

Abstract
The escalating problem of obesity and its multiple metabolic and cardiovascular complications threatens the health and longevity of humans throughout the world. The etiology of obesity and one of its chief complications, insulin resistance, involves the participation of multiple distinct organs and cell types. From the brain to the periphery, cell intrinsic and intercellular networks converge to stimulate and propagate increases in body mass and adiposity, as well as disturbances of insulin sensitivity. This Review focuses on the roles of the cadre of innate immune cells, both those that are resident in metabolic organs and those that are recruited into these organs in response to cues elicited by stressors such as overnutrition and reduced physical activity. Beyond the typical cast of innate immune characters invoked in the mechanisms of metabolic perturbation in these settings, such as neutrophils and monocytes/macrophages, these actors are joined by bone marrow-derived cells such as eosinophils and mast cells and the intriguing innate lymphoid cells (ILCs), which are present in the circulation and in metabolic organ depots. Upon high fat feeding or reduced physical activity, phenotypic modulation of the cast of plastic innate immune cells ensues, leading to the production of mediators that affect inflammation, lipid handling and metabolic signaling. Further, their consequent interactions with adaptive immune cells, including myriad T cell and B cell subsets, compound these complexities. Notably, many of these innate immune cell-elicited signals in overnutrition may be modulated by weight loss, such as that induced by bariatric surgery. Recently, exciting insights into the biology and pathobiology of these cell-type specific niches are being uncovered by state-of-the-art techniques such as single-cell RNA sequencing. This Review considers the evolution of this field of research on innate immunity in obesity and metabolic perturbation, as well as future directions.
Keywords: obesity, innate immunity, metabolic perturbation, insulin resistance, diabetes
Introduction
The incidence of obesity, a chronic, non-communicable disease, continues to increase in the United States and world-wide; the associated co-morbidities are substantial.1 Consequences of obesity, such as type 2 diabetes (T2D), cardiovascular disease (CVD), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), musculoskeletal disorders and some types of cancers significantly impact morbidity, quality of life, and mortality in this disease.1, 2 The complexity of obesity is underscored by multiple observations indicating that perturbations in both the brain and the periphery contribute to this disorder. Roles for adipocyte- and immune cell-intrinsic properties, together with their cross-talk and intercellular communications, characterize the complex milieus brewing in the adipose tissue depots challenged with high fat diets (HFDs), reduced physical activity, reduced energy expenditure and adipocyte expansion in incipient and established obesity. Further, the innate immune cells linked to obesity are not simply confined to the periphery; rather, innate immune cells of the central nervous system (CNS), such as astrocytes and microglia, also contribute to the immunometabolic consequences of overnutrition and obesity. Our understanding of these complex interactions is ever-evolving.
This Review will chronicle the evolution of this field, with focus on historical and recent insights into innate immune cells in the periphery and within the CNS, and their contributions to obesity and metabolic perturbations, such as insulin resistance, the harbinger of T2D. Only by delineating a time-, adipose and metabolic organ depot- and cell type-specific roadmap for protective vs. maladaptive pathways in overnutrition and obesity may effective and safe therapeutic targets be unveiled to target innate immune cell functions gone awry in these disorders.
“Inflammation” and Obesity
In 1993, Hotamisligil, Shargill, and Spiegelman published a seminal study in which they showed that Tnf (gene encoding Tumor Necrosis Factor-α) mRNA transcripts were significantly increased in the adipose tissue of T2D mice and rats, but not in the adipose tissue of streptozotocin-induced diabetic mice (a model of type 1 diabetes, T1D).3 In those T2D models, the upregulation of Tnf preceded the development of significant hyperglycemia. In fa/fa rats (model of T2D), the administration of soluble TNFα receptor (TNFR)-immunoglobulin G (IgG) chimeric protein did not affect glucose levels, but resulted in the need to administer significantly more glucose to the treated vs. vehicle rats in order to maintain euglycemia during a hyperinsulinemic euglycemic clamp study; those findings suggested that the treated animals were more insulin responsive.3 It was deduced from those studies that TNFα contributed to impaired sensitivity to insulin in the T2D models. Subsequent to this discovery, efforts focused on identifying the immune cell composition in the adipose tissue depots in lean vs. obese animal models and human subjects. Experiments testing these concepts placed the early spotlight on the macrophage of the innate immune system.
Macrophages & Complex Roles in Metabolic Perturbation
Macrophage Content in Obese Adipose Tissue
Two reports published in the Journal of Clinical Investigation in 2003 associated increased macrophage content in adipose tissue with obesity.4, 5 Ferrante’s group showed that in multiple murine adipose tissue depots, obesity was accompanied by a rise in macrophage content (characterized by the macrophage marker F4/80), which correlated with body mass index (BMI) and adipocyte size.4 Transcriptomic profiling of adipose tissue showed that approximately 1,000 genes were associated with obesity and body mass; a substantial number of these genes were linked to inflammation.4 Ferrante and colleagues showed that the majority of TNFα expression in obese adipose tissue was accounted for by adipose tissue macrophages (ATMs).4 In human obesity, using CD68 to mark macrophages, they showed that obese adipose tissue also displayed increased macrophage content compared to lean adipose tissue.4 In subsequent work, Ferrante’s group showed that deficiency of Ccr2 (gene that encodes C-C Motif Chemokine Receptor 2) or its pharmacological antagonism in mice fed a HFD resulted in reduced food intake, reduced weight gain and improved insulin sensitivity, in parallel with reduced ATM content.6 In a parallel study, Chen’s group demonstrated increased macrophage content and expression of inflammation-related genes in the adipose tissue of obese mice, which preceded rises in circulating insulin levels.5 These authors treated T2D ob/ob mice with the insulin-sensitizer rosiglitazone for 28 days; in rosiglitazone- vs. vehicle-treated mice, white adipose tissue (WAT) displayed a significant reduction in mRNA levels of Adam8, Ccl3, Itgam, Emr1 and Cd68, suggestive of decreased inflammation.5 Of note, the Ferrante and Chen groups both showed that whereas macrophage content in adipose tissue was affected by obesity, neutrophil content was not altered, at least under the conditions employed in their studies.4, 5 Hence, collectively, these studies suggested that macrophage infiltration in adipose tissue contributed to insulin resistance in obesity.
Early studies attempting to classify the inflammatory polarization of macrophages as “inflammatory M1” vs “anti-inflammatory M2” oversimplified the complex and plastic biology of these cells. Nevertheless, it is now clear that beyond differences in the numbers of macrophages in lean vs. obese adipose tissue, their functional properties differ as well. For example, in lean adipose tissue, ATMs may serve as resident sentinels, facilitating lipid buffering, production of anti-inflammatory cytokines such as IL4 and IL10, and pro-remodeling mechanisms, such as removal of dead adipocytes; processes favoring insulin sensitivity.7–10 However, in obesity, with increasing adipocyte size, processes are set in motion that favor recruitment of monocytes/macrophages derived from the bone marrow and the production of proinflammatory cytokines such as TNFα, generation of reactive oxygen species (ROS), and impaired clearance of dead adipocytes; processes overall favoring insulin resistance.6, 11 Interestingly, in obesity, beyond lipolysis as a means to stimulate macrophages, intercellular communication via adipocyte-released exosome-type structures may trigger macrophage differentiation in the adipose tissue, thereby priming these cells to adaptively manage the excess lipid content.12, 13 If overwhelmed, however, these processes may tip the balance to a proinflammatory ATM phenotype in which insulin resistance may be facilitated.
Beyond recruitment of monocytes/macrophages in obese adipose tissue, roles for immune cell retention factors, such as the guidance cue molecule, Netrin-1, have been suggested. Upregulation of Netrin-1 was observed in ATMs in obese mice and humans; hematopoietic deletion of Ntn1 reduced adipose tissue inflammation by stimulating macrophage emigration from the adipose tissue; in parallel, insulin sensitivity was restored in mice.14 In a separate study, using Ntn1 floxed mice, myeloid-specific deletion resulted in a small decrease in adiposity in the HFD-fed mice. Single-cell RNA-sequencing (RNA-seq) of CD45+ myeloid cells revealed that upon deletion of Ntn1 in this myeloid population, a significant attrition of ATMs was noted, especially in the resident macrophage population, which was accompanied by improved insulin sensitivity in HFD-fed mice compared with the floxed control mice fed HFD.15 Deletion of myeloid Ntn1 also regulated molecules linked to lipid handling, such as fatty acid uptake, intracellular transport of lipid, lipid droplet formation and lipolysis.15 Also, a decrease in proinflammatory eicosanoids was noted upon the deletion of myeloid Ntn1.15
In obesity, a particular subset of CD11C-expressing cells, suggested to be dendritic cells (DC), was shown to populate the adipose tissue, thereby triggering processes that further propagate inflammatory mechanisms, with predicted links to insulin resistance.16–18 Consistent with the premise that these cells are proinflammatory and facilitate insulin resistance, ablation of CD11C+ DCs in obese animals improved insulin sensitivity.18 Further, in the above-noted study on myeloid deletion of Ntn1, the population of CD45+ cells expressing CD11C was also reduced, in parallel with improved metabolic function.15
Despite the compelling evidence linking macrophages and a proinflammatory switch to obesity and insulin resistance, other studies have uncovered findings suggesting that a broader perspective is necessary to untangle these complex and chronic perturbations in metabolic dysfunction.
Macrophages Adjust their Metabolic Signatures in Obesity – It’s Not just the Numbers
A variety of unbiased proteomic, transcriptomic and metabolomic strategies have been employed to track the origins of adipose inflammation. Kratz and colleagues utilized plasma membrane proteomics approaches in both human and murine obese vs. lean subjects. They found that classical markers of the “M1” macrophage inflammatory state did not define the ATMs in obesity, either in humans or wild-type (WT) male mice fed a HFD.19 The authors hypothesized that a “metabolically-activated” macrophage might better classify the macrophages embedded in a high glucose/high fat environment. In obesity, cell surface markers indicative of altered lipid metabolism, such as ABCA1, CD36 and PLIN2 were increased compared to the lean state, and although TNFα and IL1β were increased in obese ATMs, markers of the alternatively-activated “M2” ATMs, such as CD163, CD206, and TGFβ1 were suppressed. Interestingly, although inflammatory mediators did not stimulate the development of the metabolically-activated macrophage, p62 and PPARγ were shown to trigger their development and, mechanistically, these mediators drove overall suppression of the inflammatory signatures in the ATMs.19
Robblee and colleagues used a transcriptomic approach in bone marrow-derived macrophages (BMDMs) exposed to saturated fatty acids and showed that saturated but not unsaturated fatty acids activated the endoplasmic reticulum (ER) stress sensor, IRE1α (inositol-requiring enzyme 1 α), which subsequently activated the NLRP3 inflammasome to drive expression of proinflammatory genes.20 These authors also stimulated BMDMs with lipopolysaccharide (LPS) as a control and found that the transcriptomic profiles were quite different, in that the saturated fatty acids induced only 8% of the genes that were induced by LPS. These findings suggest that there were fundamental differences in the regulation of genes linked to discrete inflammatory triggers.20 For example, in that study, prolonged treatment of the BMDMs with saturated fatty acids resulted in downregulation of a number of mRNA signatures linked to inflammatory responses. Collectively, this study suggested the critical point that although saturated fatty acid treatment of BMDMs regulated the inflammasome, the inflammatory mediators and time course of expression were overall distinct from those induced by the classical proinflammatory stimulus, LPS. Indeed, these data affirm that “macrophage inflammation” is quite complex and, at least in part, directly impacted by the initiating cues and stimuli.
In a study specifically addressing the primary metabolic routes used by ATMs in the lean vs. the obese state, and how the metabolic pathways contributed to the inflammatory signatures of the ATMs, Boutens and colleagues retrieved ATMs (F4/80+ sorted cells) and showed that obesity was associated with a distinct metabolic rewiring that favored glycolysis, in parallel with higher levels of Hif1a (hypoxia inducible factor-1 α), but the inflammatory signatures in the obese ATMs were not identical to those of the classical “M1”-like macrophages.21 Rather, using adipose tissue cultures, they found the secreted mediators from this tissue, which did not appear to be leptin or lactate, contributed to the rewiring of these ATMs and their metabolic and inflammatory states.21 An unexpected observation emerged from that study, however, as myeloid-specific deletion of Hif1a did not affect the proinflammatory state of the ATMs after 8 weeks of HFD feeding, as for example, levels of IL6 were identical in the adipose tissue from both the Hif1a myeloid-deleted and control mice.21 In that work, however, the Lysm cre recombinase strategy was used, which would have deleted Hif1a in other cells beyond macrophages, such as neutrophils and microglia. It thus cannot be excluded that non-macrophage/ATM effects contributed to these findings.
In a recent study, Petrus and colleagues examined the releasate from the WAT of 52 obese and 29 non-obese control female human subjects and discovered that levels of glutamine were significantly lower in the obese vs. the lean state, but that there were no significant differences in serum levels of glutamine, thereby reinforcing the importance of the local milieu in mechanisms driving obesity.22 Upon examining the BMI of the subjects, the authors found a strong inverse association between BMI and the levels of adipose tissue glutamine, which were related to fat mass and adipocyte size. To assess if glutamine exerted effects on ATM inflammation, WT mice were injected with glutamine while consuming a standard low fat diet; although glutamine had no effect on body weight, levels of proinflammatory genes in the epididymal WAT, Ccl2, Emr1 and Cd68 were significantly lower and levels of Adipoq (adiponectin) were significantly higher.22 Glutamine was linked to suppression of proinflammatory genes in human adipocytes; mechanistically, glutamine reduced glycolysis and reduced uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) levels. UDP-GlcNAc is the substrate for the post-translational modification O-linked β-N-acetylglucosamine (O-GlcNAc) mediated by the enzyme O-GlcNAc transferase.22
Recently, Drareni and colleagues reviewed the evidence supporting that some of the key transcription factors that regulate macrophage-mediated inflammation in obesity appear to include NF-κB; PPARα and PPARβ; PPARγ; STATs, such as STAT1 and STAT6; AP1; Interferon regulatory factors (IRFs), such as IRF5; Liver X receptors; glucocorticoid receptors (GCs); and Hypoxia inducible factor-1α.23 Given the diversity of this group of molecules, it is likely that multiple factors, such as the stage of obesity and the presence or absence of T2D (hyperglycemia) may dictate the dominant transcription factors linked to regulation of macrophage-mediated inflammation in discrete settings.
Macrophages & Further Considerations for a Broader View in Obesity and Metabolic Perturbation
Collectively, these data suggest that multiple factors, including an inextricable link between macrophage metabolism, inflammation and the degree of insulin sensitivity must be considered when probing the specific macrophage proteome, transcriptome and/or metabolome species that are operating in obesity.
First, the concept of a positive linear correlation between body mass and adipose tissue macrophage content is not without exception. It was shown that in the earliest days after caloric restriction (CR) to induce weight loss in obese mice, ATM content actually rose in the immediate time frame (approximately days 1-7 of CR), that is, leaner mice displayed higher ATM content. Notably, however, this rise in ATM content did not include increasing content of CD11C+ cells. Lipolysis was identified to be a key process triggering increased ATM content in the adipose tissue through promotion of macrophage migration.24 This early phase was followed by a gradual reduction in ATM content with ongoing and sustained weight loss in the chronic phase.24 In separate work, Weinstock and colleagues exposed mice to HFD feeding and, therefore, obesity for 24 weeks (HFD/24 weeks), followed by retrieval of visceral adipose tissue (VAT) for single-cell RNA seq. An additional group of mice underwent 24 weeks of HFD feeding followed by a switch to CR, in which the mice received 70% of the HFD for a further 2 week time course (HFD/CR). The HFD/CR mice demonstrated an approximately 12% loss of body weight with a 25% decrease in VAT mass during that time.25 Consistent with previous reports, the HFD/CR mice displayed significantly more crown-like structures (CLS) in VAT than the HFD/24 weeks mice. Although the majority of the CD45+ cells/clusters in the HFD/24 weeks group were representative of macrophages (51%), it was found that DCs, T cells, NK cells, monocytes and B cells represented 14%, 11%, 9%, 8% and 6% of the leukocytes, respectively. What was the effect of weight loss? In the HFD/CR group, the authors reported that CR restored T regulatory cells (Tregs) levels to those of lean mice. A major macrophage subpopulation in obese VAT, which expressed genes relevant to lipid binding and metabolism, became more similar to that of lean VAT after HFD/CR.25 Finally, in the HFD/CR group, unique clusters of phagocytic macrophages, activated macrophages, B cells and resident macrophages were observed, which were not similar to either the HFD/24 weeks or the lean group VAT. Hence, in the lean state, obesity or in HFD/CR, macrophages may exert a diverse range of functions in the adipose tissue, ranging from adaptive to tissue-damaging properties. Interestingly, HFD/CR contributes to reversal of many but not all of these changes in myeloid cells, along with the appearance of novel non-lean-like functional clusters.25
Aleman and colleagues tested these concepts in 10 female human subjects undergoing approximately 10% loss of body weight (and approximately 3.8% loss of fat mass) over an average of 46 days on account of consumption of a very low calorie diet (800 kcal/day).26 In that study, the authors found on repeat subcutaneous adipose tissue (SAT) biopsy that compared to baseline, CLS increased after weight loss, in parallel with reduced markers of metabolic and inflammatory perturbation, such as levels of glucose, high sensitivity C-reactive protein (CRP) and decreased circulating free fatty acids, glycerol, 25 hydroxyvitamin D and urinary isoprostane M.26 Hence, in human subjects as well, a disconnect between ATM/CLS and inflammation and metabolic perturbation was evident after weight loss.
Second, in line with the above concepts, the degree of ATM content does not necessarily align with inflammatory gene expression. Global or myeloid (bone marrow transplantation studies) deletion of Ager (the gene encoding the receptor for advanced glycation end products (RAGE)) resulted in reduced “FBC” content (F4/80, CD11B+, and CD11C+ cells) in perigonadal adipose tissue (PGAT) compared to WT mice fed HFD, in parallel with improved insulin sensitivity.27 However, PGAT levels of multiple “proinflammatory” genes, Tnf, Ccl2, and Nos2 were not reduced upon deletion of Ager; in contrast, in both experimental settings, global or myeloid deletion of Ager was associated with trends to or significantly higher levels of “anti-inflammatory” genes, Cd203d, Dc-Sign, Arg1, Il10 Cd163 compared to the WT control animals.27
Third, a key test of concept regarding roles for proinflammatory forces and, specifically, for TNFα in obesity-associated insulin resistance, was not met with definitive conclusions in human subjects. In a study published in 1996, Ofei and colleagues randomized 21 obese T2D subjects to either CDP571, a humanized monoclonal anti-TNFα antibody, or saline vehicle. Subjects received one dose of the treatments; the half-life of the antibody was said to render effective plasma levels throughout the 4-week observation period. At the end of that time, compared to baseline, there were no differences in levels of fasting glucose, insulin or C-peptide. In an insulin sensitivity test, the percentage rates of glucose clearance per minute were not affected in the subjects by CDP571.28 In a subsequent study in 2000 by Paquot and colleagues, 7 patients with obesity and varying degrees of glucose intolerance underwent two consecutive hyperinsulinemic, euglycemic clamp studies in which they received vehicle in the first study and then, in the second study, a single dose of Ro 45-2081 recombinant human TNF receptor:Fc fusion protein. In both cases, the clamp assays were performed 48 hours after the injections. The authors reported that insulin-mediated glucose disposal and glucose metabolic clearance rate did not differ in the subjects by treatment.29 Finally, based on the premise that the earlier studies might have been confounded by the short duration of treatment, Stanley and colleagues randomized 40 obese subjects with metabolic syndrome to receive either placebo or the TNFα inhibitor etanercept (Enbrel) over a 6 month period; 34 subjects completed the study. At baseline, the etanercept group was significantly younger than the placebo group. Although fasting glucose levels at end of study were significantly improved in the etanercept group vs. placebo, there were no treatment-dependent differences in fasting insulin, 2-hour insulin or HOMA-IR (Homeostatic Model Assessment of Insulin Resistance).30 Levels of high molecular weight adiponectin, however, were higher in the etanercept- vs. placebo-treated group.30 Hence, firm evidence for improved insulin sensitivity by etanercept treatment was not shown in this study; it is notable, however, that hyperinsulinemic, euglycemic clamp studies were not performed in those subjects.
Hence, the results of these reports suggested that in contrast to the findings in mouse models, TNFα blockade did not incontrovertibly affect insulin sensitivity in obese human subjects with evidence of metabolic dysfunction. In this context, possible roles for other cell types and processes in obesity, insulin resistance and T2D needed to be considered.
Neutrophils: Among the Early Responders in Obesity
Within the first few days of HFD feeding, the migration of neutrophils into adipose tissue commences.31 In obesity, neutrophils may produce cytokines and chemokines, such as TNFα and MCP1 (CCL2), thereby igniting processes that promote monocyte/macrophage infiltration into the adipose tissue.32, 33 In human subjects, akin to the observations made with respect to ATMs, at 4 weeks after bariatric surgery, the neutrophil content in obese adipose tissue continued to rise to 15-20 fold above that at the time of surgery and this persisted for months post-surgery, despite improvements in overall metabolic function.34
It is speculated that in obesity and metabolic dysfunction, one of the functions of neutrophils is the production of a serine protease elastase, which has the potential to cleave insulin receptor substrate 1 (IRS1), a mechanism to reduce insulin sensitivity in adipocytes.31, 35 The effects of exercise on elastase expression in obese mice was studied. Mice were fed a standard chow or HFD from 4 to 20 weeks of age and randomized to either a sedentary group or exercise group, five days/week. Exercise training reduced adipose neutrophil content, neutrophil elastase activity and markers of inflammation.36 However, the authors did not report in that study if these changes were accompanied by improvements in insulin sensitivity.36
Recently, neutrophil extracellular traps (NETs), which play roles in cell-cell communication mechanisms involving neutrophils, have been studied for their potential roles in obesity. NETs are structures derived from neutrophils; these cells bear a DNA scaffold that is dotted with nuclear, granular or cytosolic proteins.37 Beyond roles in anti-microbial functions, NETs may also contribute to the pathogenesis of chronic diseases, such as atherosclerosis.38 Interestingly, NETs also contain neutrophil elastase, and it was thus hypothesized that these structures may play roles in obesity. Braster and colleagues fed WT mice a 60% HFD and concomitant with the introduction of diet, mice were treated with Peptidyl Arginine Deiminase 4 inhibitor Cl-amidine; a compound that blocks NET release. The authors observed no differences in immune cell infiltration into the adipose tissue or liver and there was no effect on insulin resistance observed in these studies.39 Others treated mice with HFD-induced obesity with Cl-amidine or with DNAse, and found that although neither affected body mass or glucose/insulin sensitivity, they were associated with improved endothelial function and reduced inflammation.40 In human subjects, studies tested the effects of obesity and weight loss on NETs. In obese patients undergoing gastric band surgery, weight loss was associated with decreased neutrophil NETs and decreased evidence of neutrophil activities; interestingly, the authors speculated that such changes might affect the ability to respond to infections, as neutrophil chemotactic accuracy, which was impaired before surgery, was not affected by weight loss.41 In an observational study, D’Abbondanza and colleagues followed 73 patients with morbid obesity, 55 healthy subjects, and 21 subjects with severe coronary artery disease. NETs levels were higher in the obese vs. control group and correlated with BMI, waist and hip variables, glyco-metabolic variables and systolic blood pressure.42 After sleeve gastrectomy, there were no definitive changes in NETs.42 Collectively, the results of these studies suggest that more work will be required to determine if and at what time points during obesity and/or weight loss that NETs contribute to glucose and insulin sensitivity.
Mast Cells
Mast cells, like neutrophils, are bone marrow-derived cells and play seminal roles in immediate responses to stress, as evidenced by the eclectic set of mediators that they produce, including biogenic amines (e.g., histamine and serotonin), enzymes (e.g., proteases), cytokines, lipid metabolites (e.g., leukotrienes and prostaglandins), ATP, and neuropeptides.43 Studies in mice with obesity have shown increased mast cell content in obese vs. lean adipose tissue.44 Further, in human subjects, higher levels of serum mast cell-specific tryptase have been demonstrated in obese vs. lean individuals.45
Mechanistic evidence of links of mast cells to the pathogenesis of obesity and metabolic perturbation emerged from studies by Liu and colleagues. These researchers used mice deficient in c-kit, a growth factor that is essential for mast cell differentiation and survival.46 Compared to their controls, c-kit null mice gained significantly less weight on a HFD, with significant reductions in WAT depots, in parallel with improved glucose tolerance and increased energy expenditure.46 In other experiments, administration of the mast cell stabilizer, disodium cromoglycate (DSCG), attenuated body weight gain only in the WT but not the mast cell-deficient mice; mast cell content in the WAT was not affected, suggesting that the agent’s mechanism of action was via effects on mast cell actions.46 Other work used c-kit-independent means to induce deficiency of mast cells to test the role of these cells in diet-induced obesity. Using Cpa3Cre+ mast cell-deficient mice, which are WT for c-kit and also display reduction in basophils, it was shown that feeding HFD resulted in no differences in body mass or adiposity compared with control mice; glucose and insulin intolerance were also not altered in these mice.47 Given these divergent findings, it was important that these concepts were tested in a distinct model of metabolic stress, cold-induced thermogenesis and treatment with β-adrenergic stimuli.
Finlin and colleagues exposed lean and obese human subjects to repeated cold exposure and performed Nanostring array analysis on the SAT; regardless of BMI, mast cell tryptase and CCL26 (a chemokine for mast cells) were upregulated in the cold. Upon cold challenge (repeated cold exposure to unilateral thigh), in both lean and obese subjects, increased mast cell degranulation was evident; however, only in lean but not obese subjects, increases in mast cell content in the SATs were observed.48 In vitro analysis linked these observations to norepinephrine-stimulated mast cell degranulation.
Zhang and colleagues studied these concepts in mouse models; they employed the c-kit deficient mice described above49 and found that when stimulated by norepinephrine, the metabolic rate was enhanced in these mice compared to controls.49 Mast cell reconstitution in those mice (in the SAT) reversed these effects of norepinephrine.49 The results of these findings linked mast cells to suppression of SAT browning in mice, revealing the complex functions of these cells and their diverse released mediators in obesity and in thermogenesis.
Collectively, although the findings linking mast cells to obesity may be model-dependent, they nevertheless underscore that distinct experimental modalities should be employed, where feasible, to test hypotheses regarding roles for specific cell types in metabolic dysfunction.
Eosinophils – Flipping the switch to anti-inflammatory / protective roles for innate immune cells in obesity
Eosinophils, like monocytes, neutrophils and mast cells are also bone marrow-derived cells.50 To respond to allergic stimulation, differentiation mediated by Interleukin-5 (IL5) is critical and roles for IL3 and GM-CSF (granulocyte-macrophage colony stimulating factor) have also been described. Upon terminal differentiation, eosinophils in the circulation may then infiltrate target tissues in response to chemokines, such as eotaxin.51 Activated eosinophils may release mediators to aid in allergic responses and, akin to neutrophils, eosinophils may release EETs or eosinophil extracellular traps. Eosinophils exert anti-parasite and anti-viral roles.50
Early evidence linking eosinophils to adipose tissue homeostasis emerged from mouse models and showed that the number of eosinophils in adipose tissue in obesity was significantly reduced compared to that observed in the lean state. It was also shown that these reductions in eosinophil numbers in adipose tissue might have been linked to obesity, as, when mice lacked eosinophils, they were more prone to diet-induced obesity and glucose intolerance when fed a HFD. Further, hypereosinophilic mice were protected from diet-induced obesity.52 However, despite these compelling associative studies, it was shown that simply artificially buoying the number of eosinophils in the adipose tissue of obese mice to homeostatic levels by administering IL5 failed to improve insulin signaling or to boost energy expenditure.53 However, in IL5-transgenic mice, whose local tissues were long bathed in IL5, their basally elevated numbers of eosinophils rendered the mice protected from diet-induced obesity, adiposity and glucose intolerance.52 These findings highlight potential confounding issues that might arise in experiments probing acute vs. chronic administration of cytokines or other inflammatory mediators vs. chronic transgene-mediated expression in discrete mouse models.
Do eosinophils play roles in weight loss? Bolus and colleagues showed that in mice with HFD-induced obesity, reduced adipose tissue eosinophils could be restored by weight loss.54 In weight loss (switching mice from HFD to low fat diet), the eosinophil content in adipose tissue rose, in parallel with reduced ATM content, reduced inflammation, and improved tissue remodeling.54 However, these authors reported that eosinophils might exert unique effects in distinct tissues, as levels of eosinophils did not vary in the liver during various phases of weight gain and weight loss.
Studies considering cross-talk between eosinophils and other cells within the adipose tissue suggested that one function of eosinophils is to maintain adipogenic maturation.55 Eosinophils also contribute to the development of alternatively-activated “M2”-like macrophages via IL4 and IL13-dependent mechanisms.56 It was shown that mice deficient in Ccr2 displayed increased numbers of eosinophils, which was sustained and further increased during obesity mediated by HFD.57 This increase in numbers of eosinophils in all WAT depots was not observed in other organs such as liver or bone marrow. In that study, there were no differences in insulin sensitivity, body mass, or adiposity between the Ccr2 null and WT mice, perhaps suggesting that the specific milieu in which eosinophils are born and thrive and migrate is not typically devoid of Ccr2, which undoubtedly will exert distinct effects that were not accounted for in that study. Indeed, those authors reported that eosinophils in that model were present in the CLS, typically associated with macrophage accumulation around dead/dying adipocytes.57
In this context, innate lymphoid cells (ILCs) and ILC2, in particular, have intimate relationships with eosinophils and alternatively-activated macrophages. The three major members of the ILC family (ILC1, ILC2, and ILC3) have been linked to adipose tissue inflammation and provide a relatively recent lens into adipose inflammation.
Innate Lymphoid Cells – Key Roles in Inflammation and Metabolism
Innate lymphoid cells (ILCs) lack adaptive antigen receptors and are the innate counterparts of T lymphocytes.58–62 ILC function begins in development and they contribute to tissue homeostasis in the intestine, lung and adipose tissues.60 Earlier classification placed ILCs into three major groups. Recently, Vivier and colleagues suggested that these complex tissue resident cells ILCs be divided into five major subsets of the following: ILC1s and NK cells, which respond to intracellular pathogens and tumors and secrete molecules such as IFNγ and perforin; ILC2s, which respond to parasites and allergens and secrete molecules such as IL4, IL5, IL13 and IL9; lymphoid tissue inducer (LTi) cells, which are mesenchymal organizer cells linked to the formation of secondary lymphoid structures and secrete molecules such as RANK, TNFα, IL17 and IL22; and ILC3s, which respond to extracellular microbes such as bacteria and fungi and secrete molecules such as IL22, IL17, GM-CSF and lymphotoxin.60 In functional terms relevant to T cells, ILCs contribute to distinct types of immunity (Type 1, Type 2 and Type 3; See Reference 63). Specifically, NK/ILC1s contribute to Type 1 immunity; ILC2s contribute to Type 2 immunity; and ILC3s contribute to Type 3 immunity.60–62 Increasingly, there is evidence linking ILCs to obesity. It has been suggested that one of the important functions of ILCs in adipose tissue relates to the regulation and “polarization” of macrophage inflammation.64–66 In the sections to follow, we summarize what is currently known regarding these cells in obesity and metabolic disorders.
NK cells and ILC1s
In human and murine subjects, research has been performed examining the number and activation state of NK cells and the relationship to obesity.67 It is established that ILC1s are present in both human and murine adipose tissue and using parabiosis techniques, it was shown that ILC1s are predominantly tissue resident in mouse models.64 Lynch and colleagues found that compared to lean controls, obese patients had fewer circulating NK cells and cytotoxic T cells. Among the obese subjects specifically, metabolically healthy obese had higher levels of NK cells and cytotoxic T cells compared to the unhealthy obese independent of age and BMI; the NK cells were also less activated in the metabolically healthy vs. the unhealthy obese subjects.68 In a study in children, Tobin and colleagues found that circulating NK cells were reduced in obese vs. lean children, which inversely correlated with BMI and insulin resistance and the NK cells in the obese children were more activated than those from lean children.69
Other studies examined NK cells in the adipose tissue itself. O’Rourke and colleagues examined stromal vascular fractions (SVF) from VAT and SAT from obese human subjects undergoing bariatric surgery. VAT demonstrated higher IFNγ mRNA transcript levels, and NK cells, which constitutively express IFNγ, were also higher in the VAT vs. SAT; SVF-derived NK cells expressed IFNγ on activation, which was associated with TNFα expression by macrophages.70 These findings suggest depot-dependent differences in NK cell content in obese adipose tissue. The altered profiles of NK cells in obesity may be affected by weight loss. Diet and exercise-induced weight loss over a three month period in obese individuals resulted in increased production of IFNγ along with increased NK cell functions.71 Barra and colleagues showed that 6 weeks of high intensity interval training increased NK cell frequency in both obese women and in mice; in mice, this change in NK cells was associated with a reduced lung tumor burden, suggesting that the NK cells were more functionally competent.72
Wang and colleagues examined the effect of obesity on ILC1s; in their study, they characterized these cells as Lin−CD45+ CD127+ CD117−CRTH2−NKP44− and showed that the number of circulating and omental adipose tissue ILC1s was higher in obese vs. lean individuals.73 The adipose tissue ILC1 content positively correlated with adipose tissue fibrosis in these subjects. In obese subjects who underwent weight loss surgery, at the three months follow-up point, the numbers of circulating ILC1s were reduced compared to the pre-operative levels and the reduction of ILC1s correlated with the reduction in fasting blood glucose levels and HbA1C, suggesting that higher levels of the ILC1s were associated with poorer metabolic health.73
Wang and colleagues tested these concepts in a mouse model. They adoptively transferred adipose ILC1s (Lin−NK1.1+NKP46+T-bet+Eomes−DX5−)66 from HFD-fed WT mice into Prkdc−/−IL2rg−/− mice and fed them HFD for an additional 4 weeks. Compared to controls not receiving the ILC1s (phosphate buffered saline, PBS), there were no differences in body weight but those mice receiving the ILC1s were more glucose intolerant and showed more IFN-γ-dependent interstitial fibrosis in the VAT.73 Further, the transfer of ILC1s resulted in more CD11C+ cells and more CLS vs. PBS. Since it was shown that IL12 was required for proliferation of the adipose ILC1s during HFD-induced obesity, inhibition of IL12 using antibodies was tested in Rag1−/− mice fed a HFD. Compared to controls, there were no differences in body weight or fasting glucose, but the number of adipose ILC1s and CD11C+ cells was reduced in anti-IL12-treated mice, as was adipose fibrosis.73
In another study, O’Sullivan and colleagues performed adoptive transfer of purified adipose iNK or ILC1 from HFD-fed WT mice into Rag2−/−Il2rg−/− mice maintained on a HFD for 12 weeks. In both cases, the number of “M1”-type macrophages was higher in the adipose tissue of the recipients, but especially upon the adoptive transfer of ILC1s. Further, in both iNK or ILC1 transfers, the recipients were more glucose and insulin intolerant vs. controls, which was independent of the amount of weight gained on the HFD.66
Wensveen and colleagues also linked NK cells to adipose tissue inflammation and the induction of insulin resistance in obesity; the authors showed that in mice fed a HFD, obesity resulted in upregulation of the ligands of NK-cell activating receptor (NCR1) on adipocytes, which stimulated an increase in NK cell proliferation and their production of IFNγ; which, in turn, led to an increase in proinflammatory macrophage content in the adipose tissue along with exacerbation of insulin resistance.67 These investigators also induced deficiency of NK cells, NCR1 or IFNγ and showed that the accumulation of proinflammatory macrophages was greatly reduced, in parallel with reduced insulin resistance.67 Furthermore, Lee and colleagues tested the role of NK cells in obesity using two major strategies.65 First, they depleted NK cells with neutralizing antibodies or by their genetic ablation in E4bp4(+/−) mice (E4bp4 is essential for NK cell development74), and found improvements in insulin resistance and decreased numbers of ATM and adipose tissue inflammation. Second, when they expanded NK cells through IL15 administration or by reconstitution of NK cells into E4bp4(−/−) mice, they found that upon HFD feeding, ATM numbers and adipose tissue inflammation increased, in parallel with increased insulin resistance.65
Boulenouar treated mice with anti-asialo GM1 antibody to deplete NK cells and found increased numbers of ATMs that were more “M2”-like in nature; mice deficient in Klrk1 (lacking a major NK cell-activating receptor (or mice deficient in perforin (Prf1), which is an important pore-forming cytolytic protein), also showed increased ATMs that were “M2”-like in nature in diet-induced obesity.64
Collectively, these data suggest that NK cells and ILC1s have less impact on HFD-induced obesity but are more relevant for the metabolic consequences of HFD and obesity, potentially through modulation of macrophage survival and their pro- vs. anti-inflammatory properties. From the above studies, IFNγ was shown to be a leading candidate mediating the effects of these cells on macrophage inflammatory polarization. Studies in human subjects argue that the situation in long-term human obesity may be more complex and critically dependent on the time course (onset, duration, and/or reversal) of obesity, as well as on whether or not obesity was associated with metabolic consequences, such as T2D.
ILC2s
ILC2s are resident cells that mediate Type 2 immune functions in response to varied types of cues that cull dietary, metabolic, circadian, neural and mechanical inputs.75 In adipose tissue, these cells are defined as Lin—c-KIT+ Sca1+ immune cells that are highly expressed in human and murine adipose tissues.32 These cells express GATA3 and are characterized by their ability to produce Type 2 T helper cytokines, specifically IL5 and IL13 and, in adipose tissue, they also express the IL33 receptor.32, 76 Studies in mice revealed that with prolonged HFD feeding, the numbers of ILC2s and eosinophils decrease in VAT/epididymal VAT.77, 78 Similar observations were made in human subjects, in which it was found that the abdominal SAT of obese subjects revealed a decreased frequency of ILC2s compared to the lean controls.78 These considerations suggested that ILC2s might play adaptive/protective roles in HFD-induced obesity.
Two studies reported that ILC2s may regulate adipocyte differentiation to drive beige fat growth.78–80 Using gain- and loss-of-function approaches, Lee and colleagues showed that IL33-mediated activation of ILC2s promoted the growth of functional beige fat in mice under thermoneutral conditions. These authors showed that the activation of ILC2s stimulated the proliferation of bipotential adipocyte precursors and their subsequent commitment to the beige fat lineage.80 In a different study, Brestoff and colleagues discovered an additional mechanism by which ILC2s contributed to beiging of fat; they found that ILC2s produce methionine-enkephalin peptides, which act directly on adipocytes, the consequences of which include the upregulation of UCP1 expression in vitro and beiging in vivo.78, 79
In addition to ILC2 effects on adipocytes, ILC2s may also exert their effects in adipose tissue via modulating the functions of other immune cells, particularly eosinophils and “M2”-like ATMs. IL4 and IL13, secreted by ILC2s, are critical regulators of eosinophil migration and their beneficial activities in adipose tissue in HFD feeding.52, 77 Treatment of mice with IL25 increased ILC2 and invariant (i)NKT cells in obesity to impact weight loss and modulate glucose homeostasis.81 Hams and colleagues showed that treatment of obese mice with recombinant helminth-derived Schistosoma mansoni egg-derived ω1 (ω1) stimulates the recruitment /accumulation of ILC2s, eosinophils and alternatively activated macrophages (“M2”-like) into adipose tissue, which resulted in improved metabolism.82 In HFD-induced obese mice, treatment with the helminth S. mansoni and helminth egg-derived antigens reduced obesity, adiposity and insulin intolerance and increased type 2 immune responses in WAT, together with increased eosinophils and “M2”-like alternatively activated macrophages.83 Furthermore, ILC2s may directly affect the properties of Tregs; IL-33 activates ILC2s and Treg cells in adipose tissue in homeostasis, but also after helminth infection or treatment with IL2. However, ILC2-intrinsic IL33 activation was necessary for the accumulation of Treg cells and was independent of ILC2 type 2 cytokines, but partially dependent on direct co-stimulatory interactions via ICOSL-ICOS (inducible T cell CoStimulator).84
In turn, other cell types may modulate the functions of ILC2s in obesity. For example, ILC1-derived IFNγ production may repress the activation of ILC2s in adipose tissue, as IFNγ inhibited the activation of ILC2s and, therefore, Treg cell accumulation by IL33 in adipose tissue in HFD-induced obesity.84
In addition to the different types of ILCs, the specific tissues distinct from adipose tissue in which ILC2s reside may also have impact on adipose tissue ILC2s. Sasaki and colleagues recently reported the results of a comprehensive study in which they made a number of key observations on lymphocytes and ILCs in obesity. First, they showed that mice devoid of all lymphocytes (Il2−/−/Rag2−/−) were resistant to HFD-induced obesity. Second, they showed that when bone marrow cells from Rag2−/− mice (which lacked only the acquired immune cells) were transplanted into the Il2−/−/Rag2−/− mice, the benefits on protection from HFD-induced obesity were lost, specifically indicating roles for ILCs in these processes.85 Third, these authors found that the ILC2 effects on obesity resided in the small intestine, as the adoptive transfer of ILC2s from the small intestine and not the adipose tissue restored the development of HFD-induced obesity in the Il2−/−/Rag2−/− mice.85 They traced the specific mechanisms by transcriptional profiling of the small intestine- vs. adipose-derived ILC2s and found that the former expressed higher levels of IL2 than the latter; they implicated IL2 in the pathogenesis of diet-induced obesity by showing that blockade of IL2 signaling attenuated weight gain and reduced the populations of both ILC2s and ILC3s in the small intestine.85 These findings solidify that ILC2s are imbued with extensive plasticity and flexibility, not only on account of the host stress to which they are responding, but, also dependent on the tissue organ/depot in which they reside.
Taken together, these considerations indicate roles for ILC2s in obesity and suggest that their functions may be more complex than previously appreciated. More research is needed using gain- and loss-of-function strategies to unravel their complex stress condition- and tissue-dependent roles in obesity.
ILC3s
ILC3s regulate “type 3 immunity”, which is regulated in part by the production of mediators such as IL22, IL17, lymphotoxin and GM-CSF.60 ILC3s require the nuclear hormone retinoic acid receptor-related orphan receptor γt (RORγt) as a major regulatory factor for their development and function.86 Studies in mouse models of HFD suggested that ILC3 content increased in the adipose tissue with obesity.87 Wu and colleagues88 determined that in overweight children with asthma, there were higher frequencies of ILC3s, determined by expression of Lin−CD127+IL-23R+ cells89 in the peripheral blood of obese vs. lean asthmatic children, in parallel with higher mRNA levels of IL17 and IL22.88 However, there were no correlations between the frequencies of ILC3s and the expression of IL17A, and IL22 and the severity of asthma. The authors deduced from these results that childhood obesity was an independent factor associated with the elevated levels of ILC3s and these two key cytokines.88
Studies in animal models to examine the potential roles of ILC3s and, specifically, their associated mediators in diet-induced obesity are now coming to light. For example, Upadhyay and colleagues showed that mice devoid of Ltbr or lymphotoxin, which lacked IL23 and IL22, were resistant to HFD-induced obesity.90 Roles for IL22 were studied in other models. For example, in Winnie mice, which are predisposed to colitis, and display impaired intestinal barriers, upon HFD, treatment with IL22 improved the integrity of the intestinal barrier.91 In other studies, Hasnain and colleagues examined the cytokines that affected glucose intolerance through pancreatic beta cell stress in T2D and reported that in obese mice, IL22 administration modulated oxidative stress regulatory genes in pancreatic islets, suppressed ER stress and inflammation, promoted secretion of insulin and fully restored glucose homeostasis, which was followed by restitution of insulin sensitivity.92 In other studies testing IL22, Wang and colleagues showed that mice deficient in Il22 displayed increased metabolic abnormalities upon HFD feeding; in contrast, treatment of T2D db/db mice or obese HFD-fed mice with IL22 resulted in reversal of hyperglycemia and insulin resistance.93 Hence, in such studies, it is important to distinguish effects of full body deletion of a mediator such as Il22 versus the effects of its administration it adult mice. Further, it is important to note that no specific experiments have been performed, to date, to elucidate the specific role(s) of the ILC3s themselves in obesity and metabolic dysfunction. Rather, experiments have focused on the mediators produced by these cells and their roles in obesity. As these mediators may be produced by other cells, any specific roles of ILC3s in obesity require further interrogation. Furthermore, given the links between ILC3s and the lamina propria, it will also be important to discern if ILC3s modulate components of the gut microbiota, as has been shown in settings such as colitis.94–96 If so, then their putative effects in obesity might be more complex and beyond the production of mediators such as IL22.
Collectively, the discovery of ILCs and, more recently, their complex roles in obesity, underscore the potentially powerful influence of these cells in tipping the balance between inflammatory and metabolism-perturbing properties vs. anti-inflammatory and metabolo-protective mechanisms in obesity and, in particular, in the adipose tissue depots.
Beyond the cadre of peripheral cell types that regulate the response to HFDs and obesity, the innate immune cells of the brain, the microglia and astrocytes, also may contribute to obesity and its metabolic consequences. In the section to follow, we consider the roles of the microglia and astrocytes in obesity and metabolism.
The Innate Immune Cells of the Central Nervous System (CNS), Microglia & Astrocytes and their links to obesity and metabolism
Astrocytes and microglia are considered the innate immune cells of the CNS; their cell intrinsic and intercellular communications with other immune cells, such as T cells, and other cell types within the CNS, such as neurons, oligodendrocytes and vascular cells contribute to shaping the organism’s response to nutrient and metabolic stresses.97–99 Evidence supporting roles for these cells in obesity and its metabolic consequences continue to be uncovered; these findings highlight the complexities involved in solving the problem of obesity.
Microglia and Astrocytes: Origins and the Effects of Obesity
Astrocytes are glial cells of the CNS that are of ectodermal/neuroepithelial origin.100 Among their major roles in maintaining homeostasis and support to neurons in the CNS is the regulation of ions, such as potassium, chloride and calcium; pH; water transport and neurotransmitters, such as glutamate, GABA (γ-amino butyric acid), adenosine and monoamines.100 Astrocytes participate in extensive cell-cell communications in the CNS with neurons, vascular cells and microglia, as examples. Microglia are the yolk sac-derived primary myeloid/macrophages of the CNS;101–103 recent evidence suggests extensive region-to-region heterogeneity of microglia transcriptomic signatures in the CNS, as well as sexual dimorphism.104 The implications of both of these facets of microglia biology/pathobiology are not yet understood.
In obesity, there is growing evidence that both astrogliosis and microgliosis characterize this state.105 In rodents fed a HFD, increased hypothalamic astrocytosis was observed, in parallel with increased blood vessel diameter, suggestive of changes in the blood-brain barrier (BBB) properties.105 In this context, astrocytes envelop blood vessels in the CNS and their products and properties may affect vascular function. For example, altered expression of astrocyte connexins may directly impact the integrity of the BBB.106 Others showed that HFD feeding in rats resulted in astrocytosis and that chronic leptin exposure affected hypothalamic neuron-astrocyte interactions.107 Gao and colleagues showed that HFD increased the total number of arcuate nucleus microglia in the hypothalamus with evidence of increased microglial activity.108 Furthermore, akin to the often-cited controversies regarding whether or not peripheral immune cells infiltrate the brain in neurodegenerative disorders, so, too, in HFD feeding and obesity has this question been raised. Indeed, multiple studies suggest that in HFD conditions, peripheral immune cells may enter the CNS and, perhaps, contribute to the overall “inflammatory” state.109, 110 For example, Buckman and colleagues used a GFP+ bone marrow chimera model to show that feeding mice a HFD resulted in an approximately 30% increase in the number of GFP+ cells in the CNS when compared to the control diet mice; more than 80% of the GFP+ cells in the CNS bore CD45+ CD11B+ signature, indicating that these GFP+ cells were analogous to microglia and macrophages.109 Indeed, given that studies have suggested alterations in the BBB in obesity,111–115 it is plausible that the obese state favors the influx of peripheral inflammatory cells. Interestingly, these effects of HFD may differ between male and female organisms.111 Furthermore, even if peripheral immune cells themselves do not infiltrate the CNS in HFD feeding and obesity, their released mediators/factors may cross the BBB to exert paracrine effects on glial and/or neuronal properties. Finally, evidence is accruing that HFD affects the gut microbiome, the consequences of which may feed back into the brain to alter neuroinflammatory states.116–119 It is important to note, however, that not all studies demonstrated links between the gut microbiome, obesity and neuroinflammation.120, 121 In summary, evidence suggests that HFD feeding and obesity are associated with a neuroinflammatory state, at least in part characterized by astrocytosis and/or microgliosis. Whether or not peripheral immune cell infiltration into the CNS in these conditions is substantial and, if so, if such infiltration contributes to the overall neuroinflammatory state remains unclear. As well, potential roles for alterations in the gut microbiome and the changes in neurotransmitters, for example, likely contribute as well to the overall adverse consequences of HFD feeding and obesity. In order to settle these questions, as well as those of the role of the gut microbiome in neuroinflammation, well-controlled, sex-matched, diet-matched and time course- synchronized experiments, including a consideration of the role of circadian rhythms, as examples, will need to be designed for testing in multiple different species (eg, mice and rats and other mammals) and strains, using standardized protocols for microbiome and metabolite assessments.
Diving into the Transcriptional Regulators of Neuroinflammation in Obesity and HFD Feeding
In the CNS, neurons, glia and other cell types express the transcription factor, NF-κB.122 Its roles in both homeostasis and disease processes have been described. For example, in neurons, NF-κB signaling contributes to synaptic plasticity and learning and memory; in glia, NF-κB signaling is important for responses to injury and immune challenge, as well as for glutamate clearance and regulation of metabolism.122 In pathological settings, however, activation of NF-κB may unleash its pro-injury side, contributing to chronic inflammation, failure of repair, and loss of tissue functions. Numerous studies have tested roles for NF-κB in obesity and HFD feeding, which have unveiled its potentially deleterious effects in these settings.
Yan and colleagues showed evidence of excess TGFβ activity in the CNS in obesity and aging. Brain TGFβ caused an increase in glucose intolerance and hepatic glucose production, which was localized in part to astrocytes using astrocyte-specific glial fibrillary acidic protein (GFAP)-driven genetic approaches in mice models.123 Further, excess brain TGFβ induced an ER stress response in the hypothalamus, which resulted in accelerated mRNA decay of an inhibitor of NF-κB, IκBα, which thus unleashed a neuroinflammatory response in obesity.123
Andre and colleagues showed that feeding excess fats and excess carbohydrates to mice resulted in obesity and, in the arcuate nucleus of the hypothalamus, a rapid and significant increase in the number of microglia.124 These researchers attributed these effects to the expansion of the microglia pool in the hypothalamus. When they administered AraC (arabinofuranosyl cytidine), an anti-mitotic agent which blocked proliferation, they observed reduced food intake, and reduced body weight gain and adiposity. In parallel, this treatment also prevented the rise in microglia number and activation and reduced the expression of TNFα in the microglia, along with reduced NF-κB activation. Further, AraC resulted in restoration of leptin sensitivity in the hypothalamus.124
Douglass and colleagues employed tamoxifen/temporal regulation for deletion of the NF-κB essential cofactor, namely IKKβ in astrocytes in mice fed a HFD.125 When IKKβ was deleted in astrocytes prior to the HFD feeding, there were no effects on body weight or glucose intolerance compared to control animals. However, mice treated with HFD for 6 weeks prior to the tamoxifen treatment (i.e., deletion of IKKβ in mice with established obesity), the mice bearing astrocyte deletion of IKKβ revealed a protection from further gain of body weight, reduced food intake, increased energy expenditure, reduced glucose intolerance and insulin resistance, and reduced hypothalamic inflammation and astrocytosis.125 Hence, these data suggesting time-dependent roles for astrocyte NF-κB signaling in obesity unveil the complexities of mediating mechanisms at various stages of early obesity, progressive obesity and weight loss.
Zhang and colleagues tested the physiological roles of astrocyte NF-κB and found that astrocytic process plasticity and IKKβ/NF-κB are critical to the central control of blood glucose, blood pressure, and body weight in homeostasis and in chronic overnutrition states.126 Hence, even in physiological settings, the actions of NF-κB in astrocytes may favor metabolic perturbation in body mass and glucose tolerance.
Valdearcos and colleagues also addressed the role of NF-κB in microglia in obesity.127 These authors used PLX5622, a CSF1R inhibitor, to selectively deplete microglia from the CNS but not macrophages from the adipose tissue and showed that such depletion of microglia protected the mice from HFD-induced body weight gain and reduced total body fat mass, but not lean mass.127 There were no effects of PLX5622 in mice fed a standard chow. To test if microglia NF-κB contributed to these effects, the authors used tamoxifen-inducible Cx3cr1 ERT2 mice to specifically delete IKKβ from the microglia. HFD was then begun 4 weeks after tamoxifen; deletion of IKKβ resulted in reduced gain of body mass, reduced food intake, no change in energy expenditure, reduced microgliosis and reduced microglial activation, the latter effects were localized to the microglia of the mediobasal hypothalamus (MBH).127 When these authors deleted A20 in microglia, which is an inhibitor of NF-κB, they found that the benefits of reducing microglia NF-κB in HFD feeding were reversed, thereby illustrating the importance of microglia NF-κB in obesity in HFD using two distinct genetic approaches.127 Finally, using multiple tracking experiments, Valdearcos and colleagues showed that peripheral myeloid cells that are recruited to the MBH in HFD feeding contribute to the observed microgliosis and that this recruitment of the peripheral myeloid cells is controlled by the resident microglia of the MBH.127
It is important to note that other transcriptional regulatory factors, such as STAT3, AKT and JNK signaling have been implicated in glial inflammation in HFD feeding and obesity;128 the aforementioned experiments testing NF-κB illustrate the importance of this factor in these processes. It will be important, therefore, to consider in the experimental design of these studies the hierarchical dependence, or independence, of NF-κB with these distinct pathways in the immunometabolic responses spurred by glia in HFD feeding and obesity. This is especially relevant since generalized inhibition of these canonical signaling cascades, which play important roles in health and homeostasis, may be a double-edged sword in the armamentarium of obesity-tackling therapeutic agents.
Emerging lessons on adipose tissue immune cell content/functions in obesity - before and after significant weight loss
This Review focuses on innate immune cells in obesity and its metabolic consequences. Of note, not discussed here are the roles of adaptive immune cells, such as T cells, B cells and their multiple subsets, as well as Natural Killer T cells (NKT cells) and others in obesity and metabolic dysfunction. Thus, a key challenge that emerges is how to integrate these multiple and distinct innate immune cell types and their functions into a hierarchical schema for understanding the mechanisms underlying obesity and metabolic dysfunction. In this context, insights from studies of weight loss may be a good place to gather hints as to what cell types/mediators are actually playing pathogenetic roles. Hence, in this context, single-cell RNA-seq of adipose tissue depots may be a beneficial strategy to accomplish these goals. Data reported to date are restricted to but a few publications; however, with more work, especially considering the time course of obesity as well as “before and after” bariatric surgery and other means to induce significant weight loss, the pieces of this complex puzzle may begin to come together. For starters, Vijay and colleagues recently published the results of single-cell RNA-seq from multiple depots (SVF VAT and SAT) of 25 adipose tissue samples (12 VAT and 13 SAT) from 14 human subjects with obesity undergoing bariatric surgery. Although there are no post-operative sample analyses provided in this report, the results, nevertheless, provide state-of-the-art and novel insights into cell type specifications in human obese adipose tissue.129 The authors identified that 34% of the cells from their merged clusters expressed markers consistent with different immune cell populations; they subsetted these immune cell groupings to identify 14 new clusters of immune cells (termed IS1-IS14), which were present in both VAT and SAT depots. IS1, IS4, I6 and IS8 clustered together largely and bore transcriptome signatures reminiscent of NK and T cells (including naïve T cells and activated T cells). IS2, IS3, IS7, IS9 and IS12 were identified as ATMs; the cell types within these clusters expressed varied amounts of CD68 and were also shown to express genes linked to lipid metabolism in obesity. IS2, in particular, bore signatures relevant to “metabolically-activated macrophages.” IS5 and IS10 were identified as CD16− monocytes, which bore high expression of S100A8, S100A9 and S100A12.129 Notably, these S100s, especially S100A12, are ligands of RAGE and toll-like receptors (TLRs).130–132 S100s are considered as damage-associated molecular pattern molecules (DAMPs), which may be released by stressed cells and, thereby, amplify and perpetuate pro-damage responses, at least in part through interactions with receptors such as RAGE and TLRs, which have been described as pattern-associated molecular pattern receptors or PAMPs.133–135 Vijay and colleagues further showed that IS5 are most likely representative of DCs in these populations. Finally, they linked IS11 to B cells and noted that the remaining cluster, IS14, revealed mixed signatures of various cell types.129 Overall, this data set provides a promising starting point for which next steps include expansion of cell type-specific markers to capture a broader swath of cell types, such as the eclectic ILC1s, ILC2s and ILC3s; examination of lean subjects; examination of peripheral blood; and, where feasible, examination of SAT, at least, in the “before and after” bariatric surgery scenarios.
Indeed, for obese patients, bariatric surgery has induced significant weight loss and improvements in such metabolic and cardiovascular parameters as glycemic control, serum lipid levels, blood pressure, and renal function.136 Although the underlying mechanisms are complex and relate, in part, to the presence or absence of T2D and the type of surgical intervention employed to induce weight loss, irrespective of these factors, studies are now beginning to report the effects of weight loss on immune cell content and properties in the peripheral circulation and in the accessible adipose tissue depots.
Poitou and colleagues performed bulk RNA-seq on adipose tissue from 22 obese women before and at three months after bariatric surgery (15 Roux-en-Y gastric bypass and 7 adjustable gastric banding).137 In these women, significant reductions in body mass occurred in all 22 of them, along with improvement in multiple metabolic markers. Using a FDR (false discovery rate) of 5%, the authors reported that 7.6% of genes (1214 total) were differentially expressed post-surgery. There were trends in greater numbers of differentially-expressed genes in the Roux-en-Y vs. the adjustable gastric banding procedures.137 Gene Set Enrichment Analyses (GSEA) revealed that some of the top hits in the upregulated group were in the following categories “Ribosomes,” “mRNA splicing,” “IFNγ signaling,” and “antigen processing/presentation.” In the down-regulated group, significant hits included “ECM organization,” “biosynthesis of cholesterol,” “biosynthesis of unsaturated fatty acids,” “packaging telomere ends,” “metabolism of porphyrin and chlorophyll,” and “metabolism of starch and sucrose.” Specifically, among immune cell modules, first, a macrophage module was highly conserved post- vs. pre-operatively with the exception of two down-regulated genes, DUSP2 and CD300C. Second, within a T cell module, there were differences in genes involved in T cells signaling such as CXCR1, CXCR2, GPR97, CCR7 and IL7R. Third, neutrophil-mediated inflammation was reduced post-operatively (reflective of changes in S100A8, S100A12, CD300E, VNN2, TUBB1 and FAM65B). Finally, 19 genes linked to the interferon signaling pathways were highly preserved in the post-operative vs. the pre-operative state, except for DDX60.137 The authors concluded that overall improvements in the inflammatory state in the post-operative condition might provide insight into novel targets for therapeutic interventions.
In other studies supportive of these general conclusions, Wang and colleagues showed that ILC1s were reduced in the peripheral circulation post-bariatric surgery;73 Latorre and colleagues showed that TLR3 as well as other members of the TLR family were reduced in SAT post-operatively vs. baseline;138 and Nijhuis and colleagues showed that multiple markers of neutrophil activation were reduced in the periphery after bariatric surgery.139
Perspectives, Gaps and Next Steps
In summary, innate immune cells play important roles in metabolic homeostasis, perturbation and resolution in the adipose tissue depots. The innate immune cell contribution to these processes is not confined to the periphery, but extends to the brain, in which cells such as microglia and astrocytes respond to peripheral cues in overnutrition and attenuated physical activity, thereby contributing to obesity and metabolic dysfunction. Tables 1 and 2 depict representative pioneering studies described in this Review with respect to role for innate immune cells in metabolic perturbation in human subjects and animal models, respectively.
Table 1.
Representative Human Subject Studies
| Human subjects | Experimental Methods | Major Findings | Reference |
|---|---|---|---|
| Macrophages | |||
| Healthy lean, overweight, and obese subjects were fed a liquid formula diet of 40% fat (corn oil), 45% carbohydrate (glucose polymer), and 15% protein (casein hydrolysate). | CD68 marker was used to determine the macrophage content in lean vs. obese human adipose tissues. | Adipose tissue macrophage numbers increase in obesity and participate in inflammatory pathways that are activated in adipose tissues of obese individuals. | 4 |
| Adipose tissue from human obese and non-obese subjects was collected by abdominoplasty and laparoscopic intra-abdominal surgeries. | Samples were analyzed by proteomic, gene expression and flow-cytometric approaches. | Cell surface markers indicative of altered lipid metabolism and inflammatory markers were increased in adipose tissue macrophages of obese subjects compared to the lean controls. Markers of classical activation are absent on ATMs from obese humans. | 19 |
| 52 obese and 29 non-obese control female human subjects were recruited and their abdominal subcutaneous white adipose tissue (WAT) biopsies and serum samples were collected. | Metabolome released from subcutaneous abdominal WAT in 81 obese and non-obese women was assessed for glutamine levels (measured in the releasate and in the serum). | In WAT, glutamine was downregulated in obesity. However, there were no differences in the levels of serum glutamine between the groups. Glutamine was linked to suppression of proinflammatory genes in human adipocytes. | 22 |
| 10 female human subjects underwent weight loss by consumption of a very low calorie diet . | Subcutaneous adipose tissue (SAT) biopsy samples were obtained and stained for CLS and markers of metabolic and inflammatory perturbation, such as levels of glucose, high sensitivity C-reactive protein (hsCRP). | CLS increased after weight loss and markers of metabolic and inflammatory perturbation, such as levels of glucose, hsCRP, circulating fatty acids and glycerol were decreased. | 26 |
| Obese T2D subjects were treated with either CDP571, a humanized monoclonal anti-TNFα antibody, orrecombinant human TNF receptor: Fc fusion protein or TNFα inhibitor etanercept. | Insulin sensitivity test and/or hyperinsulinemic, euglycemic clamp assays were performed. | TNFα blockade did not incontrovertibly affect insulin sensitivity in obese human subjects. | 28–30 |
| Neutrophils | |||
| Patients with obesity who underwent bariatric/metabolic surgery were recruited. | Fasting blood and subcutaneous abdominal adipose tissue were obtained before (n=14), at one month (n=9), and 6-12 months (n=14) after bariatric/metabolic surgery from subjects who were not on insulin or anti-diabetes medication. | The neutrophil content in obese adipose tissue continued to rise to 15-20 fold above that at the time of surgery and this persisted for months post-surgery, despite improvements in overall metabolic function. | 34 |
| Twenty-three obese patients undergoing gastric band surgery were recruited to a longitudinal intervention study, along with non-obese, healthy gender- and age-matched controls. | Peripheral blood neutrophil (PBNs) were isolated by density centrifugation and a comprehensive analysis of PBN function was undertaken at various stages of the patients’ bariatric surgical care pathway. | Obese patients exhibited exaggerated PBN activity in response to various stimuli, with higher reactive oxygen species (ROS) generation and release of pro-inflammatory cytokines and lower PBN extracellular trap (NET) formation. PBN chemotactic accuracy was also impaired prior to surgery. Weight loss was associated with normalized NET production and lower ROS production and cytokine release relative to healthy controls. | 41 |
| Seventy-three patients with morbid obesity and 55 healthy subjects, and 21 subjects with severe coronary artery disease were recruited before and after bariatric surgery. | Anthropometric parameters, peripheral blood pressure, biochemical and serum analysis at the enrollment and at twelve months after surgery were measured along with assessment of plasmatic levels of MPO-DNA complexes by ELISA. | NETs levels were higher in obese than in controls and correlated with the anthropometric variable (BMI, waist, hip), glyco-metabolic variables and systolic blood pressure. After surgery, there were no definitive changes in NETs. | 42 |
| Mast cells | |||
| A general population of 1,216 persons, aged 15-69 years, was recruited. | Mast cell-specific serum tryptase was determined using ImmunoCAP Tryptase assay. | Higher levels of serum mast cell-specific tryptase were demonstrated in obese vs. lean individuals. | 45 |
| Human subjects were recruited from the Lexington, KY area in the summer; mean temperature 20-24 °C. | Baseline biopsies of thigh adipose were performed, the subjects then applied an icepack to one leg for 30 minutes each day for 10 consecutive days, and thigh biopsies were performed on the cold treated leg and the contralateral leg. Nanostring ncounter multiplex system was performed along with immunohistochemical analysis of the mast cell marker, anti-tryptase. | Regardless of BMI, mast cell tryptase and CCL26 (a chemokine for mast cells) were upregulated in the cold. Upon repeated cold exposure to unilateral thigh, in both lean and obese subjects, increased mast cell degranulation was evident; however, only in lean but not obese subjects, increases in mast cell content in the SATs were observed. | 48 |
| NK cells and ILC1s | |||
| 120 consecutive male and female obese subjects and 11 lean healthy controls were recruited | Blood samples were collected and BMI and blood pressure were measured. Peripheral blood mononuclear cells were prepared | Compared to lean controls, obese patients had fewer circulating NK cells and cytotoxic T cells | 68 |
| A cohort of 90 children (45 obese/45 non-obese) aged between 6 and 17 years was recruited. | Blood samples were collected for Peripheral blood mononuclear cells and various immune cell population was measured. Clinical assessments and biochemical measurements were performed. | Circulating NK cells were reduced in obese vs. lean children, which inversely correlated with BMI and insulin resistance and the NK cells in the obese children were more activated than those from lean children. | 69 |
| Stromovascular cell fractions (SVFs) from VAT and SAT from obese humans undergoing bariatric surgery were studied in vitro. | Transcriptional profiling, flow cytometric phenotyping, enzyme-linked immunosorbent assay and intracellular cytokine staining were performed. | VAT demonstrated higher IFNγ mRNA transcript levels, and NK cells, which constitutively express IFNγ, were also higher in the VAT vs. SAT; SVF-derived NK cells expressed IFNγ on activation, which was associated with TNFα expression by macrophages. | 70 |
| Thirty-two healthy adults with obesity were divided into control and experimental groups. Participants in the experimental group performed a 3-month program of exercise training and nutrition. | Peripheral blood mononuclear cells were analyzed by flow cytometry and Western blot assay. | Diet and exercise-induced weight loss over a three month period in obese individuals resulted in increased production of IFNγ, along with increased NK cell functions. | 71 |
| A total of 85 individuals, including 49 obese patients underwent laparoscopic Roux-en-Y gastric bypass (RYGB) surgery, and 36 age- and sex-matched non-obese non-T2D controls received elective abdominal surgery (e.g., hernia or hemangioma resection) were enrolled. | Peri-umbilical adipose tissue was collected during surgery and blood samples were collected before and 3 months after surgery. | The number of circulating and omental adipose tissue ILC1s was higher in obese vs. lean individuals and higher levels of the ILC1s were associated with poorer metabolic health. | 73 |
| ILC2s | |||
| Subcutaneous white adipose tissue (S-WAT) from the abdominal region was obtained from lean and obese subjects. | ILC2 was measured from all subjects by flow cytometry and their frequencies were also compared for all characteristics | The abdominal SAT of obese subjects revealed a decreased frequency of ILC2s compared to the lean controls | 78 |
| ILC3s | |||
| Fifty asthmatic children who were diagnosed with mild to severe asthma based on commonly-accepted clinical laboratory guidelines were recruited for the study and their blood samples were collected. | PBMCs were isolated from the blood and the levels of ILC3 was measured by flow cytometry and qPCR. | Overweight children with asthma had higher frequencies of ILC3s in the peripheral blood vs. lean asthmatic children, in parallel with higher mRNA levels of IL17 and IL22. | 88 |
| Twenty-five subcutaneous and visceral adipose tissues from 14 human obese subjects undergoing bariatric surgery were collected along with tissue from healthy non-obese subjects. | Stromal vascular fraction was isolated from mature adipocytes and subjected to single-cell RNA sequencing. | 34% of the cells expressed markers consistent with different immune cell populations, specifically of NK cells, T-cells and macrophages that were indicative of “metabolically-activated macrophages.” | 129 |
| Twenty-two obese women before and at three months after bariatric surgery were included. | Adipose tissues were collected during and three months after surgery and were subjected to bulk RNA-sequencing. | Significant reductions in body mass occurred in all 22 subjects, along with improvement in multiple metabolic markers; macrophages, T-cells and neutrophil signatures showed overall improvements in the inflammatory state in the post-operative condition. | 137 |
Table 2.
Representative Animal Subject Studies
| Animal model | Experimental Methods | Major Findings | Reference |
|---|---|---|---|
| Inflammation and obesity | |||
| Obese (ob/ob) mice, Diabetic (db/db) mice and Zucker (fa/fa) rats. | Tnf (gene encoding Tumor Necrosis Factor-α) mRNA transcripts were measured in adipose tissue of T2D mice and rats, and in streptozotocin-induced diabetic mice (a model of type 1 diabetes, T1D). | TNF-α mRNA transcripts were significantly increased in the adipose tissue of T2D mice and rats, but not in the adipose tissue of streptozotocin-induced T1D mice. In T2D mice, the upregulation of Tnf preceded the development of significant hyperglycemia. TNFα contributed to impaired sensitivity to insulin in the T2D models. | 3 |
| Macrophages | |||
| Obesity was induced in C57BL/6J mice by a high-fat diet (diet induced obesity). | F4/80 was used to determine the macrophage content in lean vs. obese adipose tissues in mice. | Obese adipose tissue displayed increased macrophage content compared to lean adipose tissue and TNFα was increased in obese adipose tissue with increased macrophage content. | 4 |
| T2D (ob/ob) obese mice | T2D (ob/ob) mice were treated with rosiglitazone for 28 days. mRNA levels of inflammatory markers were measured. | Rosiglitazone- vs. vehicle-treated mice showed decreased inflammation. | 5 |
| Netrin1fl/fl LysMCre+/− (Ntn1Δmac) mice were studied. | Ntn1 was deleted in myeloid cells and subjected to high fat feeding. Single cell RNA-sequencing of CD45+ myeloid cells was performed. | Single-cell RNA-sequencing (RNA-seq) of CD45+ myeloid cells revealed that upon deletion of Ntn1, a significant attrition of ATMs was noted. Increased insulin sensitivity, improved lipid handling and metabolic function and decreased proinflammatory eicosanoids were observed. | 15 |
| C57BL/6J and Hif1αfl/fl LysMCre−/− were subjected to high fat feeding. | High-fat feeding of wild-type mice and myeloid-specific Hif1α−/− mice was used to examine the role of hypoxia-inducible factor-1α (HIF-1α) in ATMs of obese adipose tissue. | Transcriptome analysis and real-time flux measurements showed that HIF-1α did not alter the proinflammatory state of the ATMs after 8 weeks of HFD feeding. | 21 |
| C57BL/6J mice on a chow diet received daily intraperitoneal injection of glutamine (1 g/kg body weight) or PBS (20 mL/kg body weight) for 14 days. | qPCR analyses of Glul mRNA expression in eWAT samples was performed. | Glutamine had no effects on body weight but levels of proinflammatory genes in the eWAT were significantly lower and levels of Adipoq (adiponectin) gene were significantly higher. | 22 |
| C57BL/6J mice were fed high-fat diet and their average daily food intake was measured for a month. When they reached 40-41g of weight, caloric restriction (CR) was initiated. | Abdominal subcutaneous, epididymal, perirenal, and mesenteric adipose tissues were analyzed for macrophage content by qPCR and immunohistochemistry for F4/80. | In the earliest days after CR to induce weight loss in obese mice, ATM content rose in the immediate time frame (approximately days 1-7 of CR), that is, leaner mice displayed higher ATM content and Lipolysis was identified to be the key process triggering increased ATM content in the adipose tissue through the process of increased macrophage migration. | 24 |
| C57BL/6J mice were fed high-fat diet for 24 weeks and then switched to CR @ 70% of high-fat diet for two weeks. | VAT was subjected to single-cell RNA sequencing and stained for crown-like structures (CLS) by immunohistochemistry. | A major macrophage subpopulation in obese VAT expressed genes relevant to lipid binding and metabolism and the HFD/CR mice displayed significantly more CLS in VAT. | 25 |
| C57BL/6J mice and mice with global or myeloid specific deletion of Ager (gene encoding RAGE) were fed high-fat diet. | Insulin sensitivity, the FBC content and the levels of pro and anti-inflammatory markers in the eWAT were measured. | The Ager deleted mice showed improved Insulin sensitivity, with decreased FBC content and higher levels of anti-inflammatory markers in the eWAT. | 27 |
| Neutrophils | |||
| C57BL/6J mice were fed a chow or HFD from 4 to 20 weeks of age and randomized to either a sedentary group or exercise group five days/week. | Neutrophil content, neutrophil elastase activity and markers of inflammation were measured in the adipose tissue. | Exercise training reduced adipose neutrophil content, neutrophil elastase activity and markers of inflammation. | 36 |
| C57BL/6J mice were fed a 60% HFD treated with Peptidyl Arginine Deiminase 4 inhibitor Cl-amidine; a compound that blocks neutrophil extracellular traps (NET) release. | Flow cytometry of adipose tissue and liver, immunohistological analysis and glucose and insulin tolerance tests were performed. | No differences in immune cell infiltration into the adipose tissue or liver were noted and there was no effect on insulin resistance. | 39 |
| Mast cells | |||
| C57BL/6 mice were fed a high-fat diet for 20 weeks. | Prevalence and distribution of dead adipocytes, mast cells, macrophages (F4/80), and apoptotic cells were measured in the eWAT along with gene and/or protein expression of several adipocytokines, F4/80, mMCP6 and cleaved caspase-3. | Increased mast cell content in obese vs. lean adipose tissue was observed. | 44 |
| WT (C57BL/6 and WBB6F1/J) and c-Kit deficient, KitW-sh/W-sh mice were fed a western diet for 12-13 weeks following which the mast cell stabilizer, disodium cromoglycate (DSCG) was administered. | Body weight, glucose tolerance and energy expenditure and the weight of the WAT depots were measured. | The c-Kit deficient mice on a western diet gained less weight with significant reductions in WAT depots, in parallel with improved glucose tolerance and increased energy expenditure. Administration of DSCG, attenuated body weight gain only in the WT but not the mast cell-deficient mice; mast cell content in the WAT was not affected. | 46 |
| Kit-dependent B6.KitW/Wv and Kit-independent B6.Cpa3Cre/+ mast-cell-deficient mouse strains were fed either chow diet or high-fat diet for 16 weeks. | Body weight, glucose tolerance, energy expenditure and adiposity were measured. | No differences in body mass or adiposity vs. control mice were observed and glucose and insulin intolerance were not altered in these mice. | 47 |
| WT and Kitw-sh/w-sh, c-Kit deficient mice at 12-weeks of age were injected with norepinephrine to stimulate thermogenesis and mast cells were reconstituted by bone marrow transplant. | Body weight, core body temperature, and indirect calorimetry were measured. | When stimulated by norepinephrine, the metabolic rate was enhanced in these mice compared to controls. Mast cell reconstitution in those mice (in the SAT) reversed these effects of norepinephrine. | 49 |
| Eosinophils | |||
| WT (C57BL/6) and Il4-deficient male mice were subjected to chow diet or high-fat feeding for 15-24 weeks. | Body weight, glucose tolerance and insulin sensitivity were measured. | Number of eosinophils in adipose tissue in obesity was significantly reduced compared to that observed in the lean state and when mice lacked eosinophils, they were more prone to diet-induced obesity and glucose intolerance. | 52 |
| C57BL/6J mice fed a high fat diet (HFD) were simultaneously given recombinant interleukin-5 (rIL5) for 8 weeks to increase adipose tissue (AT) eosinophils. | Weight gain, AT inflammation, glucose, lipid, and mixed-meal tolerance, AT insulin signaling, energy substrate utilization, energy expenditure, and white AT beiging capacity were measured. | Eosinophils were increased ~3-fold in AT of obese HFD-fed mice treated with rIL5, and thus were restored to levels observed in lean healthy mice. However, there were no significant differences in rIL5-treated mice among the comprehensive set of metabolic assays, despite the increased AT eosinophils. | 53 |
| C57BL/6J mice were either fed a chow diet or a high fat diet (HFD) for 12 weeks and later switched to chow diet to induce weight loss for up to 6 weeks. | Body weight, individual tissue weights, and the levels of adipose eosinophil and macrophages were measured. | Weight loss (switching mice from HFD to low fat diet) led to increased eosinophil content in adipose tissue, in parallel with reduced ATM content, reduced inflammation, and improved tissue remodeling. | 54 |
| Male WT (C57BL/6J) and CCR2 deficient (CCR2−/−) mice were subjected to high-fat diet. | Insulin sensitivity, body mass, or adiposity and the number of eosinophils in different tissues were measured. | No differences in insulin sensitivity, body mass, or adiposity were observed. Increase in numbers of eosinophils in all WAT depots was observed with no differences in other organs such as liver or bone marrow. | 57 |
| NK cells and ILC1s | |||
| WT (C57BL/6), Rag1 deficient Rag1−/−, IFNγ deficient Ifng−/− and Prkdc−/−IL2rg−/− (mice lacking mature T, B, and natural killer lymphocytes) were fed either a chow diet or high-fat diet. | In adoptive transfer experiments, adipose ILC1s were purified by flow cytometry from C57BL/6 or Ifng−/− mice fed a HFD and transferred into Prkdc−/−IL2rg−/− mice on HFD. After HFD feeding for 4 weeks, recipient peripheral organs were harvested, and adoptively transferred cells were analyzed by flow cytometry. | Compared to controls not receiving the ILC1s, there were no differences in body weight but those mice receiving the ILC1s were more glucose intolerant and showed more IFN-γ-dependent interstitial fibrosis in the VAT with more CD11C+ cells and more CLS vs. PBS. | 73 |
| WT (C57BL/6) and Ifng−/− mice were fed high-fat diet for 12 weeks . | In adoptive transfer experiments, purified adipose iNK or ILC1 from HFD-fed WT and Ifng−/− mice were transferred into Rag2−/−Il2rg−/− mice and further subjected to high-fat feeding for 2 weeks. Recipient peripheral organs were harvested and analyzed by flow cytometry 2 or 4 weeks after final transfer and additional HFD feeding. | In both cases, the number of “M1”-type macrophages was higher in the adipose tissue of the recipients, but especially upon the adoptive transfer of ILC1s. In both iNK or ILC1 transfers, the recipients were more glucose and insulin intolerant vs. controls, which was independent of the amount of weight gained on the HFD. | 66 |
| WT (C57BL/6) and E4bp4+/− mice were fed high-fat diet for 8 weeks and injected with NK cell neutralizing antibody to deplete NK cell population. To expand NK cells in vivo, mice fed chow diet or HFD for 6 weeks were intraperitoneally injected with IL-15. For the reconstitution experiments E4bp4−/− homozygous knockout mice were used. | To reconstitute E4bp4−/− homozygous knockout mice with NK cells, splenic NK cells were isolated from C57BL/6 WT mice that had been fed HFD for 8 weeks by negative sorting using a mouse NK cell isolation kit were used. | When NK cells were depleted with neutralizing antibodies or using E4bp4+/− mice, improvements in insulin resistance and decreased numbers of ATM and adipose tissue inflammation were observed. When NK cells were expanded through IL15 administration or by reconstitution of NK cells into E4bp4−/− mice, ATM numbers and adipose tissue inflammation increased, in parallel with increased insulin resistance. | 65 |
| WT (C57BL/6) were fed high-fat diet for 8 weeks and injected with NK cell neutralizing antibody to deplete NK cell population. For other experiments Klrk1−/− (mice lacking a major NK cell-activating receptor) and Prf1−/− (mice lacking perforin) knockout mice were subjected to high-fat feeding. | Adipose tissues were harvested for M1/M2 macrophage markers. | There were increased numbers of Adipose tissue macrophages that were more “M2”-like in nature in both models. | 64 |
| ILC2s | |||
| WT (C57BL/6) and IL33-deficient Il33−/− mice were subjected to prolonged high-fat diet feeding and administered carrier-free recombinant murine IL-33. | Visceral adipose tissues were collected and the numbers of ILC2s and eosinophils were measured. | With prolonged HFD feeding, the numbers of ILC2s and eosinophils decrease in VAT/epididymal VAT. | 78 |
| WT (C57BL/6) mice and heterozygous Red5 (Il5Red5/+, R5) mice were maintained at thermoneutrality and administered various doses of IL-33 for over 8 days. | Subcutaneous WAT was collected at the end of the study and the levels of UCP1 expression was measured by immunoblotting technique. | IL33-mediated activation of ILC2s promoted the growth of functional beige fat in mice under thermoneutral conditions. | 80 |
| WT (C57BL/6), IL1RL1-deficient (Il1rl1−/−) Il5tdtomato-cre/+ and Il33−/−mice were used. | IL33 was induced by either helminth infection or treatment with IL2 on these mice and the levels of ILC2s and Tregs were measured by flow cytometry. | IL-33 activates ILC2s and Treg cells in adipose tissue in homeostasis, but also after helminth infection or treatment with IL2 and ILC2-intrinsic IL33 activation was necessary for the accumulation of Treg cells. | 84 |
| Male WT ( C57BL/6), Rag2−/−, and Il2rg−/− Rag2−/− mice were fed a normal chow diet or high-fat diet (HFD) and analyzed over a period of several weeks. | Body weight, glucose and insulin tolerance tests and indirect calorimetry were measured in addition to bone marrow transplantation and gut microbiome studies. | Mice devoid of all lymphocytes (Il2rg−/− Rag2−/−) were resistant to HFD-induced obesity; bone marrow transplant indicated roles for ILCs in protection from diet induced obesity; the ILC2 effects on obesity resided in the small intestine. | 85 |
| ILC3s | |||
| Male WT ( C57BL/6), Rag2−/− and leptin deficient Lepob/ob (ob/ob) mice were fed either chow diet or high-fat diet. | Lung and adipose tissues were harvested and the presence of ILC3 was detected by flow cytometry. | ILC3 content increased in the adipose tissue with obesity. | 87 |
| Wild type (WT) C57BL/6 and Winnie mice were fed either a chow diet or a high-fat diet and administered recombinant IL-22. | Body weights of mice and diarrhea and rectal bleeding scores were recorded weekly. | Winnie mice, which are predisposed to colitis, displayed impaired intestinal barriers, upon HFD. However, treatment with IL22 improved the integrity of the intestinal barrier. | 91 |
| Wild type (WT) C57BL/6 mice, were fed either a chow diet or a high-fat diet and injected with control Ig or IL22. | Glucose and insulin tolerance tests were performed and pancreatic islets were isolated and cultured overnight for rt-qPCR analysis. | In obese mice, IL22 administration modulated oxidative stress regulatory genes in pancreatic islets, suppressed ER stress and inflammation, promoted secretion of insulin and fully restored glucose homeostasis, which was followed by restitution of insulin sensitivity. | 92 |
| Wild type (WT) C57BL/6, T2D (db/db) mice and IL-22r1 deficient mice fed a high-fat diet were used. | Body weights of mice were monitored and glucose and insulin tolerance tests were performed. | Mice deficient in Il22 displayed increased metabolic abnormalities upon HFD feeding; in contrast, treatment of T2D (db/db) mice or obese HFD-fed mice with IL22 resulted in reversal of hyperglycemia and insulin resistance. | 93 |
| Microglia | |||
| Wild type (WT) C57BL/6 and Lepob/ob (ob/ob) and fed either a chow diet or a high fat diet and were treated with leptin for 5 days. | Body weights of mice were monitored and brains were collected at the end of the study for qPCR and immunohistochemistry. | HFD increased the total number of arcuate nucleus microglia in the hypothalamus with evidence of increased microglial activity. | 108 |
| Wild type (WT) C57BL/6 and Global green fluorescent protein (C57BL/6-Tg (CAG-GFAP)) mice on C57BL/6 background were used. | Bone marrow from GFP mice were transplanted into WT recipients and fed either high-fat diet or chow diet following which adipose tissues and brain were extracted for qPCR and flow cytometry . | Feeding mice a HFD resulted in an approximately 30% increase in the number of GFP+ cells in the CNS when compared to the control diet; more than 80% of the GFP+ cells in the CNS bore CD45+ CD11B+ signature, indicating that these GFP+ cells were analogous to microglia and macrophages. | 109 |
| Wild type (WT) C57BL/6 and Tlr4−/− mice were used. | Mice were injected with either vehicle or TGF-β1 in the brain by intracerebroventricular surgery and fed either chow diet or a high-fat diet and their body weights were monitored and glucose and insulin tolerance tests were performed. | Brain TGF-β1 caused an increase in glucose intolerance and hepatic glucose production, which was localized in part to astrocytes using astrocyte-specific glial fibrillary acidic protein (GFAP)-driven genetic approaches. | 123 |
| Wild type (WT) C57BL/6 were used. | All mice were subjected to intracerebroventricular surgery and injected with artificial or aCSF containing 4.5 μg/μL BrdU with or without AraC 15 μg/μL and maintained on either chow diet or switched to an HFD. | Feeding excess fats and excess carbohydrates to mice resulted in obesity and, in the arcuate nucleus of the hypothalamus, a rapid and significant increase in the number of microglia was observed. | 124 |
| GFAP expressing GFAP/CAIkkβ+/− mice and their wild-type (WT) littermates were fed either high-fat diet or chow diet. | Body weights of mice were monitored and glucose and insulin tolerance tests were performed. | Astrocytic process plasticity and IKKβ/NF-κB are critical to the central control of blood glucose, blood pressure, and body weight in homeostasis and in chronic overnutrition states. | 126 |
| Wild type (WT) C57BL/6 fed either control or high-fat diet containing PLX5622. | PLX5622, a CSF1R inhibitor, selectively depleted microglia from the CNS but not macrophages from the adipose tissue and the body mass and composition were measured . | Depletion of microglia protected the mice from HFD-induced body weight gain and reduced total body fat mass, but not lean mass. | 127 |
Recent research has underscored that a cadre of innate immune cell types, both cell-intrinsic and intercellular communication mechanisms, drive the formation of a range of pro-inflammatory vs. anti-inflammatory mediators, whose expression appears to relate to the degree of, or protection from, metabolic perturbation. Hence, the expression of individual cytokines may not be ascribed to a single innate immune cell type. Figure 1 summarizes these concepts with respect to the consequences of overall innate immune cell function and their cross-talk with adaptive immune cells in a key organ affected by metabolic perturbation, the adipose tissue. Specifically, Figure 1A illustrates that homeostatic mediators, such as IL10, IL5, IL4, and IL13, produced by a diverse set of innate immune cells, override proinflammatory and pro-damage forces in the lean and metabolically healthy state; thus, the functions of adaptive immune cells, such as Tregs, may prevail. Beneficial interactions with the immune cells of the brain, skeletal muscle, small intestine (and the microbiome) and the liver join forces to block obesity and foster insulin sensitivity. In overnutrition and decreased physical activity, however, Figure 1B illustrates that the innate immune cells seem to become part of the problem! Specifically, when the adipose tissue depots assume a pro-DAMP signature, the innate immune cells produce mediators that block homeostasis and amplify pro-damage and pro-metabolic perturbants, such as IL6, IL1β, TNFα, MCP1, NOS2, IFNγ and IL2. Together with cell-intrinsic and intercellular cross-talk with innate immune cells of the brain, small intestine, skeletal muscle and liver, the environment becomes one of pro-damage inflammation and insulin resistance. Upon weight loss, Figure 1C, there is evidence of reducing DAMPs, and suppression of the pro-damage mediators with return of at least some of the homeostatic factors. The content of innate immune cells fluctuates; intriguingly, in the early phases of weight loss, for example, ATM content increases, perhaps in an effort to phagocytose and engulf dying adipocytes and their sequelae. More, though, needs to be discovered about the effects of weight loss on immune cells in the brain, gut, liver and skeletal muscle. Certainly, however, even in weight loss, seemingly unexpected things happen. For example, as discussed above, in weight loss, the IFN signaling pathways in the adipose tissue are not down-regulated. Such considerations reinforce that the chief function of the innate immune system is to react promptly to acute damage, such as infection or trauma. To summarize, the depictions in Figure 1A–B–C suggest that it is unlikely that a particular cytokine or inflammatory mediator is produced exclusively by one type of innate immune cell. Rather, the balance of the products of these cells, heavily influenced by the overall microenvironment, emerges from cell-intrinsic and intercellular communications, not solely with other innate immune cells, but also with those of the adaptive immune system, vascular cells and adipocytes.
Figure 1. Obesity and Multi-Organ Metabolism: Roles of Innate Immunity in Lean, Homeostatic State, Obesity and Weight Loss.
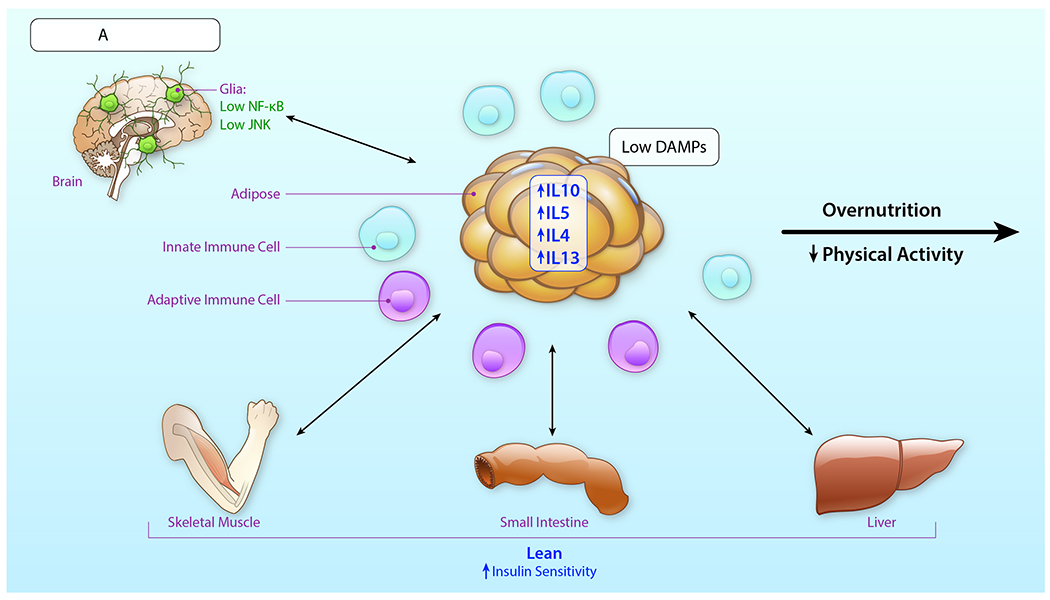
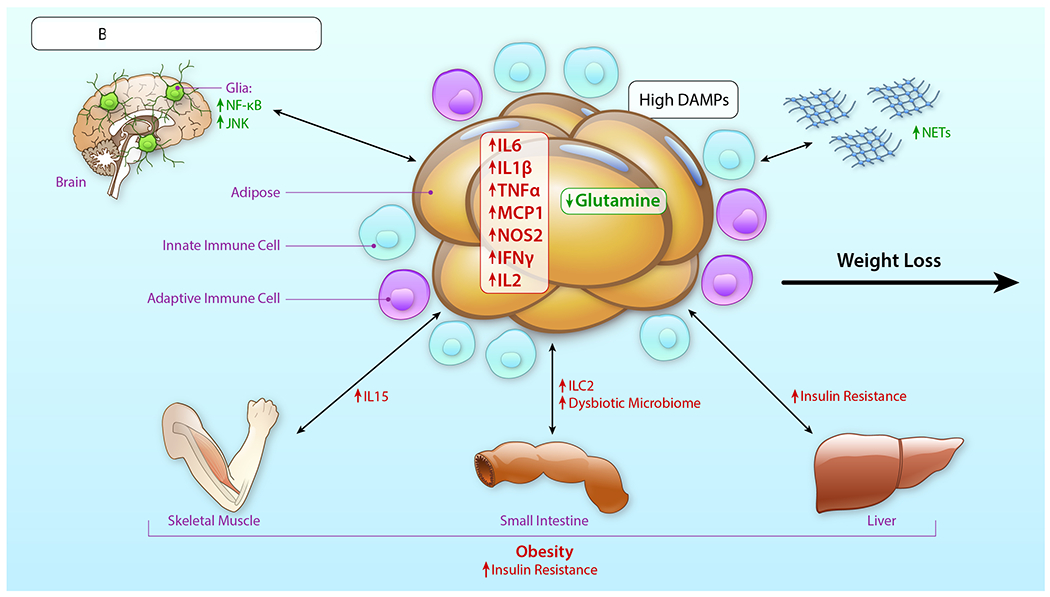
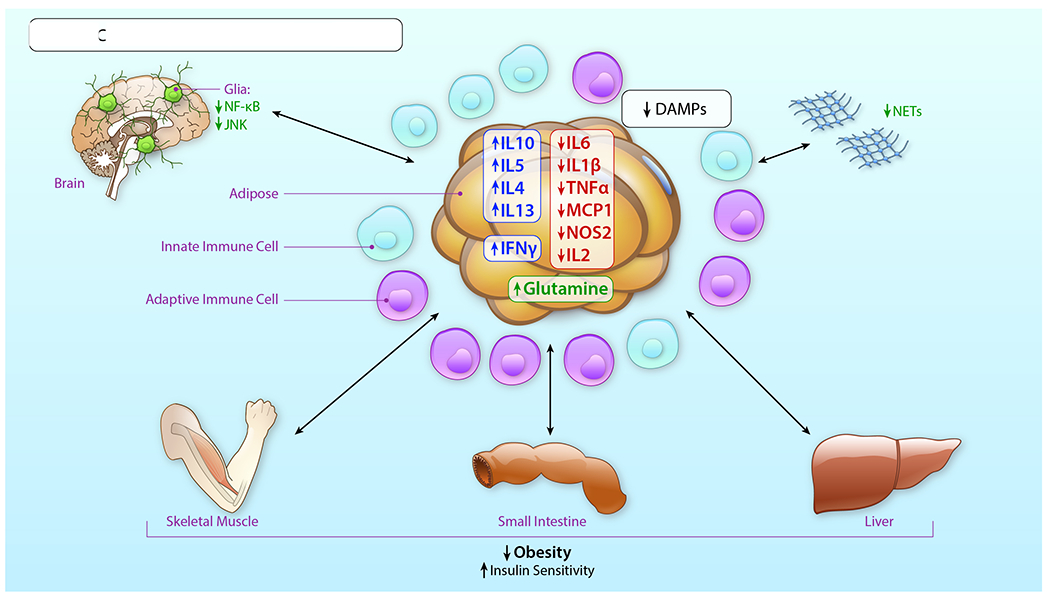
This Review considers the driving roles for innate immune cells in the adipose tissue in the lean state (A), obesity (B) and upon weight loss (C) with respect to the mediators produced by these cells, their cross-talk with the adaptive immune system as well as the influence of intercellular communications with the brain, skeletal muscle, the gut, particularly the small intestine, and the liver. As these inflammatory mediators are most likely produced/released by multiple different types of innate immune cells, as well as cells of the adaptive immune system, this Figure does not attribute roles for one cell type to expression of specific mediators in the overall environment of metabolic perturbation triggered by obesity.
In these contexts, the discovery of ILCs and their roles in tissue resident functions is likely to exert significant impact on our understanding of innate immunity and the connection to adaptive immune networks in adipose tissue and other metabolic organs. The findings that these cells contribute to Th1-, Th2- and Th3-like type immunity bridges the classically termed “innate” and “adaptive” immune cell denominations. Hence, it is not surprising that in adipose tissue, these cells may cross talk with their neighbors (other immune cells, adipocytes and vascular cells, to name a few), thereby influencing the net balance output of proinflammatory, anti-inflammatory, profibrotic and metabolism-perturbing species. More surprising, though, is the possibility that ILCs in distinct organs, such as the intestine, may influence the biology and pathobiology of ILCs in adipose tissue.85
Collectively, these exciting advances in the presence and roles of these diverse immune cell actors in the adipose tissue depots and in other metabolic organs support the very likely notion that it is not just about TNFα! Indeed, recent insights from RNA-seq suggested that classical pathways, such as type 1 interferons, may well be preserved after metabolically-beneficial weight loss, suggesting potential duality of this pathway in the “good” vs. “bad” effects on immunity and metabolism.137 Hence, it is not surprising that Wieser and colleagues showed that adipocyte expression of IFN receptors in HFD feeding was endogenously protective.140 Results such as these underscore that the innate (and adaptive) immune systems evolved to protect and defend; even if these functions go awry in chronic disease, they nevertheless remain staunchly relevant for distinct forms of acute injury to the organism, such as infection or wounding.
How do we integrate such complex data into a hierarchical scheme to delineate where, when and how to intervene safely in combatting the ever-growing epidemic of obesity? Some suggestions to glean as much insightful information as possible from our experiments include: (1) meticulous consideration and reporting of the type of obesity-provoking diets in use; (2) the study of both male and female subjects in both animal model and human studies; (3) varied time courses of diet induction, that is, the study of onset and interruption of obesity; (4) reporting of the vivarium conditions – pathogen free vs. conventional, as such considerations may affect multiple factors, including the microbiome; (5) reporting of the vivarium temperatures, as well as consideration of study performance at room temperature and at thermoneutrality, where feasible; (6) reporting of genetic background; the study of multiple backgrounds such as in rodents may be of value; and (7) reporting the age of the animals. Lessons learned from murine manipulation studies must be acknowledged as well: (1) multiple means to test roles of a specific cell type need to be considered; for example, it is unlikely that modulation of a single transcription factor will impact solely a single cell type; (2) adoptive transfers and lethal irradiation/transplantation need to consider the effect of the homing and niche re-settling nuances imbued by the distinct groups of transferred cells; (3) global and embryonic deletion approaches for hypothesis-testing models, such as in mice, need to be considered carefully, with respect to the possibility that cells of interest, which developed and matured in a gene-specific-deleted environment, may demonstrate altered epigenetic imprinting, and, therefore, they (and their precursors) may respond uniquely to various insults during maturity; (4) in using cell-type specific approaches, the use of temporally-regulated gene modulation may be useful to avoid developmental confounding issues; (5) the careful inclusion of controls when using tamoxifen or doxycycline, and the study of cre and floxed controls, for example, in gene deletion models must be considered; (6) meticulous consideration of off-target effects in all modes of gene modulation; (7) the testing of animals of multiple age ranges, including young, middle-aged and aged, where possible, given the plethora of effects of aging on adipose tissue, immune cell biology and metabolism;141 and (8) where feasible and practical, the use of pharmacological agents to stimulate or antagonize pathways of interest to provide independent means of support in experimental paradigms.
Finally, techniques such as single-cell RNA-seq and single-nucleus RNA-seq are unveiling the promise of broad-scale and unbiased approaches for many settings, including obesity. However, it will be essential to ensure that bioinformatics approaches are enabled to capture the full gamut of innate (and adaptive) immune cells that populate the metabolic organs in homeostasis and in disease. We look forward to the many new discoveries using these and other state-of-the-art approaches as we enter this new decade of obesity research.
Acknowledgements
The authors gratefully acknowledge the expert assistance of Ms. Latoya Woods in the preparation of this manuscript.
Sources of Funding
The authors acknowledge support from the U.S. Public Health Service 1P01HL131481 and 1R01DK109675.
Non-standard Abbreviations and Acronyms
- AraC
arabinofuranosyl cytidine
- ATM
adipose tissue macrophage
- BBB
blood-brain barrier
- BMDM
bone marrow-derived macrophage
- BMI
body mass index
- CLS
crown-like structures
- CNS
central nervous system
- CR
caloric restriction
- CRP
C-reactive protein
- CVD
cardiovascular disease
- DAMPs
damage-associated molecular patterns
- DC
dendritic cell
- DSCG
disodium cromoglycate
- EETs
eosinophil extracellular traps
- ER
endoplasmic reticulum
- FBC
F4/80, CD11B+, and CD11C+
- FDR
false discovery rate
- GABA
γ-amino butyric acid
- GC
glucorticoid receptor
- GFAP
glial fibrillary acidic protein
- GM-CSF
granulocyte-macrophage colony stimulating factor
- GSEA
Gene Set Enrichment Analyses
- HFD
high fat diet
- HIF-1α
hypoxia inducible factor-1 α
- HOMA-IR
Homeostatic Model Assessment of Insulin Resistance
- ICOS
Inducible T cell Costimulator
- Ig
immunoglobulin
- IL
interleukin
- ILC
innate lymphoid cell
- IRE1α
inositol-requiring enzyme 1 α
- IRF
interferon regulatory factor
- IRS1
insulin receptor substrate 1
- LPS
lipopolysaccharide
- LTi
lymphoid tissue inducer cells
- MBH
mediobasal hypothalamus
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NCR1
NK-cell activating receptor
- NETs
neutrophil extracellular traps
- NK
Natural killer cell
- NKT
Natural killer T cells
- O-GlcNAc
O-linked β-N-acetylglucosamine
- PAMP
pattern-associated molecular pattern
- PBS
phosphate buffered saline
- PGAT
perigonadal adipose tissue
- RAGE
receptor for advanced glycation end products
- RNA-seq
RNA-sequencing
- RORγt
retinoic acid receptor-related orphan receptor γt
- ROS
reactive oxygen species
- SAT
subcutaneous adipose tissue
- SVF
stromal vascular fraction
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TLR
toll like receptor
- TNFα
tumor necrosis factor alpha
- TNFR
tumor necrosis factor receptor
- Treg
T regulatory cells
- UDP-GlcNAc
uridine diphosphate N-acetylglucosamine
- VAT
visceral adipose tissue
- WAT
white adipose tissue
- WT
wild type
Footnotes
Disclosures
None
References
- 1.Purnell JQ. Definitions, Classification, and Epidemiology of Obesity In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000. [PubMed] [Google Scholar]
- 2.Abdelaal M, le Roux CW, Docherty NG. Morbidity and mortality associated with obesity. Ann Transl Med. 2017;5:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. [DOI] [PubMed] [Google Scholar]
- 4.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr., CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricardo-Gonzalez RR, Red Eagle A, Odegaard JI, Jouihan H, Morel CR, Heredia JE, Mukundan L, Wu D, Locksley RM, Chawla A. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2010;107:22617–22622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. [DOI] [PubMed] [Google Scholar]
- 11.Okuno Y, Fukuhara A, Hashimoto E, Kobayashi H, Kobayashi S, Otsuki M, Shimomura I. Oxidative Stress Inhibits Healthy Adipose Expansion Through Suppression of SREBF1-Mediated Lipogenic Pathway. Diabetes. 2018;67:1113–1127. [DOI] [PubMed] [Google Scholar]
- 12.Flaherty SE 3rd, , Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonyak MA, Lukey MJ, Cerione RA. Lipid-filled vesicles modulate macrophages. Science. 2019;363:931–932. [DOI] [PubMed] [Google Scholar]
- 14.Ramkhelawon B, Hennessy EJ, Menager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, Miller G, McPherson R, Rayner KJ, Moore KJ. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma M, Schlegel M, Brown EJ, Sansbury BE, Weinstock A, Afonso MS, Corr EM, van Solingen C, Shanley LC, Peled D, Ramasamy R, Schmidt AM, Spite M, Fisher EA, Moore KJ. Netrin-1 Alters Adipose Tissue Macrophage Fate and Function in Obesity. Immunometabolism. 2019;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Tian J, Tian X, Tang X, Rui K, Tong J, Lu L, Xu H, Wang S. Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS One. 2014;9:e92450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, Geletka L, Meyer KA, O’Rourke RW, Lumeng CN. Adipose Tissue Dendritic Cells Are Independent Contributors to Obesity-Induced Inflammation and Insulin Resistance. J Immunol. 2016;197:3650–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, Volmer R, Iwawaki T, Koliwad SK. Saturated Fatty Acids Engage an IRE1alpha-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016;14:2611–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrus P, Lecoutre S, Dollet L, Wiel C, Sulen A, Gao H, Tavira B, Laurencikiene J, Rooyackers O, Checa A, Douagi I, Wheelock CE, Arner P, McCarthy M, Bergo MO, Edgar L, Choudhury RP, Aouadi M, Krook A, Ryden M. Glutamine Links Obesity to Inflammation in Human White Adipose Tissue. Cell Metab. 2019; 10.1016/j.cmet.2019.11.019 [DOI] [PubMed] [Google Scholar]
- 23.Drareni K, Gautier JF, Venteclef N, Alzaid F. Transcriptional control of macrophage polarisation in type 2 diabetes. Semin Immunopathol. 2019;41:515–529. [DOI] [PubMed] [Google Scholar]
- 24.Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, Ferrante AW, Jr. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120:3466–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstock A, Brown EJ, Garabedian ML, Pena S, Sharma M, Lafaille J, Moore KJ, Fisher EA. Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism. 2019;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alemán JO, Iyengar NM, Walker JM, Milne GL, Da Rosa JC, Liang Y, Giri DD, Zhou XK, Pollak MN, Hudis CA, Breslow JL, Holt PR, Dannenberg AJ. Effects of Rapid Weight Loss on Systemic and Adipose Tissue Inflammation and Metabolism in Obese Postmenopausal Women. J Endocr Soc. 2017;1:625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, Patel PR, Benoit VM, Yan SF, Li H, Friedman RA, Kim JK, Ramasamy R, Ferrante AW Jr., Schmidt AM. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63:1948–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes. 1996;45:881–885. [DOI] [PubMed] [Google Scholar]
- 29.Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. 2000;85:1316–1319. [DOI] [PubMed] [Google Scholar]
- 30.Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, Grinspoon SK. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. 2011;96:E146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kane H, Lynch L. Innate Immune Control of Adipose Tissue Homeostasis. Trends Immunol. 2019;40:857–872. [DOI] [PubMed] [Google Scholar]
- 33.Trim W, Turner JE, Thompson D. Parallels in Immunometabolic Adipose Tissue Dysfunction with Ageing and Obesity. Front Immunol 2018;9:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagman DK, Larson I, Kuzma JN, Cromer G, Makar K, Rubinow KB, Foster-Schubert KE, van Yserloo B, Billing PS, Landerholm RW, Crouthamel M, Flum DR, Cummings DE, Kratz M. The short-term and long-term effects of bariatric/metabolic surgery on subcutaneous adipose tissue inflammation in humans. Metabolism. 2017;70:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, Jenkins KM, Beaulieu KA, Mouded M, Frank SJ, Wong KK, Shapiro SD. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16:219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawanishi N, Niihara H, Mizokami T, Yada K, Suzuki K. Exercise training attenuates neutrophil infiltration and elastase expression in adipose tissue of high-fat-diet-induced obese mice. Physiol Rep. 2015;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 38.Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, Kaplan MJ. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braster Q, Silvestre Roig C, Hartwig H, Beckers L, den Toom M, Doring Y, Daemen MJ, Lutgens E, Soehnlein O. Inhibition of NET Release Fails to Reduce Adipose Tissue Inflammation in Mice. PLoS One. 2016;11:e0163922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Wang Q, Venugopal J, Wang J, Kleiman K, Guo C, Eitzman DT. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci Rep. 2018;8:4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts HM, Grant MM, Hubber N, Super P, Singhal R, Chapple ILC. Impact of Bariatric Surgical Intervention on Peripheral Blood Neutrophil (PBN) Function in Obesity. Obes Surg. 2018;28:1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Abbondanza M, Martorelli EE, Ricci MA, De Vuono S, Migliola EN, Godino C, Corradetti S, Siepi D, Paganelli MT, Maugeri N, Lupattelli G. Increased plasmatic NETs by-products in patients in severe obesity. Sci Rep. 2019;9:14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bais S, Kumari R, Prashar Y, Gill NS. Review of various molecular targets on mast cells and its relation to obesity: A future perspective. Diabetes Metab Syndr. 2017;11 Suppl 2:S1001–s1007. [DOI] [PubMed] [Google Scholar]
- 44.Altintas MM, Rossetti MA, Nayer B, Puig A, Zagallo P, Ortega LM, Johnson KB, McNamara G, Reiser J, Mendez AJ, Nayer A. Apoptosis, mastocytosis, and diminished adipocytokine gene expression accompany reduced epididymal fat mass in long-standing diet-induced obese mice. Lipids Health Dis. 2011;10:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fenger RV, Linneberg A, Vidal C, Vizcaino L, Husemoen LL, Aadahl M, Gonzalez-Quintela A. Determinants of serum tryptase in a general population: the relationship of serum tryptase to obesity and asthma. Int Arch Allergy Immunol. 2012;157:151–158. [DOI] [PubMed] [Google Scholar]
- 46.Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi GP. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald HR. Hematopoietic Kit Deficiency, rather than Lack of Mast Cells, Protects Mice from Obesity and Insulin Resistance. Cell Metab. 2015;21:678–691. [DOI] [PubMed] [Google Scholar]
- 48.Finlin BS, Confides AL, Zhu B, Boulanger MC, Memetimin H, Taylor KW, Johnson ZR, Westgate PM, Dupont-Versteegden EE, Kern PA. Adipose Tissue Mast Cells Promote Human Adipose Beiging in Response to Cold. Sci Rep. 2019;9:8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, Wang X, Yin H, Zhang L, Feng A, Zhang QX, Lin Y, Bao B, Hernandez LL, Shi GP, Liu J. Functional Inactivation of Mast Cells Enhances Subcutaneous Adipose Tissue Browning in Mice. Cell Rep. 2019;28:792–803.e794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simon HU, Yousefi S, Germic N, Arnold IC, Haczku A, Karaulov AV, Simon D, Rosenberg HF. The Cellular Functions of Eosinophils: Collegium Internationale Allergologicum (CIA) Update 2020. Int Arch Allergy Immunol. 2019;181:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burgess AW, Metcalf D, Russell SH, Nicola NA. Granulocyte/macrophage-, megakaryocyte-, eosinophil- and erythroid-colony-stimulating factors produced by mouse spleen cells. Biochem J. 1980;185:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolus WR, Peterson KR, Hubler MJ, Kennedy AJ, Gruen ML, Hasty AH. Elevating adipose eosinophils in obese mice to physiologically normal levels does not rescue metabolic impairments. Mol Metab. 2018;8:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolus WR, Kennedy AJ, Hasty AH. Obesity-induced reduction of adipose eosinophils is reversed with low-calorie dietary intervention. Physiol Rep. 2018;6:e13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee EH, Itan M, Jang J, Gu HJ, Rozenberg P, Mingler MK, Wen T, Yoon J, Park SY, Roh JY, Choi CS, Park WJ, Munitz A, Jung Y. Eosinophils support adipocyte maturation and promote glucose tolerance in obesity. Sci Rep. 2018;8:9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathis D Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolus WR, Gutierrez DA, Kennedy AJ, Anderson-Baucum EK, Hasty AH. CCR2 deficiency leads to increased eosinophils, alternative macrophage activation, and type 2 cytokine expression in adipose tissue. J Leukoc Biol. 2015;98:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. [DOI] [PubMed] [Google Scholar]
- 59.Vivier E, van de Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol. 2016;17:790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. Innate Lymphoid Cells: 10 Years On. Cell. 2018;174:1054–1066. [DOI] [PubMed] [Google Scholar]
- 61.Kohlgruber A, Lynch L. Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Curr Diab Rep. 2015;15:92. [DOI] [PubMed] [Google Scholar]
- 62.Dalmas E Role of innate immune cells in metabolism: from physiology to type 2 diabetes. Semin Immunopathol. 2019;41:531–545. [DOI] [PubMed] [Google Scholar]
- 63.Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135:626–635. [DOI] [PubMed] [Google Scholar]
- 64.Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O’Connor D, Stutte S, Tavakkoli A, Winters D, Exley MA, O’Shea D, Brenner MB, von Andrian U, Lynch L. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity. 2017;46:273–286. [DOI] [PubMed] [Google Scholar]
- 65.Lee BC, Kim MS, Pae M, Yamamoto Y, Eberle D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, You J, Mosher W, Brady HJ, Shoelson SE, Lee J. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab. 2016;23:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, Sun JC. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity. 2016;45:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, Bruning JC, Mandelboim O, Polic B. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16:376–385. [DOI] [PubMed] [Google Scholar]
- 68.Lynch LA, O’Connell JM, Kwasnik AK, Cawood TJ, O’Farrelly C, O’Shea DB. Are natural killer cells protecting the metabolically healthy obese patient? Obesity (Silver Spring). 2009;17:601–605. [DOI] [PubMed] [Google Scholar]
- 69.Tobin LM, Mavinkurve M, Carolan E, Kinlen D, O’Brien EC, Little MA, Finlay DK, Cody D, Hogan AE, O’Shea D. NK cells in childhood obesity are activated, metabolically stressed, and functionally deficient. JCI Insight. 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Rourke RW, Metcalf MD, White AE, Madala A, Winters BR, Maizlin II, Jobe BA, Roberts CT Jr., Slifka MK, Marks DL. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int J Obes (Lond). 2009;33:978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jahn J, Spielau M, Brandsch C, Stangl GI, Delank KS, Bahr I, Berreis T, Wrann CD, Kielstein H. Decreased NK cell functions in obesity can be reactivated by fat mass reduction. Obesity (Silver Spring). 2015;23:2233–2241. [DOI] [PubMed] [Google Scholar]
- 72.Barra NG, Fan IY, Gillen JB, Chew M, Marcinko K, Steinberg GR, Gibala MJ, Ashkar AA. High Intensity Interval Training Increases Natural Killer Cell Number and Function in Obese Breast Cancer-challenged Mice and Obese Women. J Cancer Prev. 2017;22:260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, Chu X, Ye X, Jiang C, Wang Y, Zhang P, Zang M, Zhu D, Bi Y. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun. 2019;10:3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, Coles M, Kioussis D, Brady HJ. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol. 2009;10:1118–1124. [DOI] [PubMed] [Google Scholar]
- 75.Kim DH, Van Dyken SJ. ILC2s in High Definition: Decoding the Logic of Tissue-Based Immunity. Trends Immunol. 2020;41:7–16. [DOI] [PubMed] [Google Scholar]
- 76.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544. [DOI] [PubMed] [Google Scholar]
- 77.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, Artis D. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Flach M, Diefenbach A. Adipose tissue: ILC2 crank up the heat. Cell Metab. 2015;21:152–153. [DOI] [PubMed] [Google Scholar]
- 80.Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K, Locksley RM, Chawla A. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160:74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hams E, Locksley RM, McKenzie AN, Fallon PG. Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J Immunol. 2013;191:5349–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hams E, Bermingham R, Wurlod FA, Hogan AE, O’Shea D, Preston RJ, Rodewald HR, McKenzie AN, Fallon PG. The helminth T2 RNase omega1 promotes metabolic homeostasis in an IL-33- and group 2 innate lymphoid cell-dependent mechanism. Faseb j. 2016;30:824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hussaarts L, Garcia-Tardon N, van Beek L, Heemskerk MM, Haeberlein S, van der Zon GC, Ozir-Fazalalikhan A, Berbee JF, Willems van Dijk K, van Harmelen V, Yazdanbakhsh M, Guigas B. Chronic helminth infection and helminth-derived egg antigens promote adipose tissue M2 macrophages and improve insulin sensitivity in obese mice. Faseb j. 2015;29:3027–3039. [DOI] [PubMed] [Google Scholar]
- 84.Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, Bluestone JA, Locksley RM. Interleukin-33 and Interferon-gamma Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity. 2015;43:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sasaki T, Moro K, Kubota T, Kubota N, Kato T, Ohno H, Nakae S, Saito H, Koyasu S. Innate Lymphoid Cells in the Induction of Obesity. Cell Rep. 2019;28:202–217.e207. [DOI] [PubMed] [Google Scholar]
- 86.Satoh-Takayama N Heterogeneity and diversity of group 3 innate lymphoid cells: new cells on the block. Int Immunol. 2016;28:29–34. [DOI] [PubMed] [Google Scholar]
- 87.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu Y, Yue J, Wu J, Zhou W, Li D, Ding K, Barnie PA, Xu X, Xu H, Shi W. Obesity May Provide Pro-ILC3 Development Inflammatory Environment in Asthmatic Children. J Immunol Res. 2018;2018:1628620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Juelke K, Romagnani C. Differentiation of human innate lymphoid cells (ILCs). Curr Opin Immunol. 2016;38:75–85. [DOI] [PubMed] [Google Scholar]
- 90.Upadhyay V, Poroyko V, Kim TJ, Devkota S, Fu S, Liu D, Tumanov AV, Koroleva EP, Deng L, Nagler C, Chang EB, Tang H, Fu YX. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nat Immunol. 2012;13:947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, Denman S, Begun J, Florin TH, Perkins A, Cuiv PO, McGuckin MA, Hasnain SZ. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci Rep. 2016;6:28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, Kay TW, Thomas HE, Whitehead JP, Forbes JM, Prins JB, McGuckin MA. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. 2014;20:1417–1426. [DOI] [PubMed] [Google Scholar]
- 93.Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, Diehl L, van Bruggen N, Kolumam G, Ouyang W. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–241. [DOI] [PubMed] [Google Scholar]
- 94.Fachi JL, Secca C, Rodrigues PB, Mato FCP, Di Luccia B, Felipe JS, Pral LP, Rungue M, Rocha VM, Sato FT, Sampaio U, Clerici M, Rodrigues HG, Camara NOS, Consonni SR, Vieira AT, Oliveira SC, Mackay CR, Layden BT, Bortoluci KR, Colonna M, Vinolo MAR. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J Exp Med. 2020;217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, Tanes C, Fardus-Reid F, Swann JR, Bittinger K, Mabbott NA, Vallance BA, Willinger T, Withers DR, Hepworth MR. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J Exp Med. 2019;216:728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, Marderstein AR, Gandara J, Perez AR, Withers DR, Targan SR, Shih DQ, Scherl EJ, Longman RS. Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity. 2018;49:1077–1089.e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Macedo F, Dos Santos LS, Glezer I, da Cunha FM. Brain Innate Immune Response in Diet-Induced Obesity as a Paradigm for Metabolic Influence on Inflammatory Signaling. Front Neurosci. 2019;13:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kälin S, Heppner FL, Bechmann I, Prinz M, Tschop MH, Yi CX. Hypothalamic innate immune reaction in obesity. Nat Rev Endocrinol. 2015;11:339–351. [DOI] [PubMed] [Google Scholar]
- 100.Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiol Rev. 2018;98:239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chowen JA, Garcia-Segura LM. Microglia, neurodegeneration and loss of neuroendocrine control. Prog Neurobiol. 2019; 10.1016/j.pneurobio.2019.101720:101720. [DOI] [PubMed] [Google Scholar]
- 102.Tay TL, Carrier M, Tremblay ME. Physiology of Microglia. Adv Exp Med Biol. 2019;1175:129–148. [DOI] [PubMed] [Google Scholar]
- 103.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. [DOI] [PubMed] [Google Scholar]
- 104.Crotti A, Ransohoff RM. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity. 2016;44:505–515. [DOI] [PubMed] [Google Scholar]
- 105.Horvath TL, Sarman B, Garcia-Caceres C, Enriori PJ, Sotonyi P, Shanabrough M, Borok E, Argente J, Chowen JA, Perez-Tilve D, Pfluger PT, Bronneke HS, Levin BE, Diano S, Cowley MA, Tschop MH. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A. 2010;107:14875–14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ezan P, Andre P, Cisternino S, Saubamea B, Boulay AC, Doutremer S, Thomas MA, Quenech’du N, Giaume C, Cohen-Salmon M. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.García-Cáceres C, Fuente-Martin E, Burgos-Ramos E, Granado M, Frago LM, Barrios V, Horvath T, Argente J, Chowen JA. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology. 2011;152:1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gao Y, Ottaway N, Schriever SC, Legutko B, Garcia-Caceres C, de la Fuente E, Mergen C, Bour S, Thaler JP, Seeley RJ, Filosa J, Stern JE, Perez-Tilve D, Schwartz MW, Tschop MH, Yi CX. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia. 2014;62:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, Matlock BK, Weller K, Ellacott KL. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun. 2014;35:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang H, Graham LC, Reagan AM, Grabowska WA, Schott WH, Howell GR. Transcriptome profiling of brain myeloid cells revealed activation of Itgal, Trem1, and Spp1 in western diet-induced obesity. J Neuroinflammation. 2019;16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Contu L, Nizari S, Heath CJ, Hawkes CA. Pre- and Post-natal High Fat Feeding Differentially Affects the Structure and Integrity of the Neurovascular Unit of 16-Month Old Male and Female Mice. Front Neurosci. 2019;13:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Banks WA. The blood-brain barrier as an endocrine tissue. Nat Rev Endocrinol. 2019;15:444–455. [DOI] [PubMed] [Google Scholar]
- 113.Yamamoto M, Guo DH, Hernandez CM, Stranahan AM. Endothelial Adora2a Activation Promotes Blood-Brain Barrier Breakdown and Cognitive Impairment in Mice with Diet-Induced Insulin Resistance. J Neurosci. 2019;39:4179–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Corem N, Anzi S, Gelb S, Ben-Zvi A. Leptin receptor deficiency induces early, transient and hyperglycaemia-independent blood-brain barrier dysfunction. Sci Rep. 2019;9:2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Salameh TS, Mortell WG, Logsdon AF, Butterfield DA, Banks WA. Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS. 2019;16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Estrada JA, Contreras I. Nutritional Modulation of Immune and Central Nervous System Homeostasis: The Role of Diet in Development of Neuroinflammation and Neurological Disease. Nutrients. 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marques C, Fernandes I, Meireles M, Faria A, Spencer JPE, Mateus N, Calhau C. Gut microbiota modulation accounts for the neuroprotective properties of anthocyanins. Sci Rep. 2018;8:11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chunchai T, Thunapong W, Yasom S, Wanchai K, Eaimworawuthikul S, Metzler G, Lungkaphin A, Pongchaidecha A, Sirilun S, Chaiyasut C, Pratchayasakul W, Thiennimitr P, Chattipakorn N, Chattipakorn SC. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J Neuroinflammation. 2018;15:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bruce-Keller AJ, Salbaum JM, Luo M, Blanchard Et, Taylor CM, Welsh DA, Berthoud HR. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol Psychiatry. 2015;77:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Deshpande NG, Saxena J, Pesaresi TG, Carrell CD, Ashby GB, Liao MK, Freeman LR. High fat diet alters gut microbiota but not spatial working memory in early middle-aged Sprague Dawley rats. PLoS One. 2019;14:e0217553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang L, Jacobs JP, Lagishetty V, Yuan PQ, Wu SV, Million M, Reeve JR Jr., , Pisegna JR, Tache Y. High-protein diet improves sensitivity to cholecystokinin and shifts the cecal microbiome without altering brain inflammation in diet-induced obesity in rats. Am J Physiol Regul Integr Comp Physiol. 2017;313:R473–r486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dresselhaus EC, Meffert MK. Cellular Specificity of NF-kappaB Function in the Nervous System. Front Immunol. 2019;10:1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yan J, Zhang H, Yin Y, Li J, Tang Y, Purkayastha S, Li L, Cai D. Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat Med. 2014;20:1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Andre C, Guzman-Quevedo O, Rey C, Remus-Borel J, Clark S, Castellanos-Jankiewicz A, Ladeveze E, Leste-Lasserre T, Nadjar A, Abrous DN, Laye S, Cota D. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes. 2017;66:908–919. [DOI] [PubMed] [Google Scholar]
- 125.Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab. 2017;6:366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic Process Plasticity and IKKbeta/NF-kappaB in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab. 2017;25:1091–1102.e1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP, Koliwad SK. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017;26:185–197.e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Robb JL, Morrissey NA, Weightman Potter PG, Smithers HE, Beall C, Ellacott KLJ. Immunometabolic Changes in Glia - A Potential Role in the Pathophysiology of Obesity and Diabetes. Neuroscience. 2019; 10.1016/j.neuroscience.2019.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vijay J, Gauthier M-F, Biswell RL, Louiselle DA, Johnston JJ, Cheung WA, Belden B, Pramatarova A, Biertho L, Gibson M, Simon M-M, Djambazian H, Staffa A, Bourque G, Laitinen A, Nystedt J, Vohl M-C, Fraser JD, Pastinen T, Tchernof A, Grundberg E. Single-cell analysis of human adipose tissue identifies depot- and disease-specific cell types. Nature Metabolism. 2019;2:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. [DOI] [PubMed] [Google Scholar]
- 131.Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, Mort JS, Vogl T, Roth J, van den Berg WB, van Lent PL. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. 2012;64:1477–1487. [DOI] [PubMed] [Google Scholar]
- 132.van Lent PL, Grevers LC, Schelbergen R, Blom A, Geurts J, Sloetjes A, Vogl T, Roth J, van den Berg WB. S100A8 causes a shift toward expression of activatory Fcgamma receptors on macrophages via toll-like receptor 4 and regulates Fcgamma receptor expression in synovium during chronic experimental arthritis. Arthritis Rheum. 2010;62:3353–3364. [DOI] [PubMed] [Google Scholar]
- 133.Patel S Danger-Associated Molecular Patterns (DAMPs): the Derivatives and Triggers of Inflammation. Curr Allergy Asthma Rep. 2018;18:63. [DOI] [PubMed] [Google Scholar]
- 134.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- 135.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. [DOI] [PubMed] [Google Scholar]
- 136.Jackson HT, Anekwe C, Chang J, Haskins IN, Stanford FC. The Role of Bariatric Surgery on Diabetes and Diabetic Care Compliance. Curr Diab Rep. 2019;19:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Poitou C, Perret C, Mathieu F, Truong V, Blum Y, Durand H, Alili R, Chelghoum N, Pelloux V, Aron-Wisnewsky J, Torcivia A, Bouillot JL, Parks BW, Ninio E, Clement K, Tiret L. Bariatric Surgery Induces Disruption in Inflammatory Signaling Pathways Mediated by Immune Cells in Adipose Tissue: A RNA-Seq Study. PLoS One. 2015;10:e0125718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Latorre J, Moreno-Navarrete JM, Sabater M, Buxo M, Rodriguez-Hermosa JI, Girones J, Fort JM, Vilallonga R, Ricart W, Simo R, Fernandez-Real JM, Ortega FJ. Decreased TLR3 in Hyperplastic Adipose Tissue, Blood and Inflamed Adipocytes is Related to Metabolic Inflammation. Cell Physiol Biochem. 2018;51:1051–1068. [DOI] [PubMed] [Google Scholar]
- 139.Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring). 2009;17:2014–2018. [DOI] [PubMed] [Google Scholar]
- 140.Wieser V, Adolph TE, Grander C, Grabherr F, Enrich B, Moser P, Moschen AR, Kaser S, Tilg H. Adipose type I interferon signalling protects against metabolic dysfunction. Gut. 2018;67:157–165. [DOI] [PubMed] [Google Scholar]
- 141.Chia CW, Egan JM, Ferrucci L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ Res. 2018;123:886–904. [DOI] [PMC free article] [PubMed] [Google Scholar]