

Abstract
The elegant developmental biology experiments conducted in the 1940s by French physiologist Alfred Jost demonstrated that the sexual phenotype of a mammalian embryo depended whether the embryonic gonad develops into a testis or not. In humans, anomalies in the processes that regulate development of chromosomal, gonadal or anatomic sex result in a spectrum of conditions termed Disorders/Differences of Sex Development (DSD). Each of these conditions is rare, and understanding of their genetic etiology is still incomplete. Historically, DSD diagnoses have been difficult to establish due to the lack of standardization of anatomical and endocrine phenotyping procedures as well as genetic testing. Yet, a definitive diagnosis is critical for optimal management of the medical and psychosocial challenges associated with DSD conditions.
The advent in the clinical realm of next-generation sequencing methods, with constantly decreasing price and turnaround time, has revolutionized the diagnostic process. Here we review the successes and limitations of the genetic methods currently available for DSD diagnosis, including Sanger sequencing, karyotyping, exome sequencing and chromosomal microarrays. While exome sequencing provides higher diagnostic rates, many patients still remain undiagnosed. Newer approaches, such as whole-genome sequencing and whole-genome mapping, along with gene expression studies, have the potential to identify novel DSD-causing genes and significantly increase total diagnostic yield, hopefully shortening the patient’s journey to an accurate diagnosis and enhancing health-related quality-of-life outcomes for patients and families.
Keywords: DSD, testis development, exome, whole genome sequencing, genome mapping
Sex development
One of the most fascinating developmental processes in biology is sex development – transformation of the bipotential gonads into fully functioning testes or ovaries. In males, testicular development is initiated with the upregulation of SRY in somatic cells of undifferentiated gonads [1–3]. Expression of SRY and NR5A1 activates a cascade of genetic events, including upregulation of SOX9 in Sertoli cells leading to tubular cord formation and testicular organogenesis [4, 5]. In the absence of SRY, female-specific pathways involving genes such as RSPO1, WNT4, or NR0B1 are initiated, promoting development of the ovaries [6–8].
The developed testes or ovaries secrete hormones needed for the differentiation of the fetus into a male or a female. Hormones produced by the testes play an essential role in development of male internal (epididymis, vas deferens, seminal vesicles) and external (penis, scrotum) genitalia. Production of anti-Müllerian hormone (AMH) by Sertoli cells stimulates regression of the Müllerian structures, whereas production of testosterone and insulin-like peptide 3 by Leydig cells promote development of male internal structures and testicular descent respectively. Conversion of testosterone to dihydrotestosterone (DHT) by 5α-reductase promotes the development of the penis, scrotum and prostate. Lack of AMH and testosterone production in females allows for the continuous development of Müllerian ducts (uterus, Fallopian tubes, and upper third of vagina) and regression of Wolffian ducts is promoted by COUP-TFII [9]. Virilization of external genitalia does not occur due to absence of high levels of testosterone and DHT.
Abnormalities in sex developmental pathways may lead to Disorders/differences of Sex Development (DSD), which were defined in the 2006 Consensus Statement on Management of Intersex Disorders as “congenital conditions in which the development of chromosomal, gonadal or anatomic sex is atypical” [10]. Many of the genes identified to be important in sex development were found to be mutated in patients diagnosed with DSD. The significance of studying these disorders is not only biological, as it increases our understanding of sex differences, mechanisms of reproduction and fertility, but also human as we start to understand the complexities of societal and medical implications of human chronic conditions affecting sexual organs.
Challenges of DSD diagnosis and management
DSDs are a spectrum of chronic conditions collectively thought to affect approximately 0.5% of the population [11]. However, the incidences of discrete DSD (e.g. androgen insensitivity syndrome, 5-alpha reductase deficiency, ovotesticular DSD) are low, classifying each as “rare disease” (for a detailed description and classification of DSD conditions by etiology see [12]. Evidence-based clinical care in DSD has been hampered by a fragmented research agenda, leaving fundamental gaps in knowledge of DSD pathology. Links between treatment options and desired outcomes are poorly understood. Research is disadvantaged by small sample sizes and lack of standardization in descriptions of phenotype and medical/surgical interventions [13]. Studies examining the relationship between molecular diagnosis, gender development, and psychosocial trajectories reveal substantial unexplained variance in outcomes. The limited base of evidence is associated with significant variation in diagnostic and treatment practices within and across medical, surgical, and behavioral aspects of health, as evidenced in a recent survey conducted by the DSD-Translational Research Network (DSD-TRN) [14].
As a result, clinical management in DSD is in a state of flux with disagreements within and between healthcare providers, advocacy, and patient communities regarding what constitutes optimal care [15]. Evidence of these controversies exist in the medical literature [16], social media (e.g. http://oiiinternational.com), deliberations of human rights organizations [17, 18], the US Department of State [19] and courts of law [20]. Management of DSD is exceptionally difficult as the conditions may be associated with increased infertility, cancer, and gender dysphoria risks, as well as pervasive challenges to psychosocial adaptation for patients and families [21–25].
The emerging paradigm for clinical management recognizes the strength of focusing on an early diagnosis as well as long-term health-related quality-of-life outcomes, with patient-centered decision-making jointly by clinical teams and families. In the past few years, genetic diagnosis has made leaps with demonstrated increased diagnostic rates using exome sequencing [26] or targeted DSD panels [27–30]. With cost of next-generation sequencing decreasing and turnaround times for clinical testing now similar to those of some endocrine tests (e.g. down to 1 week for urgent exome cases at the UCLA Clinical Genomic Center; less for panels), genomic testing as a priority for DSD diagnostic clinical practice is becoming a reality [31] and is recommended by the DSD-Translational Research Network [32]. In spite of these recent advances, a large proportion of patients do not receive a firm genetic diagnosis. Emerging technologies such as whole-genome sequencing and whole-genome mapping are poised to increase diagnostic yield.
Genetics of disorders/differences of sex development
Known genetic causes of DSD include sex chromosomes aneuploidies (Turner and Klinefelter syndromes and their variants), copy number variants (CNVs) affecting open reading frames or regulatory elements (promoters/enhancers) upstream of critical dosage-sensitive genes such as SOX9, SOX3, or NR0B1 (reviewed in [33]), as well as single nucleotide variants (SNVs) in at least 75 genes involved in gonadal developmental processes and/or sex hormone biosynthesis or action [2, 3, 32, 34–37]. For the most difficult to diagnose DSD conditions, such as the non-syndromic forms of 46,XY gonadal dysgenesis, evaluation of historical data suggests that approximately 15% of are due to SRY defects [38–40], 13% to mutations in SF1/NR5A1 [41], and a few cases due to other rare genetic events (SOX9, DAX1/NR0B1,…). More recent data suggest that MAP3K1 variants may explain an additional 10-18% [26, 42, 43], still leaving nearly half of cases without a genetic diagnosis. In 46,XX individuals, the majority (80-90%) of non-syndromic Testicular DSD are explained by SRY translocations; however, only ~10% of Ovotesticular DSD have detectable Y material [44]. Recently, many genes coding for oocyte-specific transcription factors (e.g. FOXL2), folliculogenesis growth factors (e.g. BMP15), proteins that control ovarian steroidogenesis (e.g. receptor for FSH) or proteins involved in DNA replication or integrity (e.g. STAG3) have also been shown to underlie ovarian dysgenesis or premature ovarian failure, though each explain only a minority of cases and many remain unexplained [45–47].
In spite of close to 30 years of intensive research since the seminal discovery of SRY [1, 48–51] as a master sex-determining gene, the majority of patients do not receive an accurate diagnosis. This may be due in part to imperfect access to genetic testing in the clinical setting: we recently showed that 97% of undiagnosed patients in the US-based DSD-TRN registry have not exhausted clinically available diagnostic testing [32]. There are, however, likely additional genetic etiologies to be uncovered, especially in non-coding parts of the genome.
In this review, we focus on the genomic technologies used to achieve genetic diagnosis in DSD, how those technologies have helped to identify novel genes associated with DSD pathogenesis, as well as developing high-throughput technologies that could be incorporated into genetic diagnostic practice.
Chromosomal microarrays
Chromosomal microarrays (CMA) have advanced the field of genetic diagnosis as the technology provides high-resolution scanning of the entire genome. Compared with classical karyotyping techniques CMA achieves much higher precision in regard to identification of a gain or loss of genetic material. The main types of CMAs used for genetic testing are Comparative Genomic Hybridization (CGH) and Single Nucleotide Polymorphism (SNP) array. In array CGH, genomic DNA of two samples (test and control) labeled with different fluorescent tags are used to hybridize to an array of cloned genomic DNA fragments of varying sizes. The comparison of the intensities of the two fluorescent labels between test and control samples allows identification of copy-number variants (CNVs). In contrast, SNP arrays rely on identification of genetic variation via comparison of relative intensities obtained from the hybridization of a single fluorescently labeled DNA to an array of synthetic oligonucleotides to those of a pool of control samples. Microarray manufacturers have combined the two technologies to be able to not only identify gain or loss of genetic material, but also identify regions of heterozygosity and homozygosity [52].
Identification of genetic changes in genomic regions containing genes associated with DSD can help researchers pinpoint the cause or facilitate the discovery of novel genes involved in sex development. For example, deletions of NR0B1 are associated with Adrenal Hypoplasia Congenita and Hypogonadotropic Hypogonadism [53] whereas duplications cause complete gonadal dysgenesis with a female phenotype in 46,XY individuals [54]. CMA can robustly identify deletions/duplications on the X chromosome involving the NR0B1 gene, and for some patients this may lead to identification of a definitive diagnosis [55, 56]. Genetic changes identified by chromosomal microarrays can validate genes previously only identified in animal models. For example, overexpression of Sox3 in gonads of XX transgenic mice was shown to cause genetic female mice to develop as phenotypic males [57]. Screening of human SRY-negative XX males via CMA identified SOX3 duplications in several patients [57–59] suggesting that ectopic expression of the SOX3 gene in the developing gonads is sufficient to drive the male sex determination pathway.
CMAs are widely used in clinic for genetic diagnosis. In DSD, approximately 15% of the cases can be explained by the presence of CNVs (published reports of use of CMA to diagnose DSD were recently reviewed in [33]). Higher rates of diagnosis may be seen in syndromic versus isolated forms of 46,XY gonadal dysgenesis (25% vs 5.6%, respectively) [55]. In a large cohort of patients with hypospadias, cryptorchidism or ambiguous genitalia potentially clinically significant CNVs were identified in 17% of isolated and syndromic cases of hypospadias, 18% of isolated and 30% of syndromic cases of cryptorchidism, and 28% isolated and 16% syndromic cases with ambiguous genitalia [60]. Early analysis of DSD-TRN registry data showed that CMA was performed in 30% of patients, with a 14% diagnostic rate, identifying CNVs in the Y chromosome or of an entire chromosome (in a case of Klinefelter syndrome), loss of the entire Androgen Receptor gene, and one case of WAGR syndrome [32].
Routine use of CMA analysis in clinic or research could aid in identification of novel genetic variations outside of coding regions of the genome that could potentially be important, previously unrecognized, regulatory sequences for known DSD genes. For example, deletions upstream of the SOX9 gene have been identified to be causative for 46,XY gonadal dysgenesis [56, 61] whereas duplications have been associated with 46,XX Testicular and Ovotesticular DSD [62–64]. To date, microarrays have identified dozens of CNVs that may cause DSD, awaiting future research for validation [65].
Single gene testing and gene panels
Although CMA is an effective diagnostic technique for detecting chromosomal abnormalities with higher precision than traditional karyotype, it is ineffective for detection of small genetic variations. (Currently, clinical reports in the United States typically only include CNVs above 50 kb). Serial gene sequencing has been the historical method of choice, where physicians collect additional phenotypic and metabolic information via imaging and endocrine tests to inform their selection of candidate genes to be tested to identify potential mutations explaining the patient’s phenotype. Single gene testing is usually performed using Sanger sequencing, which utilizes fluorescently labeled chain-terminating dideoxynucleotides for DNA synthesis and can provide base pair resolution of fragments of up to 1000 base pairs (bp).
This practice has proven to be inefficient as phenotypes associated with DSD frequently overlap and phenotype/genotype correlations are still poor, making the candidate gene approach risky. While AR and 5ARD2 were the two most common causative genes identified by single gene testing (15 and 5 cases respectively), they also with equal frequency did not yield a genetic diagnosis (11 and 6 respectively) [32].
Single gene sequencing has gradually evolved into gene panels, which are now widely used in clinical practice for genetic diagnosis. Many disease-specific gene panels have been developed with varying number of genes tested. Although Sanger sequencing can be used for panels with a limited number of genes, panel sequencing is often accomplished by capture of targeted exons from genomic DNA using biotinylated oligonucleotide probes followed by massively parallel/next-generation sequencing. Additional Sanger sequencing (“fill in”) may be performed for genes with missing or insufficient read depth coverage to detect heterozygous variants.
Comprehensive gene panels have been designed that report high rate of diagnosis for 46,XY DSD (43% using 64 genes [27]; 45% using 56 genes [30]), as well as time-efficiency (3 days vs. 6 years to diagnosis on average) and cost-efficiency (1/3 of price) [30]. Diagnosis rates are typically lower if undiagnosed 46,XX DSD cases are included (e.g 29.5% of likely pathogenic or pathogenic variants found using a 67-gene panel [29]). Another panel of 80 genes demonstrated the superiority of the approach over single-gene testing performed on the same patient samples (28% vs. 10%) [28]. Because such targeted sequencing provides high read depth for a set of genes, it allows for detection of non-reference alleles present at very low frequencies. Such panels could therefore be useful to detect mosaics that have been hypothesized to underlie the variable phenotypic expression of DSD. However, these panels are not standardized (different providers include different genes for sequence analysis) and may not be available clinically. As these panels test a set number of genes, a limitation is their programmed obsolescence in the case of conditions with incomplete genetic understanding such as DSD, and such an approach requires re-testing as novel gene-disease associations are uncovered.
Exome sequencing
While Sanger sequencing is widely used for sequencing of DNA fragments smaller than 1 kbp in size, the emergence of massively parallel sequencing now dominates the global market. This is primarily achieved by the improvements made in imaging, microengineering, and informatics techniques that enable cheap sequencing prices and fast turnaround times. The major platform providing NGS data utilizes flowcells covered with millions of surface-bound oligonucleotides that allow attachment and parallel sequencing of hundreds of millions of independent short fragments (100-300 bp); sequences are then aligned to a reference genome for variant discovery.
The utilization of exome sequencing has transformed the clinical genetic diagnostic capabilities for undiagnosed rare disorders. Exome sequencing covers approximately 95% of the RefSeq protein-coding regions of the genome, which currently harbor 80-90% of known disease-causing variants [66]. The sequencing method is similar to those used in gene panels, exon capture followed by NGS and validation of low quality mutations via Sanger. The primary advantage of exome sequencing is its ability to provide the sequence information of all known protein coding regions of the genome, which are analyzed simultaneously for variant identification. In addition to looking for mutations in known genes involved in sex development, it has the potential for discovery of novel DSD genes. (It has been estimated that, to date, research exome sequencing has allowed for the identification of ~160 new disease genes each year [67]). Since all of the exons are sequenced, data can easily be reanalyzed as new gene associations are reported, a practice that was shown to increase diagnostic rate [68].
Exome sequencing identifies 21,000 variants on average in each case when compared to reference human genome [69]. For rare diseases such as DSD, variants with global alternate allele frequency of higher than 1% are excluded from downstream analysis as being population polymorphisms and thus likely benign. However, with increasing numbers of sequenced healthy control samples in public datasets (such as ExAC/gnomAD [70]), researchers and clinicians increasingly focus on not only global allele frequencies, but also on population-specific frequencies to accurately determine the variant frequency in the patient’s own ethic background. Limited representativity of certain ethnic backgrounds in reference databases is a clear cause of variant calling error and a severe limitation of the use of genetic diagnosis in underserved populations (e.g. [71]). It is most effective to perform exome sequencing of the patient as part of a trio, with phenotyped parents allowing to identify inherited and de novo variants. This dramatically reduces the number of variants to manually curate, thus reducing time to diagnosis [72] and increased reported diagnostic capabilities by almost 50% (from 22% to 31%) when compared to proband-only cases [69].
Limitations in exome sequencing diagnostic efficacy include incomplete gene coverage, platform variability, difficulties of variant interpretation, and ethical issues linked to incidental findings. Coverage of exonic sequences is not complete and some regions are poorly detected. It has been estimated that ~400 known exonic disease-causing variants (reported in the Human Genome Mutation Database) are not detected by current clinical exome methods [73]. This can be improved with enrichment kits and is likely to improve with future technological advances. For example, coverage for DSD genes dramatically improved in 2015 with the v.3 iteration of exon capture for the Illumina platform. Previously, only half of the set of known DSD-related genes had 100% coverage, with particularly poor capture for AMH (60%) and SOX3 (78%). Now 83% of the main 78 genes are covered at 100%, with another seven with at least 97% [32].
The characteristic short reads of next-generation sequencing may result in alignment difficulties, leading to false-positive and false-negative variant calling, which is compounded when errors are found in the reference genome. A recent study warned that “special caution is needed in variant calling” in about 12% of all coding exons [73]. Short-read sequencing coupled with indexing of long DNA fragments may alleviate some of these difficulties by tracking the locations of the short reads and bioinformatically correcting the mapping positions. On the other hand, novel long-read sequencing technologies, such as those developed by Pacific Biosciences (PacBio) or Oxford Nanopore Technologies, allow for an unbiased view of individual DNA molecules reaching hundreds of kilo bases in length for de novo genome assembly and variant calling.
Once variants have been identified, they are typically filtered via utilization of disease-specific gene lists containing previously published genes associated with the disease. The variants are then classified into pathogenic (previously reported in humans as the recognized cause of the disorder) or likely pathogenic (previously unreported in a known human DSD gene and of a type expected to cause the disorder) following American College of Medical Genetics and Genomics (ACMG) guidelines [74]. In addition, variants of unknown significance (VUS) in non-clinical genes with a known function related to the condition of the patient may be reported with the expectation that these might help guide the ordering physician’s further imaging and endocrine exploration.
The interpretation of variants is the most labor-intensive (an estimated 30 minutes per variant by manual curation [72]) part of the exome process. It is also possibly the most variability-prone because it relies on knowledge of the human interpreter. The ability of exome sequencing to provide an accurate genetic diagnosis heavily relies on the existence of current and accurate gene/phenotype data sets, such as those curated by OMIM (omim.org) or Orphanet (orpha.net), or the repositories of human variants such as ClinVar (ncbi.nlm.nih.gov/clinvar) or HGMD (hgmd.cf.ac.uk). Use of in silico filtering of variants using data sets reduces analysis time and can help standardize the process. A recent study comparing existing data sets for this purpose showed that highest accuracy was obtained by using data sets curated by the Human Phenome Ontology (HPO) that uses information from both the OMIM and Orphanet databases to generate custom disease-specific HPO-standardized phenotype-based data sets [72]. This automated variant calling however is limited to genes known to be associated with the patient’s condition and doesn’t exploit the gene discovery capacity of exome sequencing. To optimize variant calling as well as gene discovery, the Clinical Genomic Center at UCLA, one of the first US academic centers to offer clinical exome sequencing, has implemented, in addition to automated pipelines, a Genomic Data Board weekly meeting, where ordering clinicians, technicians of the sequencing laboratory, researchers, board-certified geneticists and genetic counselors meet to review identified variants and examine ethical concerns [75].
The early adoption of exome sequencing by the academic institutions in the United States has helped to identify the genetic diagnosis in approximately 30% of cases for whom the traditional molecular diagnostic methods such as single-gene testing or panels were uninformative [69, 76], a yield that has proven remarkably similar across countries, sequencing platforms, and conditions studied [77–81]. In many cases the identified genetic diagnosis provided guidance for medical treatment and management. A publication by our laboratory illustrates that, when performed early, exome sequencing is capable to not only increase the diagnostic yields in DSD, but also guide clinical management of patients [26]. In a cohort of 46,XY DSD cases, we identified pathogenic and likely pathogenic variants in 25% of patients. In many cases, we identified rare causative variants in genes that were not available for clinical testing via gene panels or single gene sequencing.
The early genetic diagnosis helped guide clinical management in several cases. Patients with male-typical levels of testosterone and female external genitalia generally suffer from some form of androgen insensitivity. In many cases these patients harbor a mutation in AR, the androgen receptor [82]; however, patients with varying degrees of virilization (partial androgen insensitivity) often do not [83]. Exome sequencing identified compound heterozygous and homozygous mutations in the HSD17B3 gene causing HSD17B3 deficiency [26]. These two cases were initially misdiagnosed in clinic as partial androgen insensitivity syndrome. Increase in production of testosterone at puberty put these patients at risk of virilization and early genetic diagnosis provided key benefits in regard to treatment options.
Fewer genes are known to be associated with 46,XX DSD compared to 46,XY DSD. Using exome sequencing, we and others identified a novel heterozygous p.Arg92Trp missense variant in the NR5A1 gene that causes isolated non-syndromic 46,XX testicular/ovotesticular DSD [84]. Typically mutations in NR5A1 are associated with 46,XY DSD, testicular dysgenesis and infertility in men. 46,XX women with NR5A1 mutations generally have primary ovarian insufficiency. Therefore NR5A1 would not have been selected as a likely candidate for testing in clinic via single gene sequencing or gene panels, resulting in a missed diagnosis. In this case exome sequencing allowed for an unbiased gene mutation analysis of all genes involved in DSD. The authors were able to show that the p.Arg92Trp mutation in the DNA-binding region of NR5A1 is associated with variable degree of testicular development in children with a 46,XX karyotype.
An ethical conundrum has been linked to the capacity of exome sequencing to identify variants in genes beyond those known to be associated with the condition for which the patient is consulting: what to do about incidental findings, those variants that may signal a life-threatening condition in the patient, may be medically actionable, but are not directly related to DSD? Different strategies have been adopted by different regulatory instances [72]. The American ACMG maintains a list of genes (currently 59) in which pathogenic variants should be sought and reported to the patient, a controversial mandate that was amended to offer the option to opt out (ACMG 2017) [85]. The Canadian College of Medical Genetics recommends against searching for these [86]. The European Society of Human genetics recommends the use of in silico panels (complete exome capture, but analysis restricted to a set of relevant genes) in the clinical setting to decrease the chance of inadvertently finding variants unrelated to the condition [87]. The Royal College of Pathologists of Australasia cautions that, in view of the ongoing debate and unlikely immediate resolution, much attention should be given to accurately informing the patient of what will and will not be disclosed [88]. There may be challenges in the context of real-life clinical settings, as it was reported that appropriate informed consent would take up to 6 hours and appropriate result delivery up to 5 hours [89].
Although it is evident that the utilization of next-generation sequencing increases the rate of DSD diagnosis there are still many cases where pathogenic mutations are not identified. For example, in our cohort of 46,XY DSD patients where diagnosis was established in approximately 1/3 of the cases, more than half of the patients contained hundreds of variants of unknown significance (VUS). While the VUS do not provide a definitive diagnosis, the mutation information can be leveraged for identification of novel genes involved in sex development.
Animal models of human DSD phenotypes can be utilized for identification of novel gene variants previously not associated with DSD. We have used B6-YPOS mice as a model for 46,XY DSD with gonadal dysgenesis and undervirilization to identify the genetic diagnosis in cases where exome sequencing had been uninformative (no pathogenic/likely pathogenic variants identified [90]. The B6-YPOS model is a strain of mice in which the presence of a Y chromosome originating from a M. domesticus Poschiavinus strain (YPOS) on a C57BL/6J (B6) background (B6-YPOS) results in disrupted testicular development and female genital phenotype [91]). B6-YPOS mice develop ovaries or ovotestes instead of testes. All B6-YPOS show delay in expression of Sry in the developing gonad [92], thereby jeopardizing the short 6-hr window when spatiotemporally appropriate levels of Sry expression and the receptive microenvironment in somatic cells on which Sry acts need to converge [93]. The use of B6-YPOS animals as a model for human DSD provides valuable information towards the identification of novel genes involved in sex development, mutations in which could lead to anomalies in gonadal development. We have identified 15 novel candidate genes for DSD by correlating abnormal gonadal gene expression between B6-YPOS and wild type males with the genes in which VUS were identified in 46,XY DSD patients [90].
Mutations identified in the novel candidate genes could potentially be causative and are awaiting validation via in vitro and in vivo models. The complexity of validation of VUS and candidate genes is compounded by the large scale of data sets generated by exome sequencing. There is a need for accelerated validation of such variants in animal, cellular or molecular models. For example, one of the candidate genes in our data set, Adamts16 (A Disintegrin And Metalloproteinase with Thrombospondin Type 1 Motif, 16) has been shown to be associated with cryptorchidism and sterility in rats after targeted disruption of Adamts16 gene [41]. We identified several mutations in Adamts16, one of which was located in the peptidase domain of the protein and was predicted to be damaging by in silico algorithms, suggesting a possible effect on the enzymatic function, but full validation of pathogenicity is incomplete.
When studying complex disorders such as DSD it is important to note that even though the mouse models can be extremely beneficial for identification of novel candidate genes for a given phenotype, they still do not provide the full spectrum of gene expression/interactions that occur during human sex development. Nevertheless, the use of animal models narrows the interpretive gap for next-generation sequencing by correlating human sequence variants with transcriptome variation in a model organism.
Whole-Genome Sequencing
The implementation of exome sequencing in clinical diagnostic practice allowed for increased diagnostic yields in almost every disease category; however, clear molecular diagnosis was identified in only approximately 30% of all cases [75, 81]. One could suspect that a substantial portion of these missed diagnoses is due to the fact that exome sequencing only captures coding regions of DNA, which accounts for a very small portion of the entire human genome.
Whole-genome sequencing (WGS) has the capability to identify single-nucleotide variants and small insertions/deletions (INDELs) with high precision not only in the exons, but also throughout the entire genome, in regions that may be involved in gene regulation. Because it doesn’t involve targeted capture, WGS has been shown to provide more uniform read depth across the genome and sequencing quality parameters than exome sequencing [94]. It is therefore more reliable than exome sequencing even for variants in the protein-coding regions of the genome. For example, a 2015 report calculated that WGS was able to detect 3% (~650) of coding variants that had been missed by exome sequencing [95]. Additionally, WGS has the potential to identify genetic aberrations larger than 1 kbp in size defined as structural variants (SV). SVs are an important category of DNA variations observed in the human genome.
WGS has now been pioneered throughout the world and reports of its clinical efficacy over existing methods are starting to be published (e.g. [96]). Its limitations are as staggering as its potential, as the daunting task of identifying relevant pathogenic variants among the millions of variants found in each genome is severely limited by our current inability to interpret the pathogenicity of most structural variants [97, 98].
NGS short reads ranging from 100-300 bp can accurately identify intergenic and intronic SNVs as well as small INDELs. However, the correct alignment of the reads to the reference genome is compromised in regions that are highly repetitive preventing the discovery of SVs. Many programs have been developed to call SV specifically from short-read NGS data using a combination of read-depth, read-pair, split-read methods [99]. The number and type of SVs identified by each of the tools vary significantly due to limitations of the methods. This leads to large variations of false discovery rates, sensitivity rates as wells as low concordance rates between tools [100, 101]. The most common method for detection of SVs in clinical practice is CMA. However, this method is primarily geared towards identification of structural variants such as insertions or deletions that result in a gain or loss of genetic material, and balanced translocations or inversions are not effectively called by CMAs.
Whole-Genome Mapping
A method that has been proposed to be useful in conjunction with WGS is Next Generation genome Mapping (NGM, developed by Bionano Genomics), which utilizes high-molecular-weight DNA for de novo genome assembly. Genome mapping relies on fluorescent labeling at specific sequence motifs of megabase size DNA fragments that allow for the construction of scaffolds into two haploid genomes of an individual. Single strand breaks are created by a nicking endonuclease, where Taq polymerase inserts fluorescent nucleotides (Fig. 1).
Fig. 1. DNA labeling for next-generation genome mapping (NGM).

The DNA-labeling workflow is divided into four consecutive steps. First, the high-molecular-weight DNA is nicked with an endonuclease of choice (Nt.BspQI or Nb.BssSI, New England BioLabs/Bionano Genomics), which introduces single-strand nicks throughout the genome. Second, Taq polymerase (NEB) recognizes these sites and replaces several nucleotides with fluorescently tagged nucleotides added to the solution. Third, the two ends of the DNA are ligated together using Taq DNA ligase (NEB). Fourth, the DNA backbone is stained with DNA Stain (Bionano Genomics). Figure originally published in Barseghyan et al. 2017, Genome Medicine 9:90. DOI 10.1186/s13073-017-0479-0. Reproduced here courtesy of publisher BioMed Central’s policy of free sharing of its open access articles.
Individual molecules of labeled DNA are then straightened and imaged in nanochannels to detect the pattern of nicking sites (Fig. 2) prior to de novo genome assembly. NGM allows detection of large insertions and deletions, inversions, translocations, as well as more complex SVs [102, 103].
Fig. 2. Chip nanochannel structure and DNA loading for next-generation genome mapping (NGM).
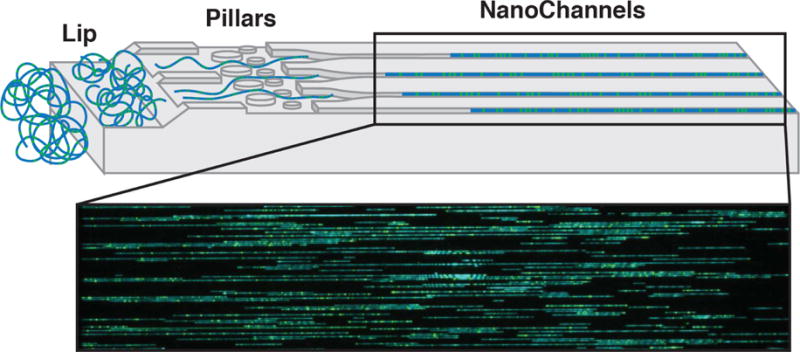
The labeled double-stranded DNA is loaded into two flow cells (Irys or Saphyr, Bionano Genomics). The applied voltage concentrates the coiled DNA at the lip (left). Later, DNA is pushed through pillars (middle) to uncoil/straighten, then into nanochannels (right). DNA is stopped and imaged in the nanochannels. Blue - staining of DNA backbone. Green - fluorescently labeled nicked sites.
Figure originally published in Barseghyan et al. 2017, Genome Medicine 9:90. DOI 10.1186/s13073-017-0479-0. Reproduced here courtesy of publisher BioMed Central’s policy of free sharing of its open access articles.
SVs are identified by differences in the alignment profiles of the de novo assembled genome maps against the publicly available human genome reference. Failure of contiguous alignment to the reference indicates the presence of putative SVs. NGM has been used by researchers for de novo genome assemblies who demonstrated that its utilization can facilitate accurate positional placement of single nucleotide sequences throughout the diploid human genome [104].
We first investigated whether we could validate the capability of NGM to be used for providing a clinical diagnosis in a cohort of patients diagnosed with Duchenne muscular dystrophy (DMD) [105]. We demonstrated that NGM could identify both heterozygous and homozygous SVs within the DMD locus ranging from 13 kbp to 5.1 Mbp. The potential of this technology to sensitively identify SVs may offer substantial advantages over CMA currently used for clinical diagnosis. In addition to SVs such as duplications, deletions or translocations resulting in a gain or loss of genetic material, NGM is capable of detecting balanced inversions and translocations as well as much smaller kilobase-size SVs. The major limitation of the NGM technology is its inability to assess base-level sequence information leading to low sensitivity for SV below 1.5 kbp size range and large, 3-5 kbp error rate in regards to true locations of SV breakpoints. Nevertheless, in comparison with NGS, NGM provides higher sensitivity for large structural variants with better false positive and false negative rates [100, 101, 106]. The turnaround times and associated costs for genome mapping are similar to genome sequencing indicating the possibility of clinical adoption.
We will now apply this NGM technology to undiagnosed DSD samples, first looking for SVs in known DSD genes. Fig. 3 shows the example of a compound heterozygous deletion identified in intron 5 of the WWOX gene in a 46,XY patient with gonadal dysgenesis. A multi-exon deletion resulting in abnormal in frame exon 5-exon 9 splicing was found in a 46,XY patient with bilateral immature testes and malignant germ cells [107]. Mouse wwox knock-out mice show gonadal abnormalities and loss of heterozygosity of WWOX has been reported to be associated with poor prognosis in ovarian and other cancers. Intron 5 of WWOX is a large intron that contains a newly described long non-coding RNA on the antisense strand, RP11-190D6.2. Expression of this RNA is testis-specific (GTEx data, gtexportal.org [108]) and its over-expression or knock-down was shown to correlate with expression of WWOX [109].
Fig. 3. Next-generation genome mapping (NGM) identifies two small deletions in intron 5 of the WWOX gene in a 46,XY DSD patient.
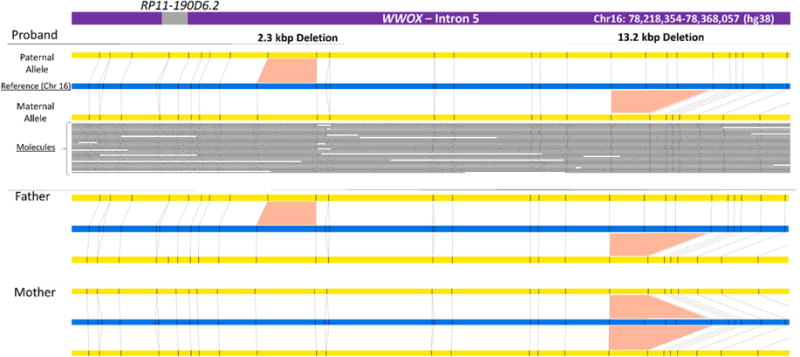
Top panel: assembly of the proband’s contigs (yellow) generated using nicking endonuclease Nb.BssSI (sites are shown as small black vertical bars), aligned to the reference genome (blue). The individual DNA molecules used to assemble the bottom allele are shown as grey lines and black dots corresponding to linearized DNA and nicking sites respectively. The genomic locations of the region displayed and of the long non-coding RNA RP11-190D6.2 (in the GRCh38/hg38 human genome assembly reference map) are shown in the top purple bar representing the WWOX gene. The middle and bottom panels show the maps of the father and mother respectively, which helped to identify parent of origin for the proband’s alleles. Similar results were obtained with a different endonuclease, Nt.BspQI (not shown). The 2.3 kbp and 13.2 kbp deletions were present in respectively 1.3% and 15% of the reference maps in the Bionano database of 150 healthy controls.
The top panel in Fig. 3 shows the contigs generated for the two alleles of the proband (in yellow) aligned to the reference (in blue) for this region of chromosome 16. The small black vertical bars in the contigs indicate the restriction sites where the DNA was nicked and fluorescently labeled. The individual molecules of various lengths used to create the contigs are shown below, with the nicking site pattern clearly visible. Deviation to this pattern signals the presence of a SV. In the proband, NGM identified two deletions (2.3 kbp and 13.2 kbp) within intron 5 upstream of RP11-190D6.2. For the 2.3 kbp deletion, the “bottom” allele is normal, showing alignment of the nicking sites to the reference, while the “top” allele shows a portion of DNA missing between two adjacent nicking sites. The 13.2 kbp deletion removes 3 restriction sites in the bottom allele that are present in the reference (blue) allele.
The technology also allowed to assign phase to the variants. Examination of the parental samples showed that the mother was homozygous and father heterozygous carrier of the larger deletion, which is present in ~15% of a control population of 150 individuals (bionanogenomics.com). The father was also a carrier of the smaller 2.3 kbp deletion (present in 1.3% of the control population). The long non-coding RNA RP11-190D6.2 is not deleted in this patient or in the published case, although its expression (and that of WWOX) may still be affected. However, in this case, the deletions are unlikely to explain the phenotype as the proband’s father (46,XY) is also compound heterozygous for the same deletions.
Long-read DNA mapping technologies allow for visualization of DNA structure that complements short-read sequencing technologies. NGM has the capacity to identify pathogenic variants related to transposons and other repetitive regions in the human genome. The interpretation of variants identified in non-coding regions is currently the biggest challenge of both genome sequencing and mapping approaches. However, as sample sizes in databases such as gnomAD, DGV and GTEx continue to increase, researchers will be able to relate genotypes and phenotype with higher precision allowing to solve larger fraction of undiagnosed genetic diseases. Integration of NGM and NGS technologies in the diagnosis of DSD, in combination with other technologies, promises to help further our understanding of how mutations in gene regulatory elements cause genetic disease.
Future directions
The developing genetic diagnostic practice is geared towards multi-platform integration analysis for identification of the underlying genetic cause. In the near future, the whole genome next-generation sequencing and mapping technologies will become widely used for genetic diagnosis. When combined, it becomes possible to survey almost all possible types of genetic variations such as SNVs, INDELs, CNV, large deletions/insertions, inversions and translocations. Implementation of these technologies early in clinic would provide greater diagnostic specificity, shorter turnaround times and guidance towards patient management. Moreover, better genotype and phenotype classification will allow for more precise risk assessments and outcome predictions.
Next-generation sequencing (NGS) increases the yield of DSD diagnosis
Mouse models can aid in variant classification
Innate methodological limitations of NGS do not allow for sensitive identification of structural variants
Next-generation mapping (NGM) accurately identifies structural variants throughout the genome
NGM coupled with whole-genome sequencing could facilitate identification of variants ranging from a single nucleotides to large structural variants
Acknowledgments
The authors were funded in part by the DSD-Translational Research Network. Funded by grant # R01 HD068138 from the Eunice Kennedy Shriver NICHD Platform for Basic and Translational Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Hayk Barseghyan, Email: hbarseghya@childrensnational.org.
Emmanuèle C. Délot, Email: edelot@childrensnational.org.
Eric Vilain, Email: evilain@childrensnational.org.
References
- 1.Koopman P, Sinclair A, Lovell-Badge R. Of sex and determination: marking 25 years of Randy, the sex-reversed mouse. Development. 2016;143(10):1633–1637. doi: 10.1242/dev.137372. [DOI] [PubMed] [Google Scholar]
- 2.Eggers S, Sinclair A. Mammalian sex determination-insights from humans and mice. Chromosome Res. 2012;20(1):215–238. doi: 10.1007/s10577-012-9274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ono M, V, Harley R. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol. 2013;9(2):79–91. doi: 10.1038/nrendo.2012.235. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Zheng M, Lau YF. The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Rep. 2014;8(3):723–733. doi: 10.1016/j.celrep.2014.06.055. [DOI] [PubMed] [Google Scholar]
- 5.Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453(7197):930–934. doi: 10.1038/nature06944. [DOI] [PubMed] [Google Scholar]
- 6.Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, Guerra L, Schedl A, Camerino G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet. 2006;38(11):1304–1309. doi: 10.1038/ng1907. [DOI] [PubMed] [Google Scholar]
- 7.Jordan BK, Mohammed M, Ching ST, Delot E, Chen XN, Dewing P, Swain A, Rao PN, Elejalde BR, Vilain E. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet. 2001;68(5):1102–1109. doi: 10.1086/320125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbaro M, Oscarson M, Schoumans J, Staaf J, Ivarsson SA, Wedell A. Isolated 46,XY gonadal dysgenesis in two sisters caused by a Xp21.2 interstitial duplication containing the DAX1 gene. J Clin Endocrinol Metab. 2007;92(8):3305–3313. doi: 10.1210/jc.2007-0505. [DOI] [PubMed] [Google Scholar]
- 9.Zhao F, Franco HL, Rodriguez KF, Brown PR, Tsai MJ, Tsai SY, Yao HH. Elimination of the male reproductive tract in the female embryo is promoted by COUP-TFII in mice. Science. 2017;357(6352):717–720. doi: 10.1126/science.aai9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee PA, C P, Houk SF, Ahmed IA, Hughes S. International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine, and E. the European Society for Paediatric, Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118(2):e488–500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- 11.Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A, Dalke KBaratz, Liao LM, Lin-Su K, Looijenga LH, 3rd, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S, D.S.D.U.C. Global Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm Res Paediatr. 2016;85(3):158–180. doi: 10.1159/000442975. [DOI] [PubMed] [Google Scholar]
- 12.Delot E, Vilain E. Disorders of Sex Development. In: Barbieri Robert, Strauss Jerome, Gargiulo Antonio., editors. Yen & Jaffe’s Reproductive Endocrinology. Elsevier; Philadelphia, PA: 2018. [Google Scholar]
- 13.Sandberg DE, Callens N, Wisniewski AB. Disorders of Sex Development (DSD): Networking and Standardization Considerations. Horm Metab Res. 2015;47(5):387–393. doi: 10.1055/s-0035-1548936. [DOI] [PubMed] [Google Scholar]
- 14.Rolston AM, Gardner M, van Leeuwen K, Mohnach L, Keegan C, Delot E, Vilain E, Sandberg DE, D.S.D.T.R.N.A. members of the, and A. Advisory Network Accord Disorders of sex development (DSD): Clinical service delivery in the United States. Am J Med Genet C Semin Med Genet. 2017;175(2):268–278. doi: 10.1002/ajmg.c.31558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adam MP, Vilain E. Emerging issues in disorders/differences of sex development (DSD) Am J Med Genet C Semin Med Genet. 2017;175(2):249–252. doi: 10.1002/ajmg.c.31564. [DOI] [PubMed] [Google Scholar]
- 16.Diamond M, Garland J. Evidence regarding cosmetic and medically unnecessary surgery on infants. J Pediatr Urol. 2014;10(1):2–6. doi: 10.1016/j.jpurol.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 17.“I Want to Be Like Nature Made Me.” Medically Unnecessary Surgeries on Intersex Children in the US. Human Rights Watch. 2017 [Google Scholar]
- 18.Méndez JE. Report of the Special Rapporteur on torture andother cruel, inhuman or degrading treatment or punishment. United Nations Human Rights. 2013 A/HRC/22/53. [Google Scholar]
- 19.Nauert H. In Recognition of Intersex Awareness Day. U.S Department of State; 2017. Press Release. [Google Scholar]
- 20.Ghorayshi A. A Landmark Lawsuit About An Intersex Baby’s Genital Surgery Just Settled For $440,000. BuzzFeed News; 2017. [Google Scholar]
- 21.Abaci A, Catli G, Berberoglu M. Gonadal malignancy risk and prophylactic gonadectomy in disorders of sexual development. J Pediatr Endocrinol Metab. 2015;28(9–10):1019–1027. doi: 10.1515/jpem-2014-0522. [DOI] [PubMed] [Google Scholar]
- 22.Arboleda VA, Sandberg DE, Vilain E. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol. 2014;10(10):603–615. doi: 10.1038/nrendo.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guercio G, Rey RA. Fertility issues in the management of patients with disorders of sex development. Endocr Dev. 2014;27:87–98. doi: 10.1159/000363633. [DOI] [PubMed] [Google Scholar]
- 24.Sandberg DE, M T. A Noncategorical Approach to the Psychosocial Care of Persons with DSD and Their Families. In: Kreukels B, Steensma T, de Vries A, editors. Gender Dysphoria and Disorders of Sex Development. Focus on Sexuality Research; 2014. [Google Scholar]
- 25.Slowikowska-Hilczer J, Hirschberg AL, Grinten HClaahsen-vander, Reisch N, Bouvattier C, Thyen U, Kettenis PCohen, Roehle R, Kohler B, Nordenstrom A, dsd LG. Fertility outcome and information on fertility issues in individuals with different forms of disorders of sex development: findings from the dsd-LIFE study. Fertil Steril. 2017;108(5):822–831. doi: 10.1016/j.fertnstert.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Baxter RM, V, Arboleda A, Lee H, Barseghyan H, Adam MP, Fechner PY, Bargman R, Keegan C, Travers S, Schelley S, Hudgins L, Mathew RP, Stalker HJ, Zori R, Gordon OK, Ramos-Platt L, Pawlikowska-Haddal A, Eskin A, Nelson SF, Delot E, Vilain E. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J Clin Endocrinol Metab. 2015;100(2):E333–344. doi: 10.1210/jc.2014-2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM, Tan TY, Cameron F, Werther G, Hutson J, O’Connell M, Grover SR, Heloury Y, Zacharin M, Bergman P, Kimber C, Brown J, Webb N, Hunter MF, Srinivasan S, Titmuss A, Verge CF, Mowat D, Smith G, Smith J, Ewans L, Shalhoub C, Crock P, Cowell C, Leong GM, Ono M, Lafferty AR, Huynh T, Visser U, Choong CS, McKenzie F, Pachter N, Thompson EM, Couper J, Baxendale A, Gecz J, Wheeler BJ, Jefferies C, MacKenzie K, Hofman P, Carter P, King RI, Krausz C, Ravenswaaij-Arts CMvan, Looijenga L, Drop S, Riedl S, Cools M, Dawson A, Juniarto AZ, Khadilkar V, Khadilkar A, Bhatia V, Dung VC, Atta I, Raza J, Chi NThiDiem, Hao TK, Harley V, Koopman P, Warne G, Faradz S, Oshlack A, Ayers KL, Sinclair AH. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016;17(1):243. doi: 10.1186/s13059-016-1105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan Y, Zhang X, Wang L, Wang R, Huang Z, Sun Y, Yao R, Huang X, Ye J, Han L, Qiu W, Zhang H, Liang L, Gu X, Yu Y. Diagnostic Application of Targeted Next-Generation Sequencing of 80 Genes Associated with Disorders of Sexual Development. Sci Rep. 2017;7:44536. doi: 10.1038/srep44536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JH, Kang E, Heo SH, Kim GH, Jang JH, Cho EH, Lee BH, Yoo HW, Choi JH. Diagnostic yield of targeted gene panel sequencing to identify the genetic etiology of disorders of sex development. Mol Cell Endocrinol. 2017;444:19–25. doi: 10.1016/j.mce.2017.01.037. [DOI] [PubMed] [Google Scholar]
- 30.Ozen S, Onay H, Atik T, Solmaz AE, Ozkinay F, Goksen D, Darcan S. Rapid Molecular Genetic Diagnosis with Next-Generation Sequencing in 46,XY Disorders of Sex Development Cases: Efficiency and Cost Assessment. Horm Res Paediatr. 2017;87(2):81–87. doi: 10.1159/000452995. [DOI] [PubMed] [Google Scholar]
- 31.Barseghyan H, Delot E, Vilain E. New genomic technologies: an aid for diagnosis of disorders of sex development. Horm Metab Res. 2015;47(5):312–320. doi: 10.1055/s-0035-1548831. [DOI] [PubMed] [Google Scholar]
- 32.Delot EC, Papp JC, Workgroup DTG, Sandberg DE, Vilain E. Genetics of Disorders of Sex Development: The DSD-TRN Experience. Endocrinol Metab Clin North Am. 2017;46(2):519–537. doi: 10.1016/j.ecl.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Croft B, Ohnesorg T, Sinclair AH. The Role of Copy Number Variants in Disorders of Sex Development. Sex Dev. 2017 doi: 10.1159/000481896. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed SF, I, Hughes A. The genetics of male undermasculinization. Clin Endocrinol (Oxf) 2002;56(1):1–18. doi: 10.1046/j.1365-2265.2002.01430.x. [DOI] [PubMed] [Google Scholar]
- 35.Arboleda VAF, A A, Vilain E. Disorders of Sex Development. In: Weiss RE, Refetoff S, editors. Genetic Diagnosis of Endocrine Disorders. 2010. pp. 227–243. ScienceDirect. [Google Scholar]
- 36.Auchus RJ, Miller WL. Defects in androgen biosynthesis causing 46,XY disorders of sexual development. Semin Reprod Med. 2012;30(5):417–426. doi: 10.1055/s-0032-1324726. [DOI] [PubMed] [Google Scholar]
- 37.Buonocore F, Achermann JC. Human sex development: targeted technologies to improve diagnosis. Genome Biol. 2016;17(1):257. doi: 10.1186/s13059-016-1128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cameron FJ, Sinclair AH. Mutations in SRY and SOX9: testis-determining genes. Hum Mutat. 1997;9(5):388–395. doi: 10.1002/(SICI)1098-1004(1997)9:5<388::AID-HUMU2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins JR. Mutational analysis of SRY in XY females. Hum Mutat. 1993;2(5):347–350. doi: 10.1002/humu.1380020504. [DOI] [PubMed] [Google Scholar]
- 40.Veitia R, Ion A, Barbaux S, Jobling MA, Souleyreau N, Ennis K, Ostrer H, Tosi M, Meo T, Chibani J, Fellous M, McElreavey K. Mutations and sequence variants in the testis-determining region of the Y chromosome in individuals with a 46,XY female phenotype. Hum Genet. 1997;99(5):648–652. doi: 10.1007/s004390050422. [DOI] [PubMed] [Google Scholar]
- 41.Ostrer H. 46,XY Disorder of Sex Development and 46,XY Complete Gonadal Dysgenesis. In: Pagon RA, et al., editors. GeneReviews(R) Seattle WA: 1993. [Google Scholar]
- 42.Pearlman A, Loke J, Le Caignec C, White S, Chin L, Friedman A, Warr N, Willan J, Brauer D, Farmer C, Brooks E, Oddoux C, Riley B, Shajahan S, Camerino G, Homfray T, Crosby AH, Couper J, David A, Greenfield A, Sinclair A, Ostrer H. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet. 2010;87(6):898–904. doi: 10.1016/j.ajhg.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Granados A, V, Alaniz I, Mohnach L, Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M, Keegan CE. MAP3K1-related gonadal dysgenesis: Six new cases and review of the literature. Am J Med Genet C Semin Med Genet. 2017;175(2):253–259. doi: 10.1002/ajmg.c.31559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delot EC, Vilain EJ. Nonsyndromic 46,XX Testicular Disorders of Sex Development. In: Adam MP, et al., editors. GeneReviews((R)) Seattle WA: 1993. [PubMed] [Google Scholar]
- 45.Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, Kaskikari R, Sankila EM, Lehvaslaiho H, Engel AR, Nieschlag E, Huhtaniemi I, de la Chapelle A. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82(6):959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- 46.Caburet S, V, Arboleda A, Llano E, Overbeek PA, Barbero JL, Oka K, Harrison W, Vaiman D, Ben-Neriah Z, Garcia-Tunon I, Fellous M, Pendas AM, Veitia RA, Vilain E. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014;370(10):943–949. doi: 10.1056/NEJMoa1309635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapman C, Cree L, Shelling AN. The genetics of premature ovarian failure: current perspectives. Int J Womens Health. 2015;7:799–810. doi: 10.2147/IJWH.S64024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berta P, Hawkins JR, Sinclair AH, Taylor A, Griffiths BL, Goodfellow PN, Fellous M. Genetic evidence equating SRY and the testis-determining factor. Nature. 1990;348(6300):448–450. doi: 10.1038/348448A0. [DOI] [PubMed] [Google Scholar]
- 49.Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351(6322):117–121. doi: 10.1038/351117a0. [DOI] [PubMed] [Google Scholar]
- 50.Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346(6281):240–244. doi: 10.1038/346240a0. [DOI] [PubMed] [Google Scholar]
- 51.Vilain E, Jaubert F, Fellous M, McElreavey K. Pathology of 46,XY pure gonadal dysgenesis: absence of testis differentiation associated with mutations in the testis-determining factor. Differentiation. 1993;52(2):151–159. doi: 10.1111/j.1432-0436.1993.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 52.Sund KL, Rehder CW. Detection and reporting of homozygosity associated with consanguinity in the clinical laboratory. Hum Hered. 2014;77(1–4):217–224. doi: 10.1159/000362448. [DOI] [PubMed] [Google Scholar]
- 53.Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, Bardoni B, Guioli S, Zehetner G, Rabl W, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372(6507):672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 54.Bardoni B, Zanaria E, Guioli S, Floridia G, Worley KC, Tonini G, Ferrante E, Chiumello G, McCabe ER, Fraccaro M, et al. A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet. 1994;7(4):497–501. doi: 10.1038/ng0894-497. [DOI] [PubMed] [Google Scholar]
- 55.Ledig S, Hiort O, Scherer G, Hoffmann M, Wolff G, Morlot S, Kuechler A, Wieacker P. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod. 2010;25(10):2637–2646. doi: 10.1093/humrep/deq167. [DOI] [PubMed] [Google Scholar]
- 56.White S, Ohnesorg T, Notini A, Roeszler K, Hewitt J, Daggag H, Smith C, Turbitt E, Gustin S, van den Bergen J, Miles D, Western P, Arboleda V, Schumacher V, Gordon L, Bell K, Bengtsson H, Speed T, Hutson J, Warne G, Harley V, Koopman P, Vilain E, Sinclair A. Copy number variation in patients with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS One. 2011;6(3):e17793. doi: 10.1371/journal.pone.0017793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, Rogers N, Knower K, Rowley L, Eyre H, Rizzoti K, McAninch D, Goncalves J, Slee J, Turbitt E, Bruno D, Bengtsson H, Harley V, Vilain E, Sinclair A, Lovell-Badge R, Thomas P. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. 2011;121(1):328–341. doi: 10.1172/JCI42580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bagli DJ, Thomas P, Chitayat D. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A. 2012;158A(7):1759–1764. doi: 10.1002/ajmg.a.35390. [DOI] [PubMed] [Google Scholar]
- 59.Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, Merico M, Manolakos E, Parada-Bustamante A, Castro A, Radi O, Camerino G, Brusco A, Sabaghian M, Sofocleous C, Forzano F, Palumbo P, Palumbo O, Calvano S, Zelante L, Grammatico P, Giglio S, Basly M, Chaabouni M, Carella M, Russo G, Bonaglia MC, Zuffardi O. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. 2015;23(8):1025–1032. doi: 10.1038/ejhg.2014.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tannour-Louet M, Han S, Corbett ST, Louet JF, Yatsenko S, Meyers L, Shaw CA, Kang SH, Cheung SW, Lamb DJ. Identification of de novo copy number variants associated with human disorders of sexual development. PLoS One. 2010;5(10):e15392. doi: 10.1371/journal.pone.0015392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lecointre C, Pichon O, Hamel A, Heloury Y, Michel-Calemard L, Morel Y, David A, Le Caignec C. Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am J Med Genet A. 2009;149A(6):1183–1189. doi: 10.1002/ajmg.a.32830. [DOI] [PubMed] [Google Scholar]
- 62.Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. 2011;364(1):91–93. doi: 10.1056/NEJMc1010311. [DOI] [PubMed] [Google Scholar]
- 63.Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, Thomas S, Bruland O, David M, Nicolino M, Labalme A, Sanlaville D, Callier P, Malan V, Huet F, Molven A, Dijoud F, Munnich A, Faivre L, Amiel J, Harley V, Houge G, Morel Y, Lyonnet S. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. 2011;48(12):825–830. doi: 10.1136/jmedgenet-2011-100255. [DOI] [PubMed] [Google Scholar]
- 64.Vetro A, Ciccone R, Giorda R, Patricelli MG, Della Mina E, Forlino A, Zuffardi O. XX males SRY negative: a confirmed cause of infertility. J Med Genet. 2011;48(10):710–712. doi: 10.1136/jmedgenet-2011-100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amarillo IE, Nievera I, Hagan A, Huchthagowder V, Heeley J, Hollander A, Koenig J, Austin P, Wang T. Integrated small copy number variations and epigenome maps of disorders of sex development. Hum Genome Var. 2016;3:16012. doi: 10.1038/hgv.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Genomes Project, C. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boycott KM, Rath A, Chong JX, Hartley T, Alkuraya FS, Baynam G, Brookes AJ, Brudno M, Carracedo A, den Dunnen JT, Dyke SOM, Estivill X, Goldblatt J, Gonthier C, Groft SC, Gut I, Hamosh A, Hieter P, Hohn S, Hurles ME, Kaufmann P, Knoppers BM, Krischer JP, Macek M, Jr, Matthijs G, Olry A, Parker S, Paschall J, Philippakis AA, Rehm HL, Robinson PN, Sham PC, Stefanov R, Taruscio D, Unni D, Vanstone MR, Zhang F, Brunner H, Bamshad MJ, Lochmuller H. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am J Hum Genet. 2017;100(5):695–705. doi: 10.1016/j.ajhg.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19(2):209–214. doi: 10.1038/gim.2016.88. [DOI] [PubMed] [Google Scholar]
- 69.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CG, Dipple KM, Grody WW, Vilain E, Nelson SF. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA. 2014 doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, C. Exome Aggregation Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Petrovski S, Goldstein DB. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 2016;17(1):157. doi: 10.1186/s13059-016-1016-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kernohan KD, Hartley T, Alirezaie N, C. Care4Rare Canada. Robinson PN, Dyment DA, Boycott KM. Evaluation of exome filtering techniques for the analysis of clinically relevant genes. Hum Mutat. 2017 doi: 10.1002/humu.23374. [DOI] [PubMed] [Google Scholar]
- 73.Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet. 2017 doi: 10.1111/cge.13190. [DOI] [PubMed] [Google Scholar]
- 74.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE, A.L.Q.A.C. Molecular Subcommittee of the ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10(4):294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 75.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CG, Dipple KM, Grody WW, Vilain E, Nelson SF. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dong Y, Yi Y, Yao H, Yang Z, Hu H, Liu J, Gao C, Zhang M, Zhou L, Asan, Yi X, Liang Z. Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med Genet. 2016;17:23. doi: 10.1186/s12881-016-0286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu HM, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CL, Tang S. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578–586. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 79.Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, Bedford HM, Bernard G, Bernier FP, Brais B, Bulman DE, Warman Chardon J, Chitayat D, Deladoey J, Fernandez BA, Frosk P, Geraghty MT, Gerull B, Gibson W, Gow RM, Graham GE, Green JS, Heon E, Horvath G, Innes AM, Jabado N, Kim RH, Koenekoop RK, Khan A, Lehmann OJ, Mendoza-Londono R, Michaud JL, Nikkel SM, Penney LS, Polychronakos C, Richer J, Rouleau GA, Samuels ME, Siu VM, Suchowersky O, Tarnopolsky MA, Yoon G, Zahir FR, F.C. Consortium, C. Care4Rare Canada. Majewski J, Boycott KM. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89(3):275–284. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, Twist G, Noll A, Heese BA, Zellmer L, Atherton AM, Abdelmoity AT, Safina N, Nyp SS, Zuccarelli B, Larson IA, Modrcin A, Herd S, Creed M, Ye Z, Yuan X, Brodsky RA, Kingsmore SF. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gottlieb B, Beitel LK, Trifiro MA. Androgen Insensitivity Syndrome. In: Pagon RA, et al., editors. GeneReviews(R) Seattle WA: 1993. [PubMed] [Google Scholar]
- 83.Lek N, Miles H, Bunch T, Pilfold-Wilkie V, Tadokoro-Cuccaro R, Davies J, Ong KK, Hughes IA. Low frequency of androgen receptor gene mutations in 46 XY DSD, and fetal growth restriction. Arch Dis Child. 2014;99(4):358–361. doi: 10.1136/archdischild-2013-305338. [DOI] [PubMed] [Google Scholar]
- 84.Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, Buonocore F, Barseghyan H, Bingham N, Rosenfeld JA, Mulukutla SN, Jain M, Burrage L, Dhar S, Balasubramanyam A, Lee B, U.D.N. Members of. Dumargne MC, Eozenou C, Suntharalingham JP, Silva Kde, Lin L, Bignon-Topalovic J, Poulat F, Lagos CF, McElreavey K, Achermann JC. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. 2016;25(16):3446–3453. doi: 10.1093/hmg/ddw186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 86.Boycott K, Hartley T, Adam S, Bernier F, Chong K, Fernandez BA, Friedman JM, Geraghty MT, Hume S, Knoppers BM, Laberge AM, Majewski J, Mendoza-Londono R, Meyn MS, Michaud JL, Nelson TN, Richer J, Sadikovic B, Skidmore DL, Stockley T, Taylor S, Karnebeek Cvan, Zawati MH, Lauzon J, Armour CM, G. Canadian College of Medical The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position Statement of the Canadian College of Medical Geneticists. J Med Genet. 2015;52(7):431–437. doi: 10.1136/jmedgenet-2015-103144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, Howard HC, Cambon-Thomsen A, Knoppers BM, Meijers-Heijboer H, Scheffer H, Tranebjaerg L, Dondorp W, Wert GMde, Public E, C. Professional Policy Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013;21(6):580–584. doi: 10.1038/ejhg.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Massively Parallel Sequencing Implementation Guidelines. The Royal College of Pathologists of Australasia; 2015. [Google Scholar]
- 89.Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med. 2011;13(6):499–504. doi: 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- 90.Barseghyan HS, Zadikyan A, Almalvez M, Segura M, Eskin EE, Bramble A, Arboleda MS, Baxter VA, Nelson R, Délot NF, Harley EC, Vilain VE. Identification of Novel Candidate Genes for 46,XY Disorders of Sex Development (DSD) using a C57BL/6J-YPOS Mouse Model. Biol Sex Differ. 2018 doi: 10.1186/s13293-018-0167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eicher EM, Washburn LL, Whitney JB, 3rd, Morrow KE. Mus poschiavinus Y chromosome in the C57BL/6J murine genome causes sex reversal. Science. 1982;217(4559):535–537. doi: 10.1126/science.7089579. [DOI] [PubMed] [Google Scholar]
- 92.Bullejos M, Koopman P. Spatially dynamic expression of Sry in mouse genital ridges. Dev Dyn. 2001;221(2):201–205. doi: 10.1002/dvdy.1134. [DOI] [PubMed] [Google Scholar]
- 93.Hiramatsu R, Matoba S, Kanai-Azuma M, Tsunekawa N, Katoh-Fukui Y, Kurohmaru M, Morohashi K, Wilhelm D, Koopman P, Kanai Y. A critical time window of Sry action in gonadal sex determination in mice. Development. 2009;136(1):129–138. doi: 10.1242/dev.029587. [DOI] [PubMed] [Google Scholar]
- 94.Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics. 2014;15:247. doi: 10.1186/1471-2105-15-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, Shang L, Boisson B, Casanova JL, Abel L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112(17):5473–5478. doi: 10.1073/pnas.1418631112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, Nalpathamkalam T, Pellecchia G, Yuen RKC, Szego MJ, Hayeems RZ, Shaul RZ, Brudno M, Girdea M, Frey B, Alipanahi B, Ahmed S, Babul-Hirji R, Porras RB, Carter MT, Chad L, Chaudhry A, Chitayat D, Doust SJ, Cytrynbaum C, Dupuis L, Ejaz R, Fishman L, Guerin A, Hashemi B, Helal M, Hewson S, Inbar-Feigenberg M, Kannu P, Karp N, Kim R, Kronick J, Liston E, MacDonald H, Mercimek-Mahmutoglu S, Mendoza-Londono R, Nasr E, Nimmo G, Parkinson N, Quercia N, Raiman J, Roifman M, Schulze A, Shugar A, Shuman C, Sinajon P, Siriwardena K, Weksberg R, Yoon G, Carew C, Erickson R, Leach RA, Klein R, Ray PN, Meyn MS, Scherer SW, Cohn RD, Marshall CR. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genom Med. 2016;1 doi: 10.1038/npjgenmed.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feero WG. Clinical application of whole-genome sequencing: proceed with care. JAMA. 2014;311(10):1017–1019. doi: 10.1001/jama.2014.1718. [DOI] [PubMed] [Google Scholar]
- 98.Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP, Pakdaman N, Ormond KE, Caleshu C, Kingham K, Klein TE, Whirl-Carrillo M, Sakamoto K, Wheeler MT, Butte AJ, Ford JM, Boxer L, Ioannidis JP, Yeung AC, Altman RB, Assimes TL, Snyder M, Ashley EA, Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311(10):1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tattini L, D’Aurizio R, Magi A. Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front Bioeng Biotechnol. 2015;3:92. doi: 10.3389/fbioe.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Ye K, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO, Genomes P. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.English AC, Salerno WJ, Hampton OA, Gonzaga-Jauregui C, Ambreth S, Ritter DI, Beck CR, Davis CF, Dahdouli M, Ma S, Carroll A, Veeraraghavan N, Bruestle J, Drees B, Hastie A, Lam ET, White S, Mishra P, Wang M, Han Y, Zhang F, Stankiewicz P, Wheeler DA, Reid JG, Muzny DM, Rogers J, Sabo A, Worley KC, Lupski JR, Boerwinkle E, Gibbs RA. Assessing structural variation in a personal genome-towards a human reference diploid genome. BMC Genomics. 2015;16:286. doi: 10.1186/s12864-015-1479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mak AC, Lai YY, Lam ET, Kwok TP, Leung AK, Poon A, Mostovoy Y, Hastie AR, Stedman W, Anantharaman T, Andrews W, Zhou X, Pang AW, Dai H, Chu C, Lin C, Wu JJ, Li CM, Li JW, Yim AK, Chan S, Sibert J, Dzakula Z, Cao H, Yiu SM, Chan TF, Yip KY, Xiao M, Kwok PY. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics. 2016;202(1):351–362. doi: 10.1534/genetics.115.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jaratlerdsiri W, Chan EKF, Petersen DC, Yang C, Croucher PI, Bornman MSR, Sheth P, Hayes VM. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget. 2017;8(14):23588–23602. doi: 10.18632/oncotarget.15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seo JS, Rhie A, Kim J, Lee S, Sohn MH, Kim CU, Hastie A, Cao H, Yun JY, Kim J, Kuk J, Park GH, Kim J, Ryu H, Kim J, Roh M, Baek J, Hunkapiller MW, Korlach J, Shin JY, Kim C. De novo assembly and phasing of a Korean human genome. Nature. 2016;538(7624):243–247. doi: 10.1038/nature20098. [DOI] [PubMed] [Google Scholar]
- 105.Barseghyan H, Tang W, Wang RT, Almalvez M, Segura E, Bramble MS, Lipson A, Douine ED, Lee H, Delot EC, Nelson SF, Vilain E. Next-generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017;9(1):90. doi: 10.1186/s13073-017-0479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hastie Alex R, E TL, Wing Andy, Pang Chun, Zhang Xinyue, Andrews Warren, Lee Joyce, Liang TiffanyY, Wang Jian, Zhou Xiang, Zhu Zhanyang, Anantharaman Thomas, Zdzakula Zeljko, Bocklandt Sven, Surti Urvashi, Saghbini Michael, Austin Mike, Borodkin Mark, Holmlin R Erik, Cao Han. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. 2017 doi: 10.1101/102764. [DOI]
- 107.White S, Hewitt J, Turbitt E, van der Zwan Y, Hersmus R, Drop S, Koopman P, Harley V, Cools M, Looijenga L, Sinclair A. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur J Hum Genet. 2012;20(3):348–351. doi: 10.1038/ejhg.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, Guan P, Korzeniewski GE, Lockhart NC, Rabiner CA, Rao AK, Robinson KL, Roche NV, Sawyer SJ, Segre AV, Shive CE, Smith AM, Sobin LH, Undale AH, Valentino KM, Vaught J, Young TR, Moore HM, G.T. Consortium A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank. 2015;13(5):311–319. doi: 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tong W, Yang L, Yu Q, Yao J, He A. A new tumor suppressor lncRNA RP11-190D6.2 inhibits the proliferation, migration, and invasion of epithelial ovarian cancer cells. Onco Targets Ther. 2017;10:1227–1235. doi: 10.2147/OTT.S125185. [DOI] [PMC free article] [PubMed] [Google Scholar]