

Supplemental Digital Content is available in the text
Keywords: hepatocellular carcinoma, lncRNA, prognosis, signature
Abstract
Primary hepatic carcinoma is 1 of the most common malignant tumors globally, of which hepatocellular carcinoma (HCC) accounts for 85% to 90%. Due to the high degree of deterioration and low early detection rate of HCC, most patients are diagnosed when they are already in the middle and advanced stages, and the prognosis are always poor.
RNA sequencing data from the cancer genome atlas was used to explore differences in lncRNA expression profiles. LncRNA was extracted by gdcRNAtools in R package. Multivariate cox analysis was performed on the screened lncRNAs. The relationship between the lncRNA model and prognosis as well as clinical characteristics of patients with HCC was analyzed. Finally, a predictive nomogram in the the cancer genome atlas cohort was established and verified internally
Based on the RNA sequencing survival analysis, a 9- lncRNAs prognosis model, including TMCC1-AS1, AC008892.1, AL031985.3, L34079.2, U95743.1, KDM4A-AS1, SACS-AS1, AC005534.1, LINC01116 was established. The 9-lncRNA prognosis model was a reliable tool for predicting prognosis of HCC, and the nomogram of this prognosis model could help clinicians to choose personalized treatment for HCC patients
This model was significant to complement clinic characteristics of HCC and to promote personalized management of patients, it also provided a new idea for researches on the prognosis of HCC.
1. Introduction
Hepatocellular carcinoma (HCC) is 1 of the most common liver malignant tumor. It is reported to be the fifth most common cancer and the third most common cancer-related cause of death worldwide.[1,2] Due to difficulties in precise diagnosis at an early stage, only 20% of HCC patients could obtain curative treatment through liver transplantation, surgical resection, or ablation treatment.[3] Although some progress has been made in diagnosis and treatment, the 5-year survival rate of HCC in all stages is still lower than 30% due to postoperative recurrence and other reasons.[4] Comprehensive genomic research shows that the role of RNA has attracted widespread attention. Many potential valuable RNAs need to be identified to improve the clinical outcome of HCC patients. However, there are still few specific biomarkers that can be used to show therapeutic effects, and prognostic factors are essential for the treatment of HCC. Therefore, there is a pressing need for molecular screening of HCC biomarkers, to improve prognosis of HCC and reduce the mortality rate.
In humans, more than 85% of the genome is transcribed. Non-coding RNA (ncRNA), which refers to RNA that does not encode a protein, is a new type of transcript that is encoded by the genome. Although not translated, ncRNAs play important roles in various cellular and physiological functions.[5] Non-coding RNAs are divided into 2 groups based on their size. One group is short RNAs with less than 200 nucleotides, such as miRNA, siRNA, snoRNA. The other group is long non-coding RNAs with more than 200 nucleotides. Long non-coding RNAs play a key role in regulating chromatin dynamics, gene expression, growth, differentiation, and development.[6] It is now fully recognized that more than 75% of the human genome is functional and encodes a large number of ncRNAs.[7] Increasing evidence also have indicated that lncRNAs are involved in transcriptional regulation, epigenetic gene regulation, and the pathogenesis of many diseases including cancer.[8,9] Imbalanced lncRNA has been reported to be associated with tumorigenesis and metastasis of HCC.[10] Therefore, constructing a prognostic expression profile of lncRNA in hepatic carcinoma is extremely significant to clarify the mechanism of its development, and further to improve the diagnosis and prognosis.
In this study, RNA sequencing (RNA-seq) data from HCC in the cancer genome atlas (TCGA) was used to explore differences in lncRNA expression profiles between HCC and adjacent livers, and to identify potential genetic biomarkers. Based on the RNA-seq survival analysis, a 9- lncRNAs prognosis model, including TMCC1-AS1, AC008892.1, AL031985.3, L34079.2, U95743.1, KDM4A-AS1, SACS-AS1, AC005534.1, LINC01116 was established. Previous studies had reported that lncRNA TMCC1-AS1 was an independent prognostic factor for patients with HCC.[11,12] LINC01116 is closely related to the occurrence, development and prognosis of many types of cancers such as bladder cancer, ovarian cancer, and osteosarcoma.[5,13,14] Other key lncRNAs have been rarely studied in tumor related researches. The relationship between the lncRNA model and prognosis as well as clinical characteristics of patients with HCC was analyzed. Finally, a predictive nomogram in the TCGA cohort was established and verified internally. Overall, this prognostic model and nomogram might help to predict prognostic of HCC patients.
2. Methods
2.1. Downloading and preprocessing of transcriptome data
RNA-seq count files of liver cancer patients were downloaded from TCGA. The data set is called TCGA-LIHC. Non-cancer and non-cancerous tissue samples were excluded, and finally 412 samples were obtained for subsequent analysis, including 371 patient samples and 50 control samples.
2.2. Extraction and difference analysis of lncRNA
The gdcRNAtools toolkit has been widely used in the construction of ceRNA networks. The count files from TCGA include not only regular mRNA data, but also lncRNA expression data, so lncRNA was extracted by gdcRNAtools in R first, then difference analysis between lncRAN in patients and controls was performed, cutoff value was set as | logFC | > 1 & FDR < 0.05.
2.3. Screening of lncRNAs related to prognosis
Clinical information of corresponding patients were downloaded, samples without overall survival time were excluded, and finally 365 samples were included in this study. First, the samples were randomly divided into training set (N = 184) and test set (N = 181), according to 1:1. Then lncRNAs related to prognosis were identified by 3 criteria.
-
(1)
lncRNAs with p < 0.01 were screened out by univariate cox analysis.
-
(2)
Based on robust likelihood-based survival modeling, key lncRNAs were identified by the R language package rbsurv (A forward selection was employed to generate a series of lncRNA models and the optimal model was then selected by using the criterion of minimal AIC (Table 1)).
-
(3)
Multivariate cox analysis was performed on the screened lncRNAs.
Table 1.
Survival-associated lncRNA signature screening using forward selection.
2.4. Construction of lncRNA prognosis model
Finally 9 lncRNAs were identified, and a risk score calculation formula, as follows, was established.
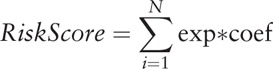 |
where N is the number of genes, exp was the expression value of gene, and coef was the coefficient of lncRNA in the multi Cox regression analysis.
2.5. Evaluation and verification of model
Survival curve of the test set was calculated using the same formula to verify the robustness of this model, and to further evaluate its stability.
3. Results
3.1. Differential expression analysis of lncRNA
14,388 lncRNAs were extracted by gdcRNAtools.R package, then difference analysis between lncRNAs in patients and controls was performed, cutoff value was set as | logFC | > 1 & FDR < 0.05. 3116 lncRNAs that differentially expressed were screened out. The volcanic map and heat map were shown in Figure 1.
Figure 1.
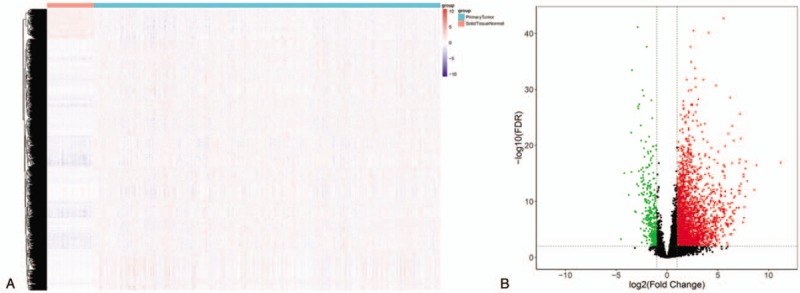
Volcanic maps and heat maps of the differentially expressed genes. A: Volcano maps of the differentially expressed genes. Red for normal samples and green for tumor samples. B: Volcano map. Green represents the down-regulated gene and red represents the up-regulated gene.
3.2. Screening of lncRNAs related to prognosis
Clinical information from 371 patients were downloaded, samples without overall survival time were excluded, and 365 samples were included in this study. Then the samples were randomly divided into training set (N = 184) and test set (N = 181), according to 1:1. Firstly, univariate cox analysis was performed on 3116 different lncRNAs, and 290 lncRNAs with P < .01 were screened out (S_Table 1, http://links.lww.com/MD/E286), then the 290 lncRNAs were analyzed using R language package rbsurv based on robust likelihood-based survival modeling, to identify the key lncRNAs. A forward selection was employed to generate a series of lncRNA models and the optimal model was then selected by using the criterion of minimal AIC (Table 1). Finally, 9 lncRNAs (TMCC1-AS1, AC008892.1, AL031985.3, L34079.2, U95743.1, KDM4A-AS1, SACS-AS1, AC005534.1, LINC01116) were screened out as signature lncRNAs which could predict the overall survival rate of HCC.
3.3. Construction of the lncRNA prognosis model
Multivariate cox analysis was performed on the 9 lncRNAs screened out, and the risk model was as follows:
Risk score = (0.082 × expression value of TMCC1-AS1) + (0.20024 × expression value of AC008892.1) + (0.45842 × expression value of AL031985.3) + (0.70212 × expression value of L34079.2) + (0.07987 × expression value of U95743.1) + (0.12007 × expression value of KDM4A-AS1) + (0.08302 × expression value of SACS-AS1) + (0.19776 × expression value of AC005534.1) + (0.2085 × expression value of LINC01116)
We used the surv cutpoint function in the survmier package to determine the best cutoff value, according to the risk score. And patients were divided into 2 categories: the high risk group and the low risk group. The survival analysis based on risk score found that patients with high and low risks could be clearly separated. The ROC curve of 5-year survival rate of the patient was calculated, and the AUC was found to be as high as 0.7454, which proved that this model had strong predictive power. The survival curve and ROC curve were shown in Figure 2, and the specific information of patients in the training set was shown in Figure 3.
Figure 2.
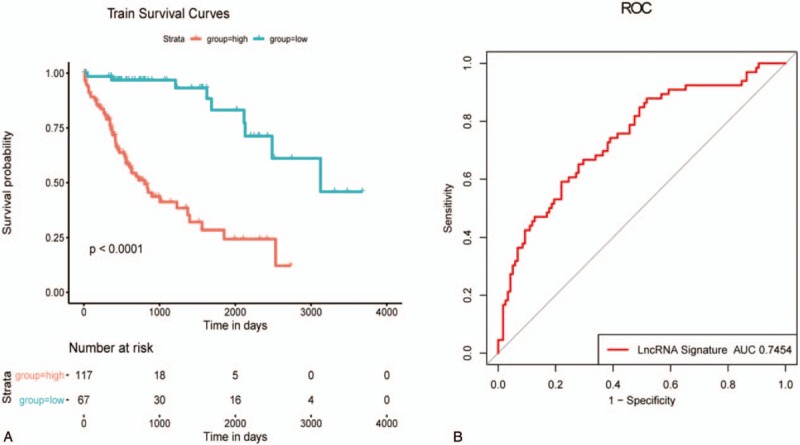
Survival curve and ROC curve in training dataset. A: Kaplan-Meier estimates of the patients’ survival using the 9-lncRNA signature. The Kaplan-Meier plots were used to visualize the patients’ survival probabilities for the low-risk versus high-risk group of patients based on the median risk score. B: Receiver operating characteristic (ROC) analysis of the sensitivity and specificity of the survival prediction by the 9-lncRNA risk score,.
Figure 3.
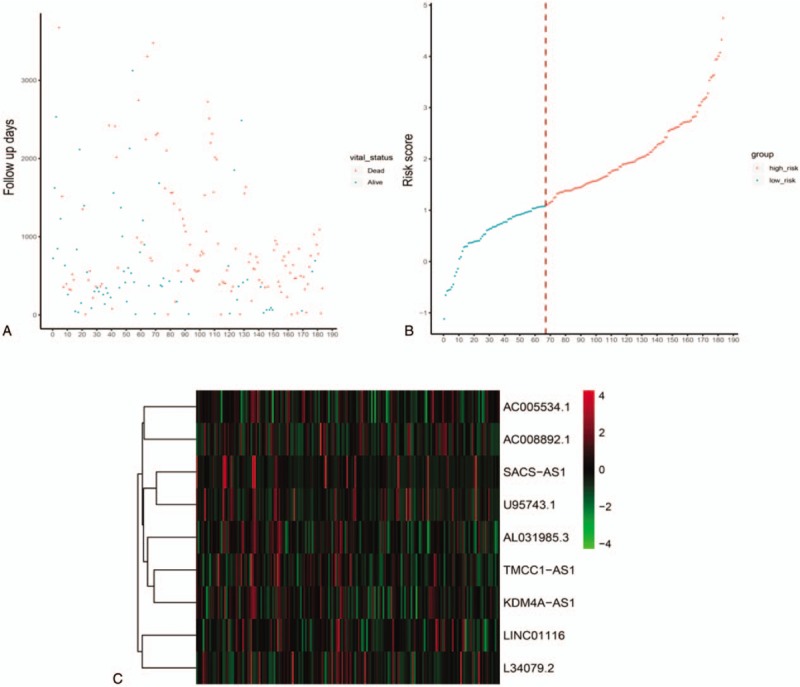
LncRNA risk score analysis of HCC patients in training dataset. A-C: The relationship between survival information, risk score, and z-score transformed expression values are shown (top-downAC008892.1, AC005534.1, SACS-AS1, U95743.1, AL031985.3, TMCC1-AS1, KDM4A-AS1, LINC0116 and L34079.2.
3.4. Evaluation of independent prognostic factors
Then gender, age, weight, pathological staging, tumor staging, inflammatory response, and risk score were included in the study. Univariate and multivariate Cox analysis were performed on the training set, and a forest plot was drawn. The results showed that risk score was an independent prognostic factor. At the same time, a nomogram model for the training data set was established, which also showed that risk score had the strongest correlation with survival time, indicating that this risk model based on 9 -lncRNAs signature had a strong predictive power in prognosis of patients with HCC (Fig. 4 A-B) (S_Table 2, Available at: http://links.lww.com/MD/E287).
Figure 4.
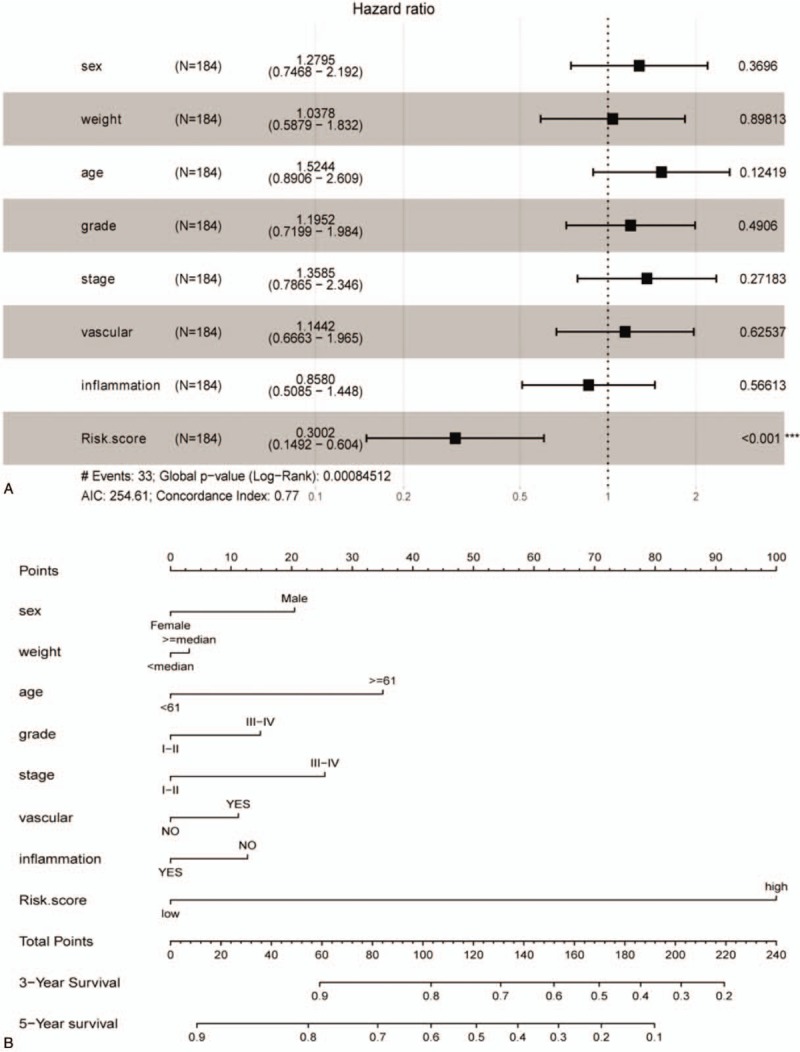
The nomogram to predict survival in training dataset. A: Multifactor analysis in training dataset. B: The nomogram for predicting proportion of patients with 3-yr and 5-yr survival.
3.5. Evaluation and validation of the model
Survival curve of the testing set was calculated using the same formula to verify the robustness of this model, and to further evaluate its stability. Results showed that they could be clearly distinguished the patient prognosis. The survival curve and specific information of patients in the testing set were shown in Figure 5. All these results indicated the reliability of our prognostic model, and it was supposed to provide new strategies for treatment.
Figure 5.
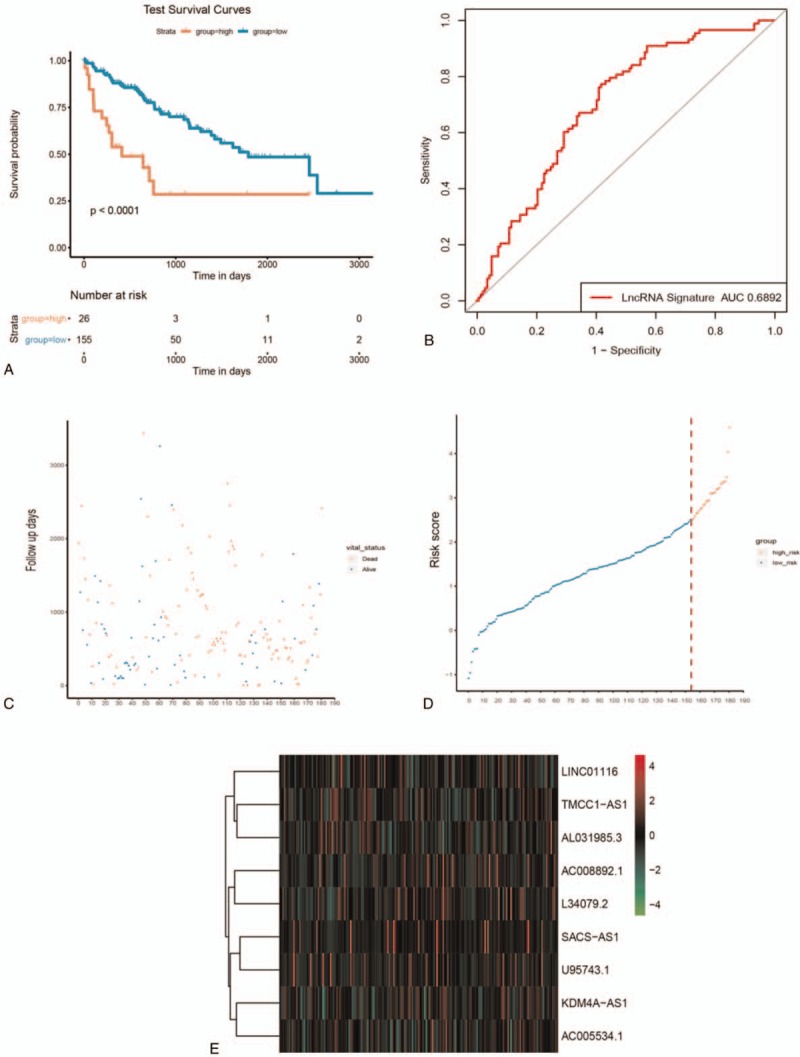
LncRNA risk score and survival analysis of HCC patients in testing dataset. A: Kaplan-Meier estimates of the patients’ survival using the 9-lncRNA signature. The Kaplan-Meier plots were used to visualize the patients’ survival probabilities for the low-risk versus high-risk group of patients based on the median risk score. B-D: the relationship between survival information, risk score, and z-score transformed expression values are shown (top-downAC008892.1, AC005534.1, SACS-AS1, U95743.1, AL031985.3, TMCC1-AS1, KDM4A-AS1, LINC0116 and L34079.2).
3.6. Model comparison
The risk model in literature[15] was selected and compared with our 9-lncRNA signature risk model. Based on the corresponding genes in this model, risk score of training set were re-calculated respectively using multivariate Cox analysis, to make the 2 models comparable. ROC of the 2 models were evaluated, and the samples were divided into high-risk group and low-risk group according to the optimal threshold. The prognostic difference in OS between the 2 groups were calculated. The ROC and KM curve of 6-lncRNA signature risk model were shown in Figure 6.
Figure 6.
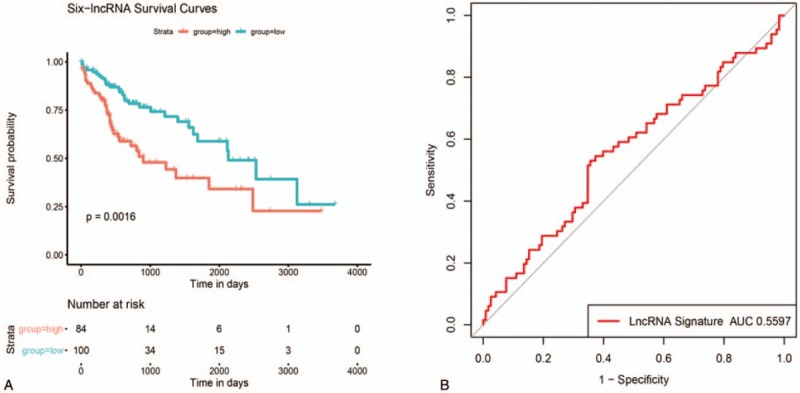
The ROC and KM curve of 6-lncRNA signature risk model. A: Survival curve of 6-lncRNA signature risk model, the horizontal axis represents survival time (d), the vertical axis represents survival probability; B: ROC curve of 6-lncRNA signature, the horizontal axis represents false positive rate, and the vertical axis represents true positive rate.
4. Discussion
HCC is 1 of the most common malignancies worldwide, causing approximately 700,000 deaths each year.[16] Multiple genetic changes are responsible for the progression of HCC, so exploring the molecular mechanisms of HCC is essential for the diagnosis, treatment, and prognosis of HCC.[17] LncRNA is transcribed by RNA polymerase II/III, showing epigenetic characteristics common to protein-encoding genes, usually spliced by spliceosome and having a poly A tail.[18] Physiologically, lncRNA is involved in many levels of gene expression regulation, from transcriptional and translational regulation to controlling mRNA stability and protein degradation,[19] further affecting cell proliferation, differentiation, and related biological behaviors.[20] In recent years, LncRNA has received increasing attention for its function in human diseases including cancer. Aberrantly expressed lncRNAs can indicate certain stages of cancer progression and can predict early cancer progression or effectively maintain tumor-related signaling pathways.[21] Recent reports from lncRNA suggest that they can be used as novel biomarkers and therapeutic targets.[22,23] Therefore, this study established a prognostic model based on lncRNA in HCC to help improve the diagnosis and predict the prognosis of HCC.
In this study, 3116 differentially expressed lncRNAs in HCC were screened out, and 290 lncRNAs were identified related to the prognosis of HCC by univariate Cox analysis. Then 9 key lncRNAs were identified based on the robust likelihood survival model, and a risk model was established by multivariate Cox analysis. The survival analysis based on risk score showed that patients with high expression had a significantly worse prognosis than the low expression group. The 5-year survival ROC curve showed that the AUC was as high as 0.7454, which proved that this model had a strong predictive power.
Previous studies showed that HCC patients with higher levels of lncRNA TMCC1-AS1 had shorter overall survival (OS) time. Qiu-Jie Zhao et al constructed a 5-lncRNA model significant related to prognosis, based on the lncRNA expression profiles of 370 HCC patients from TCGA, of which TMCC1-AS1 played an important role.[11,12] Chu A found that U95743.1 was closely related to overall survival of gastric cancer patients infected with H. pylori.[24]
Nai qiang Zhu et al constructed a lncRNA-miRNA-mRNA competitive endogenous RNA network through a bioinformatics method, and verified that SACS-AS1 was related to the survival of bladder cancer patients. These results provided a new perspective for researches on lncRNA-related ceRNA network in bladder cancer, thus shedding light on the development of diagnostic biomarkers and therapeutic targets.[25]
Among the 9 lncRNAs screened out, LINC01116 is the most studied at present. Y.N. Fang et al. found that the expression of LINC01116 in epithelial ovarian cancer (EOC) tissues was higher than that in adjacent tissues, and EOC patients with high expression of LINC01116 had significantly worse DFS and OS than patients with low expression. In addition, overexpression of LINC01116 promotes the malignant progression of ovarian cancer by up-regulating Bcl-2 and down-regulating Caspase-3 and Caspase-9 in EOC cells.[13]
B. Zhang et al identified an important lncRNA-miRNA-mRNA pathway involved in LINC01116 in osteosarcoma through bioinformatics analysis, and verified that LINC01116 promoted viability and migration of osteosarcoma cells through IL6R in vitro and in vivo tests.[14]
HB. Hu et al screened the differentially expressed LINC01116 in the public database Gene Expression Omnibus. In vitro and in vivo experiments showed that LINC01116 was highly expressed in breast cancer and related to the overall survival, tumor size, and tumor metastasis (TNM) staging, suggesting that LINC01116 may be a new prognostic biomarker for breast cancer. These previous findings to some extent illustrate the significance of the key lncRNAs in this study in the development of some cancers.[26]
Gender, age, weight, pathological staging, tumor staging, inflammatory response, and risk score were also included in the study. Univariate and multivariate Cox analysis were performed on the training set, and a forest plot was drawn. The results showed that risk score was an independent prognostic factor. A nomogram model for the training set was established, which also showed that risk score had the strongest correlation with survival time. Internal verification of the TCGA cohort showed the same results, indicating that this risk model based on 9 lncRNAs could perform a reliable and satisfactory prediction on prognosis of patients with HCC.
We chose a 6-lncRNA signature risk model developed by Gu JX[15] to be compared with our 9-lncRNA signature, to evaluate the predictability of our model for HCC samples. Risk score of the training set were re-calculated based on the genes in the 6-lncRNA signature risk model, by multivariate Cox analysis. And the ROC of the 2 models were evaluated. Results showed that our 9-lncRNA signature model performed better, the accuracy of the diagnosis based on these 9 lncRNAs was qualified enough to be provided as a solution for molecular diagnosis in HCC.
As far as we know, the biomarkers of these 9 genes have not been studied before and they would provide new ideas for the diagnosis and prognosis of HCC. Pathological staging is a key prognostic determinant for oncologists and HCC patients. However, patient with the same stage of cancer may have different clinical outcomes, indicating that current clinical staging system is not reasonable enough to precisely distinguish the prognosis. The current pathological diagnosis is based entirely on the anatomy of the disease and the staging system, which cannot fully reflect the biological heterogeneity of patients with liver cancer. These problems may affect the prediction accuracy of traditional systems in patients with liver cancer. Our nomograms combine information from different genetic and HCC clinical data to better predict patient outcomes.
5. Conclusion
In summary, a 9-lncRNA prognostic risk model was innovatively established in HCC, based on data from TCGA in this study. Results showed that the 9-lncRNA prognosis model was a reliable tool for predicting prognosis of HCC, and the nomogram of this prognosis model could help clinicians to choose personalized treatment for HCC patients. However, large-scale prospective studies are still needed to further evaluate the robustness of this model before clinical application, and the underlying biological mechanisms related to this model should be studied in depth as well.
Acknowledgments
Thank numerous individuals participated in this study
Author contributions
Conceptualization: Benyuan Deng, Zhongwu Liu.
Data curation: Min Yang, Zhongwu Liu.
Formal analysis: Benyuan Deng.
Methodology: Min Yang, Ming Wang.
Project administration: Zhongwu Liu.
Software: Benyuan Deng.
Validation: Ming Wang.
Writing – original draft: Benyuan Deng.
Writing – review and editing: Benyuan Deng, Min Yang, Zhongwu Liu.
Footnotes
Abbreviations: HCC = hepatocellular carcinoma, lncRNA = Long non-coding Ribonucleic Acid, RNA = Ribonucleic Acid, RNA-seq = RNA sequencing, TCGA = the cancer genome atlas.
How to cite this article: Deng B, Yang M, Wang M, Liu Z. Development and validation of 9-long Non-coding RNA signature to predicting survival in hepatocellular carcinoma. Medicine. 2020;99:21(e20422).
The data used to support the findings of this study are available from the corresponding author on reasonable request
Ethics approval and consent to participate was not applicable.
The authors have no funding and conflicts of interest to disclose.
Supplemental digital content is available for this article.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.md-journal.com).
References
- [1].Raza A, Sood GK. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World J Gastroenterol 2014;20:4115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen Y, Hu W, Lu Y, et al. A TALEN-based specific transcript knock-down of PIWIL2 suppresses cell growth in HepG2 tumor cell. 9-lncRNA signature predicting liver cancer prognosis 2014;47:448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schwabe RF, Wang TC. Targeting liver cancer: first steps toward a miRacle? Cancer Cell 2011;20:698–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chapman BC, Paniccia A, Hosokawa PW, et al. Impact of facility type and surgical volume on 10-year survival in patients undergoing hepatic resection for hepatocellular carcinoma. J Am Coll Surg 2017;224:362–72. [DOI] [PubMed] [Google Scholar]
- [5].Beaver LM, Kuintzle R, Buchanan A, et al. Long noncoding RNAs and sulforaphane: a target for chemoprevention and suppression of prostate cancer. The Journal of nutritional biochemistry 2017;42:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bhan A, Mandal SS. LncRNA HOTAIR: a master regulator of chromatin dynamics and cancer. Biochim Biophys Acta 2015;1856:151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sanfilippo PG, Hewitt AW. Translating the ENCyclopedia Of DNA Elements Project findings to the clinic: ENCODE's implications for eye disease. Clin Exp Ophthalmol 2014;42:78–83. [DOI] [PubMed] [Google Scholar]
- [8].Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009;136:629–41. [DOI] [PubMed] [Google Scholar]
- [9].Karlsson O, Baccarelli AA. Environmental Health and Long Non-coding RNAs. Curr Environ Health Rep 2016;3:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang Y, Chen L, Gu J, et al. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat Commun 2017;8:14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao QJ, Zhang J, Xu L, et al. Identification of a five-long non-coding RNA signature to improve the prognosis prediction for patients with hepatocellular carcinoma. World J Gastroenterol 2018;24:3426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cui H, Zhang Y, Zhang Q, et al. A comprehensive genome-wide analysis of long noncoding RNA expression profile in hepatocellular carcinoma. Cancer Med 2017;6:2932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fang YN, Huang ZL, Li H, et al. LINC01116 promotes the progression of epithelial ovarian cancer via regulating cell apoptosis. Eur Rev Med Pharmacol Sci 2018;22:5127–33. [DOI] [PubMed] [Google Scholar]
- [14].Zhang B, Yu L, Han N, et al. LINC01116 targets miR-520a-3p and affects IL6R to promote the proliferation and migration of osteosarcoma cells through the Jak-stat signaling pathway. Biomed Pharmacother 2018;107:270–82. [DOI] [PubMed] [Google Scholar]
- [15].Gu JX, Zhang X, Miao RC, et al. Six-long non-coding RNA signature predicts recurrence-free survival in hepatocellular carcinoma. World J Gastroenterol 2019;25:220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- [17].Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 2013;13:123–35. [DOI] [PubMed] [Google Scholar]
- [18].Ozsolak F. Third-generation sequencing techniques and applications to drug discovery. Expert Opin Drug Discov 2012;7:231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Silva A, Bullock M, Calin G. The clinical relevance of long non-coding RNAs in cancer. Cancers 2015;7:2169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tian T, Gong Z, Wang M, et al. Identification of long non-coding RNA signatures in triple-negative breast cancer. Cancer Cell Int 2018;18:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov 2011;1:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Braconi C, Patel T. Non-coding RNAs as therapeutic targets in hepatocellular cancer. Curr Cancer Drug Targets 2012;12:1073–80. [PMC free article] [PubMed] [Google Scholar]
- [23].Xu D, Yang F, Yuan JH, et al. Long noncoding RNAs associated with liver regeneration 1 accelerates hepatocyte proliferation during liver regeneration by activating Wnt/beta-catenin signaling. Hepatology 2013;58:739–51. [DOI] [PubMed] [Google Scholar]
- [24].Chu A, Liu J, Yuan Y, et al. Comprehensive analysis of. aberrantly expressed ceRNA network in gastric cancer with and without H pylori infection. J Cancer 2019;10:853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhu N, Hou J, Wu Y, et al. Integrated. analysis of a competing endogenous RNA network reveals key lncRNAs as potential prognostic biomarkers for human bladder cancer. Medicine (Baltimore) 2018;97:e11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hu HB, Chen Q, Ding SQ. LncRNA LINC01116 competes with miR-145. for the regulation of ESR1 expression in breast cancer. Eur Rev Med Pharmacol Sci 2018;22:1987–93. [DOI] [PubMed] [Google Scholar]