

Summary
Cancer cells display metabolic plasticity to survive stresses in the tumor microenvironment. Cellular adaptation to energetic stress is coordinated in part by signaling through the liver kinase B1 (LKB1)-AMP-activated protein kinase (AMPK) pathway. Here, we demonstrate that miRNA-mediated silencing of LKB1 confers sensitivity of lymphoma cells to mitochondrial inhibition by biguanides. Using both classic (phenformin) and newly developed (IM156) biguanides, we demonstrate that elevated miR-17∼92 expression in Myc+ lymphoma cells promotes increased apoptosis to biguanide treatment in vitro and in vivo. This effect is driven by the miR-17-dependent silencing of LKB1, which reduces AMPK activation in response to complex I inhibition. Mechanistically, biguanide treatment induces metabolic stress in Myc+ lymphoma cells by inhibiting TCA cycle metabolism and mitochondrial respiration, exposing metabolic vulnerability. Finally, we demonstrate a direct correlation between miR-17∼92 expression and biguanide sensitivity in human cancer cells. Our results identify miR-17∼92 expression as a potential biomarker for biguanide sensitivity in malignancies.
Keywords: biguanide, microRNA, LKB1, AMPK, Myc, lymphoma, metabolic vulnerabilities, energy-sensing, OXPHOS inhibitor
Graphical Abstract
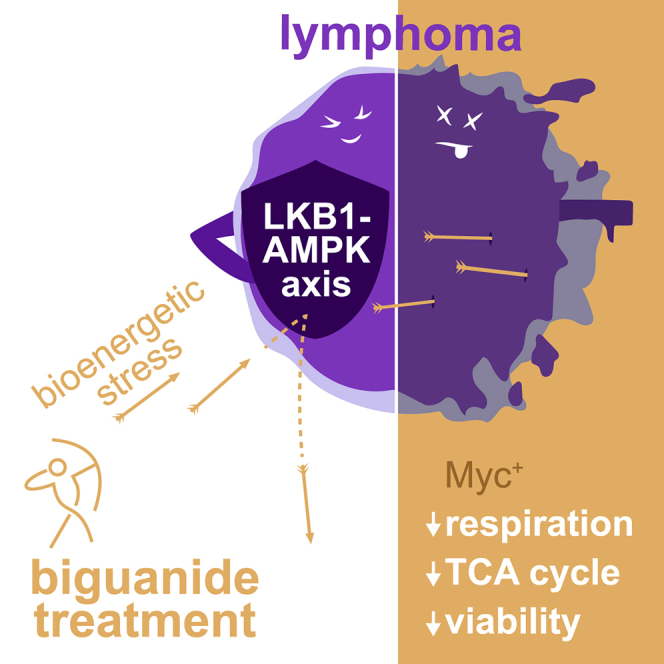
Highlights
IM156 is a newly developed biguanide with more potency than phenformin
IM156 inhibits mitochondrial respiration to cause bioenergetic stress in lymphoma
miR-17-dependent silencing of LKB1 reduces adaptation to bioenergetic stress
miR-17∼92 expression correlates with biguanide sensitivity in human cancer cells
Izreig et al. show that miR-17∼92 expression in cancer cells sensitizes them to biguanide treatment by disrupting bioenergetic stability. These data implicate miR-17∼92 expression as a potential biomarker for biguanide sensitivity in hematological malignancies and possibly solid tumors.
Introduction
A major driver of metabolic reprogramming in cancer is the c-Myc proto-oncogene (Myc), a transcription factor that broadly regulates the expression of genes involved in anabolic metabolism and cellular bioenergetics.1 Oncogene activation or tumor suppressor inactivation promotes changes in cellular metabolism that facilitate transformation and tumor cell proliferation.2 However, the increased metabolic activity stimulated by transformation imposes additional stresses on tumor cells, such as nutrient depletion and redox imbalance, which must be countered for tumors to survive and grow. The liver kinase B1 (LKB1)-AMP-activated protein kinase (AMPK) pathway contributes to tumor cell survival by promoting the cellular sensing of and adaptation to such bioenergetic stress.3 Inactivation of LKB1 in cancer, conversely, promotes anabolic metabolic reprogramming at the expense of metabolic flexibility.4, 5, 6, 7 In cells lacking LKB1, including non-small cell lung cancer (NSCLC), AMPK activation is reduced and cells are more sensitive to metabolic stresses induced by nutrient limitation or inhibitors of oxidative phosphorylation (OXPHOS).8,9 This raises the possibility of using OXPHOS inhibitors to target cancers with reduced bioenergetic capacity and/or defects in energy-sensing control systems. Potent OXPHOS inhibitors,10 putative inhibitors of mitochondrial protein synthesis,11,12 and enhancers of the degradation of respiratory chain proteins13,14 are under study as antineoplastic agents.
Biguanides represent another class of drugs proposed to target cellular bioenergetics in tumor cells. The most well-known biguanide is metformin, a drug commonly used to treat type 2 diabetes. Metformin reduces the activity of mitochondrial complex I, thereby limiting mitochondrial ATP production and imposing bioenergetic stress on cells.15,16 This leads to reduced hepatic gluconeogenesis and reduced hyperglycemia in type 2 diabetes. However, if adequate drug levels can be achieved in other cell types, lineage-specific consequences of energetic stress are observed, including cytostatic or cytotoxic effects on cancer cells.17 Older retrospective studies18 reported that diabetics treated with metformin had reduced cancer risk and better cancer prognosis relative to diabetic patients not taking metformin,18 generating interest in repurposing biguanides as anticancer agents.17 However, further pharmaco-epidemiologic studies19,20 failed to confirm these findings, and early randomized clinical trials of metformin in advanced cancer have shown no survival benefit in pancreatic cancer21 and only minor benefit in lung cancer.22 Pharmacokinetic factors, such as the requirement for the active transport of metformin by the organic cation 1 (OCT1) transporters, may account in part for the discrepancies between preclinical models and clinical trials with respect to the anticancer effects of metformin. Nevertheless, many in vivo cancer models demonstrate significant in vivo antineoplastic activity of biguanides,6,23, 24, 25, 26, 27, 28, 29 raising the possibility that biguanides with better bioavailability and toxicity profiles may have clinical utility.
Important in the clinical development of OXPHOS inhibitors as antineoplastic drugs is the selection of subsets of cancers that are particularly sensitive to metabolic stress. Preclinical work by Shackelford et al.8 demonstrated that biguanides, specifically phenformin, could be effective as single agents for LKB1-deficient KRAS mutant NSCLC, in keeping with the role of LKB1 in adaptation to energetic stress. While the mutation of LKB1 is found in ∼20%–30% of NSCLCs, we hypothesized that biguanide-sensitive cancers can be extended to those with increased expression of MYC, which we have previously reported promotes translational suppression of LKB1 via the microRNA (miRNA) miR-17∼92.30 In this study, using loss- and gain-of-function models of miR-17∼92, we demonstrate that elevated miR-17∼92 expression, specifically the seed family miR-17 and -20, confers increased sensitivity of mouse and human lymphoma cells to apoptosis induced by two biguanides—phenformin and the newly developed biguanide IM156. As single agents, both phenformin and IM156, but not metformin, extended the survival of mice bearing miR-17∼92-expressing lymphomas. These results suggest that miR-17∼92 could function as a biomarker for biguanide sensitivity in cancer.
Results
IM156 Is a Newly Developed Biguanide That Inhibits Mitochondrial Respiration
The limited bioavailability of metformin and its dependence on OCT1 for cellular uptake potentially limit its applicability in the treatment of cancer.31 We investigated the biological properties of phenformin and the newly developed biguanide IM156, which are more hydrophobic and therefore potentially more bioavailable to cells than metformin (Figure 1A). To test the impact of these biguanides on tumor cell respiration, we acutely treated Myc-dependent mouse lymphoma cells (Eμ-Myc cells) with either metformin, phenformin, or IM156 and assessed changes in the oxygen consumption rate (OCR) using the Seahorse XF96 extracellular flux analyzer. Across a range of concentrations, phenformin and IM156 decreased OCR (Figure 1B), with IM156 exhibiting greater potency than phenformin and metformin at equal concentrations. IM156 was more effective than phenformin at reducing cellular ATP production at equal concentrations, correlating with the effect of IM156 on oxidative phosphorylation (Figure 1C). These data are consistent with IM156 functioning as a more potent inhibitor of mitochondrial respiration than phenformin.
Figure 1.

IM156 Is a Newly Developed Biguanide That Inhibits Mitochondrial Respiration
(A) Chemical structure of the biguanides metformin, phenformin, and IM156.
(B) Dose-dependent reduction of the OCR of Eμ-Myc+ lymphoma cells by metformin, phenformin, and IM156. Data were derived from the maximal point of OCR reduction after 30 min of treatment with the indicated biguanide and expressed relative to untreated cells at the same time point. Data represent mean ± SEM for biological replicates (n = 4–6 per drug per concentration).
(C) Mitochondrial ATP production rate of Eμ-Myc+ lymphoma cells after incubation with 100 μM of either vehicle, phenformin (Phen), or IM156. Data represent mean ± SD for biological replicates (n = 8–9 per group).
(D) Dose-dependent inhibition of substrate oxidation by electron transport chain complexes in IM156-treated purified bovine mitochondrial membranes. NADH oxidation was measured for complex I, III, and IV activity (dark blue), and succinate oxidation was measured for complex II, III, and IV activity (gray) over the indicated concentrations of IM156. Data represent mean ± SD for technical replicates (n = 3–4 per group). See also Figure S1A.
(E) Dose-dependent reduction of OCR by IM156 in HEK293T cells expressing empty vector (Ctrl) or yeast NDI1 (NDI1). Data were derived from the maximal point of OCR reduction after 30 min of treatment with IM156 and expressed relative to the maximal OCR at the same time point and group. Data represent mean ± SD for technical replicates (n = 10 per concentration per group).
(F) Immunoblot for phosphorylated (pAMPK, T172) and total AMPKα following treatment with vehicle or equivalent doses of phenformin (100 μM) or IM156 (10 μM) for 2 h. Actin is shown as a loading control.
(G) Viability of Eμ-Myc+ lymphoma cells following 48 h treatment with phenformin or IM156, comparing 10 different doses. EC50 for each treatment is indicated. Data represent mean ± SEM for biological replicates (n = 3 per drug per concentration).
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To confirm that the effect of IM156 on mitochondrial respiration was through complex I inhibition, we treated purified bovine mitochondrial membranes with IM156. IM156 treatment decreased complex I-dependent NADH oxidation in a significant, dose-dependent manner, with a lower half-maximal inhibitory concentration (IC50) (2.2 μM) than previously reported for phenformin (340 μM),16 while succinate oxidation (via complex II) was minimally affected at equivalent doses of IM156 (Figure 1D). This minimal decrease in succinate oxidation could be an effect of IM156 on any of the complexes II, III, or IV, but could also be an effect of the drug on the coupled assay enzymes fumarase and malic enzyme. IM156 also reduced NADH oxidation in purified complex I in a dose-dependent manner (Figure S1A). To further confirm the specificity of IM156 for complex I, we assessed the IM156-mediated inhibition of OCR in cells expressing NDI1, a yeast NADH dehydrogenase that is resistant to biguanides.28 NDI1 expression rescued the effects of IM156 on cellular respiration, promoting an ∼100-fold shift in the IC50 of IM156 (Figure 1E). These data support that IM156 blocks OXPHOS through the inhibition of complex I and does so more potently than phenformin.
We next determined whether phenformin and IM156 similarly activate the energy sensor AMPK, as depletion of cellular ATP by biguanide treatment is a known trigger for AMPK activation.7 Given the difference in potency of phenformin and IM156, we used doses (100 and 10 μM, respectively) that produced an equivalent ∼50% decrease in OCR from baseline after a 2-h treatment (Figures S1B and S1C). A 2-h treatment with these concentrations of phenformin or IM156 yielded similar levels of activating AMPK phosphorylation at T-172 (Figure 1F). Lastly, we treated Eμ-Myc+ lymphoma cells with a range of concentrations of either phenformin or IM156. Based on cell viability measurements, IM156 exhibited higher potency and induced lymphoma cell death at lower concentrations than phenformin (half-maximal effective concentration [EC50] of 12 μM for IM156 compared to 62 μM for phenformin; Figure 1G).
miR-17∼92 Sensitizes Lymphoma Cells to Apoptosis by Biguanides
Previously, we demonstrated that the oncogenic miRNA cluster miR-17∼92 is required for Myc-dependent metabolic reprogramming in lymphoma and does so in part through the translational suppression of LKB1.30 We examined whether the expression of miR-17∼92 alters the sensitivity of lymphoma cells to biguanide treatment. We used Eμ-Myc B cell lymphoma cells harboring floxed miR-17∼92 alleles, which allowed us to study the effect of the conditional deletion of miR-17∼92 in the presence of constitutive Myc expression.32 Eμ-Myc lymphoma cells deleted for miR-17∼92 (Δ/Δ) were more resistant to phenformin treatment than their isogenic counterparts expressing miR-17∼92 (fl/fl) (Figure 2A). Phenformin treatment actively induced apoptosis in miR-17∼92-expressing Eμ-Myc lymphoma cells as shown by the presence of active (cleaved) caspase-3 (Figure 2B). Levels of caspase-3 cleavage were markedly reduced in Eμ-Myc lymphoma cells lacking miR-17∼92 (Figure 2B).
Figure 2.

miR-17∼92 Sensitizes Lymphoma Cells to Apoptosis by Biguanides
(A) Viability of Ctrl (fl/fl) and miR-17∼92-deficient (Δ/Δ) Eμ-Myc+ lymphoma cells untreated (Ctrl, gray) or treated with 100 μM phenformin (Phen, blue) for 48 h. Data represent mean ± SD for biological replicates (n = 3 per group).
(B) Immunoblot for active (cleaved) caspase-3 in cells treated as in (A). Actin is shown as a loading control.
(C) Dose of phenformin (left) or IM156 (right) required to achieve 50% decrease in cell viability (EC50) in biguanide-sensitive Eμ-Myc+ lymphoma cells expressing control (Ctrl) or miR-17∼92 (+17∼92) expression vectors. Cell viability was measured 48 h post-biguanide treatment. See also Figures S1B and S1C.
(D) Viability of control (Ctrl) or miR-17∼92-expressing (+17∼92) Raji cells after 48 h treatment with 100 μM phenformin. Histogram shows representative staining for cell death using fluorescent viability dye (FVD), which is quantified at right. Data represent mean ± SD for biological replicates (n = 3 per group).
(E) Dose of phenformin (left) or IM156 (right) required to achieve a 50% decrease in cell viability (EC50) in biguanide-resistant Raji cells expressing control (Ctrl) or miR-17∼92 (+17∼92) expression vectors following 48 h of treatment with biguanide.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Since miR-17∼92 is recurrently amplified in lymphoma,33,34 we next tested whether an increased copy number of miR-17∼92 was sufficient to increase the sensitivity of lymphoma cells to biguanides. To test this, we generated Eμ-Myc lymphoma cells and Raji lymphoma cells, a human Burkitt’s lymphoma cell line known to display low MYC levels,30 with ectopic expression of the entire miR-17∼92 polycistron (hereafter denoted as +17∼92). Eμ-Myc lymphoma cells overexpressing miR-17∼92 were significantly more sensitive than control cells when treated with either phenformin or IM156 (Figures 2C and S1B). miR-17∼92 overexpression led to a 10-fold shift in the EC50 of Eμ-Myc cells to IM156 treatment (2 μM versus 24 μM). Similar results were observed in Raji cells engineered to express higher levels of miR-17∼92 (Figures 2D, 2E, and S1C). Although Raji cells were less sensitive to biguanide treatment, with EC50 values ranging from 90 μM to 3.5 mM as compared to Eμ-Myc+ EC50 values ranging from 2 to 62 μM (Figures S1B and S1C), the overexpression of miR-17∼92 still conferred a ∼3-fold increase in sensitivity to biguanides compared to control cells. These data indicate that elevated miR-17∼92 enhances the sensitivity of lymphoma cells to biguanide treatments and that Myc and miR-17∼92 synergize to increase sensitivity to biguanide treatment.
We next asked whether the enhanced sensitivity to biguanides observed in vitro extended to in vivo models. Nude mice harboring either control or miR-17∼92-overexpressing Eμ-Myc cells were administered phenformin or IM156 in their drinking water ad libitum, and survival was tracked compared to tumor-bearing mice administered regular drinking water. While biguanide treatment of mice bearing control lymphoma cells produced no discernible survival benefit (Figure 3A), both phenformin and IM156 significantly prolonged the lifespan of mice bearing aggressive miR-17∼92-overexpressing tumors (Figure 3B). Our observations indicate that phenformin and IM156 can act as single agents to extend the survival of mice bearing tumors with elevated miR-17∼92 expression, with elevated miR-17∼92 expression functioning as a predictor of sensitivity to biguanides.
Figure 3.

Biguanide Treatment Selectively Impairs the Growth of miR-17∼92-Expressing Lymphoma Cells In Vivo
(A and B) Kaplan-Meier curves for the viability of nude mice injected with 1 × 106Myc+/Ctrl (A) or Myc+/+17∼92 (B) lymphoma cells. Mice were provided with untreated water (Ctrl, n = 10), 0.9 mg/mL phenformin (Phen, n = 8), or 0.8 mg/mL IM156 (IM156, n = 10) ad libitum following intravenous tumor cell injection.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
miR-17∼92 Confers Sensitivity to Biguanides through the Suppression of LKB1
We next explored the mechanism by which elevated miR-17∼92 expression confers biguanide sensitivity in lymphoma cells. NSCLC cells that have lost LKB1 display increased sensitivity to phenformin.8 We previously identified LKB1 as a target of gene silencing by miR-17∼92 via miR-17- and -20-dependent suppression of LKB1 protein expression.30 Deletion of miR-17∼92 in Eμ-Myc cells (Δ/Δ cells) increased LKB1 protein levels (Figure 4A), while over-expression of miR-17∼92 reduced LKB1 levels (Figure 4B). Increased miR-17∼92 expression resulted in reduced AMPK phosphorylation (Figure 4B), which downregulated Ser-792 phosphorylation of Raptor (Figure 4C) and thus enhanced mammalian target of rapamycin complex 1 (mTORC1) signaling, as evidenced by the increased phosphorylation of rS6 and 4EBP phosphorylation in miR-17∼92-expressing cells (Figure 4C). The deletion of miR-17∼92 in Eμ-Myc cells (Δ/Δ cells) was sufficient to confer a survival advantage in response to IM156 treatment (Figure 4D). Reducing LKB1 expression in Δ/Δ cells using small hairpin RNAs (shRNAs) targeting LKB130 restored biguanide sensitivity in Δ/Δ cells to match that observed in lymphoma cells expressing miR-17∼92 (Figure 4D). Similarly, lymphoma cells expressing miR-17∼92 but lacking expression of miR-17 and -20 were resistant to IM156 treatment (Figure 4E).
Figure 4.

miR-17∼92 Confers Sensitivity to Biguanides through the Suppression of LKB1
(A) Immunoblot of LKB1 and actin protein levels in Ctrl (fl/fl) and miR-17∼92-deficient (Δ/Δ) Eμ-Myc+ lymphoma cells.
(B and C) Immunoblot of AMPK pathway (B) and mTORC1 pathway (C) activation in control (Ctrl) or miR-17∼92-expressing (+17∼92) Eμ-Myc+ lymphoma cells. AMPK activation was determined by measuring total and phosphorylated (p-AMPK, T172) AMPKα. mTORC1 activity was assessed by measuring levels of total and phosphorylated ribosomal S6 protein (rS6, S235/236), 4EBP (T37/46), and Raptor (S792).
(D) Viability of the following Eμ-Myc+ lymphoma cells: Ctrl (fl/fl), miR-17∼92-deficient (Δ/Δ), and miR-17∼92-deficient expressing shRNAs targeting LKB1 (Δ/Δ+shLKB1/2). Cells were treated with 10 μM IM156 for 48 h and viability assessed by flow cytometry. Data represent mean ± SD for biological replicates (n = 3 per group).
(E) Viability of Eμ-Myc+ lymphoma cells expressing miR-17∼92 (+17∼92) or miR-17∼92 lacking the miR-17 and -20 seed families (+17∼92Δ(17+20)). Cells were treated with vehicle (Ctrl) or 10 μM IM156 for 48 h and viability assessed by flow cytometry. Data represent mean ± SD for biological replicates (n = 3 per group).
(F) Immunoblot of total and phosphorylated (p-AMPK, T172) AMPKα levels in control (Ctrl) and miR-17∼92-expressing (+17∼92) Eμ-Myc+ lymphoma cells following a 2-h treatment with vehicle (–), 100 μM phenformin (+Phen), or 10 μM IM156 (+IM156).
(G) Schematic of transcriptional differences in miRNA between control (fl/fl) and miR-17∼92 expressing Eμ-Myc+ lymphoma cells. miR-17 and -20 are responsible for the repression of LKB1, which increases biguanide sensitivity.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Given that an increased copy number of miR-17∼92 leads to LKB1 repression in lymphoma cells (Figure 4B), we tested whether AMPK activation was differentially engaged downstream of either phenformin or IM156 treatment. Lymphoma cells overexpressing miR-17∼92 displayed reduced AMPK phosphorylation following phenformin or IM156 treatment compared to control cells (Figure 4F), suggesting uncoupling of the LKB1-AMPK axis to metabolic stress (Figure 4G).
Biguanide Treatment Affects Central Carbon Metabolism in miR-17∼92-Expressing Myc+ Lymphoma Cells
We next characterized the effect of IM156 treatment on lymphoma cell bioenergetics using a Seahorse XF96 extracellular flux analyzer. Lymphoma cells overexpressing miR-17∼92 exhibit higher basal metabolic rates, both in terms of glycolytic rate (extracellular acidification rate [ECAR]) and respiration (OCR), than control cells (Figure 5A). This corresponded to higher overall rates of ATP production by miR-17∼92-expressing lymphoma cells compared to controls, which was due largely to increased glycolytic ATP production (Figure 5B). miR-17∼92-expressing lymphoma cells also displayed a larger bioenergetic scope than control cells (Figure 5C). The bioenergetic profile of both cell types was significantly altered by IM156 treatment, characterized by a decrease in mitochondrial ATP production (JATP ox) and an increase in glycolytic ATP production (JATP gly) (Figure 5C). IM156 reduced OXPHOS-derived ATP production in both cell types by >50% (Figure 5C).
Figure 5.

Biguanide Treatment Affects Central Carbon Metabolism in miR-17∼92-Expressing Lymphoma Cells
(A) ECAR and OCR of control (Ctrl) or miR-17∼92-expressing (+17∼92) Eμ-Myc+ lymphoma cells. Data represent mean± SD for biological replicates (n = 12 per group).
(B) ATP production rates (JATP) for Eμ-Myc+ lymphoma cells expressing control (Ctrl) or miR-17∼92 (+17∼92) expression vectors. Seahorse experiments were performed under standard cell culture media conditions (Glc, 25 mM; Gln, 2 mM). JATPtotal is the sum of the glycolytic (JATPgly) and OXPHOS (JATPox) ATP production rates. Data represent mean ± SDs for biological replicates (n = 12 per group) (Ctrl, n = 8; Ctrl+IM156, n = 8; +17∼92, n = 11; +17∼92+IM156, n = 12).
(C) Bioenergetic capacity plot of Eμ-Myc+ lymphoma cells expressing control (Ctrl) or miR-17∼92 overexpression (+17∼92) vectors following a 2-h treatment with vehicle (gray) or IM156 (purple). Rectangles define the maximum bioenergetic space of each cell type.
(D) Heatmap showing relative metabolite abundances for control (Ctrl) or miR-17∼92-expressing (+17∼92) Eμ-Myc+ lymphoma cells following a 2-h treatment with vehicle (Ctrl), phenformin (Phen), or IM156 (n = 3 per group).
(E) Fractional enrichment of U-[13C]-glucose-derived lactate in miR-17∼92-expressing Eμ-Myc+ lymphoma cells following a 2-h treatment with vehicle (Ctrl), phenformin (Phen), or IM156. Data represent mean ± SD for biological replicates (n = 3 per group).
(F) Fractional enrichment of U-[13C]-labeled glutamine in miR-17∼92-expressing Eμ-Myc+ lymphoma cells following a 2-h treatment with vehicle (Ctrl), phenformin (Phen), or IM156. Data represent mean ± SD for biological replicates (n = 3 per group).
(G) Fractional enrichment of U-[13C]-glucose-derived citrate and U-[13C]-glutamine-derived citrate and aspartate in miR-17∼92-expressing Eμ-Myc+ lymphoma cells, following a 2-h treatment with vehicle (Ctrl), phenformin (Phen), or IM156. Data represent mean ± SD for biological replicates (n = 3 per group).
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
We next examined the impact of biguanide treatment on metabolite dynamics in lymphoma cells. Consistent with their increased bioenergetic profile (Figures 5A–5C), miR-17∼92-expressing lymphoma cells displayed an increased abundance of metabolites involved in glycolysis (e.g., pyruvate, lactate), the tricarboxylic acid (TCA) cycle (e.g., citrate, succinate, fumarate, malate), and amino acid metabolism (Figure 5D). Biguanide treatment had a minimal effect on amino acid levels in either cell type (Figure 5D). However, levels of TCA cycle intermediates were markedly reduced in miR-17∼92-expressing cells upon treatment with phenformin or IM156 (Figure 5D). This may be the result of the stalled processing of TCA cycle intermediates due to NADH buildup following complex I inhibition, as previously suggested.35 This was not due to an inhibition of glucose or glutamine uptake, as 13C-tracing experiments revealed no change in glucose trafficking to lactate (Figure 5E) or glutamine uptake (Figure 5F) in the presence of phenformin or IM156, consistent with previous findings using phenformin.36 The lack of increased intracellular 13C-glucose-derived lactate in phenformin- or IM156-treated cells may be due to increased export into the culture medium, as suggested by the shift in glycolytic ATP production in IM156-treated cells (Figure 5C). Biguanide treatment blunted 13C-glucose and 13C-glutamine entry into the TCA cycle, as evidenced by the reduced conversion of glucose and glutamine to citrate (Figure 5G). 13C-glutamine-derived production of aspartate, a key intermediate for protein and nucleotide biosynthesis and tumor cell growth,35,36 was reduced by 60%–65% following a 2-h administration of phenformin or IM156 (Figure 5H). These data suggest that although miR-17∼92 enhances central carbon metabolism and bioenergetic potential in lymphoma cells, these pathways are uniquely sensitive to biguanide treatment.
miR-17 and -20 Expression Correlates with Biguanide Sensitivity in Human Lymphoma Cells
Having defined the downstream effects of biguanides on the metabolism and viability of miR-17∼92-expressing Myc+ lymphoma cells, we expanded our analysis to test biguanide sensitivity in a panel of human lymphoma cell lines. Ten verified MYC+ lymphoma cell lines were used for this analysis: SU-DHL-4, OCI-Ly7, Jeko-1, Rec-1, Karpas 1718, OCI-Ly-1, OCI-Ly-2, OCI-Ly-3, OCI-Ly-8, and OCI-Ly-18. For each cell line, we measured levels of both the primary (pri-miR) and mature (miR) forms of miR-17 and miR-20 and determined the EC50 for cell viability to phenformin or IM156. From these data, we observed a negative correlation between the relative level of miR-17 and -20 expressed in cells and the dose of biguanide required to achieve EC50 (Figures 6A, 6B, and S2; Table S1), with cells displaying the highest miR-17 and -20 expression showing the greatest sensitivity to biguanide treatment. Levels of pri-miR-17 were a better predictor of biguanide sensitivity than mature miR-17 (Figures 6A and 6B). IM156 was more potent than phenformin in inducing cell death in human lymphoma cells, similar to that observed in Eμ-Myc+ lymphoma cells. These data demonstrate miR-17 expression as a biomarker for biguanide sensitivity.
Figure 6.

miR-17 and -20 Expression Correlates with Biguanide Sensitivity in Human Lymphoma Cells
(A and B) EC50 for phenformin (Phen, left) and IM156 (right) correlated to the relative levels of pri-miR-17 (A) and mature miR-17 (B) transcripts for 10 human lymphoma cell lines. Linear regression (dotted line) is shown for both drugs with shaded 95% confidence interval.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
Myc is abnormally expressed in many human cancers; however, the development of small molecule Myc inhibitors has been a persistent challenge,37 prompting consideration of alternative approaches to Myc targeting. Here, we report that a downstream target of Myc-driven metabolism, miR-17∼92, while increasing bioenergetic capacity of tumor cells also increases sensitivity to apoptosis induced by biguanides. Using both classic (phenformin) and newly developed (IM156) biguanides, we demonstrated that human and mouse lymphoma cells with elevated miR-17∼92 expression were highly sensitized to biguanide treatment. As single agents, both phenformin and IM156 were effective at extending the survival of mice bearing miR-17∼92-expressing lymphomas. In addition, we observed a statistical correlation between miR-17 levels in human lymphoma cells and sensitivity to biguanide treatment. Our data provide rationale for the clinical investigation of miR-17∼92 expression level as a potential biomarker for biguanide sensitivity in cancer.
Enhanced Myc activity leads to the overexpression of miR-17∼92, which in turn lowers LKB1 expression. This leads to enhanced tumor growth associated with increased glycolysis and oxidative phosphorylation, but it also reduces the capacity of lymphoma cells to activate the LKB1-AMPK system to overcome the energetic stress imposed by biguanides. These cells are then susceptible to apoptosis upon biguanide treatment, as both phenformin and IM156 inhibit oxidative phosphorylation, and the LKB1-AMPK system cannot be activated to respond to this inhibition. Another important function of LKB1-AMPK signaling is the surveillance of mitochondrial integrity.38 The potential impact of miR-17∼92 on mitochondrial homeostasis and changes in response to biguanide treatment remains to be determined.
Our results address gaps in knowledge related to the mechanism by which LKB1 loss induces biguanide sensitivity. Previous work indicates that phenformin treatment increases reactive oxygen species (ROS) levels in cells with reduced LKB1 expression,8 leading to oxidative damage and preferential cell death of LKB1 null tumor cells. We show here that glucose- and glutamine-dependent fueling of the TCA cycle was inhibited in miR-17∼92-expressing lymphoma cells responding to biguanide treatment. This fueling is a key node for the production of reducing equivalents for ATP production and biosynthetic intermediates for growth (e.g., citrate, aspartate). Our data are consistent with observations that reducing mitochondrial aspartate production in tumor cells can impair cell viability, due in part to diminished purine and pyrimidine biosynthesis.39,40 Reduced biosynthetic metabolism, combined with reduced mitochondrial NADH production and an inability to activate AMPK signaling, appear to sensitize miR-17∼92-expressing lymphoma cells to metabolic stress. Consistent with this, biguanide treatment, particularly IM156, slowed lymphoma cell growth in vitro and extended the lifespan of tumor-bearing mice.
There has been increased interest in the clinical use of biguanides for cancer therapy. Encouraging preclinical data17 have led to >100 clinical trials of metformin for indications in oncology,21 but the results of randomized trials have been disappointing. The lack of clinical benefit of metformin in trials reported to date does not necessarily imply that biguanides as a class have no activity. Pharmacokinetic factors may account at least in part for the discrepancies between preclinical models and clinical trials with respect to the antineoplastic activity of metformin. The hydrophilic nature of metformin dictates that cellular uptake is dependent on OCT1 transporters before gaining access to mitochondria.31 This inherently limits the action of metformin to those cells expressing OCT1, which can vary between tumors.41 The biguanide phenformin displays greater bioavailability and antineoplastic activity than metformin,31,41,42 but its clinical use has been limited due to the risk of lactic acidosis, especially in patients with compromised renal or liver function.43 These results justify the investigation of novel biguanides with better pharmacokinetic and pharmacodynamic properties than metformin in neoplastic tissue as a strategy for OXPHOS inhibition for cancer treatment.
We identified IM156 from a library of ≥1,000 biguanides as a potential anticancer drug candidate.44 Our data indicate that IM156 is a more potent OXPHOS inhibitor than metformin or phenformin. IM156 may have advantages over metformin as it is a more potent, hydrophilic biguanide that we hypothesize is more bioavailable in neoplastic tissues. IM156 is presently being evaluated in a Phase I clinical trial (NCT 032 72256) in patients with advanced solid tumors and lymphoma. Our work here explored the use of IM156 as a monotherapy for cells with reduced LKB1-AMPK signaling, but combination therapy with other metabolic targets may increase efficacy with this agent. Recent reports have demonstrated synthetic lethality in tumor cells when metformin is combined with the pharmacological inhibition of monocarboxylate transporters (MCTs),45,46 due in part to ATP depletion following the loss of NAD+ regeneration when lactate cannot be exported by MCT1 or MCT4. Inhibition of aspartate synthesis by the malate-aspartate shuttle enzyme Got1 also synergizes with biguanides to enhance cancer cell death.40
Our research adds to prior evidence8,47 that clinical trials of biguanides or other OXPHOS inhibitors are likely to show clinical activity if evaluated in patients with cancers that are particularly sensitive to energetic stress, such as those with elevated expression of MYC or miR-17∼92 and/or decreased function of LKB1. Although LKB1 inactivation is relatively common in NSCLC,7 it is not common among other malignancies. In contrast, MYC is among the most commonly dysregulated oncogenes in both hematologic malignancies and solid tumors. Because of the paucity of viable treatments targeting MYC-driven cancers, linking the downstream MYC target miR-17∼92 to the inhibition of LKB1 may identify a cohort of patients without LKB1 mutations who are particularly sensitive to biguanide treatment, opening avenues for the treatment of these cancers.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-human/mouse beta-actin (1:2000 dilution) | Cell Signaling | Cat # 4967 |
| Rabbit polyclonal anti-human/mouse 4E-BP (1:1000 dilution) | Cell Signaling | Cat # 9452 |
| Rabbit polyclonal anti-human/mouse 4E-BP (phospho-T36/47) (1:1000 dilution) | Cell Signaling | Cat # 9459 |
| Rabbit polyclonal anti-human/mouse rS6 (pS235/236) (1:1000 dilution) | Cell Signaling | Cat # 2211 |
| Mouse monoclonal anti-human/mouse rS6 (54D2) (1:1000 dilution) | Cell Signaling | Cat # 2317 |
| Rabbit monoclonal anti-human/mouse Raptor (24C12) (1:1000 dilution) | Cell Signaling | Cat # 2280 |
| Rabbit polyclonal anti-human/mouse Raptor (pS792) (1:1000 dilution) | Cell Signaling | Cat # 2083 |
| Rabbit polyclonal anti-human/mouse AMPKα (1:1000 dilution) | Cell Signaling | Cat # 2532 |
| Rabbit monoclonal anti-human/mouse AMPKα (pT172) (40H9) (1:1000 dilution) | Cell Signaling | Cat # 2535 |
| Mouse monoclonal primary anti-human/mouse/rat LKB1 (Ley 37D/G6) (1:2500 dilution) | Santa Cruz Biotechnology | Cat # sc-32245 |
| Rabbit polyclonal anti-human/mouse Caspase3 (1:1000 dilution) | Cell Signaling | Cat # 9662 |
| Biological Samples | ||
| Bos taurus (bovine) heart | C. Humphries & Sons | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Metformin hydrochloride | Toronto Research Chemicals | Cat # M258815 |
| phenformin | Toronto Research Chemicals | Cat # P296900 |
| sucralose | Sigma | Cat # 69293-100G |
| 4-OHT | Sigma | Cat # H7904-5MG |
| XF Base Medium Minimal DMEM without Phenol Red | Agilent | Cat # 103335-100 |
| IMDM GlutaMAX | Thermo Fisher | Cat # 3198-022 |
| Fetal bovine serum | Wisent-Bioproducts | Cat # 080-450 |
| Penicillin-streptomycin | Wisent-Bioproducts | Cat # 450-201-EL |
| L-Glutamine | Biochrom AG | Cat # M11-004 |
| 2-mercaptoethanol | GIBCO | Cat # 21985-023 |
| RPMI | Wisent | Cat # 350-000 |
| puromycin | BioShop Canada | Cat # PUR333.25 |
| butanol | Sigma-Aldrich | Cat # B7906-500ML |
| hydrochloric acid, 6N | Fisher Scientific | Cat #SA56-4 |
| sodium dicyanamide | Sigma-Aldrich | Cat # 178322-25G |
| phenylamine | Sigma-Aldrich | Cat # 242284-500ML |
| trypan blue | LifeTech (Biobar) | Cat # 15250-061 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat # 65-0865-14 |
| [U-13C]-glucose | Cambridge Isotopes | Cat # CLM-1396 |
| FCCP | Cayman | Cat # 15218 |
| Oligomycin | Sigma-Aldrich | Cat # 1404-19-9 |
| Rotenone | Cayman | Cat # 13955 |
| Antimycin A | Sigma Aldrich | Cat # A8674 |
| Poly-D-lysine hydrobromide | Sigma Aldrich | Cat # P6407 |
| Lipofectamine 3000 | Thermo Fisher | Cat # L3000001 |
| Trizma | Sigma Aldrich | Cat # T1503 |
| sucrose | Sigma Aldrich | Cat # S0389 |
| horse heart cytochrome c | Sigma Aldrich | Cat # C2506 |
| NADH | Sigma Aldrich | CAS # 606-68-6 |
| Malic enzyme | Jones and Hirst53 | N/A |
| Fumarase | Jones and Hirst53 | N/A |
| potassium sulfate (KSO4) | Sigma Aldrich | Cat # P0772 |
| magnesium sulfate (MgSO4) | VWR | Cat # 2312982 |
| sodium succinate | Sigma Aldrich | Cat # S2378 |
| NADP+ | Sigma Aldrich | Cat # N0585 |
| 3-((3-cholamidopropyl)dimethylammonium)-1-propanesulfonate (CHAPS) | Santa Cruz Biotechnology | Cat # sc-29088A |
| soy bean asolectin | Avanti Polar Liquids | Cat # 541601G |
| decylubiquinone (DQ) | Santa Cruz Biotechnology | Cat # sc-358659 |
| DMSO | Fisher Bioreagents | Cat # BP231-100 |
| Glucose ≥ 99,5% D(+) | Roth | Cat # HN06.1 |
| [13C1]-glutamine | Cambridge Isotopes | Cat # CLM-3612-PK |
| [U-13C]-glucose | Cambridge Isotopes | Cat # CLM-1396 |
| methanol | VWR | CAMX0475-1 |
| D-myristic acid | Sigma Aldrich | Cat # 366889-1G |
| methoxyamine hydrochloride | Sigma Aldrich | Cat # 226904-5G |
| pyridine | Sigma Aldrich | Cat # 270970-100ML |
| N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA) | Sigma Aldrich | Cat # 394882-5ML |
| CHAPS buffer (10 mM Tris-HCl, 1 mM MgCl2, 1 mM EGTA, 0.5 mM CHAPS, 10% glycerol, and 5 mM NaF) | (Faubert et al., 2014)4 | N/A |
| AMPK lysis buffer, RIPA buffer | Cell Signaling | Cat # 9806S |
| QIAzol lysis reagent | QIAGEN | Cat # 79306 |
| HBSS | Wisent | Cat # 311-510-CL |
| Critical Commercial Assays | ||
| Seahorse XFe96 FluxPak | Agilent Technologies | Cat # 102416-100 |
| miRNeasy Mini kit | QIAGEN | Cat # 217004 |
| miScript II RT kit | QIAGEN | Cat # 218160 |
| SensiFAST SYBR Lo-Rox Mix | Bioline | Cat # BIO-92005 |
| miScript primer assays | QIAGEN | Cat # 218300 |
| Dual-Glo Luciferase assay kit | Promega | Cat # E2920 |
| Experimental Models: Cell Lines | ||
| Eμ-Myc lymphoma cells | Izreig et al.30 | N/A |
| Eμ-Myc Cre-ERT2+; miR-17∼92fl/fl lymphoma cells | Mu et al.32 | N/A |
| Raji lymphoma cells | Izreig et al.30 | N/A |
| 293T, human embryonic kidney (HEK) cells | ATCC | Cat # CRL-3216 |
| Ink4a-null MEF feeder cells | Izreig et al.30 | N/A |
| Experimental Models: Organisms/Strains | ||
| CD-1 nude mice, Crl:CD1-Foxn1nu | Charles River Laboratories | Cat # CR:086 |
| Recombinant DNA | ||
| Stk11 shRNA (LMP backbone): 5′- AGGTCAAGATCCTCAAGAAGAA −3′ | Izreig et al.30 | N/A |
| Yeast NDI1 | Wheaton et al.28 | N/A |
| Software and Algorithms | ||
| FlowJo 9.9.5 | FlowJo LLC | https://www.flowjo.com |
| GraphPad Prism V6 or V7 | GraphPad Software | https://www.graphpad.com |
| El-Maven | Elucidata Inc | https://elucidatainc.github.io/ElMaven/ |
| Bioenergetics Calculations Spreadsheet | Mookerjee et al.50 | https://russelljoneslab.vai.org/tools |
| Mass isotopomer distribution for TCA cycle intermediates was determined using a custom algorithm developed at McGill University | McGuirk et al.54 | N/A |
| Other (Mouse Diets) | ||
| Teklad global soy protein-free extruded rodent diet | Envigo | Cat # 2020X |
Resource Availability
Lead Contact
Additional information and request for resources and reagents should be directed to and will be made available by the Lead Contact, Russell G. Jones (russell.jones@vai.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. The biguanide IM156 is the intellectual property of ImmunoMet Therapeutics. Requests for ImmunoMet Therapeutics reagents can be made through the Lead Contact.
Data and Code Availability
All data associated with this study are available in the main text or the supplementary materials. A spreadsheet to assist in converting OCAR and ECAR Seahorse data into oxidative and glycolytic ATP production rates is available at https://russelljoneslab.vai.org/tools. This study did not generate any code.
Experimental Model and Subject Details
Mice
CD-1 nude mice were purchased from Charles River Laboratories (Wilmington, MA). All animals had ad libitum access to food and water and were maintained at 22–24°C on a 12-hour light/12-hour dark cycle with 5 mice per cage. Mice were bred and maintained under specific pathogen-free conditions at McGill University under approved protocols. For tumor xenograft assays, water bottles carrying 1.2% sucralose, 0.9 mg/mL phenformin +1.2% sucralose, or 0.8 mg/mL IM156 + 1.2% sucralose were provided for ad libitum consumption. Mice were observed daily for health status until clinical displays of disease, such as weight loss and poverty of movement, met IACUC criteria for euthanasia at which point mice were euthanized. Experiments were performed on female mice between 8 and 20 weeks of age.
Cell lines
HEK293 cells were obtained from ATCC. Expression constructs for NDI1 were a generous gift of Dr. Navdeep Chandel.28 The generation of Eμ-Myc Cre-ERT2+; miR-17∼92fl/fl lymphoma cells has been described previously.32 Deletion of miR-17∼92 was achieved by culturing Eμ-Myc Cre-ERT2+;miR-17∼92fl/fl cells with 250 nM 4-OHT for four days,32 followed by subcloning 4-OHT-treated cells to isolate cells deficient for miR-17∼92. Eμ-Myc cells were cultured on a layer of irradiated Ink4a null MEF feeder cells in DMEM and IMDM medium (50:50 mix) supplemented with 10% fetal bovine serum (FBS), 20000 U/mL penicillin, 7 mM streptomycin, 2 mM glutamine, and β-mercaptoethanol. Raji cells were cultured in RPMI medium supplemented with 10% FBS, 20000 U/mL penicillin, 7 mM streptomycin, and 2 mM glutamine. Cells were grown at 37°C in a humidified atmosphere supplemented with 5% (v/v) CO2. Retroviral-mediated gene transfer into lymphoma cells was conducted as previously described.30 Briefly, lymphoma cells were transduced via spin infection, followed by culture in 4 μg/mL puromycin for four days, and subsequent subcloning by limiting dilution. miR-17∼92 constructs have been previously described.32 Knockdown of Stk11 via shRNA (sequence: 5′- AGGTCAAGATCCTCAAGAAGAA −3′) has been described.30
Method Details
Synthesis of IM156
IM156 was synthesized as previously reported.48 In brief, pyrrolidine was dissolved in butanol before concentrated hydrochloric acid was added and stirred for 30 min at 0°C. Sodium dicyanamide was then added and stirred for 24 h under reflux. After completion of the reaction was confirmed, the adduct—N1-pyrollidine cyanoguanidine—was purified. Next, phenylamine was dissolved in butanol before concentrated hydrochloric acid was added and stirred for 30 min at room temperature. The N1-pyrollidine cyanoguanidine prepared above was then added and stirred for 6 h under reflux. This mixture was concentrated under reduced pressure then dissolved in a 6 N hydrochloric acid/methanol solution before adding ethyl acetate. The precipitate—IM156A HCl—was filtered and dried under reduced pressure. IM156.HCl can be converted to free form by adding sodium hydroxide. The free form can be converted to the acetate salt form—IM156A—by adding acetic acid.
Cell proliferation and viability assays
Cells were seeded at a density of 1 × 105 cells/mL in 3.5 cm dishes, and cell counts determined via trypan blue exclusion using a TC20 Automated Cell Counter (Biorad). For viability measurements, cells were stained with Fixable Viability Dye eFluor 780 (eBioscience) and analyzed using a Gallios flow cytometer (Beckman Coulter, Fullerton, CA) and FlowJo software (Tree Star, Ashland, OR).
Seahorse XF96 Respirometry and metabolic assays
Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) using established protocols.49 In brief, lymphoma cells (7.5 × 104 per well) were plated in poly-D-lysine-coated XF96 Seahorse plates in 140 μL of unbuffered DMEM containing 25 mM glucose and 2 mM glutamine, followed by centrifugation at 500xg for five minutes. XF assays consisted of sequential mix (3 min), pause (3 min), and measurement (5 min) cycles, allowing for determination of OCR and ECAR every 8 min. Following four baseline measurements, 20 μL of untreated media, phenformin, or IM156 were injected into respective wells, and OCR and ECAR tracked over time. We converted ECAR and OCR measurements into glycolytic and oxidative ATP production rates based on protocols developed by Mookerjee and Brand.50 This method allows us to directly compare the contribution of glycolysis and OXPHOS to ATP production while correcting for the contribution of glycolysis and TCA cycle metabolism to ECAR. Tools to complete this analysis are available for download at https://russelljoneslab.vai.org/tools.
HEK293 cells were transfected with vectors expressing empty vector (Ctrl) or yeast NADH dehydrogenase NDI1 using Lipofectamine 3000 according to manufacturer’s instructions. Transfected HEK293 cells were seeded at a density of 30,000 cells/well in poly-D-Lysine-coated Seahorse plates. Oxygen consumption rates were recorded by using Seahorse XF96 bioanalyzer in the presence of serial dilution of IM156 as described above.
Complex I and mitochondrial membrane kinetic measurements
Mitochondrial membranes51 and purified complex I52 were prepared from Bos taurus (bovine) heart. All assays were performed at 32°C in a SpectraMax 96-well plate reader and maximal rates determined by linear regression. NADH:O2 oxidoreduction by membranes was measured using 5 μg/mL membranes in 10 mM Tris-HCl (pH 7.4), 250 mM sucrose, and 0.15 μM horse heart cytochrome c (Sigma-Aldrich Ltd.) with 200 μM NADH and monitored using ε340–380(NADH) = 4.81 mM−1cm−1. Succinate:O2 oxidoreduction was measured using a coupled enzyme assay using 300 ug/mL Malic enzyme and 60 ug/mL Fumarase both prepared as described.53 Activity was measured in the same buffer as above with the addition of 1 mM KSO4 and 2 mM MgSO4 using 5 μg/mL membranes with 5 mM succinate and 2 mM NADP+. This was monitored using ε340–380(NADPH) = 4.81 mM−1 cm−1 as described previously.53 NADH:decylubiquinone oxidoreduction by purified complex I was measured using 0.5 μg/mL complex I in 20 mM Tris-HCl (pH 7.2), 0.15% 3-((3-cholamidopropyl)dimethylammonium)-1-propanesulfonate (CHAPS, Merck Chemicals Ltd), and 0.15% soy bean asolectin (Avanti Polar Lipids), with 200 μM decylubiquinone (DQ, Santa Cruz Biotechnology) and 200 μM NADH, and monitored as above. IM156A was added from a DMSO stock and control experiments included DMSO.
GC-MS analysis of 13C-labeled metabolites
Cellular metabolites were extracted and analyzed by GC-MS using previously described protocols.4,30 Eμ-Myc cells (3-5 × 106 per 3.5 cm dish) were incubated for 2 hours in untreated, 100 μM phenformin, or 10 μL IM156 medium containing 10% dialyzed FBS and U-[13C]-glucose or U-[13C]-glutamine (Cambridge Isotope Laboratories). Cells were washed twice with normal saline, then lysed in ice-cold 80% methanol and sonicated. For GC-MS analysis, D-myristic acid (750 ng/sample) was added to metabolite extracts as an internal standard prior to drying samples by vacuum centrifugation with sample temperature controlled at −4°C (LabConco). Dried extracts were dissolved in 30 μL methoxyamine hydrochloride (10 mg/ml) in pyridine and derivatized as tert-butyldimethylsilyl (TBDMS) esters using 70 μL N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA). An Agilent 5975C GC-MS equipped with a DB-5MS+DG (30 m x 250 μm x 0.25 μm) capillary column (Agilent J&W, Santa Clara, CA, USA) was used for all GC-MS experiments, and data collected by electron impact set at 70 eV. A total of 1 μL of derivatized sample was injected per run in splitless mode with inlet temperature set to 280°C, using helium as a carrier gas with a flow rate of 1.5512 mL/min (rate at which D27-myristic acid elutes at 17.94 min). The quadrupole was set at 150°C and the GC/MS interface at 285°C. The oven program for all metabolite analyses started at 60°C held for 1 min, then increasing at a rate of 10°C/min until 320°C. Bake-out was at 320°C for 10 min. Sample data were acquired in scan mode (1–600 m/z). Mass isotopomer distribution for TCA cycle intermediates was determined using a custom algorithm developed at McGill University54. After correction for natural 13C abundances, a comparison was made between non-labeled (12C) and 13C-labeled abundances for each metabolite. Metabolite abundance was expressed relative to the internal standard (D-myristic acid) and normalized to cell number.
Immunoblotting and Quantitative Real-Time PCR
Lymphoma cell lines were subjected to SDS-PAGE and immunoblotting using CHAPS and AMPK lysis buffers as previously described.55 Primary antibodies against β-actin, 4EBP (total, phospho-T36/47), rS6 (total and p S235/236), Raptor (total and pS792), AMPKα (total and phospho-T172), and Caspase-3 were obtained from Cell Signaling Technology (Danvers, MA). Primary antibody against LKB1 (Ley 37D/G6) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). HRP-conjugated secondary antibodies from Cell Signaling Technologies were used to detect primary antibodies, and immobilized proteins detected by chemiluminescence using Pearce ECL Substrate (ThermoFisher Scientific). For qPCR quantification of mature miRNAs, Qiazol was used to isolate RNA, miRNEasy Mini kit was used to purify miRNAs and total mRNA, and cDNA was synthesized using the miScript II RT kit (QIAGEN). Quantitative PCR was performed using the SensiFAST SYBR Hi-ROX kit (Bioline) and an AriaMX Real Time PCR system (Agilent Technologies). miScript primer assays (QIAGEN) were used to detect mature miRNAs of the miR-17∼92 cluster, with miRNA expression normalized relative to U6 RNA levels.
Tumor xenograft assays
Lymphoma cells were resuspended in HBSS at a concentration of 5 × 106 cells/mL, and 106 cells/200 μL were injected intravenously into CD-1 nude mice (Charles River). Water bottles carrying 1.2% sucralose, 0.9 mg/mL phenformin +1.2% sucralose, or 0.8 mg/mL IM156 + 1.2% sucralose were provided for ad lib consumption.
Quantification and Statistical Analysis
Statistics were determined using paired Student’s t test, ANOVA, or Log-rank (Mantel–Cox) using Prism software (GraphPad) unless otherwise stated. Data are calculated as the mean ± SEM for biological triplicates and the mean ± SD for technical replicates unless otherwise stated. Statistical significance is represented in figures by: ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001.
Additional Resources
Spreadsheet (to assist in converting OCAR and ECAR Seahorse data into glycolytic and oxidative ATP production rates based on protocols developed by Mookerjee and Brand50): https://russelljoneslab.vai.org/tools.
Acknowledgments
We thank Dr. Judy Hirst for her collaboration with ImmunoMet Therapeutics and guidance of the experiments. We thank members of the Jones laboratory and colleagues at ImmunoMet Therapeutics for advice regarding the manuscript. We acknowledge Drs. Ralph DeBerardinis and Navdeep Chanel for providing essential reagents. We acknowledge technical assistance from the Metabolomics and Flow Cytometry Core facilities at the VAI and McGill/GCRC. The GCRC Metabolomics Core Facility is supported by grants from the Canadian Foundation for Innovation (CFI), Canadian Institutes of Health Research (CIHR), and the Terry Fox Research Institute (TFRI). We acknowledge salary support from the McGill Integrated Cancer Research Training Program (to S.I.), the Fonds de la Recherche du Québec – Santé (FRQS; to S.I. and T.F.D.), and the CIHR (to R.G.J.). This research has been supported by grants from the CIHR (MOP-142259, to R.G.J., and MOP-123352, to T.F.D.) and funding from ImmunoMet Therapeutics. In addition, this work was supported by the Medical Research Council (MC_UU_00015/2).
Author Contributions
S.I., A.G., I.K., H.R.B., A.O.D., L.M.D., S.M.K.-G., G.B., T.F.D., M.N.P., and R.G.J. created the experimental design and executed it. R.C.L., M.D.M., N.A.J., and S.Y. provided the essential reagents. S.I., R.D.S., and R.G.J. conducted the bioinformatics and data analysis. S.I., A.G., A.O.D., G.B., D.A., R.D.S., T.F.D., I.K., H.R.B., and R.G.J. performed the data interpretation. S.I., K.S.W., M.S.R., S.Y., M.N.P., and R.G.J. wrote and edited the manuscript.
Declaration of Interests
R.G.J. and M.N.P. serve on the Scientific Advisory Board of ImmunoMet Therapeutics. M.S.R. is a consultant to ImmunoMet Therapeutics. S.Y. is an employee of ImmunoMet Therapeutics. R.G.J. and ImmunoMet Therapeutics hold a provisional patent for the use of IM156 for treatment of malignancy based on miR-17∼92, Myc, or LKB1 expression.
Published: May 19, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100014.
Supplemental Information
References
- 1.Stine Z.E., Walton Z.E., Altman B.J., Hsieh A.L., Dang C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBerardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Momcilovic M., Shackelford D.B. Targeting LKB1 in cancer - exposing and exploiting vulnerabilities. Br. J. Cancer. 2015;113:574–584. doi: 10.1038/bjc.2015.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faubert B., Vincent E.E., Griss T., Samborska B., Izreig S., Svensson R.U., Mamer O.A., Avizonis D., Shackelford D.B., Shaw R.J., Jones R.G. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker S.J., Svensson R.U., Divakaruni A.S., Lefebvre A.E., Murphy A.N., Shaw R.J., Metallo C.M. LKB1 promotes metabolic flexibility in response to energy stress. Metab. Eng. 2017;43(Pt B):208–217. doi: 10.1016/j.ymben.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Algire C., Amrein L., Bazile M., David S., Zakikhani M., Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30:1174–1182. doi: 10.1038/onc.2010.483. [DOI] [PubMed] [Google Scholar]
- 7.Shackelford D.B., Shaw R.J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shackelford D.B., Abt E., Gerken L., Vasquez D.S., Seki A., Leblanc M., Wei L., Fishbein M.C., Czernin J., Mischel P.S., Shaw R.J. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., DePinho R.A., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molina J.R., Sun Y., Protopopova M., Gera S., Bandi M., Bristow C., McAfoos T., Morlacchi P., Ackroyd J., Agip A.A. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018;24:1036–1046. doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- 11.Jhas B., Sriskanthadevan S., Skrtic M., Sukhai M.A., Voisin V., Jitkova Y., Gronda M., Hurren R., Laister R.C., Bader G.D. Metabolic adaptation to chronic inhibition of mitochondrial protein synthesis in acute myeloid leukemia cells. PLoS One. 2013;8:e58367. doi: 10.1371/journal.pone.0058367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baccelli I., Gareau Y., Lehnertz B., Gingras S., Spinella J.F., Corneau S., Mayotte N., Girard S., Frechette M., Blouin-Chagnon V. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell. 2019;36:84–99.e8. doi: 10.1016/j.ccell.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Ishizawa J., Zarabi S.F., Davis R.E., Halgas O., Nii T., Jitkova Y., Zhao R., St-Germain J., Heese L.E., Egan G. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell. 2019;35:721–737.e9. doi: 10.1016/j.ccell.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrzejewski S., Gravel S.-P., Pollak M., St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges H.R., Jones A.J.Y., Pollak M.N., Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollak M. Overcoming Drug Development Bottlenecks With Repurposing: Repurposing Biguanides to Target Energy Metabolism for Cancer Treatment. Nat. Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 18.Evans J.M., Donnelly L.A., Emslie-Smith A.M., Alessi D.R., Morris A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bensimon L., Yin H., Suissa S., Pollak M.N., Azoulay L. The use of metformin in patients with prostate cancer and the risk of death. Cancer Epidemiol. Biomarkers Prev. 2014;23:2111–2118. doi: 10.1158/1055-9965.EPI-14-0056. [DOI] [PubMed] [Google Scholar]
- 20.Suissa S., Azoulay L. Metformin and the risk of cancer: time-related biases in observational studies. Diabetes Care. 2012;35:2665–2673. doi: 10.2337/dc12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kordes S., Pollak M.N., Zwinderman A.H., Mathôt R.A., Weterman M.J., Beeker A., Punt C.J., Richel D.J., Wilmink J.W. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 22.Arrieta O., Barrón F., Padilla M.S., Avilés-Salas A., Ramírez-Tirado L.A., Arguelles Jiménez M.J., Vergara E., Zatarain-Barrón Z.L., Hernández-Pedro N., Cardona A.F. Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.2019.2553. Published online September 5, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birsoy K., Possemato R., Lorbeer F.K., Bayraktar E.C., Thiru P., Yucel B., Wang T., Chen W.W., Clish C.B., Sabatini D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buzzai M., Jones R.G., Amaravadi R.K., Lum J.J., DeBerardinis R.J., Zhao F., Viollet B., Thompson C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 25.Dupuy F., Griss T., Blagih J., Bridon G., Avizonis D., Ling C., Dong Z., Siwak D.R., Annis M.G., Mills G.B. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 2013;1:18. doi: 10.1186/2049-3002-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Momcilovic M., Jones A., Bailey S.T., Waldmann C.M., Li R., Lee J.T., Abdelhady G., Gomez A., Holloway T., Schmid E. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. 2019;575:380–384. doi: 10.1038/s41586-019-1715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vara-Ciruelos D., Dandapani M., Russell F.M., Grzes K.M., Atrih A., Foretz M., Viollet B., Lamont D.J., Cantrell D.A., Hardie D.G. Phenformin, But Not Metformin, Delays Development of T Cell Acute Lymphoblastic Leukemia/Lymphoma via Cell-Autonomous AMPK Activation. Cell Rep. 2019;27:690–698.e4. doi: 10.1016/j.celrep.2019.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheaton W.W., Weinberg S.E., Hamanaka R.B., Soberanes S., Sullivan L.B., Anso E., Glasauer A., Dufour E., Mutlu G.M., Budigner G.S., Chandel N.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishton R.J., Barnes C.E., Nichols A.G., Cohen S., Gerriets V.A., Siska P.J., Macintyre A.N., Goraksha-Hicks P., de Cubas A.A., Liu T. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016;23:649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izreig S., Samborska B., Johnson R.M., Sergushichev A., Ma E.H., Lussier C., Loginicheva E., Donayo A.O., Poffenberger M.C., Sagan S.M. The miR-17∼92 microRNA Cluster Is a Global Regulator of Tumor Metabolism. Cell Rep. 2016;16:1915–1928. doi: 10.1016/j.celrep.2016.07.036. [DOI] [PubMed] [Google Scholar]
- 31.Pollak M. Potential applications for biguanides in oncology. J. Clin. Invest. 2013;123:3693–3700. doi: 10.1172/JCI67232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mu P., Han Y.C., Betel D., Yao E., Squatrito M., Ogrodowski P., de Stanchina E., D’Andrea A., Sander C., Ventura A. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ota A., Tagawa H., Karnan S., Tsuzuki S., Karpas A., Kira S., Yoshida Y., Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 34.He L., Thomson J.M., Hemann M.T., Hernando-Monge E., Mu D., Goodson S., Powers S., Cordon-Cardo C., Lowe S.W., Hannon G.J., Hammond S.M. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griss T., Vincent E.E., Egnatchik R., Chen J., Ma E.H., Faubert B., Viollet B., DeBerardinis R.J., Jones R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015;13:e1002309. doi: 10.1371/journal.pbio.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janzer A., German N.J., Gonzalez-Herrera K.N., Asara J.M., Haigis M.C., Struhl K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:10574–10579. doi: 10.1073/pnas.1409844111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang C.V., Reddy E.P., Shokat K.M., Soucek L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer. 2017;17:502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gui D.Y., Sullivan L.B., Luengo A., Hosios A.M., Bush L.N., Gitego N., Davidson S.M., Freinkman E., Thomas C.J., Vander Heiden M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016;24:716–727. doi: 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segal E.D., Yasmeen A., Beauchamp M.C., Rosenblatt J., Pollak M., Gotlieb W.H. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem. Biophys. Res. Commun. 2011;414:694–699. doi: 10.1016/j.bbrc.2011.09.134. [DOI] [PubMed] [Google Scholar]
- 42.Appleyard M.V.C.L., Murray K.E., Coates P.J., Wullschleger S., Bray S.E., Kernohan N.M., Fleming S., Alessi D.R., Thompson A.M. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br. J. Cancer. 2012;106:1117–1122. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwong S.C., Brubacher J. Phenformin and lactic acidosis: a case report and review. J. Emerg. Med. 1998;16:881–886. doi: 10.1016/s0736-4679(98)00103-6. [DOI] [PubMed] [Google Scholar]
- 44.Choi J., Lee J.H., Koh I., Shim J.K., Park J., Jeon J.Y., Yun M., Kim S.H., Yook J.I., Kim E.H. Inhibiting stemness and invasive properties of glioblastoma tumorsphere by combined treatment with temozolomide and a newly designed biguanide (HL156A) Oncotarget. 2016;7:65643–65659. doi: 10.18632/oncotarget.11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamin D., Robay D., Hindupur S.K., Pohlmann J., Colombi M., El-Shemerly M.Y., Maira S.M., Moroni C., Lane H.A., Hall M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018;25:3047–3058.e4. doi: 10.1016/j.celrep.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renner K., Seilbeck A., Kauer N., Ugele I., Siska P.J., Brummer C., Bruss C., Decking S.M., Fante M., Schmidt A. Combined metabolic targeting with metformin and the NSAIDs diflunisal and diclofenac induces apoptosis in acute myeloid leukemia cells. Front. Pharmacol. 2018;9:1258. doi: 10.3389/fphar.2018.01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Momcilovic M., McMickle R., Abt E., Seki A., Simko S.A., Magyar C., Stout D.B., Fishbein M.C., Walser T.C., Dubinett S.M., Shackelford D.B. Heightening Energetic Stress Selectively Targets LKB1-Deficient Non-Small Cell Lung Cancers. Cancer Res. 2015;75:4910–4922. doi: 10.1158/0008-5472.CAN-15-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ju K.D., Kim H.J., Tsogbadrakh B., Lee J., Ryu H., Cho E.J., Hwang Y.H., Kim K., Yang J., Ahn C., Oh K.H. HL156A, a novel AMP-activated protein kinase activator, is protective against peritoneal fibrosis in an in vivo and in vitro model of peritoneal fibrosis. Am. J. Physiol. Renal Physiol. 2016;310:F342–F350. doi: 10.1152/ajprenal.00204.2015. [DOI] [PubMed] [Google Scholar]
- 49.Ma E.H., Verway M.J., Johnson R.M., Roy D.G., Steadman M., Hayes S., Williams K.S., Sheldon R.D., Samborska B., Kosinski P.A. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity. 2019;51:856–870.e5. doi: 10.1016/j.immuni.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Mookerjee S.A., Gerencser A.A., Nicholls D.G., Brand M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017;292:7189–7207. doi: 10.1074/jbc.M116.774471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharpley M.S.S., Shannon R.J.J., Draghi F., Hirst J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- 52.Fedor J.G.G., Jones A.J.Y., Di Luca A., Kaila V.R.I., Hirst J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. USA. 2017;114:12737–12742. doi: 10.1073/pnas.1714074114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones A.J.Y.J.Y., Hirst J. A spectrophotometric coupled enzyme assay to measure the activity of succinate dehydrogenase. Anal. Biochem. 2013;442:19–23. doi: 10.1016/j.ab.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGuirk S., Gravel S.P., Deblois G., Papadopoli D.J., Faubert B., Wegner A., Hiller K., Avizonis D., Akavia U.D., Jones R.G. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. 2013;1:22. doi: 10.1186/2049-3002-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faubert B., Boily G., Izreig S., Griss T., Samborska B., Dong Z., Dupuy F., Chambers C., Fuerth B.J., Viollet B. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are available in the main text or the supplementary materials. A spreadsheet to assist in converting OCAR and ECAR Seahorse data into oxidative and glycolytic ATP production rates is available at https://russelljoneslab.vai.org/tools. This study did not generate any code.