we first investigated ASM activity and found it to be demonstrable in a variety of parenchymal (colon and liver) and hematopoietic (spleen, thymus and dendritic) cells, with the highest levels in DCs (Fig. 1a), a critical CD1d-expressing antigen-presenting cell (APC) in the periphery16. Consistent with this being functionally important, flow cytometry (gating strategies in Supplementary Fig. 1) revealed a dramatic reduction of iNKT cells in the thymus, liver and spleen of adult Asm−/− mice (Fig. 1b,c and Supplementary Fig. 2a). This was associated with a perturbation in the distribution of the few remaining iNKT cells, as shown by decreased relative levels of iNKT cells that expressed CD4 and a relative enrichment in the CD4−CD8− (double-negative (DN)) iNKT cell fraction in the periphery (spleen and liver) (Supplementary Fig. 2b). The reduction in iNKT cell abundance was also associated with a decrease in absolute iNKT cell numbers (Fig. 1d). Furthermore, when PBS57-loaded CD1d-tetramer-positive cells (that is, iNKT cells) were excluded, there were no changes in the relative abundance of tetramer-negative thymic T cells (Supplementary Fig. 3a) or alterations in the distribution of CD4+, CD8+, DN or CD4+CD8+ (double-positive (DP)) thymocytes (Supplementary Fig. 3b). In the periphery, tetramer-negative T cells were unaltered in relative abundance in the livers of Asm−/− mice and slightly reduced in the spleen of these mice (Supplementary Fig. 3a) in association with minor changes in the proportion of CD4+, CD8+ and DN cells (Supplementary Fig. 3b). We also did not observe changes in the relative abundance of γδ T cells, another type of unconventional T cell, and regulatory T (Treg) cells when tetramer-positive cells were excluded from the analysis (Supplementary Fig. 3c). To rule out the possibility that the reduction in iNKT cells was secondary to a general disturbance of lysosomal function, we investigated mice just after weaning at the age of 2 weeks, when the accumulation of sphingomyelin in parenchymal tissues is known to be minimal14. In these young mice, we also observed a reduction in iNKT cells (Fig. 1e). These studies demonstrate that ASM is functionally present in immune tissues and that deficiency is associated with a specific alteration in iNKT cell homeostasis.
Fig. 1 |. Acid sphingomyelinase-deficient mice have a reduced number of iNKT cells.

a, ASM activity was measured in tissues from wild-type (WT) mice (n = 4) using a colorimetric assay in tissue lysates generated by repeated freeze-thaw cycles from the indicated tissues. The results are representative of two independent experiments. b, Representative flow cytometry of lymphocytes from spleens in WT and Asm−/− mice visualizing the number of iNKT cells as defined by a PBS57-loaded CD1d tetramer and CD3. c, Percentages of iNKT cells among lymphocytes in the thymus, spleen and liver of age- and sex-matched WT (n = 5) and Asm−/− (n = 4) mice defined by a PBS57-loaded CD1d tetramer and CD3. The results are representative of three independent experiments. d, Absolute numbers of iNKT cells in the thymus, spleen and liver of age- and sex-matched WT (n = 5) and Asm−/− (n = 5) mice defined by a PBS57-loaded CD1d tetramer and TCRβ. The results are representative of three independent experiments. e, Representative flow cytometry of lymphocytes from spleens in WT and Asm−/− mice at 2 weeks of age visualizing the number of iNKT cells as defined by a PBS57-loaded CD1d tetramer and CD3. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by a two-sided Student’s t-test. *P < 0.001, **P < 0.0001.
ASM is a major regulator of iNKT cell function in vivo.
To determine whether ASM deficiency affects iNKT cell function in vivo, we examined the responses of Asm−/− mice in three different iNKT cell-dependent disease models. First, systemic α-galactosylceramide (α-GalCer) administration, a model of iNKT cell-mediated hepatitis, resulted in reduced serum levels of alanine transaminase (ALT) and cytokines including interleukin (IL)-4 and interferon (IFN)-γ in Asm−/− mice compared to wild-type mice (Fig. 2a). Second, we observed that delayed type hypersensitivity induced by oxazolone, a model for skin allergy that is dependent on iNKT cell activation17, was reduced in Asm−/− mice, as defined by weight loss after skin sensitization (Fig. 2b) and ear swelling following oxazolone re-challenge (Fig. 2c). Lastly, we examined concanavalin A (ConA) hepatitis, a model of hepatic injury driven by direct (CD1d-independent) activation of iNKT cells18. In this model, tissue damage (Fig. 2d,e) and damage-associated elevations of serum transaminases (Fig. 2f) were dramatically reduced in Asm−/− mice, accompanied by a reduction in the expression of iNKT cell-associated cytokines, such as IL-4 after 6 h and IFN-γ after 24 h, in the serum (Fig. 2f). Such diminished in vivo responses to α-GalCer, oxazolone and ConA in Asm−/− mice are consistent with the decreased levels of iNKT cells and/or impaired presentation of agonistic CD1d-restricted antigens. Together, these models document the importance of ASM in regulating iNKT cell-dependent inflammation.
Fig. 2 |. NKT cell-mediated disease models are affected by ASM deficiency.

a, Age- and sex-matched WT and Asm−/− mice were injected with 2 μg of α-GalCer intraperitoneally (i.p.). Cytokine levels in serum were determined 4 h and 24 h after injection and ALT levels in serum were determined 24 h after injection. Results are representative of three independent experiments. b, Oxazolone was used to sensitize WT and Asm−/− mice on the skin at day 0. The mice were followed daily with weight measurements and the values reflect the weight relative to the starting weight. Results are representative of three independent experiments. c, Five days after the skin sensitization, the mice received a re-challenge by application of oxazolone to the ear. Ear swelling was measured by a sensitive micrometer and the lines indicate the increase in ear thickness. d, Quantification of necrotic areas in WT and Asm−/− mice injected with ConA. The results represent pooled results from three independent experiments. e, Representative hematoxylin and eosin (H&E)-stained tissue sections from WT and Asm−/− mice 24 h after injection with ConA. The black bar indicates 200 μm. Results are representative of three independent experiments. f, Age- and sex-matched WT and Asm−/− mice were injected with ConA. Cytokine and ALT levels were determined in serum 6 and 24 h after injection. The results represent pooled results from three independent experiments. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by a two-sided Student’s t-test (a), two-way ANOVA with Bonferroni’s correction for multiple comparisons (b and c) or a two-sided Mann-Whitney U-test (d and f). *P < 0.05, **P < 0.01, ***P < 0.001.
ASM deficiency leads to altered lipid antigen presentation and thymic iNKT cell selection.
We hypothesized that the reduction in iNKT cells observed in Asm−/− mice was due to reduced presentation of CD1d-restricted antigens involved in the positive selection of iNKT cells in the thymus. In contrast to classical MHC class I- and class II-restricted T cells, which are positively selected by cortical thymic epithelial cells, iNKT cells are selected by CD1d-expressing thymocytes4. We observed that, although CD1d expression was unaffected by ASM deficiency in young mice and only slightly reduced in the thymus of adult mice (Supplementary Fig. 4), thymocytes from both adult and young (2-week-old) Asm−/− mice compared to wild-type thymocytes led to reduced IL-2 release by co-cultured iNKT hybridomas in response to CD1d-restricted presentation of endogenous (self-) and exogenous (α-GalCer) antigens (Fig. 3a). Of note, similar cell-surface expression of CD1d in young Asm−/− and wild-type mice suggests that defects in CD1d-restricted antigen presentation in the absence of ASM are not an indirect consequence of altered CD1d trafficking, as the latter typically affects CD1d cell-surface expression19,20. Further, the iNKT hybridomas used are ASM proficient, which rules out iNKT cell-intrinsic defects due to the lack of ASM as the cause of reduced activation. Inhibition with 19G11, a monoclonal antibody directed against CD1d, confirmed that iNKT cell activation in these assays was CD1d dependent (Supplementary Fig. 5a).
Fig. 3 |. Lipid antigen presentation by thymocytes and DCs from ASM-deficient mice is reduced and bone marrow transfer restores iNKT cell levels in Asm−/− mice.
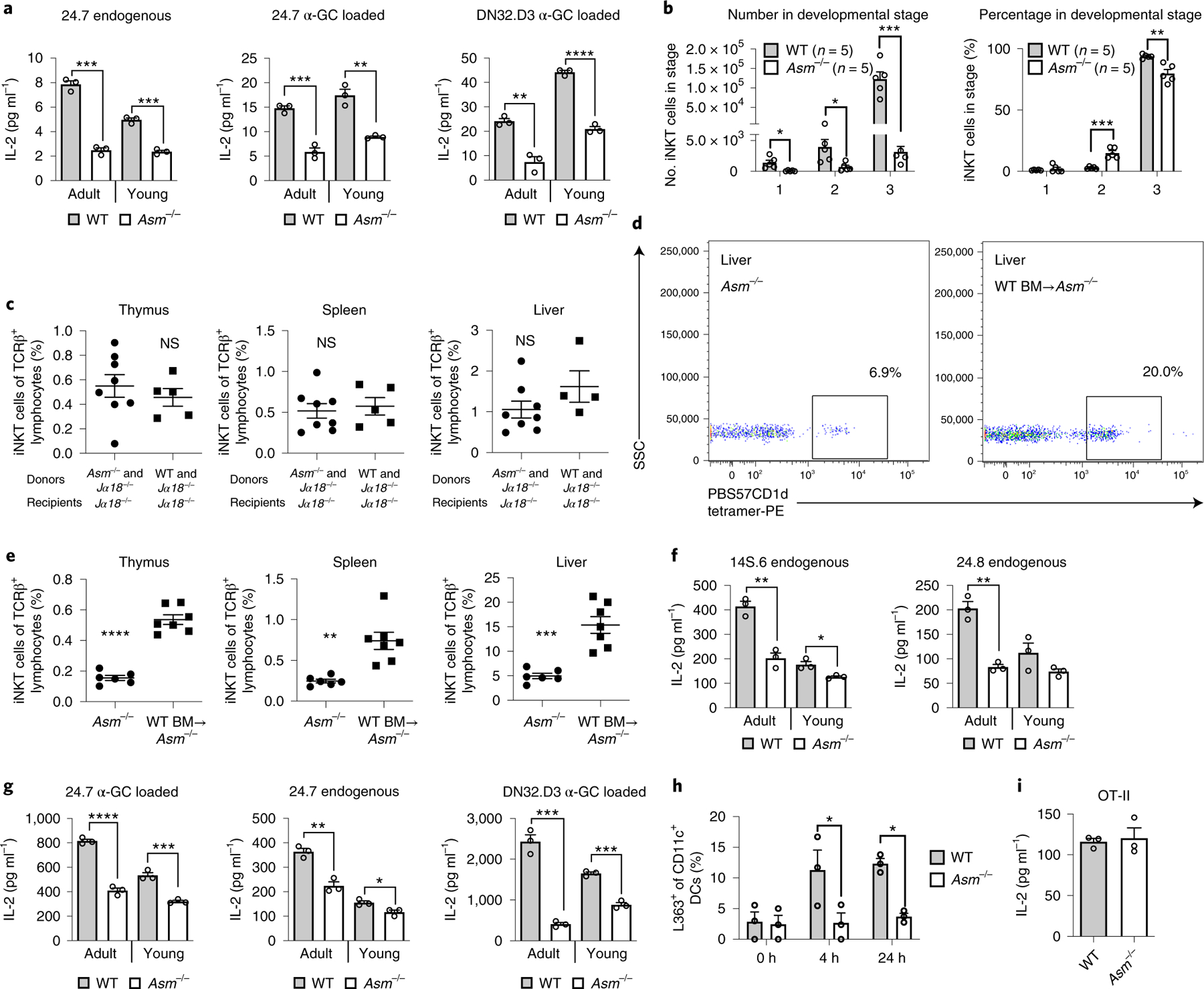
a, Thymocytes were incubated with α-GalCer (α-GC) for 4 h or left untreated, followed by addition of the indicated iNKT hybridomas. Thymocytes from both young (2-week-old) and adult Asm−/− mice were used as indicated. b, The graphs show absolute (left) and relative (right) numbers of iNKT cells in different thymic developmental stages in Asm−/− and WT mice. Stage 1 was defined as CD24loCD44loNK1.1lo, stage 2 as CD24loCD44hiNK1.1lo and stage 3 as CD24loCD44hiNK1.1hi. The results are representative of two independent experiments. c, Bone marrow (BM) chimeras were made by the transfer of a mixture of Jα18−/− and WT (n = 5) or Asm−/− (n = 8) BM into irradiated Jα18−/− recipients. The graphs demonstrate the percentage of cells positive for PBS57-loaded CD1d tetramer among TCRβ-positive cells (iNKT cells) 3 months after BM transfer. The results are representative of two independent experiments. d,e, Bone marrow (BM) chimeras were made by transfer of WT CD45.1+ BM to Asm−/− mice (n = 7) in comparison to non-irradiated Asm−/− mice not receiving BM (n = 6). The dot plots (d) show representative plots from the livers of a non-irradiated Asm−/− mouse not receiving BM and an Asm−/− mouse receiving WT BM. The graphs (e) demonstrate the percentage of cells positive for PBS57-loaded CD1d tetramer among TCRβ-positive cells (iNKT cells) 3 months after BM transfer. The results represent pooled results from three independent experiments. f,g, CD11c+ DCs were extracted from spleens with magnetic beads and co-cultured with the indicated NKT hybridomas. Before co-culture, the DCs in 24.7 α-GC loaded and DN32.D3 α-GC loaded were loaded with α-GC for 4 h (g). DCs from both young (2-week-old) and adult Asm−/− mice were used as indicated. h, CD11c+ DCs were extracted from spleens and incubated with α-GC for 4 or 24 h and stained with an antibody recognizing α-GC bound to CD1d in three technical replicates. The results are representative of two independent experiments. i, DCs from WT and Asm−/− mice were loaded with ovalbumin and cultured with ovalbumin-reactive T cells from OT-II mice. In all co-culture experiments (a,f,g and i), IL-2 levels were measured in three independent wells 20–24 h after addition of the indicated NKT hybridomas or T cells, and all these results are representative of three independent experiments. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by a two-sided Student’s t-test in all panels except h, where a two-way ANOVA with Bonferroni’s correction for multiple comparisons was used. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant.
Given the dramatic reduction of iNKT cells in Asm−/− mice and the defects of Asm−/− thymocytes in CD1d-restricted antigen presentation (Fig. 3a), we investigated whether ASM deficiency affects thymic development of iNKT cells. To do this, we characterized thymic iNKT cell development4. As expected from the reduction in thymic iNKT cells in Asm−/− mice (Fig. 1c,d), ASM deficiency was associated with dramatically reduced absolute numbers of iNKT cells at stages 1, 2 and 3 of thymic development (Fig. 3b, left). Within the small population of thymic iNKT cells in Asm−/− mice, the relative proportion of cells in stage 2 was increased and the proportion in stage 3 was reduced compared to wild-type mice (Fig. 3b, right). These results suggest a major bottleneck at the earliest stages of iNKT development in the thymus when ASM is absent.
To determine whether these developmental differences were due to an intrinsic defect in iNKT cells in Asm−/− mice rather than an abnormality in antigen presentation resulting in altered iNKT cell positive selection, we generated mixed-bone-marrow chimeras, in which irradiated recipient mice with homozygous deficiency in the gene encoding TCR α-chain joining region 18 (Traj18−/−; called ‘Jα18−/−’ here) received bone marrow from Jα18−/− mice, which is ASM sufficient and expresses CD1d but does not give rise to iNKT cells owing to a deficiency of the invariant TCR Jα segment21, mixed 1:1 with bone marrow from either Asm−/− or wild-type mice. In this experiment, functional CD1d was provided by Jα18−/− thymocytes, which should allow unimpaired development of iNKT cells from the precursor cells in Asm−/− bone marrow, the only source of iNKT cells in this experiment. In line with this hypothesis, iNKT cells derived from Asm−/− bone marrow were observed at similar abundance to those derived from wild-type bone marrow in the thymus, spleen and liver (Fig. 3c). These studies ruled out a cell-intrinsic defect in iNKT cells in the developing thymus of Asm−/− mice. Furthermore, reconstitution of irradiated CD45.2+ Asm−/− mice with bone marrow from wild-type ASM-expressing CD45.1+ mice restored iNKT cells (Fig. 3d,e). These studies demonstrate that iNKT cell defects in Asm−/− mice arise from the bone-marrow-derived radiosensitive compartment. In control experiments, irradiation of Asm−/− mice followed by transfer of Asm−/− bone marrow did not result in an increase in the abundance of iNKT cells (Supplementary Fig. 5b).
Similarly to thymocytes, CD11c+ DCs from the spleens of young and adult Asm−/− mice were less able to stimulate the endogenous reactivity of the non-invariant NKT cell hybridoma 14S.6 and the iNKT cell hybridoma 24.8 (ref.22) (Fig. 3f). Additionally, we observed decreased presentation of endogenous and exogenous (α-GalCer) lipid antigens to the 24.7 and DN32.D3 iNKT cell hybridomas in comparison to DCs from wild-type mice (Fig. 3g). The activation of iNKT cells was CD1d restricted and abolished by antibody-mediated blockade of CD1d (Supplementary Fig. 5c). By direct staining using a monoclonal antibody recognizing the complex of α-GalCer bound to CD1d on the cell surface, it was evident that reduced iNKT cell activation resulted from reduced loading of CD1d with α-GalCer (Fig. 3h). Finally, MHC class II-restricted antigen presentation of ovalbumin by DCs, which requires lysosomal processing of the model antigen ovalbumin, to T cells from OT-II mice23 was unperturbed in Asm−/− mice, thus excluding general defects in lysosomal antigen processing and presentation of antigens to T cells (Fig. 3i). Together, these data demonstrate that ASM deficiency limits the ability of thymocytes and DCs to load and present CD1d-associated antigens, resulting in defects in thymic development and maturation of iNKT cells.
ASM deficiency in humans is associated with reduced abundance of iNKT cells and an altered iNKT cell phenotype.
To extend these studies to humans, we investigated whether a similar defect in CD1d-restricted antigen presentation can be detected in patients with NPD-A and NPD-B who are deficient in ASM. To this end, we used lentiviral transduction to introduce human CD1d into Epstein-Barr virus (EBV)-transformed B cells from a patient with NPD-B and three healthy controls (Supplementary Fig. 6a). While CD1d expression in NPD lymphoblasts was within the range observed for controls (Supplementary Fig. 6a), the CD1d-transfected NPD lymphoblasts exhibited decreased CD1d-restricted presentation of α-GalCer to the iNKT hybridoma 58ab (ref.24) compared to similarly transfected control lymphoblasts (Fig. 4a). Given the reduced iNKT cell numbers in Asm−/− mice, we analyzed the abundance of iNKT cells in the peripheral blood of patients with NPD-A and NPD-B. While iNKT cell numbers showed variation in healthy controls, in line with previous observations25, iNKT cell frequencies in patients with NPD-A and NPD-B were significantly decreased and at the lower end of those observed in controls (Fig. 4b,c and Supplementary Table 1). The median relative abundance of iNKT cells among patients with NPD-A and NPD-B was 0.014% (interquartile range, 0.012–0.029%) compared to 0.086% (interquartile range, 0.037–0.18%) among the control subjects (P = 0.0004). In addition, the phenotype of the residual iNKT cells in patients with NPD differed from that in controls, as shown by a dramatically altered CD4+/CD8+ iNKT cell ratio and reduced expression of the maturation marker CD161 (ref.26), which is acquired through interactions with CD1d in the periphery4 (Supplementary Fig. 6b). This reduction in iNKT cells in humans with NPD is in marked contrast to observations in Gaucher’s disease27, Fabry’s disease28 and NPD type C29, all of which represent sphingolipid-dependent lysosomal storage diseases, wherein iNKT cell levels are not affected. In contrast to iNKT cells, no alterations were detected in the abundance of conventional T cells or their CD4+ and CD8+ subpopulations (Fig. 4d and Supplementary Fig. 6c). In conclusion, the iNKT cell defects observed in patients with NPD suggest a role of ASM in the regulation of human iNKT cell development, in line with the observations made in Asm−/− mice. The absence of defects in conventional T cells in patients with NPD further suggests that such alterations are specific to iNKT cells and that they do not result from general defects in lysosomal antigen processing. This is in line with unaltered MHC class II-restricted antigen presentation (Fig. 3i) and unaffected frequencies of conventional T cells (Supplementary Fig. 3a) in Asm−/− mice.
Fig. 4 |. Human ASM deficiency is associated with reduced CD1d-restricted antigen presentation to iNKT cells and a reduced level of iNKT cells.

a, EBV-transformed B cells were transduced with a lentiviral construct encoding human CD1d, loaded with α-GalCer and cultured with the 58ab hybridoma. The graph shows the mean IL-2 levels from three independent wells with cells from the indicated individuals. The results are representative of three independent experiments. b, Representative dot plots from a patient with NPD and a healthy control. The percentages indicate the percentage of iNKT cells among lymphocytes. c,d, The graphs demonstrate the level of iNKT cells (c) and T cells (d) in patients with NPD (n = 5) compared to healthy controls (n = 70). The values indicate the percentage of iNKT cells or T cells among lymphocytes. e, ASM activity in PBMCs was measured using a colorimetric assay and the iNKT cell phenotype was examined by flow cytometry in healthy controls (n = 25). The graphs show the correlation between ASM activity and the percentage of CD8+ (left) or CD161+ (right) cells among iNKT cells. Mean values are shown with error bars representing the s.e.m. in a, and the line indicates the median value in c. P values were calculated by one-way ANOVA with Bonferroni’s correction for multiple comparisons (a), a two-sided Mann-Whitney U-test (c and d) and two-sided Pearson’s correlation (e). *P < 0.05, **P < 0.01, ***P < 0.001; NS, not significant.
We also evaluated a potential correlation between ASM activity and iNKT cell abundance and function among healthy humans. To this end, we determined ASM activity in peripheral blood mononuclear cells (PBMCs) and correlated it with peripheral iNKT cell frequency and iNKT cell phenotype. While we did not observe a correlation between ASM activity in PBMCs and the abundance of iNKT cells, ASM activity was variable and positively correlated with the expression of CD161 and the relative proportion of iNKT cells expressing CD8 (Fig. 4e). These results suggest that ASM activity regulates CD1d and NKT cells in the periphery and/or the thymus in healthy human subjects under physiological conditions.
ASM deficiency leads to early accumulation of sphingomyelin, which competes with agonistic lipids for binding to CD1d.
Our results so far demonstrated that deficiency in ASM is associated with impaired presentation of CD1d-associated antigens, leading to defects in the positive selection of iNKT cells. We therefore analyzed whether changes in the lipidome contribute to the observed defects. To this end, we first examined the lipid content of the thymi and livers of 2-week-old wild-type and Asm−/− mice using mass spectrometry. The analysis of the thymus revealed an increase in sphingomyelin and dihydro (DH)-sphingomyelin levels with C24:1 and C16:0 being the most abundant species detected (Fig. 5a), whereas the levels of ceramides and DH-ceramides, the breakdown products of sphingomyelin and DH-sphingomyelin, respectively, were unaffected (Fig. 5b). In comparison, an analysis of total liver tissue exhibited a modest increase in sphingomyelin and decreased ceramide abundance in Asm−/− relative to wild-type mice (Supplementary Fig. 7). We therefore investigated whether the species of sphingomyelin that were increased in Asm−/− thymi could interfere with CD1d-mediated antigen presentation. To exclude lipid effects on the APCs themselves, we used an APC-free assay with plate-bound CD1d9, where sphingomyelin and α-GalCer directly compete for binding to CD1d. In addition, we used sphingomyelin concentrations that were within the range of the molar concentrations of sphingomyelin observed in thymocytes, calculated on the basis of the average size of a thymocyte30. We observed that sphingomyelin C24:1 inhibited the ability of α-GalCer to stimulate the DN32.D3 hybridoma in response to plate-bound CD1d, whereas sphingomyelin C16:0 possessed a detectable, albeit less pronounced, inhibitory effect (Fig. 5c). The other sphingomyelin species that were increased in Asm−/− mice made up only a small fraction compared to the levels of C24:1 and C16:0 sphingomyelin. Nevertheless, we tested the effects of the next most elevated species and observed that C24:0 and C18:0 sphingomyelin elicited similar effects in blocking α-GalCer presentation (Fig. 5d). Direct activation of the DN32.D3 hybridoma with plate-bound anti-CD3 was not influenced by the inhibitory sphingomyelin species, indicating that sphingomyelin does not directly affect iNKT cell function (Supplementary Fig. 8a). Furthermore, we confirmed, using the plate-bound CD1d assay, that ASM does not directly facilitate the loading of lipids onto CD1d (Supplementary Fig. 8b). These studies indicate that ASM deficiency is associated with accumulation of sphingomyelin species that compete with agonistic CD1d-binding lipids, thus limiting their ability to stimulate iNKT cells. As iNKT cells are selected by antigens processed in the endolysosomal pathway20,31 and presented by other thymocytes, we developed a technique for defining the lipid content of the lysosomes in thymocytes (see Methods). The identity of the purified lysosomes from thymocytes was confirmed by enrichment of lysosomal markers (Lamp1, Lamp2 and cathepsin D), but not markers for mitochondria (CoxIV), early endosomes (Rab5), nuclei (histone H3 (HisH3)) or cytosol (GAPDH and tubulin), as determined by western blot (magnetic fraction in Fig. 6a) and electron microscopy (Fig. 6b). Lipidomic analysis of the purified lysosome fractions demonstrated an increased ratio of the sphingomyelin species shown to be inhibitory for CD1d-restricted antigen presentation (Fig. 5c,d) relative to the total level of ceramide in Asm−/− compared to wild-type thymocytes of 2-week-old (Fig. 6c), 10-week-old (Fig. 6d) and 20-week-old (Fig. 6e) mice. It is notable that significantly elevated ratios of sphingomyelin species were already observed in the lysosomes of thymocytes from 2-week-old mice, which is consistent with the iNKT cell defects observed at these early stages of development, long before other features of ASM deficiency arise.
Fig. 5 |. Lipids increased in Asm−/− mice block antigen presentation.

a, Sphingomyelin levels in the thymus of 2-week-old Asm−/− (n = 2) and WT (n = 2) mice were quantified by mass spectrometry. The graphs show the mean levels of sphingomyelins and DH-sphingomyelins with carbon chains of different lengths. b, Ceramide levels in the thymus of 2-week-old Asm−/− (n = 2) and WT (n = 2) mice were quantified by mass spectrometry. The graphs show the mean levels of ceramides, DH-ceramides, hexosylceramides and DH-hexosylceramides. c,d, Plate-bound CD1d was incubated with α-GalCer and a dose range of the indicated sphingomyelin species before addition of the iNKT hybridoma DN32.D3. The graphs show the mean level of IL-2 in three independent wells in the culture supernatants 16–24 h after addition of iNKT cells. The asterisks indicate the significance levels for comparisons to cytokine secretion when no sphingomyelin was added (0 nM). The results are representative of three independent experiments. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by one-way ANOVA with Bonferroni’s correction for multiple comparisons. *P < 0.001, **P < 0.0001.
Fig. 6 |. Lipids blocking CD1d-restricted antigen presentation are increased in thymic lysosomes of 2-week-old Asm−/− mice.

a, Lysosomes were extracted from the thymi of WT and Asm−/− mice using magnetic beads. The figure shows lysosome identity for the samples by western blotting for markers of different cellular compartments in the whole tissue lysate, the magnetic-bead-purified lysosomes, the soluble non-bound material, mitochondria-enriched sediment and the non-magnetic microsomal debris. The results are representative of three independent experiments. The blot images are cropped so all markers can be visualized. b, Electron microscopy images of the purified lysosome fraction. The red arrows indicate lysosomes. The black bar represents 100 nm. The results are representative of three independent experiments. c, Sphingomyelin levels in the magnetic bead fraction were quantified by mass spectrometry. The graphs show the ratios of the indicated sphingomyelin species to the total level of ceramide in 2-week-old WT and Asm−/− mice. d,e, Ratios of the indicated sphingomyelin species to the total level of ceramide in 10-week-old (d) and 18- to 20-week-old (e) WT and Asm−/− mice. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by a two-sided Student’s t-test (c–e). *P < 0.05, **P < 0.01, ***P < 0.001. UC, ultracentrifuge; CtsD, cathepsin D; Cer, ceramide; SM, sphingomyelin.
Sphingomyelin can displace agonistic lipids from CD1d.
Our results suggested that ASM deficiency leads to early accumulation of sphingomyelin, which competes as a non-agonistic lipid with agonistic lipids for binding to CD1d. This leads to interference with CD1d-restricted positive selection of iNKT cells in the thymus and with CD1d-dependent maturation of iNKT cells in the periphery. We therefore addressed whether sphingomyelin, in addition to its ability to compete with agonistic lipids for binding to CD1d (Fig. 5c,d), can also displace agonistic lipids that are already bound to CD1d. To this end, recombinant CD1d was loaded with sulfatide (SLF), a negatively charged agonistic lipid for non-invariant NKT cells32,33, and incubated with increasing concentrations of sphingomyelin to determine whether sphingomyelin can actively displace SLF. Indeed, successful replacement of SLF was confirmed in native isoelectric focusing (IEF) gel electrophoresis by a band shift to the 0 position. Even at a 1:1 (sphingomyelin:SLF) molar ratio, sphingomyelin replaced approximately 50% of the bound SLF, similarly to what was observed with the agonistic lipid α-GalCer (Fig. 7a). When we first loaded sphingomyelin or α-GalCer onto murine ****CD1d and then added SLF, we observed less replacement of α-GalCer is the hallmark of strong iNKT cell antigens. The resolved structure agrees with the observation that, once α-GalCer is bound to CD1d it is not easily outcompeted by SLF, whereas sphingomyelin seems to interact less tightly with CD1d, supporting the notion that SLF can outcompete sphingomyelin more easily. However, the binding assay also revealed that CD1d-bound lipids can be replaced with other lipids in a dose-dependent manner. This phenomenon is drastically different from that of peptide binding to conventional MHC class I molecules, where it is difficult to replace a peptide once it is bound36.
Fig. 7 |. Mouse CD1d-sphingomyelin crystal structure.

a, Comparison using a lipid competition assay of the ability of sphingomyelin (SM) (top) and α-GalCer (α-GC) (middle) to outcompete SLF preloaded onto murine CD1d, in a dose-dependent manner, from an equimolar ratio (1:1) to a ninefold molar excess of lipid compared to murine CD1d-SLF complexes. SLF has a single negative charge and migrates on the IEF gel to position −1, whereas replacement with the uncharged lipids, sphingomyelin and α-GalCer, results in a gel shift to position 0. Preloaded sphingomyelin can be outcompeted by SLF, but α-GalCer forms a more stable interaction with CD1d and cannot be outcompeted in this in vitro assay (bottom). The results are representative of two independent experiments. b, Mouse CD1d-sphingomyelin crystal structure. An overview is shown of the CD1d (gray) and β2M (light blue) complex presenting sphingomyelin (yellow) between the α1 and α2 helices. c, 2Fo − Fc electron density drawn as a blue mesh around the lipid and contoured at 1σ. d, Hydrogen bond interactions (blue dashed lines) of CD1d residues with sphingomyelin. e, Hydrogen bond interactions (blue dashed lines) of CD1d residues with α-GalCer.
Treatment with recombinant human ASM can restore iNKT levels.
Our results raised the question of whether restoration of ASM in Asm−/− mice could reverse sphingomyelin-dependent inhibition of CD1d-restricted antigen presentation or even augment presentation by wild-type cells. As in vivo administration of rhASM37 has been shown to reverse the visceral lipid defect in Asm−/− mice38 and is under development as treatment for NPD-B, we reasoned that in vivo injection of rhASM might enhance CD1d-restricted antigen presentation and restore iNKT cell development. Importantly, it has been shown that rhASM is taken up into lysosomes37, where CD1d localizes and lipid exchange occurs39. Indeed, 1 d after rhASM administration into Asm−/− (Fig. 8a) or wild-type (Fig. 8b) mice, spleen DCs exhibited increased α-GalCer-induced iNKT cell activation compared to cells from vehicle-treated mice. Of note, the ability of rhASM to increase iNKT cell activation by wild-type DCs suggests that ASM serves as a tunable factor in determining the CD1d-dependent antigen presentation capacity of DCs under constitutive conditions. We further investigated whether rhASM treatment from birth could reverse the reduction in iNKT cell levels in Asm−/− mice. Following 2 weeks of rhASM treatment, ASM activity was significantly increased in the thymi and livers of treated Asm−/− mice (Fig. 8c) and thymic sphingomyelin abundance was reduced to that found in wild-type mice (Fig. 8d). Moreover, rhASM treatment was associated with an increase in iNKT cells (Fig. 8e,f). Together, these results demonstrate that rhASM enhances CD1d-restricted antigen presentation in wild-type and Asm−/− DCs and can partially rescue the iNKT cell defect observed in Asm−/− mice.
Fig. 8 |. Pharmacological ASM treatment in Asm−/− mice restores iNKT cells.
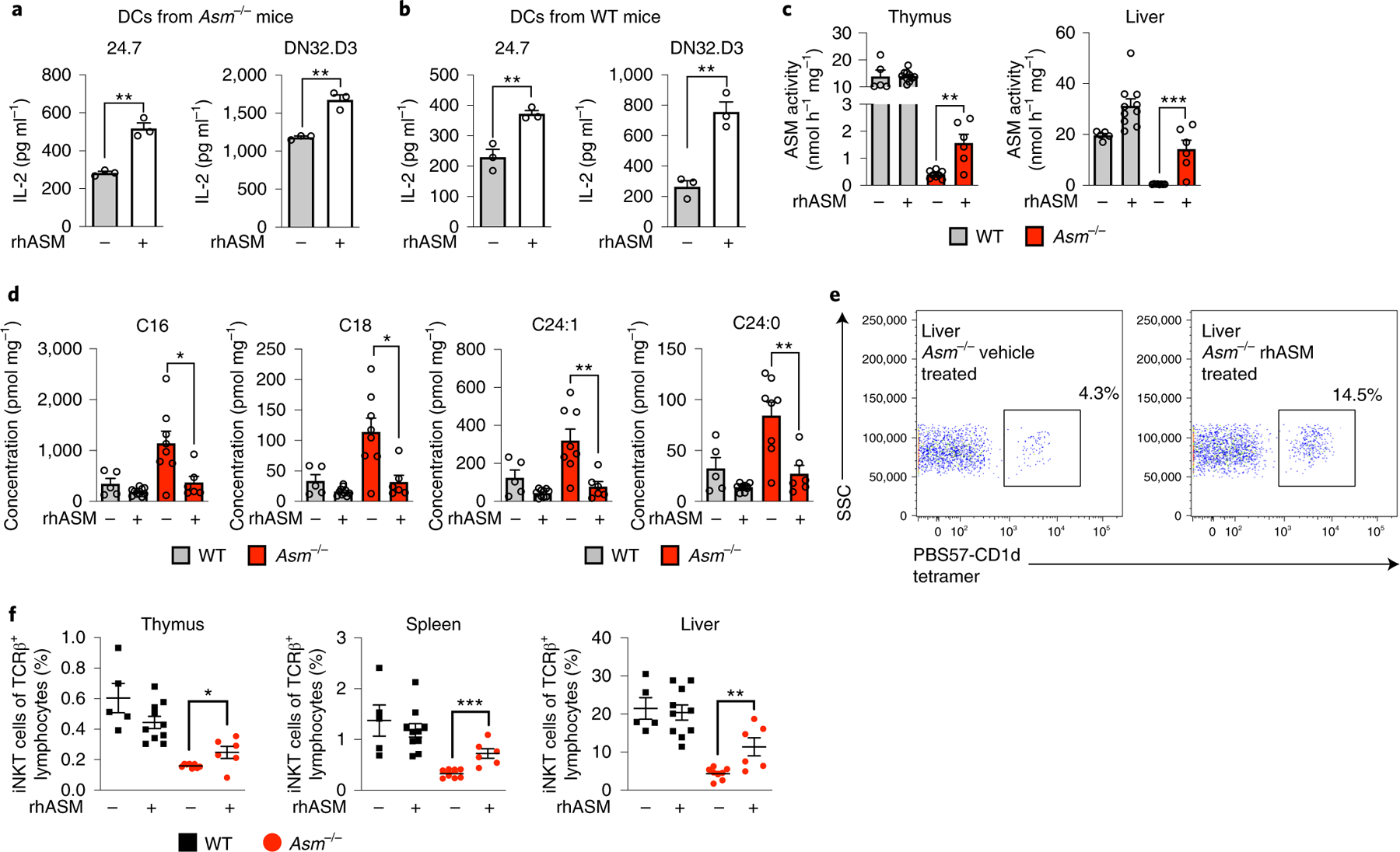
a,b, CD11c+ DCs were extracted from the spleens of Asm−/− mice (a) or WT (b) mice treated with one dose of rhASM (5 μg g−1) 12–16 h before extraction and loaded with α-GalCer for 4 h. The indicated iNKT hybridomas were added and IL-2 levels were measured in three independent wells after 20–24 h. The results are representative of three independent experiments. c, WT and Asm−/− mice were treated every other day from birth with rhASM (5 μg g−1). The graphs show the ASM activity levels in the liver and thymus of vehicle-treated and rhASM-treated Asm−/− and WT mice 2 d after the last enzyme injection. d, The graphs show the levels of the blocking sphingomyelin species as measured by mass spectrometry after treatment with rhASM. e, Representative dot plot from the livers of a vehicle-treated and an rhASM-treated Asm−/− mouse. The cells are gated on the lymphocyte population and TCRβ-positive cells. The percentages correspond to cells positive for PBS57-loaded CD1d tetramer. f, iNKT cell levels at the age of 2 weeks in the thymus, spleen and liver of mice that were treated with rhASM or vehicle as indicated in the graphs. The numbers represent the pooled results from three independent experiments with vehicle-treated (Asm−/− (n = 8) and WT (n = 5)) and rhASM-treated (Asm−/− (n = 6) and WT (n = 10)) mice (c,d and f). The hypothesis tested using the Student’s t-test was that rhASM would increase iNKT cell abundance in Asm−/− mice. In all panels, the mean values are shown with the error bars representing the s.e.m. P values were calculated by a two-sided Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
iNKT cells are a potent subset of T cells whose activation requires tight control to prevent overt autoimmunity8. While previous work has focused on the identification of CD1d-associated lipids that activate iNKT cells (agonistic lipids), lipidomic studies have revealed that the vast majority of CD1d-associated lipids are non-agonistic self-lipids, derived from abundant cellular sphingolipids and glycerophospholipids40. This raises the question of whether the abundance of such non-agonistic CD1d-associated lipids is critical for control of iNKT cell development, CD1d-restricted iNKT cell activation and iNKT cell-mediated inflammation. Here, we have addressed this question through the study of ASM, an enzyme critical for control of the abundance of sphingomyelin, a major CD1d-associated lipid that does not activate iNKT cells. Our results demonstrate that ASM, through control of the abundance of sphingomyelin, regulates the balance between activating and non-activating CD1d-associated lipids and controls iNKT cell activation and development.
These studies highlight and focus attention on the important role played by inhibitory self-lipids in guiding CD1d-restricted responses and further indicate that, in addition to exogenously derived inhibitory sphingolipids provided by the commensal microbiota41, the host employs endogenous strategies to serve a similar purpose.
The accumulation of sphingomyelin in Asm−/− mice was associated with diminished positive selection of iNKT cells in the thymus and altered maturation of iNKT cells in the thymus and the periphery4. This deficiency in iNKT cells also translated into altered responses in iNKT cell-mediated disease models. Defects in the CD1d-iNKT axis observed in Asm−/− mice were paralleled by iNKT cell defects in patients with NPD-A and NPD-B. These findings contrast with those for patients with NPD-C, a disease sharing the neurological phenotype but with a different molecular basis42, who have a normal distribution of iNKT cells29.
Given the critical role of iNKT cells in antimicrobial immunity against common respiratory pathogens such as Pneumococcus and Pseudomonas spp.43,44, iNKT cell defects are anticipated to contribute to susceptibility of patients with NPD-A and NPD-B to pneumonia, which represents the most common cause of death in these patients45. However, the relevance of our data extends far beyond individuals with NPD-A and NPD-B. As such, the correlation between ASM activity and the iNKT cell phenotype in healthy individuals, as well as the promotion of CD1d-restricted antigen presentation by rhASM in wild-type mice, suggests that ASM functions as a major regulator of iNKT cell development and functions under constitutive conditions in normal hosts. Our results may also contribute to understanding of the variability of iNKT cell levels and subsets in humans25. These observations likely apply in a similar manner to other major cellular lipids that associate with CD1d and fail to activate iNKT cells. As such, our findings suggest that iNKT cell development and immunity is tightly linked to, and controlled by, cellular lipid metabolism. This has broad implications for common immunometabolic diseases, such as non-alcoholic steatohepatitis, in which inflammation and tissue destruction are mediated by iNKT cells, and in which changes in lipid metabolism may indeed act as primary drivers of pathogenic iNKT cell activation through alterations in the balance between iNKT cell activating and non-activating lipids for iNKT cells. As such, conditions that regulate ASM levels in immune cells may have important effects on iNKT cell function2.
In conclusion, our studies pinpoint sphingomyelin as a lipid species that endogenously regulates CD1d-restricted antigen presentation by thymocytes and peripheral activation of iNKT cells in response to endogenous, as well as exogenous, stimulatory antigens. Further, we demonstrate that ASM, an enzyme localized to the lysosome, plays a critical role in regulating the levels of this important inhibitory lipid. In addition, our studies further suggest that these pathways are amenable to therapeutic manipulation, not only in humans with lipid storage diseases46, but also in circumstances wherein ASM supplementation (or blockade) may aid in promoting CD1d-restricted responses that are beneficial in the treatment of infectious diseases, autoimmunity and cancer.
Methods
Mouse models.
Asm−/− mice were described previously as a mouse model for NPD14. These mice are on a C57BL/6 background and have been backcrossed for >10 generations. The mouse line was maintained by breeding heterozygote mice (Asm+/− × Asm+/−) because Asm−/− mice develop a neurological disease at an advanced age. The litters were genotyped by PCR (conditions available upon request). Mice used in the experiments were matched by age and sex. Adult mice were defined as those >6 weeks of age. Wild-type littermates generated through Asm+/− × Asm+/− breeding were used as controls. To generate bone marrow chimeras, wild-type bone marrow from C57BL/6 mice carrying CD45.1, which we were able to trace using flow cytometry, were utilized. In the experiments with mixed-bone-marrow chimeras, bone marrow from Jα18−/− mice was mixed at a 1:1 ratio with bone marrow from Asm−/− or wild-type mice. The CD45.1+ (strain 2014) and Jα18−/− (strain 30524) mouse models are commercially available (Jackson Laboratories). OT-II mice (strain 4194) used to acquire ovalbumin-reactive T cells are also commercially available through Jackson Laboratories. The number of mice included in each individual experiment was based on experience in the laboratory with similar experiments. No animals were excluded from the reported experiments. Animals were allocated to each experimental group on the basis of genotype and age/sex without randomization, and no blinding was performed. All mice were housed in a specific-pathogen-free facility with food and water available ad libitum in accordance with the guidelines set out by the Harvard Medical Area Standing Committee on Animals and the Norwegian Food Safety Authority (Mattilsynet).
Bone marrow chimeras.
Recipient mice were irradiated two times with 600 rad 1 d before the injection of bone marrow. Bone marrow was extracted from donor mice from the femur by flushing with PBS, washing the cells and adjusting the concentration. For the mixed-bone-marrow chimeras, bone marrow from Jα18−/− and Asm−/− or wild-type mice was mixed 1:1. Then, 1 million cells were injected into the tail vein. The mice were then kept on acidic water for 4 weeks and observed for general well-being. After 3 months the engraftment and iNKT cell percentages were evaluated by flow cytometry. Engraftment was judged by CD45.1+ percentage among lymphocytes.
α-GalCer hepatitis.
Age- and sex-matched mice were injected with 2 μg of α-GalCer i.p. At 4 h after injection the mice were bled from the tail vein, and 24 h after injection the mice were killed and blood was collected by cardiac puncture. IL-4 and IFN-γ were measured in the serum by ELISA (BD Biosciences). ALT was measured by colorimetric assay (Stanbio).
Concanavalin A hepatitis.
ConA (Sigma-Aldrich) was dissolved in PBS and injected into the tail vein at a dose of 13.5 mg kg−1. Only male mice were used in these experiments. The mice were monitored hourly for well-being, and blood was sampled after 6 h. At 24 h, the mice were killed and liver and blood were sampled. IL-4 and IFN-γ were measured in the serum by ELISA (BD Biosciences). ALT was measured by a colorimetric assay (Stanbio). Liver sections were stained with H&E, and the sections were scored by a semiautomatic algorithm for necrotic areas.
Oxazolone skin painting and induced delayed type hypersensitivity.
At day 0, the abdominal skin of the mice was shaved. Then, 150 μl of 3% oxazolone dissolved in 3:1 ethanol:acetone was applied to the skin to sensitize the mice. The mice were followed daily with weight measurements. At 5 d after the skin sensitization, the mice received a re-challenge by application of 20 μl of 1% oxazolone dissolved in 1:1 acetone:sunflower oil to the ear. A sensitive micrometer was used to measure ear swelling on the following days.
Genotyping.
Mouse tails or mouse ear biopsies were digested in lysis buffer (100 mM Tris-HCl (pH 8.5), 5 mM EDTA, 0.2% SDS, 200 mM NaCl and proteinase K (Roche)) overnight at 55 °C. Genomic DNA was phenol-extracted and precipitated with isopropanol.
Patients with Niemann-Pick disease and healthy controls.
Five patients were analyzed using flow cytometry of PBMCs, including one with NPD-A (a 1-yearold) and four with NPD-B (three adults and a 10-year-old patient). In addition, 70 control subjects were studied using flow cytometry of PBMCs, consisting of 15 children and 55 adults (blood donors). The children’s control group consisted of children undergoing orthopedic surgery, without infections, underlying chronic illness or use of medication. For correlation of iNKT phenotype and ASM activity, blood samples from adult healthy controls were obtained from blood donors (using buffy coats) from the Blood Bank of Centro Hospitalar de São João, Porto, Portugal. The study was approved by the ethical committees of the participating hospitals. The participants or their legal representatives gave written consent to participate in the study. Blood samples were collected and processed within 24 h of blood draw. PBMCs were isolated by density centrifugation before staining with fluorochrome-conjugated antibodies or tetramers. The recruitment of patients with NPD to generate lymphoblasts was approved by the institutional review board at the Icahn School of Medicine. Further details on recruitment can be found in the Life Sciences Reporting Summary.
Cell culture.
The murine NKT hybridomas 24.7, 24.8, 14.S6 and DN32.D3 were maintained in DMEM supplemented by 10% FBS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 0.25 μg ml−1 amphotericin B (Antibiotic-Antimycotic, Gibco). Regular and transduced human EBV-transformed cells and the 58ab hybridoma were maintained in RPMI supplemented by 10% FBS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 0.25 μg ml−1 amphotericin B. Lentiviral transduction of EBV-transformed cells with a construct carrying human CD1D was performed as described previously25.
Immunomagnetic isolation of lysosomes.
The isolation of lysosomes from murine tissue was performed according to Fritsch et al.47. In brief, tissue was thawed in 500 μl of homogenization buffer (HB) (15 mM HEPES (pH 7.4), 250 mM sucrose, 0.5 mM MgCl2 containing complete protease inhibitor and cyanase nuclease (SERVA Electrophoresis)) followed by three rounds of careful sonication (10 s, amp 2.5 at 4 °C using a cooled cup resonator (G. Heinemann) in a total volume of 1 ml) and centrifugation for 4 min at 1,500g. The resulting supernatant was loaded on a 16% iodixanol-HB cushion and centrifuged for 1 h at 150,000g. The resulting floating fraction was aspirated carefully from the cushion and diluted to 750 μl with HB. Two micrograms of anti-Lamp1 (Santa Cruz Biotechnology, cat. no. sc-8098) was added and samples were incubated for 30 min while rolling at 4 °C. Five microliters of Protein G microbeads (Miltenyi Biotec, cat. no. 130–071-101) were added followed by another 30-min incubation. Samples were then loaded onto a HOKImag magnetic isolation device (Hoock) for organelle pulldown. The eluate was finally sedimented by centrifugation for 1 h at 20,000g and used for downstream application.
Lipidomics.
Sphingolipids were quantified by extracting them from the cells (after spiking with an internal standard cocktail from Avanti Polar Lipids, cat. no. LM-6005) with analysis by liquid chromatography, electrospray-ionization tandem mass spectrometry using an API 4000 QTrap (SCIEX), as previously described48.
SDS-PAGE and western blots.
For SDS-PAGE, sedimented organelles were resuspended in 15 μl of modified RIPA buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1% Triton X-100, 1 mM EDTA, 0.25% sodium deoxycholate) and protein concentration was determined by a bicinchoninic acid assay (Pierce). For SDS-PAGE, anyKD gels (Bio-Rad) were used and 3 μg of protein was loaded. Proteins were transferred to a polyvinylidene difluoride membrane (Carl-Roth). The membranes were blocked with 5% skim milk in TBST and incubated overnight with the primary antibody diluted from 1:500–1:5,000 in 5% skim milk. The peroxidase-conjugated secondary antibodies were incubated with the membranes for 1 h diluted 1:10,000 in 5% skim milk. Blots were developed using the ECL kit and film (GE Healthcare). The following antibodies were used: anti-Lamp1 (Southern Biotech, cat. no. 9835–01), anti-Lamp2 (Southern Biotech, cat. no. 9840–01), anti-CoxIV (Santa Cruz Biotechnology, cat. no. sc-292052) anti-HisH3 (Cell Signaling, cat. no. 4499S), anti-Rab5 (Santa Cruz Biotechnology, cat. no. sc-598), anti-cathepsin D (Millipore, cat. no. Ab2, IM16), anti-GAPDH (Proteintech, cat. no. HRP-60004) and anti-tubulin (Proteintech, cat. no. HRP-66031).
Electron microscopy.
Electron microscopy was performed as described previously47. After sedimentation, isolated lysosomes were fixed in 3% glutaraldehyde in PBS, followed by overnight washing in 0.1 M phosphate buffer. Samples were dehydrated and then embedded in araldite. Ultrathin sections were treated with uranyl acetate and lead citrate. Images were acquired on a JEM1400plus electron microscope (Jeol).
Flow cytometry.
Lymphocyte populations from various murine organs were prepared as previously described49. In brief, the spleen and thymus were macerated to obtain a single cell population and passed through a filter, and the liver was perfused retrogradely through the portal vein and macerated followed by density centrifugation. The lymphocytes were suspended in 2% FBS in PBS. To avoid nonspecific staining, the Fc receptors were blocked by using anti-mouse CD16/32 (clone 93, BioLegend, cat. no. 101302) and staining was performed with appropriate fluorochrome-conjugated monoclonal antibodies and a CD1d tetramer loaded with PBS57 (from the National Institute of Health tetramer core facility). The following antibodies were used for staining murine samples: anti-mouse CD3e (clone 145–2C11, BD Biosciences, cat. no. 553066), anti-mouse TCRβ (clone H57–597, BD Biosciences, cat. no. 553170), anti-mouse TCRβ (clone H57–597, BioLegend, cat. no. 109212), anti-mouse CD45.1 (clone A20, BioLegend, cat. no. 110728), anti-mouse CD45.2 (clone 104, BD Biosciences, cat. no. 553772), anti-mouse CD1d (clone 1B1, BD Biosciences, cat. no. 553846), anti-mouse CD11c (clone HL3, BD Biosciences, cat. no. 553801), anti-mouse CD44 (clone 1M7, BioLegend, cat. no. 103031), anti-mouse CD24 (clone M1/69, BioLegend, cat. no. 101821), anti-mouse NK1.1 (clone PK136, BioLegend, cat. no. 108724), anti-mouse CD4 (clone RM4–5, BioLegend, cat. no. 100528), anti-mouse CD8 (clone 53–6.7, BioLegend, cat. no. 100714), anti-mouse TCR γ-δ (clone UC7–13DS, BioLegend, cat. no. 107504), anti-mouse CD304 (clone 3E12, BioLegend, cat. no. 145207), anti-mouse CD1d-α-GalCer (clone L363, eBioscience, cat. no. 12–2019-82) and the murine CD1d tetramer loaded with PBS57 (from the National Institute of Health tetramer core facility). For evaluating CD1d expression on transduced EBV-lines, anti-human CD1d (clone 51.1, BioLegend, cat. no. 350308) was used. Flow cytometry data were acquired using a MACSQuant Analyser (Miltenyi Biotec) and a BD FACSVerse (BD Biosciences).
For staining of the samples from patients with NPD and healthy controls, the staining mixture was composed of anti-human CD3 (clone UCHT1, eBioscience, cat. no. 17–0038-42), anti-human CD3 (clone SK7, eBioscience, cat. no. 45–0036-42), anti-human CD4 (clone RPA-T4, eBioscience, cat. no. 25–0049-42), anti-human CD8 (clone RPA-T8, eBioscience, cat. no. 47–0088-42) and anti-human CD161 (clone HP-3G10, eBioscience, cat. no. 53–1619-42) and the human CD1d tetramer loaded with PBS57 (from the National Institute of Health tetramer core facility). Patient and control samples were acquired on a three-laser BD FACS Canto II flow cytometer using BD FACSDiva software (BD Biosciences). Estimation of absolute cell counts was performed using CountBright Absolute Counting Beads (Molecular Probes). The analyses were performed using FlowJo software (Tree Star).
Cell-based antigen-presentation assays.
DCs were extracted from the spleens of mice using CD11c selection beads (Miltenyi Biotec). Thymocytes were prepared by passing the thymus through a 40-μm strainer, followed by washing in PBS. Fifty thousand DCs or 100,000 thymocytes were seeded in 96-well flat-bottom cell culture plates as indicated in the individual experiments. The cells were either loaded with 100 ng ml−1 α-GalCer or left untreated. Unloaded antigen was washed away before addition of 50,000–100,000 of the indicated NKT hybridomas. In some experiments the CD1d monoclonal antibody 19G11 (BioXCell, cat. no. BE0000) or a corresponding isotype control (clone LTF-2, BioXCell, cat. no. BE0090) was added to the cultures. In similar assays DCs were loaded with ovalbumin and cultured with 100,000 CD4+ T cells from OT-II mice. Cytokine secretion was measured in the culture supernatants by ELISA (BD Biosciences).
Surface loading of antigens.
DCs were extracted from spleens as described above followed by incubation with 100 ng ml−1 α-GalCer. After incubation for 4–24 h, the cells were washed and stained with an anti-mouse α-GalCer-CD1d complex (clone L363, eBioscience)50. Flow cytometry data were acquired as described previously.
Histological assessment of necrosis.
H&E-stained liver sections were scanned using a Histech Panoramic Midi Slide Scanner (3DHISTECH). The resulting images were downscaled by a factor of four. To achieve reliable segmentation of necrotic versus non-necrotic tissue, Ilastik v.1.3.2 (ref.51), a toolkit for performing machine-learning-based image processing, was used. An average of three annotations were made for each segmentation class (background, necrotic and normal tissue) on each of five cropped images that represented the overall variation of morphological phenotype among the samples. We employed all features of σ = 10, and all color- and texture-based features of σ = 5. After training the random forest classifier, the entire sections of all samples were analyzed and the percentage of necrotic areas in each sample was determined.
Enzyme replacement therapy.
rhASM was produced as described previously37; briefly, human ASM was overexpressed in Chinese hamster ovary cells and purified from cell culture medium. As iNKT cell development mainly takes place at an early age, we started treatment as early as possible52. Neonatal mice from day 2 of life were injected with 5 μg g−1 rhASM i.p. every other day until they were 2 weeks old. The mice were then killed and the percentage of iNKT cells was determined by flow cytometry as described above. Samples from the liver and thymus were also collected at the same time for the determination of ASM activity and lipid content.
APC-free antigen-presentation assays.
Monomeric mouse CD1d (National Institutes of Health tetramer core facility) was coated onto 96-well cell culture plates overnight (0.25 μg per well). Unbound CD1d was thoroughly washed away followed by incubation with 100 ng ml−1 α-GalCer and a dose range of different sphingomyelin species for 16–24 h. Unbound lipids were washed off and the α-GalCer-reactive iNKT cell hybridoma DN32.D3 was added. In the experiments evaluating a direct loading effect from rhASM, a gradient of the same rhASM described above for the replacement experiments was added, along with a gradient of BSA as a negative control and saposin-B (Cusabio) as a positive control11. These experiments were performed under neutral and acidic conditions. Cytokine secretion was measured in the culture supernatants by ELISA (BD Biosciences).
Lipid competition assays.
Recombinant mouse CD1d was expressed and purified as previously reported53. CD1d was loaded with 3× molar excess of SLF (bovine brain, Avanti Polar Lipids; dissolved at 5 mg ml−1 in DMSO) in 100 mM Tris (pH 7.0) 100 mM NaCl at 37 °C for 2 h. After lipid loading, excess lipid was removed by ultrafiltration using Amicon filter cartridges (30-kDa molecular weight cut-off). CD1d-SLF complexes were then incubated overnight at room temperature with increasing concentration of either sphingomyelin (C24:1) (synthetic, Avanti Polar Lipids) or α-GalCer. Lipids were dissolved in DMSO (5 mg ml−1) and incubated with CD1d in the presence of 0.01% tyloxapol. As a control, CD1d-SLF complexes were incubated with only tyloxapol. In a reversed experiment, CD1d was first loaded with 6× molar excess of either sphingomyelin (10 mg ml−1 in DMSO) or α-GalCer (0.2 mg ml−1 in vehicle) at room temperature overnight, purified from excess lipid by ultrafiltration and incubated with 3× molar excess of SLF for 1 h at 30 °C, which is the minimum dose and time necessary for full loading of insect-cell-expressed mouse CD1d (data not shown). Four microliters (2–4 μg) of the various CD1d-lipid-loading products were analyzed using native IEF gel electrophoresis on pH 5–8 gels with a PhastSystem (GE Healthcare) and stained with Coomassie dye.
CD1d-sphingomyelin crystallization and structure determination.
CD1d was loaded with sphingomyelin as reported above. Excess lipid was removed by size exclusion chromatography using a Superdex S200 GL10/300 column (GE Healthcare). CD1d-sphingomyelin complexes were concentrated to 5 mg ml−1, and crystals were grown by sitting-drop vapor diffusion while mixing 0.5 μl of protein with 0.5 μl of precipitate (20% polyethylene glycol 3350 and 200 mM sodium malonate (pH 7.0)). Crystals were flash-cooled in crystallization solution containing 20% glycerol. Diffraction data from a single crystal were collected at beamline 7.1 of the Stanford Synchrotron Lightsource. Diffraction data were processed to 1.95 Å using HKL2000 0.98 (ref.54). The structure was determined using molecular replacement in Phaser55, followed by iterative cycles of model building in COOT v.0.8.9.2 (ref.56) and restrained refinement in REFMAC v.5.8.0158 (ref.57). Geometry restraints for sphingomyelin were obtained using the PRODRG2 server58.
ASM activity measurement.
For ASM activity measurement, tissue samples were homogenized and subjected to three freeze-thaw cycles (with freezing for at least 10 min) followed by centrifugation. The protein in the supernatant was used for subsequent assays. Protein concentrations were measured and normalized using a bicinchoninic acid assay (Pierce). The ASM activity was then measured using an ASM activity assay kit (Echelon Biosciences).
Statistical analyses.
All statistical analyses were carried out using GraphPad Prism v.8.2 (GraphPad Software). Variables were evaluated for a normal distribution either by the D’Agostino and Pearson normality test59 or by manually examining the distribution of observations. For normally distributed variables a two-sided Student’s t-test was used, whereas for variables not fulfilling this requirement, a two-sided Mann-Whitney U-test was used. Correlations were evaluated by Pearson’s r. For experiments where multiple comparisons were included, one-way ANOVA with Bonferroni’s correction for multiple comparisons was used. For time-series, a two-way ANOVA test with Bonferroni’s correction for multiple comparisons was used. A P value <0.05 was considered statistically significant. Further details on study design can be found in the Nature Research Reporting Summary.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The crystal structure is available at https://www.rcsb.org, PDB structure ID: 6CYW. The rest of the data that support the findings of this study are available from the corresponding authors upon reasonable request.
Supplementary Material
Table 1 |.
Data collection and refinement statistics for the CD1d-sphingomyelin structure
| Mouse CD1d-sphingomyelin C24:1a | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 42.1, 105.3, 106.4 |
| α, β, γ (°) | 90 |
| Resolution (Å) | 50–1.95 (2.00–1.95)b |
| Rsym | 11.9 (58.4) |
| I/σ(I) | 23.4 (2.5) |
| Completeness (%) | 97.8 (82.5) |
| Redundancy | 4.2 (3.9) |
| Refinement | |
| Resolution (Å) | 50–1.95 |
| No. reflections | 33,561 |
| Rwork/ Rfree | 22.3 / 25.6 |
| No. atoms | 3,420 |
| Protein | 2,990 |
| Ligand/ion | 112 |
| Water | 318 |
| B factors | |
| Protein | 33.4 |
| Ligand/ion | 51.1 |
| Water | 38.9 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.11 |
One crystal was used for data collection.
Values in parentheses are for the highest-resolution shell.
Acknowledgements
This work was supported by US National Institutes of Health grants HD28607 (MERIT Award) (to E.H.S.), DK044319, DK051362, DK053056, DK088199 and the Harvard Digestive Diseases Center DK034854 (to R.S.B.), the Southeastern Norwegian Health Trust, the Unger-Vetlesen Foundation, the Caroline Musæus Aarvolds fund and the Norwegian PSC Research Center (to E.M.), the European Research Council (ERC Starting Grant agreement no. 336528), the Deutsche Forschungsgemeinschaft (DFG) (ZE 814/4-1, ZE 814/7-1) and the DFG Excellence Cluster Center for Regenerative Therapies Dresden (to S.Z.), the DFG (SCHU733/14-1) (to S.S. and J.F.) and the DFG Excellence Cluster Inflammation at Interfaces Schleswig-Holstein (EXC 306) (to S.S.). This work was partially funded by FEDER funds under the Portugal 2020 partnership agreement through the Norte Portugal Regional Operational Program (Norte 2020) (Norte-01-0145-FEDER-000012). J. Bame, J. Danielson, A. Dias, M.L. Maia, S. Torquato, J. Øgaard and J. Anmarkrud are thanked for invaluable technical help. We thank the following physicians for patient and control subject recruitment in Portugal: T. Cardoso and N. Alegrete (CHS João, Porto), E. Martins and E. Silva (CH Porto, Porto), and L. Ribeiro and A. Pereira (CHU Coimbra, Coimbra). We acknowledge the blood bank of CHS João, Porto, and the National Institutes of Health tetramer core facility for provision of PBS57-loaded CD1d tetramer and CD1d monomer.
Footnotes
online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41590-019-0504-0
Competing interests
E.H.S. is a consultant for Sanofi Genzyme, and M.F.M. has received a research grant from Sanofi Genzyme.
Supplementary information is available for this paper at https://doi.org/10.1038/s41590-019-0504-0.
Peer review information Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reprints and permissions information is available at www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
under exclusive licence to Springer Nature America, Inc. 2019
References
- 1.Brigl M & Brenner MB CD1: antigen presentation and T cell function. Annu. Rev. Immunol 22, 817–890 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Brennan PJ, Brigl M & Brenner MB Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol 13, 101–117 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Godfrey DI & Berzins SP Control points in NKT cell development. Nat. Rev. Immunol 7, 505–518 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Godfrey DI, Stankovic S & Baxter AG Raising the NKT cell family. Nat. Immunol 11, 197–206 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Gapin L, Godfrey DI & Rossjohn J Natural killer T cell obsession with self-antigens. Curr. Opin. Immunol 25, 168–173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merrill AH Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev 111, 6387–6422 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan W, Kang S, Evans J & Cresswell P Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J. Immunol 182, 4784–4791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salio M, Silk JD, Jones YE & Cerundolo V Biology of CD1- and MR1-restricted T cells. Annu. Rev. Immunol 32, 323–366 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Fox LM et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 7, e1000228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith E & Schuchman E The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 22, 3419–3431 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Im JS et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity 30, 888–898 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrotta C & Clementi E Biological roles of acid and neutral sphingomyelinases and their regulation by nitric oxide. Physiology 25, 64–71 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Zeidan YH & Hannun YA The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr. Mol. Med 10, 454–466 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Horinouchi K et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat. Genet 10, 288–293 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Truman J-P, Gadban MM, Smith KJ & Hammad SM Acid sphingomyelinase in macrophage biology. Cell Mol. Life Sci 68, 3293–3305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang OO et al. CD1d on myeloid dendritic cells stimulates cytokine secretion from and cytolytic activity of Vα24JαQ T cells: a feedback mechanism for immune regulation. J. Immunol 165, 3756–3762 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Nieuwenhuis EE et al. CD1d and CD1d-restricted iNKT-cells play a pivotal role in contact hypersensitivity. Exp. Dermatol 14, 250–258 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Takeda K et al. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl Acad. Sci. USA 97, 5498–5503 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cernadas M et al. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J. Immunol 171, 4149–4155 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Chiu Y-H et al. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat. Immunol 3, 55–60 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Cui J et al. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science 278, 1623–1626 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Behar S, Podrebarac TA, Roy C, Wang C & Brenner M Diverse TCRs recognize murine CD1. J. Immunol 162, 161–167 (1999). [PubMed] [Google Scholar]
- 23.Barnden MJ, Allison J, Heath WR & Carbone FR Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol 76, 34–40 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Thedrez A et al. CD4 engagement by CD1d potentiates activation of CD4+ invariant NKT cells. Blood 110, 251–258 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Zeissig S et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J. Clin. Invest 120, 2889–2899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNab F et al. The influence of CD1d in postselection NKT cell maturation and homeostasis. J. Immunol 175, 3762–3768 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Balreira A, Lacerda L, Miranda C & Arosa FA Evidence for a link between sphingolipid metabolism and expression of CD1d and MHC class II: monocytes from Gaucher disease patients as a model. Brit. J. Haematol 129, 667–676 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Pereira CS et al. Invariant natural killer T cells are phenotypically and functionally altered in Fabry disease. Mol. Genet. Metab 108, 241–248 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Speak AO et al. Invariant natural killer T cells are not affected by lysosomal storage in patients with Niemann-Pick disease type C. Eur. J. Immunol 42, 1886–1892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salinas F, Smith L & Goodman J Cell size distribution in the thymus as a function of age. J. Cell Physiol 80, 339–345 (1972). [DOI] [PubMed] [Google Scholar]
- 31.Chiu Y-H et al. Distinct subsets of CD1d-restricted T cells recognize self-antigens loaded in different cellular compartments. J. Exp. Med 189, 103–110 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jahng A et al. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med 199, 947–957 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girardi E et al. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat. Immunol 13, 851 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zajonc DM et al. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nat. Immunol 6, 810–818 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zajonc DM et al. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med 202, 1517–1526 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sette A et al. Peptide binding to the most frequent HLA-A class I alleles measured by quantitative molecular binding assays. Mol. Immunol 31, 813–822 (1994). [DOI] [PubMed] [Google Scholar]
- 37.He X et al. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim. Biophys. Acta Prot. Struct. Mol. Enzymol 1432, 251–264 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Miranda S et al. Infusion of recombinant human acid sphingomyelinase into Niemann-Pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J 14, 1988–1995 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Bendelac A, Savage PB & Teyton L The biology of NKT cells. Annu. Rev. Immunol 25, 297–336 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Cox D et al. Determination of cellular lipids bound to human CD1d molecules. PLoS ONE 4, e5325 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An D et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.NP-C Guidelines Working Group. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab 98, 152–165 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Nieuwenhuis E et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat. Med 8, 588 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Kinjo Y et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat. Immunol 12, 966–974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGovern MM et al. Morbidity and mortality in type B Niemann-Pick disease. Genet. Med 15, 618 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Platt FM Sphingolipid lysosomal storage disorders. Nature 510, 68–75 (2014). [DOI] [PubMed] [Google Scholar]
References
- 47.Fritsch J, Tchikov V, Hennig L, Lucius R & Schütze S A toolbox for the immunomagnetic purification of signaling organelles. Traffic 20, 246–258 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Shaner RL et al. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res 50, 1692–1707 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeissig S, Olszak T, Melum E & Blumberg RS Analyzing antigen recognition by natural killer T cells. Methods Mol. Biol 960, 557–572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu K et al. Production and characterization of monoclonal antibodies against complexes of the NKT cell ligand α-galactosylceramide bound to mouse CD1d. J. Immunol. Methods 323, 11–23 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sommer C, Straehle C, Kothe U & Hamprecht FA ILASTIK: interactive learning and segmentation toolkit. Proc. 8th IEEE International Symposium on Biomedical Imaging 1, 230–233 (2011). [Google Scholar]
- 52.Olszak T et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J et al. Lipid binding orientation within CD1d affects recognition of Borrelia burgorferi antigens by NKT cells. Proc. Natl Acad. Sci. USA 107, 1535–1540 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–26 (1997). [DOI] [PubMed] [Google Scholar]
- 55.McCoy AJ, Grosse-Kunstleve RW, Storoni LC & Read RJ Likelihood-enhanced fast translation functions. Acta Crystallogr. Sect. D. Biol. Crystallogr 61, 458–464 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott W & Cowtan K Features and development of Coot. Acta Crystallogr. Sect. D. Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murshudov GN, Vagin AA & Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Schüttelkopf AW & van Aalten DMF PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D. Biol. Crystallogr 60, 1355–1363 (2004). [DOI] [PubMed] [Google Scholar]
- 59.D’Agostino R & Pearson E Tests for departure from normality. Empirical results for the distributions of b2 and √b1. Biometrika 60, 613 (1973). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure is available at https://www.rcsb.org, PDB structure ID: 6CYW. The rest of the data that support the findings of this study are available from the corresponding authors upon reasonable request.