

Introductory Paragraph
Vascular contributions to dementia and Alzheimer’s disease are increasingly recognized1–6. Recent studies have suggested that blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction7, including the early clinical stages of Alzheimer’s disease5,8–10. Apolipoprotein E4 (APOE4), the major Alzheimer’s disease susceptibility gene11–14, leads to accelerated blood-brain barrier breakdown and degeneration of brain capillary pericytes15–19 that maintain blood-brain barrier integrity20–22. Whether APOE4 cerebrovascular effects contribute to cognitive impairment remains, however, largely unknown. Here we show that APOE4 carriers (ε3/ε4 and ε4/ε4) are distinguished from non-carriers (ε3/ε3) by blood-brain barrier breakdown in the hippocampus and medial temporal lobe. This finding is apparent in cognitively unimpaired APOE4 carriers, more severe in those with cognitive impairment, but not related to cerebrospinal fluid or positron emission tomography measurements of Alzheimer’s amyloid-β or tau pathology23. Indeed, high baseline cerebrospinal fluid levels of the blood-brain barrier pericyte injury biomarker soluble platelet-derived growth factor receptor-β7,8 predicted future cognitive decline in APOE4 carriers but not in non-carriers, even after controlling for amyloid-β and tau status, and were correlated with increased activity of blood-brain barrier degrading cyclophilin A-matrix metalloproteinase-9 pathway19 in cerebrospinal fluid. Our findings suggest that blood-brain barrier breakdown contributes to APOE4 associated cognitive decline, does so independently of Alzheimer’s disease pathology, and might be a therapeutic target in APOE4 carriers.
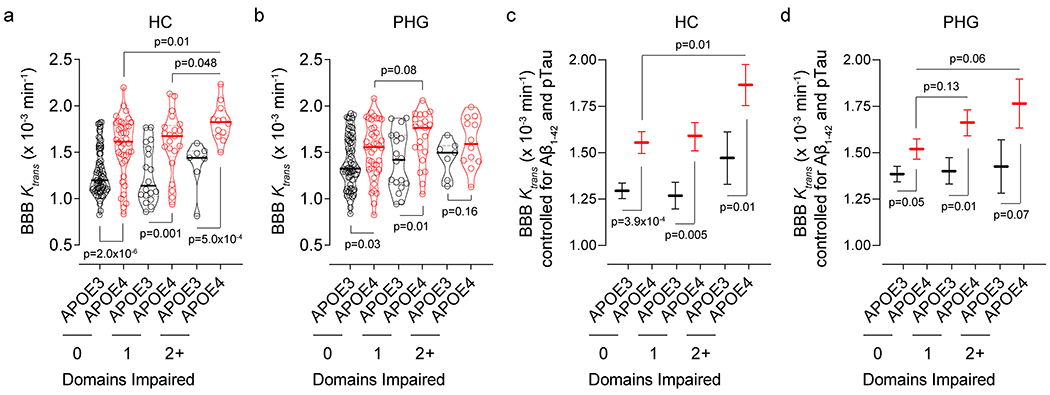
The analysis of blood-brain barrier (BBB) permeability by dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI)7,8 (Fig. 1a; see Methods) in 245 participants (Extended Data Table 1) indicated BBB breakdown in the hippocampus (HC) and parahippocampal gyrus (PHG) in cognitively normal APOE4 (ε3/ε4 and ε4/ε4) carriers compared to cognitively normal APOE3 homozygotes (ε3/ε3) both with clinical dementia rating (CDR) score of 0. The BBB breakdown in HC and PHG in APOE4 carriers further increases with cognitive impairment at CDR=0.5 (Fig. 1b–d). This increase was independent of cerebrospinal fluid (CSF) Aβ and tau changes (Fig. 1e–h), i.e., whether individuals were Aβ+ or Aβ− and pTau+ or pTau− using the accepted cut-off values7,24,25 (see Methods), where Aβ+ and pTau+ status indicates classical AD pathways23. In contrast, APOE3 carriers developed less pronounced BBB changes in HC and PHG with cognitive impairment (Fig. 1b–d). No significant BBB changes were found in other gray or white matter brain regions in APOE4 carriers compared to APOE3 homozygotes, except for increased BBB permeability in the caudate nucleus, and minor leaks in the frontal cortex and corpus callosum in cognitively normal APOE4 carriers (Extended Data Fig. 1). These findings hold when cognitive dysfunction was evaluated by neuropsychological performance (see Methods) (Extended Data Figs. 2 and 3).
Figure 1. Blood-brain barrier breakdown in the hippocampus and parahippocampal gyrus in APOE4 carriers increases with cognitive impairment independently of CSF Aβ and tau status.
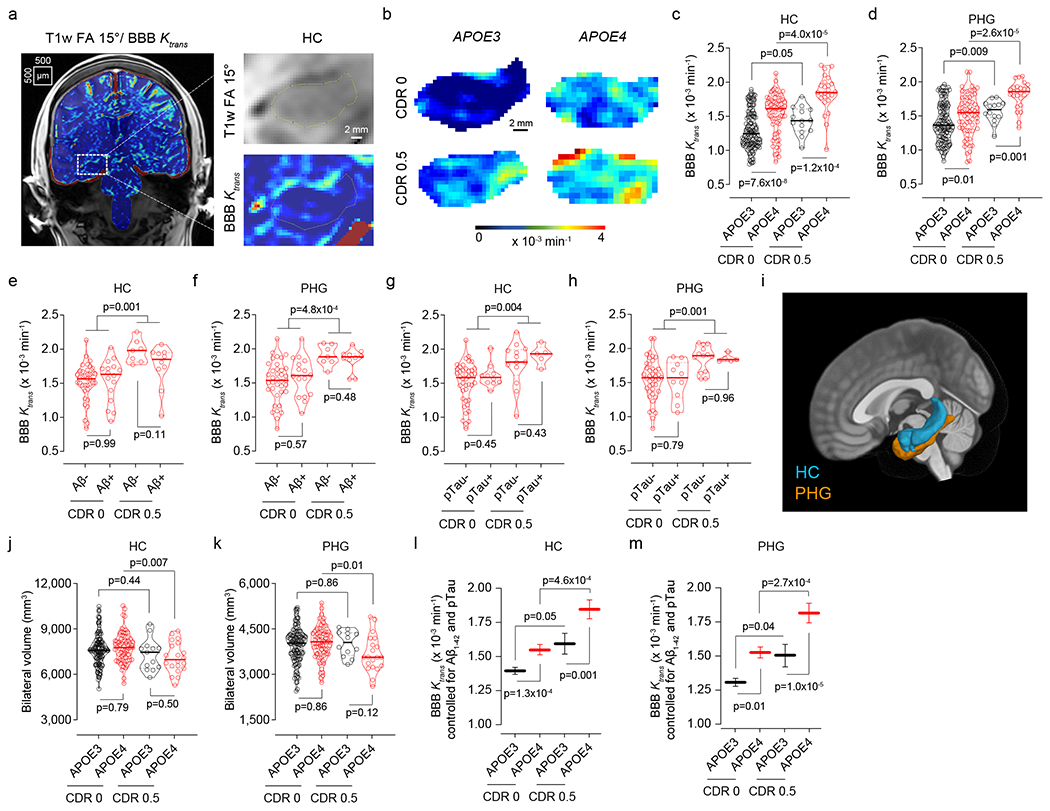
(a,b) Blood-brain barrier (BBB) permeability Ktrans maps generated by dynamic contrast-enhanced magnetic resonance imaging in the hippocampus (HC) of APOE3 homozygotes (APOE3) and APOE4 carriers (APOE4) with clinical dementia rating (CDR) score 0 or 0.5. (c,d) BBB permeability, Ktrans constant, in the HC (c) and parahippocampal gyrus (PHG, d) in CDR 0 APOE3 (black, n=128) and APOE4 (red, n=68) and CDR 0.5 APOE3 (black, n=14) and APOE4 (red, n=25) carriers. (e,f) Ktrans values in the HC (e) and PHG (f) in APOE4 carriers CDR 0 Aβ1-42 negative (Aβ-; n=37) or positive (Aβ+; n=16), and CDR 0.5 Aβ- (n=7) or Aβ+ (n=10). (g,h) Ktrans values in the HC (g) and PHG (h) in APOE4 carriers CDR 0 pTau negative (pTau−; n=42) or positive (pTau+; n=10), and CDR 0.5 pTau− (n=13) or pTau+ (n=5). (i) HC (blue) and PHG (orange) overlaid on a 3D template. (j,k) HC (j) and PHG (k) volumes in CDR 0 APOE3 (n=124) and APOE4 (n=75) and CDR 0.5 APOE3 (n=13) and APOE4 (n=20) carriers. (l,m) Ktrans (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, CSF Aβ1-42 and pTau status, and HC and PHG volumes) in the HC (l) and PHG (m) in CDR 0 APOE3 (black, HC n=125; PHG n=128) and APOE4 (red, HC and PHG n=68) and CDR 0.5 APOE3 (black, HC n=12; PHG n=14) and APOE4 (red, HC n=20; PHG n=25) carriers. In c-h, j, and k, continuous line indicates median value; dotted line indicates interquartile range. Significance by ANCOVA for main effects and post-hoc comparisons controlling for age, sex, and education. All ANCOVA omnibus tests remained significant at false discovery rate threshold of 0.05.
HC and PHG volumes decreased with cognitive impairment in APOE4, but not APOE3 carriers (Fig. 1i–k). The BBB breakdown in HC and PHG in APOE4 carriers, but not APOE3 homozygotes, remained a highly significant predictor of cognitive impairment after statistically controlling for age, sex, education, CSF Aβ and pTau status, and HC and PHG volumes, as shown by the estimated marginal means from the analysis of covariance (ANCOVA) models (Fig. 1l,m), and confirmed by logistic regression models (Supplementary Information Table 1). The BBB dysfunction (Fig. 1c,d,l,m) preceded brain atrophy (Fig. 1j,k) and was independent of systemic vascular risk factors (Extended Data Fig. 4).
Because Aβ and tau can both lead to blood vessel abnormalities and BBB breakdown3,26,27, in a subset of 74 and 96 participants (Extended Data Tables 2a,b), respectively, we studied whether BBB disruption in APOE4 carriers is downstream to amyloid and tau accumulation. Voxel-based analysis of brain uptake of amyloid by positron emission tomography (PET) indicated a substantially higher accumulation in the orbital frontal cortex (OFC) in cognitively normal APOE4 carriers compared to APOE3 homozygotes, as reported28, but failed to detect accumulation of tau tracer in either APOE4 or APOE3 carriers (Extended Data Fig. 5a–d). To determine how BBB permeability relates to amyloid and tau accumulation, we selected 5-mm thick coronal slices in regions of interest including the HC and PHG, where BBB disruption is seen first in APOE4 carriers compared to APOE3 homozygotes (Fig. 1b,d,e), OFC, where amyloid accumulation develops initially in APOE4 carriers, and inferior temporal gyrus (ITG), a region affected early by tau pathology29 (Extended Data Fig. 5b,d,e).
Brain uptake of amyloid and tau tracers (after correction for the choroid plexus off-target binding for tau tracer; see Methods and Extended Data Fig. 5f,g) indicated no difference between APOE4 and APOE3 carriers in HC, although uptake of both tracers was modestly increased (Fig. 2a,b). BBB disruption in HC in APOE4 carriers compared to APOE3 homozygotes (Fig. 2c) was consistent with our findings in the larger cohort (Fig. 1b,c). There was no difference in amyloid and tau accumulation in PHG between APOE4 carriers and APOE3 homozygotes, despite BBB disruption in APOE4 carriers (Fig. 2d–f). There was a higher amyloid accumulation in OFC in cognitively normal APOE4 compared to APOE3 carriers (Fig. 2g,h), but no changes in BBB integrity (Fig. 2g,i). There was no change in ITG tau accumulation or BBB integrity in APOE4 compared to APOE3 carriers (Fig. 2j–l). Altogether, these data suggest that BBB disruption in HC and PHG in APOE4 carriers is independent of AD pathology, and that BBB breakdown in APOE4 carriers starts in the medial temporal lobe, a region responsible for memory encoding and other cognitive functions.
Figure 2. Blood-brain barrier breakdown in APOE4 carriers is independent of amyloid and tau brain accumulation.
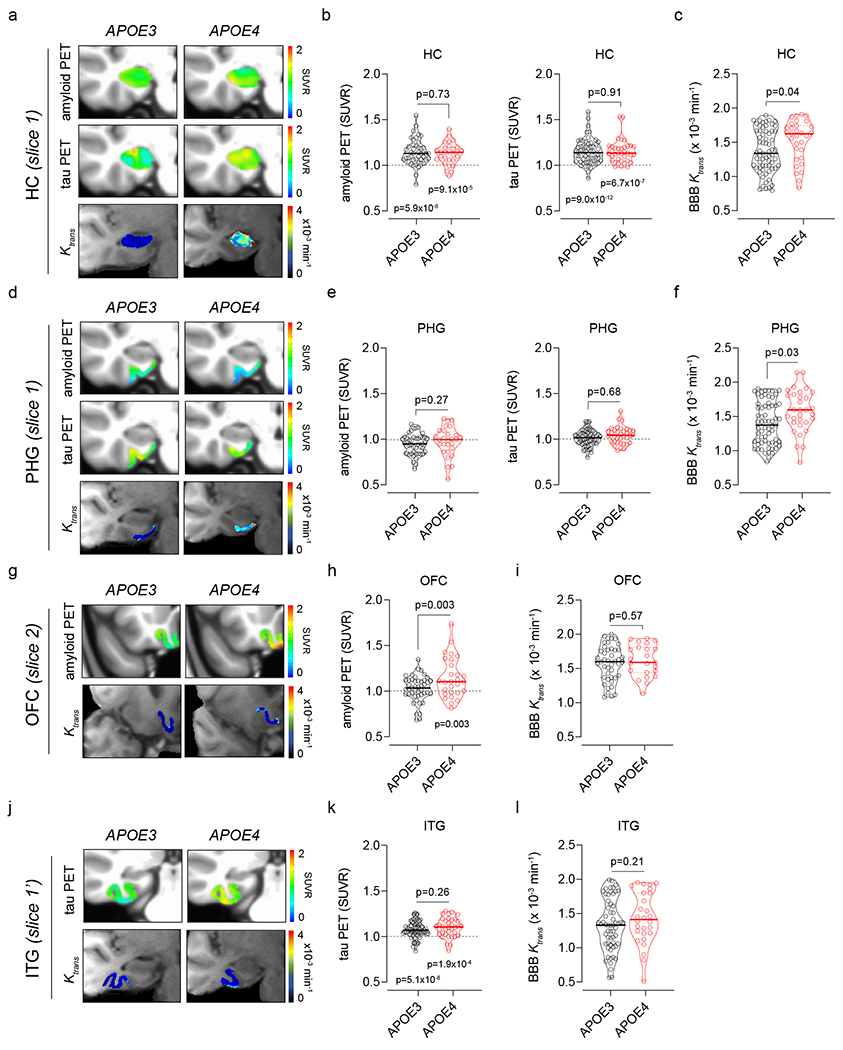
All studies were performed in individuals with clinical dementia rating score 0. (a) Representative superimposed left-hippocampus (HC) amyloid PET (top), tau PET (middle), and BBB Ktrans maps (bottom) from APOE3 (left) and APOE4 (right) carriers. (b,c) HC: amyloid and tau tracer uptake (b) and BBB Ktrans constant (c) in APOE3 (n=45, 60, and 65) and APOE4 (n=29, 37, and 31) carriers. (d) Representative superimposed left-parahippocampal gyrus (PHG) amyloid PET (top), tau PET (middle), and BBB Ktrans maps (bottom) from APOE3 (left) and APOE4 (right) carriers. (e,f) PHG: amyloid and tau tracer uptake (e) and BBB Ktrans constant (f) in APOE3 (n=45, 60, and 65) and APOE4 (n=29, 37, and 31) carriers. (g) Representative superimposed left-medial orbital frontal cortex (OFC) amyloid PET (top) and BBB Ktrans maps (bottom) from APOE3 (left) and APOE4 (right) carriers. (h,i) OFC: amyloid tracer uptake (h) and BBB Ktrans constant (i) in APOE3 (n=45 and 44) and APOE4 (n=29 and 23) carriers. (j) Representative superimposed left-inferior temporal gyrus (ITG) tau PET (top) and BBB Ktrans maps (bottom) from APOE3 (left) and APOE4 (right) carriers. (k,l) ITG: tau tracer uptake (l) and BBB Ktrans constant (l) in APOE3 (n=60 and 59) and APOE4 (n=37 and 28) carriers. In b, c, e, f, h, i, k, and l, continuous lines indicate median values and dotted lines indicate interquartile range. The BBB Ktrans constant was determined in all participants (see Extended Data Tables 2a,b) who received either both amyloid and tau tracers (n=58), only amyloid tracer (n=9) or only tau tracer (n= 29). Significance by ANCOVA for group comparisons controlling for age, sex, and education, and one-tailed t-tests for comparison of PET values to SUVR = 1.
Elevated cerebrospinal fluid (CSF) levels of soluble platelet-derived growth factor receptor-β (sPDGFRβ) in humans and animal models indicate pericyte injury linked to BBB breakdown7,8,30 and cognitive dysfunction7,30. Using a median split for visual display of the CSF sPDGFRβ baseline levels from 350 participants (see Methods), we stratified all participants into two groups, with low CSF sPDGFRβ levels (0-600 ng/mL, grey), and high sPDGFRβ levels (600-2,000 ng/mL, blue) (Fig. 3a). Our data in 146 APOE4 carriers and APOE3 homozygotes evaluated by cognitive exams over two-year intervals up to 4.5 years from baseline show that participants with higher baseline CSF sPDGFRβ levels exhibited accelerated cognitive decline on global mental status exam and global cognitive composite z-scores, which remained significant after controlling for CSF Aβ and tau status (Fig. 3b,c; Supplementary Information Table 2). When stratified by APOE status, higher baseline CSF sPDGFRβ levels in APOE4 carriers predicted cognitive decline after controlling for CSF Aβ and pTau status (Fig. 3d,e; Supplementary Information Table 3), but did not predict decline in APOE3 homozygotes (Fig. 3f,g; Supplementary Information Table 4).
Figure 3. Elevated baseline CSF levels of pericyte injury biomarker sPDGFRβ predict cognitive decline in APOE4 carriers.
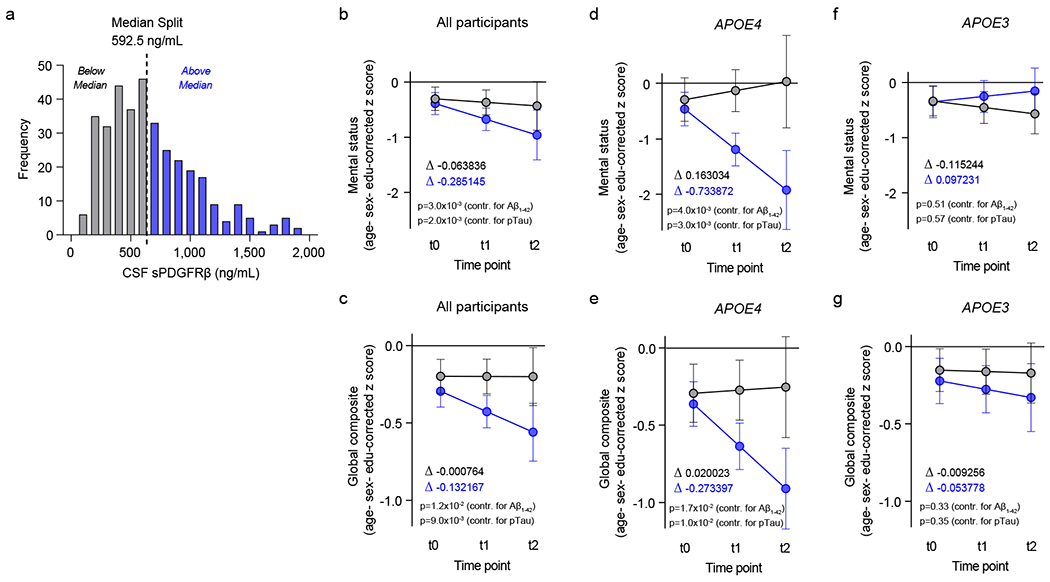
(a) Histogram frequency distribution of CSF sPDGFRβ values using median split to divide participants into two groups with high (blue, above median 600-2,000 ng/ml) and low (grey, below median; 0-600 ng/ml) baseline CSF sPDGFRβ levels. All longitudinal analyses used baseline CSF sPDGFRβ as a continuous predictor of future cognitive decline. (b-c) Linear mixed model analysis of all studied participants (n=146) followed over 2-year intervals for up to 4.5 years after baseline lumbar puncture (LP) shows that higher baseline CSF sPDGFRβ (blue) predicts greater decline in demographically-corrected mental status exam scores over time (p=0.01), which remains significant after controlling for CSF Aβ (p=0.002) and pTau (p=0.002) status (b), and in global cognitive composite scores (p=0.01), which also remains significant after controlling for CSF Aβ (p=0.017) and pTau (p=0.01) status (c). (d-e) Higher CSF sPDGFRβ (blue) in APOE4 carriers (n=58) predicts future decline in mental status exam scores (p=0.005) after controlling for CSF Aβ (p=0.004) and pTau (p=0.003) status (d), and in global cognitive composite scores (p=0.02) after controlling for CSF Aβ (p=0.02) and pTau (p=0.01) status (e). (f-g) Baseline CSF sPDGFRβ does not predict decline in APOE3 homozygotes (n=88) on either mental status (f) or global composite (g) scores regardless of CSF Aβ or pTau status. Panels b-g, for graphical depiction, separate lines indicate median split of baseline CSF sPDGFRβ (grey, below median; blue, above median). ∆ slopes provided for median split of baseline CSF sPDGFRβ groups (grey, below median; blue, above median). Time was modeled as t0 = −1 to 0.5 years post-LP, t1 = 0.5 to 2.5 years post-LP, and t2 = 2.5 to 4.5 years post-LP. The error bars are SE of the estimate. In panels b-g, significance by linear mixed model analysis; no multiple comparison correction applied. See Supplementary Information Tables 2–4 for detailed statistics.
The increase in CSF sPDGFRβ with cognitive impairment was also found on cross-sectional CDR analysis in APOE4, but not APOE3 carriers (Fig. 4a–b; Extended Data Table 3; Supplementary Information Table 5). Increased CSF levels of sPDGFRβ in APOE4 carriers correlated with increases in BBB permeability in HC and PHG (Fig. 4c,d), and elevated levels of molecular biomarkers of BBB breakdown including albumin CSF/plasma quotient, and CSF fibrinogen and plasminogen (Fig. 4e–g).
Figure 4. Elevated CSF sPDGFRβ, cyclophilin A and matrix metalloproteinase-9 in APOE4 carriers.
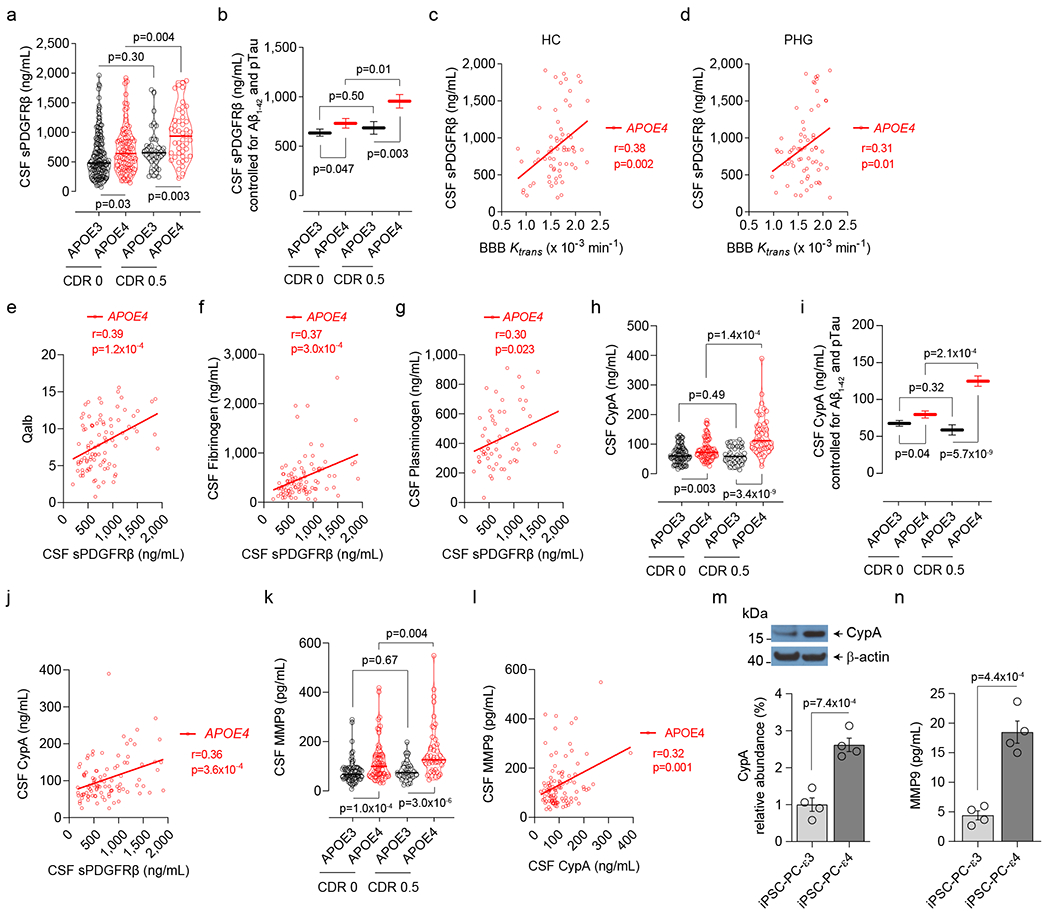
(a) CSF sPDGFRβ in CDR 0 APOE3 (black, n=152) and APOE4 (red, n=95) and CDR 0.5 APOE3 (black, n=42) and APOE4 (red, n=45) carriers. (b) CSF sPDGFRβ (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, CSF Aβ1-42 and pTau status) in CDR 0 APOE3 (black, n=152) and APOE4 (red, n=95) and CDR 0.5 APOE3 (black, n=42) and APOE4 (red, n=45) carriers. (c,d) CSF sPDGFRβ and BBB Ktrans correlation in the hippocampus (HC, n=65; c) and parahippocampal gyrus (PHG, n=65; d) in APOE4 carriers. (e-g) Correlations between CSF sPDGFRβ and albumin quotient (Qalb, n=92; e), fibrinogen (n=93; f), and plasminogen (n=57; g) in APOE4 carriers. (h) CSF cyclophilin A (CypA) in CDR 0 APOE3 (black, n=75) and APOE4 (red, n=62) and CDR 0.5 APOE3 (black, n=33) and APOE4 (red, n=45) carriers. (i) CSF CypA (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, CSF Aβ1-42 and pTau status) in CDR 0 APOE3 (black, n=75) and APOE4 (red, n=62) and CDR 0.5 APOE3 (black, n=33) and APOE4 (red, n=45) carriers. (j) CSF CypA and sPDGFRβ correlation in APOE4 carriers (n=96). (k) CSF matrix metalloproteinase-9 (MMP9) in CDR 0 APOE3 (black, n=72) and APOE4 (red, n=68) and CDR 0.5 APOE3 (black, n=33) and APOE4 (red, n=45) carriers. (l) CSF MMP9 and CypA correlation in APOE4 carriers (n=104). (m,n) CypA (m; see Extended Data Fig. 8), and secreted MMP9 in the culture medium (n), in human iPSC-derived APOE3 (ε3/ε3) and APOE4 (ε4/ε4) pericytes. Mean + SEM from four independent culture replicates. In a, h and k, continuous lines indicate median values and dotted lines interquartile range. Significance by ANCOVA for main effects and post-hoc comparisons controlling for age, sex, and education. Panels c-g, j, and l: two-tailed simple linear regression; Pearson correlation coefficient (r). Panels m and n, unpaired one-tailed Student t-test.
Next, we focused on proinflammatory cyclophilin A-matrix metalloproteinase-9 (CypA-MMP9) pathway that when activated by brain capillary pericytes in APOE4, but not APOE3, knock-in mice leads to MMP9-mediated BBB breakdown, which in turn leads to neuronal stress related to leaked blood-derived neurotoxic proteins followed by neuronal dysfunction and loss of synaptic proteins19. Activation of CypA-MMP9 pathway was also noted by brain issue analysis in degenerating brain capillary pericytes in APOE4 compared to APOE3 AD carriers16. Here, we found that living APOE4 carriers, but not APOE3 homozygotes, develop an increase in CypA CSF levels with cognitive impairment (Fig. 4h,i), which correlated with elevated CSF sPDGFRβ (Fig. 4j). APOE4, but not APOE3 carriers, had also elevated MMP9 CSF levels with cognitive impairment (Fig. 4k), which correlated with elevated CSF CypA levels (Fig. 4l), suggesting that activation of CypA-MMP9 pathway in APOE4 carriers correlates with pericyte injury similarly as shown in animal models19. There were no differences in glia, other inflammatory, and endothelial cell injury CSF biomarkers between impaired and unimpaired APOE4 and APOE3 participants, but there was an increase in neuron-specific enolase with cognitive impairment in APOE4 carriers confirming neuronal stress (Extended Data Fig. 6), consistent with HC and PHG atrophy (Fig. 1j,k).
Studies in APOE knock-in mice and mouse pericytes have shown that apoE3, but not apoE4, inhibits CypA via low-density lipoprotein receptor-related protein 1 transcriptionally, which in turn inhibits MMP9 transcriptionally19. Consistent with the mouse data, human induced pluripotent stem cells-derived APOE4 (ε4/ε4) compared to APOE3 (ε3/ε3) pericytes had substantially higher levels of CypA and secreted MMP9 (Fig. 4m,n) suggesting that apoE may control the CypA-MMP9 pathway in human pericytes in an isoform-specific manner similar as in mouse models19.
APOE4 compared to APOE3 reduced CSF Aβ1-42 and increased CSF pTau levels with cognitive impairment (Extended Data Fig. 7) as reported23, which remained significant after controlling for CSF sPDGFRβ levels (Extended Data Fig. 7). All together these findings support that Aβ and tau pathways operate independently of the BBB breakdown pathway during early stages of cognitive impairment in APOE4 carriers.
In summary, we show that 1) BBB breakdown contributes to cognitive decline in APOE4 carriers independent of AD pathology; 2) high baseline CSF levels of sPDGFRβ can predict future cognitive decline in APOE4 carriers; and 3) APOE4, but not APOE3, activates the CypA-MMP9 pathway in CSF, which may lead to accelerated BBB breakdown causing neuronal and synaptic dysfunction19. Since blockade of the CypA-MMP9 pathway in APOE4 knock-in mice restores BBB integrity followed by normalization of neuronal and synaptic functions19, one can consider that CypA inhibitors (some of which have been used in humans for non-neurological applications31) might also suppress the CypA pathway in cerebral blood vessels in APOE4 carriers that should improve cerebrovascular integrity, and the associated neuronal and synaptic deficits, slowing down cognitive impairment.
Methods
Study Participants
Participants were recruited from three sites, including the University of Southern California (USC), Los Angeles, CA, Washington University (WashU), St. Louis, MO, and Banner Alzheimer’s Institute Phoenix, AZ and Mayo Clinic Arizona, Scottsdale, AZ as a single site. At the USC site, participants were recruited through the USC Alzheimer’s Disease Research Center (ADRC): combined USC and the Huntington Medical Research Institutes (HMRI), Pasadena, CA. At the WashU site, participants were recruited through the Washington University Knight ADRC. At Banner Alzheimer’s Institute and Mayo Clinic Arizona site, participants were recruited through the Arizona Apolipoprotein E (APOE) cohort. The study and procedures were approved by the Institutional Review Boards of USC ADRC, Washington University Knight ADRC, and Banner Good Samaritan Medical Center and Mayo Clinic Scottsdale indicating compliance with all ethical regulations. Informed consent was obtained from all participants prior to study enrollment. All participants (n=435) underwent neurological and neuropsychological evaluations performed using the Uniform Data Set (UDS)32 and additional neuropsychological tests, as described below, and received a venipuncture for collection of blood for biomarker studies. A lumbar puncture (LP) was performed in 350 participants (81%) for collection of cerebrospinal fluid (CSF). The dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) for assessment of blood-brain barrier (BBB) permeability was performed in 245 participants (56%) who had no contraindications for contrast injection. Both LP and DCE-MRI were conducted in 172 participants. Among the 245 DCE-MRI participants, 74 and 96 were additionally studied for brain uptake of amyloid and tau PET radiotracers, respectively, as described below. All biomarker assays, MRI, and PET scans were analyzed by investigators blinded to the clinical status of the participant.
Participant Inclusion and Exclusion Criteria
Included participants (≥45 years of age) were confirmed by clinical and cognitive assessments to be either cognitively normal or at the earliest symptomatic stage of Alzheimer disease. A current or prior history of any neurological or psychiatric conditions that might confound cognitive assessment, including organ failure, brain tumors, epilepsy, hydrocephalus, schizophrenia, and major depression, was exclusionary. Participants were stratified by APOE genotype as APOE4 carriers (ε3/ε4 and ε4/ε4) and APOE4 non-carriers (ε3/ε3) also defined as APOE3 homozygotes who were cognitively normal or with mild cognitive dysfunction, as determined by clinical dementia rating (CDR) scores33 and the presence of cognitive impairment in one or more cognitive domains based on comprehensive neuropsychological evaluation including performance on ten neuropsychological tests assessing memory, attention/executive function, language and global cognition. For all analyses individuals with ε3/ε4 and ε4/ε4 alleles were pooled together in a single APOE4 group, as we did not find in the present cohort consisting of 82-86% ε3/ε4 and 14-18% ε4/ε4 participants (depending on the outcome measure) a significant difference between the two versus one ε4 allele for the studied parameters including the BBB permeability unidirectional transfer constant Ktrans values and sPDGFRβ CSF values (see statistical section below). Individuals have been additionally stratified by Aβ and pTau CSF analysis as either Aβ-positive (Aβ1-42+, <190 pg/mL) or Aβ-negative (Aβ1-42-, >190 pg/mL), or pTau-positive (pTau+, >78 pg/mL) or pTau-negative (pTau−, <78 pg/mL), using accepted cutoff values7,24,25.
Participants were excluded if they were diagnosed with vascular cognitive impairment or vascular dementia. Clinical diagnoses were made by neurologists and criteria included whether the patient 1) had a known vascular brain injury and 2) the clinician judged that the vascular brain injury played a role in their cognitive impairment, and/or pattern and course of symptoms. In addition to clinical diagnosis, presence of vascular lesions was confirmed by moderate-to-severe white matter changes and lacunar infarcts by fluid-attenuated inversion recovery (FLAIR) MRI and/or subcortical microbleeds by T2*-weighted MRI1. Participants were also excluded if they were diagnosed with Parkinson’s disease, Lewy body dementia or frontotemporal dementia. History of a single stroke or transient ischemic attack was not an exclusion unless it was related to symptomatic onset of cognitive impairment. Participants also did not have current contraindications to MRI and were not currently using medications that might better account for any observed cognitive impairment.
Clinical Exam
Participants underwent clinical assessments according to UDS procedures harmonized across all study sites, including clinical interview and review of any neurocognitive symptoms and health history with the participant and a knowledgeable informant. A general physical and neurologic exam was conducted. The CDR assessment was conducted in accordance with published standardization procedures, including standardized interview and assessment with the participant and a knowledgeable informant. In accordance with current diagnostic models for cognitive and biological research criteria for cognitive impairment and AD23, participants were separately stratified by cognitive impairment and AD biomarker abnormality using established cutoffs for CSF Aβ1-42 and pTau7,24,25. Cognitive impairment was determined based on global CDR score and neuropsychological impairment in one or more cognitive domains.
Vascular Risk Factors
The vascular risk factor (VRF) burden in each participant was evaluated through physical examination, blood tests, and clinical interviews with the participant and informant; history of cardiovascular disease (heart failure, angina, stent placement, coronary artery bypass graft, intermittent claudication), hypertension, hyperlipidemia, type 2 diabetes, atrial fibrillation, and transient ischemic attack or minor stroke were investigated. The total VRF burden was defined by the sum of these risk factors, as previously described7. We assigned an elevated VRF burden to individuals with 2 or more VRFs. This threshold was adopted based on previous studies showing that the presence of two or more VRFs is associated with occult cerebrovascular disease at autopsy in older adults with AD, whereas a single VRF is common and not necessarily associated with increased cerebrovascular disease in this population34,35.
Cognitive Domain Impairment Evaluation
Impairment in one or more cognitive domains was based on performance on comprehensive neuropsychological testing, using previously described neuropsychological criteria for cognitive impairment7. All participants underwent neuropsychological testing that included the UDS battery (version 2.0 or 3.0) plus supplemental neuropsychological tests at each site. Raw test scores were converted to age-, sex- and education-corrected z-scores using the National Alzheimer’s Coordinating Center (NACC) regression-based norming procedures (www.alz.washington.edu). Normalized z-scores from a total of 10 neuropsychological tests were evaluated in determining domain impairment, including three tests per cognitive domain (memory, attention/executive function and language) and one test of global cognition. Impairment in one or more cognitive domains was determined using previously described neuropsychological criteria, and was defined as a score >1 standard deviation (SD) below norm-referenced values on two or more tests within a single cognitive domain or three or more tests across cognitive domains36. Prior studies have established improved sensitivity and specificity of these criteria relative to those employing a single test score, as well as adaptability of this diagnostic approach to various neuropsychological batteries36,37. Participants were excluded from cognitive domain analyses if they had less than 90% complete neuropsychological test data (i.e., 53, 24, and 82 participants were excluded for MRI, PET, and CSF analyses, respectively). Included participants were classified as 0, 1, or 2+ based on the number of cognitive domains with two or more impaired test scores.
Test battery specifics for each UDS version and recruitment site are as follows. i) Global cognition: MMSE for UDS version 238 and MoCA for UDS version 339. ii) Memory: The Logical Memory Story A Immediate and Delayed free recall tests [modified from the original Wechsler Memory Scales – Third Edition (WMS-III)] for UDS version 2 and the Craft Stories Immediate and Delayed free recall for UDS version 3. For supplemental tests the USC participants underwent the California Verbal Learning Test – Second Edition (CVLT-II) and the Selective Reminding Test (SRT) sum of free recall trials. Norm-referenced scores for these supplemental test scores were derived from a nationally representative sample published with the test manual (CVLT-II)40 and in studies of normally aging adults (SRT). iii) Attention and executive function: The Trails A, Trails B, and Wechsler Adult Intelligence Scale - Revised (WAIS-R) Digit Span Backwards tests for UDS version 2 and the Trails A, Trails B and Digit Span Backwards tests for UDS version 3. iv) Language: The Animal Fluency, Vegetable Fluency, and Boston Naming Tests for UDS version 2 and Animal Fluency, Vegetable Fluency, and Multilingual Naming Test (MINT) for UDS version 3.
Magnetic Resonance Imaging and Analysis
The MRI data sets were obtained at Mark and Mary Stevens Neuroimaging and Informatics Institute of USC and Washington University of St. Louis. We developed a standardized high-resolution 3T MR imaging brain scan protocol. For USC site, a Siemens 3T Prisma scanner was used with a product 32-channel head receive coil and body transmit coil. For WashU site, a Siemens 3T mMR with 20-channel head coil and Siemens 3T Vida with 64-channel head coil were used. Anatomical coronal spin echo T2-weighted scans were first obtained through the hippocampi (TR/TE 8020/50 ms, NEX = 1, slice thickness 2 mm with 2 mm gap between slices, FOV = 175 x 175 mm, matrix size = 448 x 448). Baseline coronal T1-weighted maps were then acquired using a T1-weighted 3D volumetric interpolated breath-hold sequence (VIBE) sequence and variable flip angle method using flip angles of 2°, 5°, 10°, 12°, and 15°. Coronal DCE-MRI covering the hippocampi and temporal lobes were acquired using a T1-weighted 3D VIBE sequence (FA = 15°, TR/TE = 5.14/2.18 ms, NEX = 1, slice thickness 5 mm with no gap, FOV 175 x 175 mm, matrix size 320 x 320, voxel size was 0.550 x 0.550 x 5 mm3). This sequence was repeated for a total of 16 min with an approximate time resolution of 15.4 sec. Gadolinium-based contrast agent (GBCA), Gadoterate meglumine (Dotarem®, Guerbet, France) (0.05 mmol/kg), was administered intravenously into the antecubital vein using a power injector, at a rate of 3 mL/s followed by a 25 mL saline flush, 30 s into the DCE scan.
The standardization and optimization of the MRI protocol required several tests performed on a phantom. Specifically, scanner characterization and calibration sequences including B0, T1, and variable flip-angle mapping were implemented, optimized, and applied. After the achievement of good results in terms of quality control and reproducibility, we standardized and employed the same pre-contrast and dynamic T1-weighted protocols at both USC and Washington University sites. Of note, all the other MR sequences were identical too on both scanners.
In order to minimize the inter-site variability, the entire MRI protocol including the anatomical and DCE pulse sequences were 100% mirrored from one site to another. To minimize inter-site variability, the same contrast agent Gadoterate meglumine (Dotarem®) were injected to participants at the same concentration (0.05 mmol/kg). Finally, the same exact pre- and post-processing analysis pipeline was applied for both sites which includes T1 multi-FA mapping using linear fitting and Patlak-based DCE modeling using the arterial input function determined in each individual from the internal carotid artery. Applying all the above cited factors significantly limited inter-site variability.
The consistency of the results from the two sites has been additionally confirmed by our previous publication7. In brief, we performed the analysis of the combined DCE datasets from both USC and WashU sites, and additionally site-specific analysis for each of the two sites separately, which showed no statistically significant differences across sites. Recently, we invited a subset of 52 participants for an additional T1-weighted scan without contrast (using the same scanner and same MR pulse sequences) after their first DCE-MRI41 and measured both B0 and T1 values at 2-year interval. This study showed that the results were unchanged and consistent across the scans, supporting minimal intra-site variability.
Quantification of the Blood-Brain Barrier Permeability
Quantification of Regional Brain Volumes
HC and PHG morphometry were performed using the FreeSurfer (v5.3.0) software package42 (http://surfer.nmr.mgh.harvard.edu/), as previously performed7. HC and PHG were segmented using FreeSurfer Desikan-Killiany and subcortical atlases43,44. Then, regional volumes (mm3) were derived accordingly. The technical details of this procedure are described in previous publications45,46. Data processing and visualization were performed using the Laboratory of Neuro Imaging (LONI) pipeline system (http://pipeline.loni.usc.edu) and Quantitative Imaging Toolkit47–49.
Positron Emission Tomography and Analysis
The PET image acquisition was performed at the Molecular Imaging Center of USC or Mallinckrodt Institute of Radiology of WashU. Amyloid and tau PET studies were conducted using 18F-Florbetaben (FBB) or 18F-Florbetapir (FBP) and 18F-Flortaucipir (AV1451), respectively. FBB (Life Molecular Imaging, Inc.) was obtained from SOPHIE, Inc. for USC site, while FBP was provided by Eli Lilly and Company for WashU site. For all amyloid PET analysis FBP and FBB datasets were combined. AV1451 was provided by Avid Radiopharmaceuticals, Inc. for USC site and was produced by the Mallinckrodt Institute of Radiology for WashU site. A Siemens Biograph 64 PET scanner was used at USC site. At WashU site, FBP scans were acquired on a Siemens mMR and AV1451 scans were acquired on a Siemens Biograph mCT. The mCT session was used for attenuation correction of the mMR scans. Participants were injected with 300 MBq (±10%) of FBB or 370 MBq (±10%) of FBP. FBB and FBP images were acquired from 90 to 110 min and 50 to 70 min, respectively, after injection in accordance with the manufacturers’ recommendation. Individuals who participated in amyloid and tau PET studies also had their DCE-MRI scan within a 2.2 ± 0.9 and 2.1 ± 0.6 month period of their amyloid and tau PET scans, respectively.
Briefly, a computed tomography (CT) scan was performed first for attenuation correction before each PET imaging session. The downloaded PET images from FBB, FBP, and AV1451 tracers were processed by using standard uptake value maps (SUV in g/mL). All PET images were co-registered to structural high-resolution 3D T1-weighted Magnetization Prepared Rapid Acquisition Gradient Echo (MP-RAGE) MRI images using FSL-FLIRT (FMRIB’s Linear Image Registration Tool)50. FreeSurfer-segmented cerebellum was used as a reference tissue to normalize for both amyloid and tau51.
After co-registration of PET images into an anatomical reference image (MNI152 standard-space), the Statistical Parametric Mapping (SPM12) was used for group comparison in a voxel-by-voxel basis. Age at time of PET imaging session, sex, and education were introduced in a multiple regression model as covariates. Level of significance was set to p<0.001 for amyloid and p<0.005 for tau (uncorrected p values) with the minimum number of voxels (Ke) in a cluster of 50.
Additionally, given the known AV1451 off-target ligand binding in the choroid plexus (CP)52,53, which can contribute to HC regional AV1451 signal due to close proximity of CP to HC and relatively low spatial resolution of PET scans (i.e., ~6-mm voxel size), we took advantage of visualizing CP by DCE-MRI, also performed in these individuals, that allowed us to subtract contribution of the CP signal to the HC AV1451 proper signal. The following steps were employed to correct for off-target ligand binding to CP (see Extended Data Fig. 5): Step 1) HC masks were generated from the 3D T1-weighted MP-RAGE; Step 2) CP masks were generated from the T1-weighted VIBE image post-GBCA (FA = 15°) image; Step 3) HC and CP masks were overlaid; and Step 4) CP overlap with HC mask was subtracted to obtain CP-corrected HC PET signal after adding 6-mm voxel size on top of CP mask generated from DCE data. Representative images of HC AV1451 PET signal before and after applying the CP correction are shown in Extended Data Fig. 5.
We next quantified regional changes in amyloid and tau SUV ratio (SUVR) in relation to regional DCE-MRI Ktrans values in all participants stratified by APOE genotype. The regional SUVR values were taken from the FreeSurfer-segmented HC, PHG, OFC28, and ITG29. The BBB Ktrans constant (DCE-MRI) was determined in all participants (Extended Data Tables 2a,b). This includes those who received both amyloid and tau (n=58), only amyloid (n=9) or only tau (n=29).
Lumbar Puncture and Venipuncture
Participants underwent a lumbar puncture and venipuncture in the morning after an overnight fast. The CSF was collected in polypropylene tubes, processed (centrifuged at 2,000 g, 4°C, 10 min USC site; 5 min WashU site), aliquoted into polypropylene tubes and stored at −80°C until assay. Blood was collected into ethylenediaminetetraacetic acid (EDTA) tubes and processed (centrifuged at 2000 g, 4°C, 10 min USC site; 5 min WashU site). Plasma and buffy coat were aliquoted in polypropylene tubes and stored at −80°C; buffy coat was used for DNA extraction and APOE genotyping.
APOE Genotyping
DNA was extracted from buffy coat using the Quick-gDNA Blood Miniprep Kit (Catalog no. D3024, Zymo Research, Irvine, CA). APOE genotyping was performed via polymerase chain reaction (PCR)-based retention fragment length polymorphism analysis, as previously reported7.
Molecular Assays
Quantitative Western Blotting of sPDGFRβ
The quantitative Western blot analysis was used to detect sPDGFRβ in human CSF (ng/mL), as we previously reported7,8.
BBB Breakdown Biomarkers
Albumin quotient (Qalb the ratio of CSF-to-plasma albumin levels) and CSF levels of fibrinogen and plasminogen were determined using enzyme-linked immunosorbent assay (ELISA), as we previously reported7,8.
Cyclophilin A (CypA)
We developed a novel CypA assay on the Meso Scale Discovery (MSD) platform. Standard-bind 96-well plates (Catalog no. L15XA-3 / L11XA-3, MSD, Rockville, MD) were spot-coated with 5 μL per well of 40 μg/mL rabbit polyclonal cyclophilin A antibody (Catalog no. 10436-T52, Sino Biological, Wayne, PA) prepared in 0.03% Triton X-100 in 0.01 M PBS pH 7.4 solution. The plates were left undisturbed overnight to dry at room temperature. The next day, the plates were blocked with 150 μL per well of Blocking One (Catalog no. 03953-95, Nacalai Tesque, Japan) and incubated for exactly one hour with shaking. Meanwhile, samples and standards were prepared in Blocking One blocking buffer. Different concentrations ranging from 3.5-200 ng/mL of a recombinant human cyclophilin A protein (Catalog no. 3589-CAB, R&D Systems, Minneapolis, MN) were used to generate a standard curve. All CSF samples were diluted 1:3. After blocking, the plates were manually washed 3 times with 200 μL per well of wash buffer (in 0.05% Tween-20 in 0.01 M PBS pH 7.4). The prepared samples or standards were added at 25 μL per well, and the plates were incubated overnight at 4°C with shaking.
The next day, the plates were washed 3 times, and 25 μL per well of 1 μg/mL sulfo-tagged mouse monoclonal cyclophilin A detection antibody (Catalog no. ab58144, Abcam, Cambridge, MA), prepared in Blocking One. The plates were incubated for 90 min at room temperature with shaking. Next, plates were washed 4 times, then 150 μL per well of 2x Read Buffer T with surfactant (Catalog no. R92TC-3, MSD, Rockville, MD) was added and the plates were read immediately on the MSD SECTOR Imager 6000 (MSD, Rockville, MD) with electrochemiluminescence detection.
The raw readings were analyzed by subtracting the average background value of the zero standard from each recombinant standard and sample readings. A standard curve was constructed by plotting the recombinant standard readings and their known concentrations and applying a nonlinear four-parameter logistics curve fit. The CypA concentrations were calculated using the samples’ reading and the standard curve equation; the result was corrected for the sample dilution factor to arrive at the CypA concentration in the CSF samples.
Matrix Metalloproteinase-9 (MMP9)
CSF levels of MMP9 were determined using human MMP9 Ultra-Sensitive Kit from MSD (Cat. No. K151HAC).
Neuron-Specific Enolase
CSF levels of neuron-specific enolase (NSE) were determined using ELISA (Cat. No. E-80NEN, Immunology Consultant Laboratories, Portland, OR). The company no longer sells this product; thus, this analyte has been measured in a majority of participants but not in individuals that most recently enrolled in the study.
S100B
CSF levels of the astrocyte-derived cytokine, S100 calcium-binding protein B (S100B), were determined using ELISA (Cat. No. EZHS100B-33K, EMD Millipore, Billerica, MA).
Inflammatory Markers
MSD multiplex assay was used to determine CSF levels of intercellular adhesion molecule 1 (ICAM1) (Cat. No. K15198D, MSD, Rockville, MD), and interleukin-6 (IL6), IL-1β, tumor necrosis factor α (TNFα), and interferon gamma (IFNγ) (Cat. No. K15049G, MSD, Rockville, MD).
Aβ Peptides
MSD multiplex assay (Cat. No. K15200E, MSD, Rockville, MD) was used to determine CSF levels of Aβ1-42. Participants were stratified based on CSF analysis as either Aβ1-42-positive (Aβ1-42+, <190 pg/mL) or Aβ1-42-negative (Aβ-, >190 pg/mL) using the accepted cutoff values as previously reported for the MSD 6E10 Aβ peptide assay24.
Tau
Phosphorylated tau (pT181) was determined by ELISA (Cat. No. 81581, Innotest®, Fujirebio US, Inc., Malvern, PA). Participants were stratified based on CSF analysis as either pTau181-positive (pTau+, >78 pg/mL) or pTau181-negative (pTau−, <78 pg/mL), using the accepted cutoff value as previously reported25.
Human Induced Pluripotent Stem Cells (iPSCs)
iPSC lines were generated by reprogramming of APOE ε4/ε4 and APOE ε3/ε3 control and AD donor skin fibroblasts as recently reported54. Reprogramming was performed using integration-free Sendai virus vectors and passaged cells to passage 15 and confirmed normal karyotype. hiPSCs were maintained on Matrigel (Corning) in mTeSR1 (Catalog no. 85850, StemCell Technologies, Vancouver, BC, Canada) supplemented with 10 ng/mL FGF2 StemBeads (StemCultures) or mTeSR plus (StemCell Technologies) every other day.
Differentiation of iPSCs into Pericytes
Differentiation of iPSCs into pericytes was carried out as described previously55. Briefly, iPSCs were dissociated with ReLeSR (catalog no. 05872, StemCell Technologies) and seeded at 55,000 cells/cm2 in Essential 8 medium (Catalog no. A1517001, ThermoFisher, Waltham, MA, USA) supplemented with ROCK inhibitor Y-27632 (10 μM, catalog no. 72304, StemCell Technologies) on Matrigel (0.5 mg/6-well plate, Catalog no. 354230, Corning, NY, USA). After 24 hours incubation, the iPSCs were switched into STEMdiff Mesoderm Induction Medium (MIM, Catalog no. 05221, StemCell Technologies) for 5 days with daily medium change. On day 6 of MIM treatment, the cells were plated on Matrigel at 25,000 cells/cm2 in Pericyte Medium (Catalog no. 1201, ScienCell, Carlsbad, CA, USA) for an additional 7 days. The differentiated cells were dissociated with Accutase (catalog no. 07920, Stemcell Technologies). Following incubation with human PDGFRβ biotinylated antibody (Catalog no. BAF385, R&D Systems, Minneapolis, MN, USA), the cells were incubated with anti-biotin microbeads (Catalog no. 130-090-485, Miltenyi Biotec, Bergisch Gladbach, NRW, Germany) and magnetically sorted using MACS LS columns (Catalog no. 130-042-401, Miltenyi Biotec) following the manufacturer’s instructions. Sorted pericytes were plated at a density of 25,000 cells/cm2 on Matrigel-coated coverslips for immunocytochemistry analyses or ploy-L-lysine-coated six-well culture plates for Western blot analyses. Differentiated pericytes were positive for pericyte markers PDGFRβ, CD13, and NG2, and negative for endothelial marker CD31, astrocytic marker GFAP, and microglial marker CD11b.
Statistical Analyses
Prior to performing statistical analyses, we first screened for outliers using the Grubbs’ test, also called the ESD method (Extreme Studentized Deviate), applying a significance level of α=0.01 (https://www.graphpad.com/quickcalcs/grubbs1/). For each of the outliers identified, a secondary index of outlier influence was applied using the degree of deviation from the mean (greater than +/− 3 SDs)56. Using these stringent criteria, a total of 5 outliers one each in main Figure panels 1j, 1k and 2j, and one each in Extended Data Figs. 6a and 6b, were removed from analyses, as indicated in the legends of these figures. Continuous variables were also evaluated for departures from normality through quantitative examination of skewness and kurtosis, in addition to visual inspection of frequency distributions. Where departures of normality were identified, log10-transformations were applied, and distribution normalization was confirmed prior to parametric analyses. This has been done for main Figs. 4h and 4k, and Extended Data Figs. 7a, 7b, 7d, and 7e. Since use of log10-transformations accounts for any non-normality this obviated the need for outliers exclusion.
DCE-MRI Ktrans, and CSF sPDGFRβ and CypA
Regional DCE-MRI Ktrans values and CSF sPDGFRβ, CypA and MMP9 levels were compared across the entire sample stratified by APOE status. Since in APOE4 group relatively small number of participants were homozygous ε4/ε4 compared to heterozygous ε3/ε4, i.e., 14% for DCE-MRI analysis, and 18% for sPDGFRβ analysis, and initial comparisons between ε4/ε4 and ε3/ε4 carriers did not show any significant differences in regional HC and PHG DCE-MRI Ktrans values (CDR 0, pHC=0.19 and pPHG=0.54 (PHG); CDR 0.5, pHC=0.22 and pPHG=0.84) or CSF sPDGFRβ levels (CDR 0, p=0.23; CDR 0.5, p=0.47), all subsequent analyses combined APOE4 carriers (ε3/ε4 and ε4/ε4), and compared these participants to APOE3 carriers (ε3/ε3) stratified by cognitive impairment status (CDR 0 vs 0.5 and 0 vs 1 vs 2+ cognitive domains impairment using analysis of covariance (ANCOVA) with false discovery rate (FDR) correction for multiple comparisons (see details below). For CDR analyses, model covariates included age, sex, and education. Cognitive domain impairment was determined using age, sex, and education-corrected values, so these covariates were not additionally included in the analyses. Additional post-hoc ANCOVA analyses evaluated whether observed differences remained significant after stratifying APOE4 carriers by CSF Aβ1-42 and pTau status, and after statistically controlling for CSF Aβ1-42 and pTau status and regional brain volume in APOE4 non-carriers and carriers. These findings were also confirmed by hierarchical logistic regression models using the same covariates.
PET AD Biomarkers
In a subset of participants who underwent amyloid and tau PET imaging together with DCE-MRI studies, we used ANCOVA models controlled for age, sex and education to compare regional amyloid and tau ligand binding and DCE-MRI values in a set of APOE4 non-carriers and carriers within a priori regions of interest, based on prior imaging studies, to determine whether distinct regional pathologies were observed by APOE4 carrier status.
Baseline CSF sPDGFRβ as a continuous predictor of cognitive decline
For linear mixed model analysis, baseline CSF sPDGFRβ was a continuous predictor of demographically-corrected global cognitive change over 2-year follow up intervals, controlling for CSF Aβ1-42 and CSF pTau status. Global cognition was indexed by age-, sex-, and education-corrected z-scores on mental status exam (MMSE or MoCA) and as the global cognitive composite of all age-, sex-, and education-corrected neuropsychological test z-scores (see above for list of neuropsychological tests). Time was modeled with date of lumbar puncture as baseline (T0) with two follow up intervals of 2 years each (T1-2). Additional analyses confirmed all findings when time was modeled as time since baseline, with date of lumbar puncture as baseline (T0) and follow up as annual intervals (T1-n).
All longitudinal mixed models treated CSF sPDGFRβ as a continuous predictor. Although we have previously established CSF sPDGFRβ as a marker of pericyte injury7,8,57, the optimal cutoff value for abnormal CSF sPDGFRβ levels indicative of pericyte injury remains unknown. Autopsy studies are required to determine optimal in vivo biomarker cutoff values predictive of gold-standard neuropathological measures, such as studies conducted for CSF and PET markers of amyloid and tau. Given the lack of available autopsy data relating CSF sPDGFRβ to neuropathological markers of pericyte injury, we chose to divide participants by CSF sPDGFRβ values using median split for the purposes of visual display only (higher CSF sPDGFRβ was above sample median and lower CSF sPDGFRβ was below sample median). The median split was not used in statistical analyses and was only used for the purpose of visual display (see Figure 3a) for statistical parameters from analyses using CSF sPDGFRβ as a continuous predictor of cognitive decline).
Correlational Analyses
Pearson product moment correlations were used to evaluate relationships among CSF sPDGFRβ, CypA, MMP9, fibrinogen, plasminogen and hippocampal and parahippocampal BBB Ktrans levels among APOE4 carriers.
Multiple Comparison Correction and Missing Data
Given the large number of analyses, FDR-correction was applied to p-values for primary study outcomes (i.e., DCE-MRI, sPDGFRβ) evaluated in the entire sample by APOE4 carrier status and CDR status using the Benjamini-Hochberg method58 in ANCOVA and logistic regression models controlling for age, sex, education, brain volume, and CSF Aβ1-42 and pTau status (for DCE-MRI analyses). Post-hoc confirmatory analyses in participant subsets further evaluating independence of CSF and PET markers of amyloid and tau, evaluation of mechanistic markers (i.e., CypA and MMP9), and longitudinal analysis of predictive value of CSF sPDGFRβ were not multiple comparison corrected. For longitudinal data with variable follow up, we utilized linear mixed model analyses with the account for missing data via the missing at random assumption.
Extended Data
Extended Data Figure 1. Regional BBB Ktrans constant in eight additional brain regions in APOE4 carriers and non-carriers (APOE3) with CDR status 0 and 0.5.
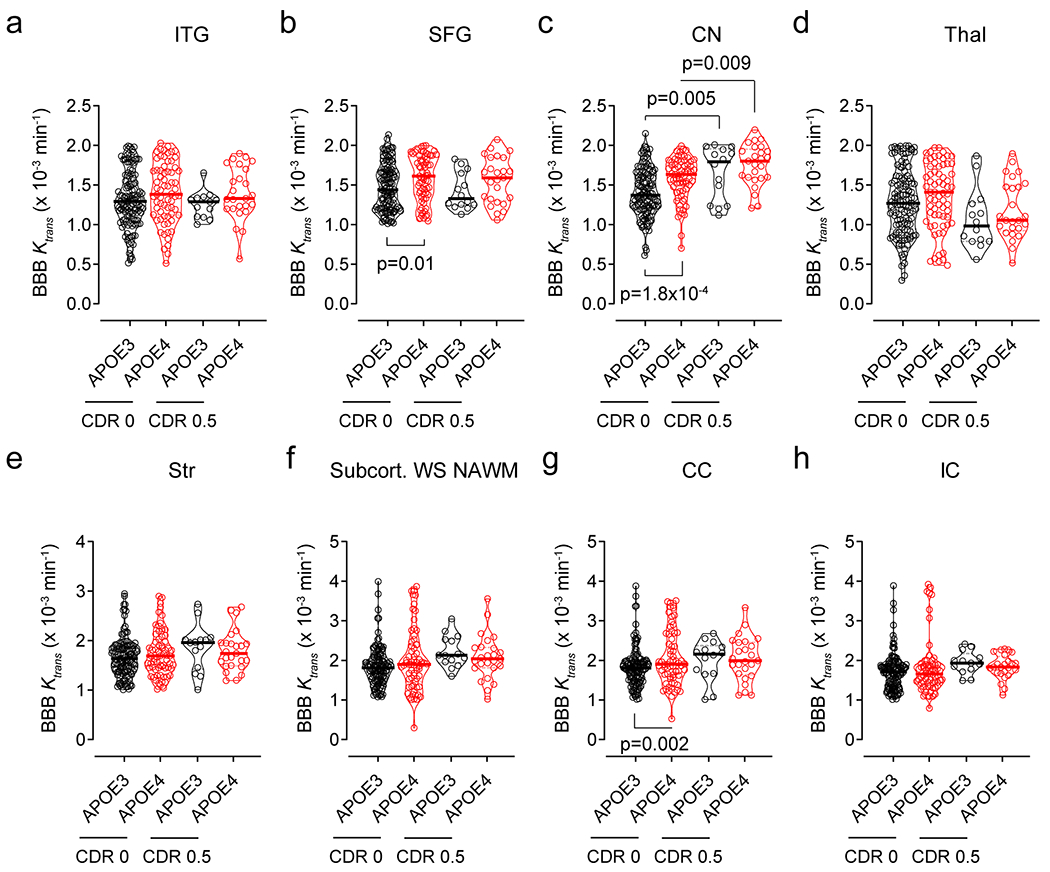
DCE-MRI BBB permeability, Ktrans constant, in the inferior temporal gyrus (ITG, a), superior frontal gyrus (SFG, b), caudate nucleus (CN, c), thalamus (Thal, d), striatum (Str, e), subcortical watershed normal-appearing white matter (Subcort. WS NAWM, f), corpus callosum (CC, g), and internal capsule (IC, h) in CDR 0 APOE3 (black, n=128) and APOE4 (red, n=68) carriers, CDR 0.5 APOE3 (black, n=14) and APOE4 (red, n=25) carriers. Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. Significance by ANCOVAs for main effects and post-hoc comparisons controlling for age, sex, and education.
Extended Data Figure 2. BBB breakdown in the hippocampus and parahippocampal gyrus in APOE4 carriers increases with cognitive domain impairment.
(a,b) DCE-MRI BBB permeability, Ktrans constant, in the hippocampus (HC, a) and parahippocampal gyrus (PHG, b) in individuals with 0 cognitive domain impaired that are APOE3 (black, n=70) and APOE4 (red, n=40) carriers, 1 cognitive domain impaired that are APOE3 (black, n=18) and APOE4 (red, n=21) carriers, and 2+ cognitive domains impaired that are APOE3 (black, n=7) and APOE4 (red, n=12) carriers. (c,d) Ktrans (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, CSF Aβ1-42 and pTau status, and HC and PHG volumes) in the HC (c) and PHG (d) in individuals with 0 cognitive domain impaired that are APOE3 (black, n=70) and APOE4 (red, n=40) carriers, 1 cognitive domain impaired that are APOE3 (black, n=18) and APOE4 (red, n=21) carriers, and 2+ cognitive domains impaired that are APOE3 (black, n=7) and APOE4 (red, n=12) carriers. Panels a and b: Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. Significance by ANCOVA for main effects and post-hoc comparisons controlling for age, sex, and education. All ANCOVA omnibus tests remained significant at false discovery rate threshold of 0.05.
Extended Data Figure 3. Regional BBB Ktrans constant in eight additional brain regions in APOE4 carriers and non-carriers (APOE3) with different degree of cognitive domain impairment.
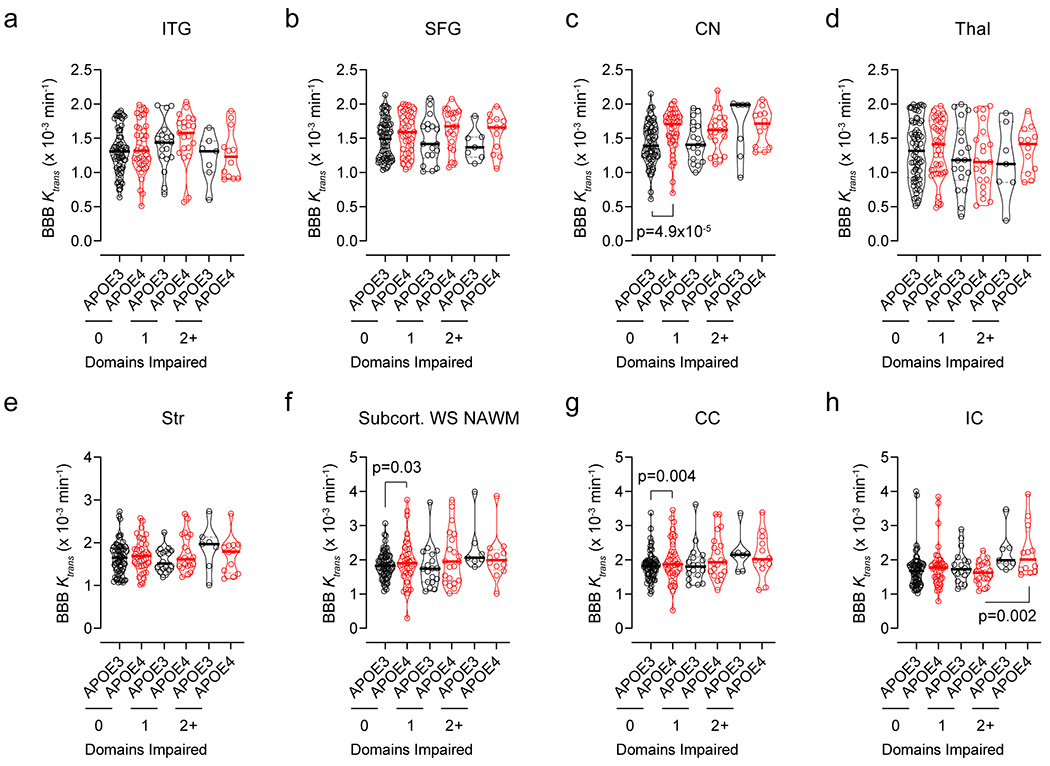
DCE-MRI BBB permeability, Ktrans constant, in the inferior temporal gyrus (ITG, a), superior frontal gyrus (SFG, b), caudate nucleus (CN, c), thalamus (Thal, d), striatum (Str, e), subcortical watershed normal-appearing white matter (Subcort. WS NAWM, f), corpus callosum (CC, g), and internal capsule (IC, h) in individuals with 0 cognitive domain impaired that are APOE3 (black, n=70) and APOE4 (red, n=40) carriers, 1 cognitive domain impaired that are APOE3 (black, n=18) and APOE4 (red, n=21) carriers, and 2+ cognitive domains impaired that are APOE3 (black, n=7) and APOE4 (red, n=12) carriers. Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. Significance tests from ANCOVAs for main effects and post-hoc comparisons controlling for age, sex, and education.
Extended Data Figure 4. Regional BBB Ktrans constant in all studied brain regions in APOE4 carriers and non-carriers (APOE3) in relation to vascular risk factors.
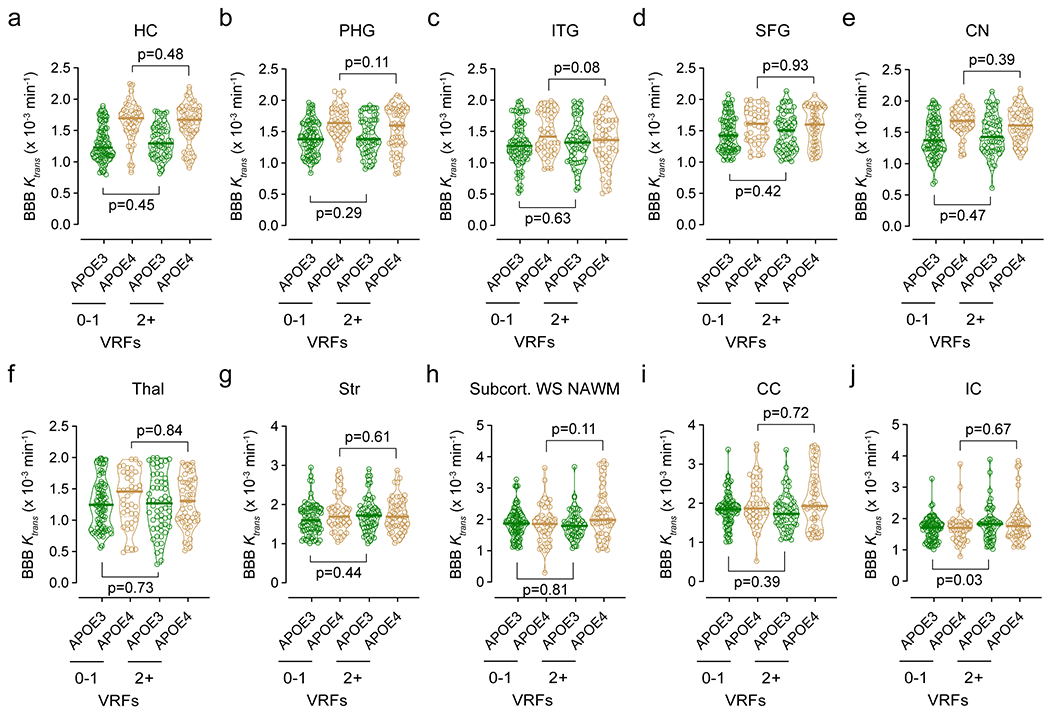
DCE-MRI BBB permeability, Ktrans constant, in the hippocampus (HC, a), parahippocampal gyrus (PHG, b), inferior temporal gyrus (ITG, c), superior frontal gyrus (SFG, d), caudate nucleus (CN, e), thalamus (Thal, f), striatum (Str, g), subcortical watershed normal-appearing white matter (Subcort. WS NAWM, h), corpus callosum (CC, i), and internal capsule (IC, j) in APOE3 (black, n=80) and APOE4 (red, n=42) carriers with 0-1 vascular risk factors (VRFs), and APOE3 (black, n=58) and APOE4 (red, n=51) carriers with 2+ VRFs. Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. Significance by ANCOVAs for main effects and post-hoc comparisons controlling for age, sex, and education (ns=non-significant).
Extended Data Figure 5. Amyloid and tau PET analysis in APOE4 carriers and correction of 18F-AV1451 off-target binding in the choroid plexus.
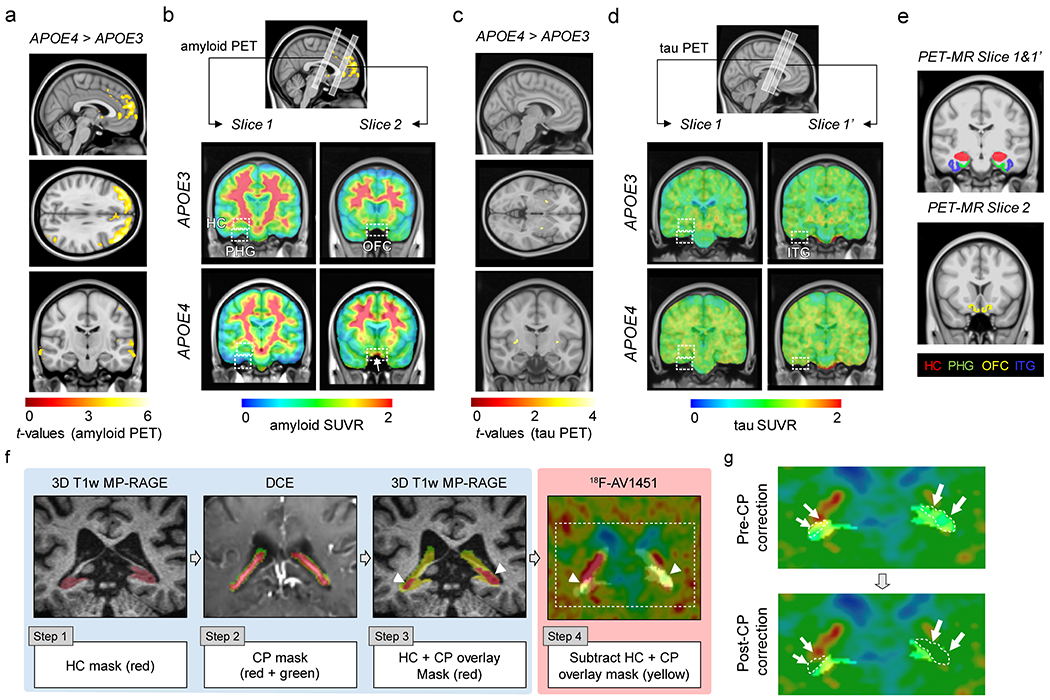
All studies were performed in individuals with clinical dementia rating score 0. Amyloid and tau PET studies were conducted using 18F-Florbetaben (FBB) or 18F-Florbetapir (FBP), and 18F-Flortaucipir (AV1451), respectively. For amyloid PET data analysis, FBP and FBB datasets were combined. (a) Uptake of amyloid tracers by the orbital frontal cortex (OFC) in APOE4 (n=29) relative to APOE3 (n=45) carriers (voxel-wise 2-sample one-tailed t-tests). (b) Representative amyloid PET Standardized Uptake Value Ratios (SUVR) maps from APOE3 homozygote (APOE3) (upper) and APOE4 carrier (APOE4) (lower). Slices 1 and 2, regions-of-interest (ROIs) for amyloid PET and BBB DCE-MRI scans (see e). Arrow, amyloid tracer uptake by OFC. The APOE3 and APOE4 representative images used FBP. (c) Uptake of tau tracer shows undetectable tau accumulation in APOE3 (n=60) or APOE4 (n=37) carriers (voxel-wise 2-sample one-tailed t-tests). (d) Representative tau PET SUVR maps from APOE3 (upper) and APOE4 (lower) carriers. Slice 1 and slice 1’, ROIs for tau PET and BBB DCE-MRI scans (see e). (e) Coronal 3D scans of regions studied in main Figure 2: hippocampus (HC; red), parahippocampal gyrus (PHG; green), medial orbital frontal cortex (OFC; yellow), and inferior temporal gyrus (ITG; blue). (f) Correction of 18F-AV1451 off-target binding in the choroid plexus: Step 1) Hippocampus (HC) masks were generated from the 3D T1-weighted MP-RAGE; Step 2) Choroid plexus (CP) masks were generated from the T1-weighted VIBE image post-GBCA (flip angle = 15°) image; Step 3) HC and CP masks were overlaid (arrowheads, red); and Step 4) CP overlap with HC masks (arrowheads, yellow) were subtracted to obtain CP-corrected HC tau PET signal after adding 6-mm voxel size on top of CP mask generated from DCE data. (g) Representative images of HC tau PET signal before (left) and after (right) applying the CP correction (arrows and white dotted lines = overlap between HC and CP).
Extended Data Figure 6. CSF biomarkers of glia and inflammatory response and endothelial and neuronal cell injury in APOE4 carriers and non-carriers (APOE3).
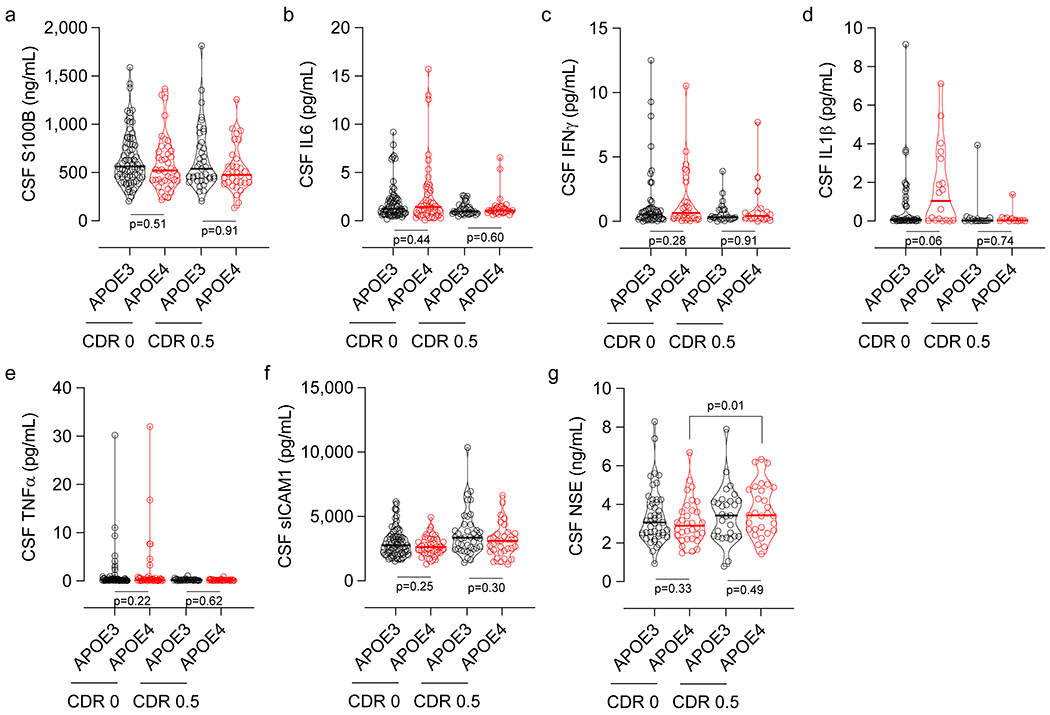
(a) CSF astrocytic S100 calcium-binding protein B (S100B) levels in CDR 0 APOE3 (black, n=77) and APOE4 (red, n=41) carriers, and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=32) carriers. (b) CSF interleukin 6 (IL6) levels in CDR 0 APOE3 (black, n=71) and APOE4 (red, n=47) carriers, and CDR 0.5 APOE3 (black, n=34) and APOE4 (red, n=32) carriers. (c) CSF interferon gamma (IFNγ) levels in CDR 0 APOE3 (black, n=54) and APOE4 (red, n=29) carriers, and CDR 0.5 APOE3 (black, n=25) and APOE4 (red, n=17) carriers. (d) CSF interleukin 1β (IL1β) levels in CDR 0 APOE3 (black, n=43) and APOE4 (red, n=18) carriers, and CDR 0.5 APOE3 (black, n=17) and APOE4 (red, n=13) carriers. (e) CSF tumor necrosis factor α (TNFα) levels in CDR 0 APOE3 (black, n=70) and APOE4 (red, n=46) carriers, and CDR 0.5 APOE3 (black, n=34) and APOE4 (red, n=32) carriers. (f) CSF soluble intercellular adhesion molecule 1 (sICAM1) levels in CDR 0 APOE3 (black, n=77) and APOE4 (red, n=40) carriers, and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=33) carriers. (g) CSF Neuron-Specific Enolase (NSE) levels in CDR 0 APOE3 (black, n=47) and APOE4 (red, n=32) carriers, and CDR 0.5 APOE3 (black, n=29) and APOE4 (red, n=29) carriers. Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. Panels a and b had one outlier each, which were removed prior to statistical analysis using methods described in Statistical Analyses section. Significance by ANCOVAs for main effects and post-hoc comparisons controlling for age, sex, and education (ns=non-significant).
Extended Data Figure 7. Lower CSF Aβ1-42 and increased pTau levels in APOE4 carriers with cognitive impairment.
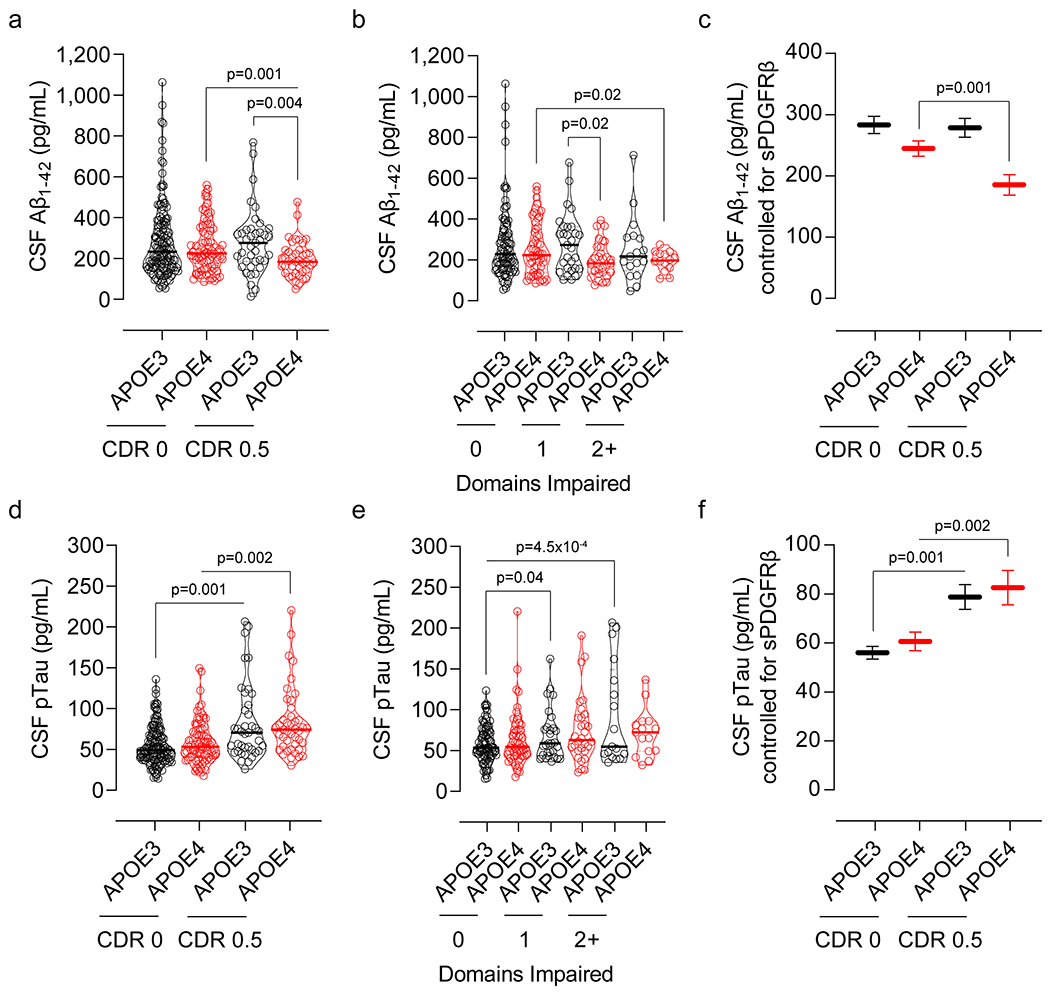
(a) CSF Aβ1-42 levels in CDR 0 APOE3 (black, n=141) and APOE4 (red, n=83) and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=41) carriers. (b) CSF Aβ1-42 levels in APOE3 (black, n=89) and APOE4 (red, n=55) carriers with 0 cognitive domain impaired, APOE3 (black, n=29) and APOE4 (red, n=31) carriers with 1 cognitive domain impaired, and APOE3 (black, n=17) and APOE4 (red, n=14) carriers with 2+ cognitive domains impaired. (c) CSF Aβ1-42 levels (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, and CSF sPDGFRβ levels) in CDR 0 APOE3 (black, n=141) and APOE4 (red, n=83) and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=41) carriers. (d) CSF pTau levels in CDR 0 APOE3 (black, n=141) and APOE4 (red, n=82) and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=43) carriers. (e) CSF pTau levels in APOE3 (black, n=89) and APOE4 (red, n=56) carriers with 0 cognitive domain impaired, APOE3 (black, n=29) and APOE4 (red, n=30) carriers with 1 cognitive domain impaired, and APOE3 (black, n=17) and APOE4 (red, n=15) carriers with 2+ cognitive domains impaired. (f) CSF pTau levels (estimated marginal means ± SEM from ANCOVA models corrected for age, sex, education, and CSF sPDGFRβ levels) in CDR 0 APOE3 (black, n=141) and APOE4 (red, n=82) and CDR 0.5 APOE3 (black, n=39) and APOE4 (red, n=43) carriers. Violin plot continuous lines indicate median values and dotted lines indicate interquartile range. CSF Aβ1-42 and pTau values were log10-transformed due to non-normal distribution prior to statistical analysis. Significance tests from ANCOVAs for main effects and post-hoc comparisons controlling for age, sex, and education.
Extended Data Figure 8. Full scans of western blots.

Full scans of western blots for CypA shown in Figure 4 panel m (top).
Extended Data Table 1.
APOE3 and APOE4 participants studied for regional blood-brain barrier permeability changes by dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI).
| APOE Genotype | APOE3 | APOE4 | APOE3 | APOE4 |
|---|---|---|---|---|
| Clinical Dementia Rating (CDR) | 0 | 0 | 0.5 | 0.5 |
| No. of participants | 130 | 76 | 14 | 25 |
| Age at MRI, years, Mean (SD) | 69.9 (7.9) | 67.3 (8.7) | 73.8 (8.3) | 69.4 (8.7) |
| Female, % | 62.3 | 57.9 | 42.9 | 56 |
| Education, years, Mean (SD) | 16.6 (2.7) | 16.7 (2.0) | 16.4 (2.5) | 17.1 (2.1) |
| Cognitive domain impairment, No. 0,1, 2+ | 78, 17, 2 | 38, 16,4 | 6, 2,6 | 6, 8,9 |
| Vascular risk factors, No. 0-1, 2+ | 77, 53 | 38, 38 | 8,6 | 10, 15 |
Extended Data Table 2.
a. APOE3 and APOE4 participants studied for regional amyloid brain accumulation by PET and blood-brain barrier permeability changes by DCE-MRI. FBB, participants who received 18F-Florbetaben; FBP, participants who received 18F-Florbetapir.
b. APOE3 and APOE4 participants studied for regional tau brain accumulation by PET and blood-brain barrier permeability changes by DCE-MRI.
| APOE Genotype | APOE3 | APOE4 |
|---|---|---|
| Clinical Dementia Rating (CDR) | 0 | 0 |
| No. of participants | 45 | 29 |
| No. of participants (FBB, FBP) | 5, 40 | 9, 20 |
| Age at amyloid PET, years, Mean (SD) | 68.4 (7.5) | 65.7 (8.8) |
| Female, % | 73.3 | 65.5 |
| Education, years, Mean (SD) | 16.7 (2.7) | 16.5 (2.1) |
| Cognitive domain impairment, No. 0,1, 2+ | 24, 4, 0 | 17, 4, 1 |
| Vascular risk factors, No. 0-1, 2+ | 23, 22 | 19, 10 |
| APOE Genotype | APOE3 | APOE4 |
| Clinical Dementia Rating (CDR) | 0 | 0 |
| No. of participants | 60 | 37 |
| Age at tau PET, years, Mean (SD) | 68.7 (7.9) | 64.0 (8.4) |
| Female, % | 66.6 | 37.8 |
| Education, years, Mean (SD) | 16.5 (2.7) | 16.6 (2.1) |
| Cognitive domain impairment, No. 0,1, 2+ | 27, 5, 1 | 15, 6, 1 |
| Vascular risk factors, No. 0-1, 2+ | 36, 24 | 23, 14 |
Extended Data Table 3.
APOE3 and APOE4 participants studied for CSF sPDGFRβ levels.
| APOE Genotype | APOE3 | APOE4 | APOE3 | APOE4 |
|---|---|---|---|---|
| Clinical Dementia Rating (CDR) | 0 | 0 | 0.5 | 0.5 |
| No. of participants | 157 | 105 | 40 | 48 |
| Age at LP, years, Mean (SD) | 70.2 (8.9) | 67.3 (9.9) | 76.5 (7.3) | 72.8 (8.1) |
| Female, % | 64 | 61 | 39 | 37 |
| Education, years, Mean (SD) | 16.5 (2.7) | 16.3 (2.3) | 15.7 (2.8) | 16.2 (2.8) |
| Cognitive domain impairment, No. 0,1, 2+ | 91,20,2 | 62, 17,2 | 9, 8, 14 | 12, 16, 15 |
| Vascular risk factors, No. 0-1, 2+ | 100, 57 | 57, 48 | 22, 18 | 18, 30 |
Supplementary Material
Acknowledgments
The work of B.V.Z. is supported by the National Institutes of Health (NIH) grant nos. R01AG023084, R01NS090904, R01NS034467, R01AG039452, 1R01NS100459, 5P01AG052350, and 5P50AG005142, in addition to the Alzheimer’s Association strategic 509279 grant, Cure Alzheimer’s Fund, and the Foundation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference no. 16 CVD 05. D.A.N. is supported by the NIH grant nos. R01AG060049, R01AG064228, P01AG052350 and P50AG016573 and Alzheimer’s Association grant nos. AARG-17–532905 and Alzheimer’s Association strategic 509279 grant. D.P.B and M.G.H. are supported by the L.K. Whittier Foundation, grant nos. P01AG052350, R01AG054434, and R01AG055770. Enrollment of participants into the WashU Knight ADRC is supported by NIH grant nos. P50AG05681 (J.C.M.), P01AG03991 (J.C.M.), and P01AG026276 (J.C.M.). E.M.R. is supported by the National Institute of Aging (NIA) grant nos. P30AG19610 and R01AG031581, in addition to the state of Arizona. Enrollment of participants into the USC ADRC is supported by NIH grant no. 5P50AG005142 (H.C.C.). Avid Radiopharmaceuticals, Inc., a wholly owned subsidiary of Eli Lilly and Company, provided doses of FBP and financial support for FBP scanning at WashU site. Avid Radiopharmaceuticals, Inc. also provided WashU site with AV1451 precursor and technology transfer for producing the tracer on site. Avid Radiopharmaceuticals, Inc. was not involved in data analysis or interpretation.
Footnotes
Competing Interests
The authors declare no competing interests.
Data Availability
All data generated and/or analyzed during this study are either included in this article (and its Supplementary Information) or are available from the corresponding author on reasonable request. Source Data for Figs. 1–4 are provided with the article.
Code Availability
All software used in this study are publicly available: Rocketship v1.2 (https://github.com/petmri/ROCKETSHIP/blob/master/dce/compare_gui.m), FreeSurfer (v5.3.0) (http://surfer.nmr.mgh.harvard.edu/), FSL-FLIRT (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FLIRT), SPM12 (https://www.fil.ion.ucl.ac.uk/spm/software/spm12/), and Quantitative Imaging Toolkit (https://cabeen.io/about/publication/cabeen2018quantitative/).
References
- 1.Wardlaw JM et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapasi A, DeCarli C & Schneider JA Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. (Berl.) 134, 171–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iadecola C The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96, 17–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iturria-Medina Y et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun 7, 11934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney MD, Sagare AP & Zlokovic BV Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol 14, 133–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweeney MD et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 15, 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nation DA et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med 25, 270–276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montagne A et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Haar HJ et al. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol. Aging 45, 190–196 (2016). [DOI] [PubMed] [Google Scholar]
- 10.van de Haar HJ et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 281, 527–535 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Roses AD Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med 47, 387–400 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 14.Genin E et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hultman K, Strickland S & Norris EH The APOE ɛ4/ɛ4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 33, 1251–1258 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halliday MR et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab 36, 216–227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salloway S et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J. Neurol. Sci 203–204, 183–187 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Zipser BD Microvascular injury and blood-brain barrier leakage in Alzheimer/’s disease. Neurobiol Aging 28, 977–986 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armulik A et al. Pericytes regulate the blood-brain barrier. Nature 468, 557–561 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Bell RD et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikolakopoulou AM et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci 22, 1089–1098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan C et al. Diagnostic Values of Cerebrospinal Fluid T-Tau and Aβ₄₂ using Meso Scale Discovery Assays for Alzheimer’s Disease. J. Alzheimers Dis. JAD 45, 709–719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roe CM et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 80, 1784–1791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montagne A, Zhao Z & Zlokovic BV Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med 214, 3151–3169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett RE et al. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 115, E1289–E1298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fouquet M, Besson FL, Gonneaud J, La Joie R & Chételat G Imaging brain effects of APOE4 in cognitively normal individuals across the lifespan. Neuropsychol. Rev 24, 290–299 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Schultz SA et al. Widespread distribution of tauopathy in preclinical Alzheimer’s disease. Neurobiol. Aging 72, 177–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miners JS, Kehoe PG, Love S, Zetterberg H & Blennow K CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res. Ther 11, 81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanciu C, Trifan A, Muzica C & Sfarti C Efficacy and safety of alisporivir for the treatment of hepatitis C infection. Expert Opin. Pharmacother 20, 379–384 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Morris JC et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis. Assoc. Disord 20, 210–216 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Morris JC The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Nation DA et al. Antemortem pulse pressure elevation predicts cerebrovascular disease in autopsy-confirmed Alzheimer’s disease. J. Alzheimers Dis. JAD 30, 595–603 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bangen KJ et al. Aggregate effects of vascular risk factors on cerebrovascular changes in autopsy-confirmed Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 11, 394–403.e1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jak AJ et al. Quantification of five neuropsychological approaches to defining mild cognitive impairment. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 17, 368–375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jak AJ et al. Neuropsychological Criteria for Mild Cognitive Impairment and Dementia Risk in the Framingham Heart Study. J. Int. Neuropsychol. Soc. JINS 22, 937–943 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weintraub S et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis. Assoc. Disord 23, 91–101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Besser L et al. Version 3 of the National Alzheimer’s Coordinating Center’s Uniform Data Set. Alzheimer Dis. Assoc. Disord 32, 351–358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delis D, Kramer J, Kaplan E & Ober B California Verbal Learning Test. (PsychCorp, 2000). [Google Scholar]
- 41.Montagne A et al. Undetectable gadolinium brain retention in individuals with an age-dependent blood-brain barrier breakdown in the hippocampus and mild cognitive impairment. Alzheimers Dement. J. Alzheimers Assoc 15, 1568–1575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischl B FreeSurfer. NeuroImage 62, 774–781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischl B et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33, 341–355 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Desikan RS et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 31, 968–980 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Fischl B & Dale AM Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. U. S. A 97, 11050–11055 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischl B, Sereno MI, Tootell RB & Dale AM High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum. Brain Mapp 8, 272–284 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dinov I et al. Neuroimaging study designs, computational analyses and data provenance using the LONI pipeline. PloS One 5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sepehrband F et al. Neuroanatomical morphometric characterization of sex differences in youth using statistical learning. NeuroImage 172, 217–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabeen RP, Laidlaw DH & Toga AW Quantitative imaging toolkit: Software for interactive 3D visualization, data exploration, and computational analysis of neuroimaging datasets. in Proceedings of the International Society for Magnetic Resonance in Medicine (ISMRM) vol. 2854 (2018). [Google Scholar]
- 50.Jenkinson M, Bannister P, Brady M & Smith S Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage 17, 825–841 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Bullich S et al. Optimal Reference Region to Measure Longitudinal Amyloid-β Change with 18F-Florbetaben PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med 58, 1300–1306 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Marquié M et al. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case. Acta Neuropathol. Commun 5, 75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mishra S et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: Defining a summary measure. NeuroImage 161, 171–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.TCW J et al. Cholesterol and matrisome pathways dysregulated in human ε4 glia. bioRxiv (2019) doi: 10.1101/713362. [DOI] [Google Scholar]
- 55.Faal T et al. Induction of Mesoderm and Neural Crest-Derived Pericytes from Human Pluripotent Stem Cells to Study Blood-Brain Barrier Interactions. Stem Cell Rep. 12, 451–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aggarwal CC Outlier analysis. (Springer, 2013). [Google Scholar]
- 57.Sagare AP, Sweeney MD, Makshanoff J & Zlokovic BV Shedding of soluble platelet-derived growth factor receptor-β from human brain pericytes. Neurosci. Lett 607, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glickman ME, Rao SR & Schultz MR False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol 67, 850–857 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.