

Abstract
Diabetes is a major risk factor for coronary heart disease (CHD). The major form of diabetes is type 2 diabetes (T2D), which is thus largely responsible for the CHD association in the general population. Recent years have seen major advances in the genetics of T2D, principally through ever-increasing large-scale genome-wide association studies (GWAS). This article addresses the question of whether this expanding knowledge of the genomics of T2D provides insight into the etiologic relationship between T2D and CHD. We will investigate this relationship by reviewing the evidence for shared genetic loci between T2D and CHD; by examining the formal testing of this interaction (Mendelian randomization studies assessing whether T2D is causal for CHD); and then turn to the implications of this genetic relationship for therapies for CHD, for therapies for T2D, and for therapies that affect both. In conclusion, the growing knowledge of the genetic relationship between T2D and CHD is beginning to provide the promise for improved prevention and treatment of both disorders.
Keywords: Genetic assocation; coronary artery disease; cerebrovascular disease; genetic epidemiology; diabetus mellitus; coronary heart disease; stroke; Genetic, assocation studies; diabetes, type 2; cerebrovascular disease/stroke
Type 2 diabetes (T2D) is the most common form of diabetes, responsible for the vast majority in Europeans and even greater proportions in other ethnic groups. T2D arises when circulating insulin levels are insufficient to maintain normal glucose levels. The central defects are insulin resistance and inadequate insulin secretion. Which of these are primary is still a matter of debate, and in fact may differ from patient to patient.1 Both phenomena are important in the development of T2D, and genetic defects predisposing to both are likely to be important contributors to the disease process. Though classically a disease of adults, children are increasingly commonly being diagnosed with T2D. T2D is a progressive disorder, with beta-cell function deteriorating over time. Thus, in the early years of diabetes, lifestyle modification and oral medications may be sufficient while in the later years, supplemental insulin therapy is often required. T2D is a worldwide epidemic.2 The incidence of diabetes (mainly diagnosed diabetes) in most countries increased from the 1990s to the mid-2000s, and has been stable or falling in recent years, depending on the country.3 It has been estimated that 1 in every 3 individuals (1 in 2 minorities) born in the U.S. in the year 2000 will develop diabetes in their lifetime,4 mainly due to the epidemic of obesity. Genome-wide association study (GWAS) findings suggest that failure of insulin secretion is the key event that initiates T2D; many diabetes loci discovered by GWAS appear to compromise insulin secretion or pancreatic beta-cell development.5 While the microvascular complications of T2D are a great cause of morbidity, mortality in diabetes is mainly driven by the macrovascular complications of coronary heart disease (CHD) and stroke. Diabetes has been long recognized as a risk factor for cardiovascular (CV) disease (CVD), conferring a 2- to 4-fold increased risk of CVD mortality.6–8 This review will explore the relationship between T2D and CHD, focusing on insights derived from genetic studies that have exploited the many findings arising from GWAS.
THE GENETICS OF T2D
Twin and family studies have long suggested a genetic component to the susceptibility to T2D. Monozygotic twin studies demonstrate very high concordance (70-90%) for T2D in twin pairs,9 yet the overall familial aggregation of T2D is not consistent with a single, simple mode of inheritance. Genetic heterogeneity is a likely explanation, as supported by GWAS findings. Lifestyle and environment are also critical to the development of T2D. The heritability of T2D has been estimated as 40-70%,10, 11 with estimates as low as 30% when including individuals with late-onset diabetes.12 The component disorders underlying diabetes also display substantial heritability. Insulin sensitivity can be directly quantified by physiologic phenotyping procedures, such as the euglycemic hyperinsulinemic clamp, the frequently sampled intravenous glucose tolerance test (FSIGT), or the insulin suppression test.13 The heritability of insulin sensitivity ranges from 37% to 55% when quantified by euglycemic clamp14–18 and 28 to 44% when quantified by FSIGT.19–21 Heritability estimates for insulin secretion range from 25% to 57% as determined from insulin measurements during oral glucose tolerance testing22–24 and 23% to 84% as determined from intravenous glucose tolerance tests.18, 20, 25–28 That T2D is a feature of several monogenic disorders (e.g., myotonic dystrophy) also supports its genetic origin as does the existence of monogenic diabetes disorders (e.g., maturity onset diabetes of the young).
Before widespread application of GWAS approaches, candidate gene analyses yielded a few confirmed susceptibility loci for T2D, including peroxisome proliferator activated receptor gamma (PPARG), the pancreatic beta-cell inwardly rectifying potassium channel Kir 6.2 (KCNJ11), and wolfram syndrome 1 (WFS1).29–32. In 2006, TCF7L2 (transcription factor 7 like 2), the locus with the strongest effect on the risk of T2D (1.4 odds for T2D per risk allele), was discovered by investigators who were following up a linkage signal found on chromosome 10 in Icelandic individuals.33
As with many other common diseases, GWAS revolutionized the genetics of T2D. GWAS is an agnostic approach that interrogates the entire genome for genetic variants (most commonly single nucleotide polymorphisms, SNPs) that are associated with a specific disease or trait.34 The first wave of GWAS in T2D consisted of five studies published in 2007, all of which were conducted in European-origin subjects.35–39 In the subsequent years, over 25 additional GWAS and GWAS meta-analyses were conducted, facilitated by increasing sample sizes, data sharing to confirm signals, and collaboration between multiple centers worldwide. By 2018, GWAS in multiple race/ethnic groups had resulted in ~140 T2D loci.40 Later that year, this number was dramatically increased to 403 distinct signals in 243 loci by a meta-analysis of 32 GWAS involving nearly 900,000 European-origin subjects in the DIAMANTE (DIAbetes, Meta-ANalysis Trans-Ethnic) Consortium.41 Considering earlier findings in other ethnic groups, this brought the total number of signals to ~460, the physiology of which is being delineated.42 Numbers will rise further with the anticipated publication of race/ethnic-specific and trans-ethnic meta-analyses by DIAMANTE. The currently available loci explain about 20% of the heritability of T2D in Europeans.
For the full potential of these findings to be realized (e.g., development of new drugs), detailed understanding of the function of T2D loci, the majority of which are in non-coding regions, will be needed. For regulatory variation, this entails identification of the gene or genes (not necessarily the nearest gene) whose expression is altered by the T2D variant and how that alteration modifies phenotype. Given the flood of new loci, this process will take time. However, as we recently described for the genetics of obesity,43 a tremendous amount can be learned from GWAS findings before functional characterization. Herein, we will focus on how GWAS results have been used to explore the relationships between diabetes and CVD.
DIABETES AND CHD
Diabetes is a risk factor for CHD, independent of traditional risk factors such as hypertension, hyperlipidemia, and tobacco smoking. Diabetes at baseline is associated with 2- to 3-fold increased rates of incident CHD, myocardial infarction (MI), and fatal CHD.44, 45 The concept of diabetes as a cardiovascular risk equivalent largely arose from a Finnish study that found that the seven-year incidence of MI in diabetic subjects with no history of prior MI was the same as that in nondiabetic subjects with a history of prior MI.46 Consistent with this, measures of insulin, insulin resistance, and the metabolic syndrome also predict future CVD events and mortality.47–56 Increased fasting insulin, insulin resistance, and beta-cell dysfunction are associated with increased carotid intima-media thickness (IMT), a marker of subclinical atherosclerosis.57, 58 Of interest, there is evidence that increased carotid IMT can predict future development of diabetes,59 suggesting a possible complex bidirectional relationship or common origins. Indeed, among patients undergoing angiography (excluding those with known diabetes), rates of newly discovered impaired glucose tolerance and diabetes correlated with the extent of coronary artery disease (CAD).60
A shared genetic basis can explain some measure of the co-occurrence of diabetes and CVD, referred to as pleiotropy or the “common soil” hypothesis.61 In individuals with normal glucose tolerance, a family history of T2D was a predictor of increased common carotid IMT, independently of other risk factors such as smoking, adiposity, lipids, fasting glucose, fasting insulin and blood pressure.62, 63 In another study of normal glucose tolerant individuals, a history of T2D in the mother or father was associated with elevated carotid IMT compared to matched controls with no parental history of diabetes; those with a history of T2D in both parents had an even higher carotid IMT than those with a single parent with T2D.64 In young adults with type 1 diabetes, a family history of T2D was an independent predictor of carotid IMT.65, 66 The clinical relevance of elevated carotid IMT in those with a family history of T2D is unknown, as such a family history has not been found to increase the risk of incident CHD events.67 Neverthess, the association sets the stage for a deeper examination using genetic variants.
One way to investigate the genetic relation between T2D and CHD is to look for coheritability, the concept that genes, possibly many genes, are shared for two traits or diseases. Classically, studies to determine coheritability, also referred to as genetic correlation, utilized twin or family studies.17 In the modern era, alternative methods utilize GWAS data to assess heritability and coheritability without family studies. Thus, one can estimate genetic correlation by examining the correlation of the entire genome-wide SNP data from GWAS between two traits. Using this strategy, Bulik-Sullivan et al. calculated genetic correlation between numerous traits and diseases, including T2D and CHD.68 They found significant positive genetic correlation between T2D and adiposity traits (BMI, extreme BMI, overweight, obese, hip circumference, waist circumference, waist-hip ratio), glycemic traits (fasting glucose, fasting insulin, HbA1c) and triglycerides. Inverse genetic correlation with T2D was observed for HDL cholesterol and birth weight. A positive genetic correlation (rg=0.385) between T2D and CAD was observed. CAD was also genetically correlated positively with BMI, triglycerides, waist circumference and waist-hip ratio and negatively with HDL cholesterol, height, and years of education. Of interest, the magnitude of genetic correlation with T2D was higher than that of the other correlates with CAD. In a Chinese population study, the genetic correlation between T2D and CHD was 0.15.69
SHARED LOCI BETWEEN T2D AND CHD
Chromosome 9p21.3
One of the earliest identified and robust shared loci lies at chromosome 9p21.3 near the CDKN2A and CDKN2B genes, identified in the earliest GWAS for T2D35, 36, 39, 70 and MI and CHD.71–73 Further studies suggested that the T2D and CHD signals in this region are independent.74, 75 Considering GWAS findings through 2013, the various SNPs associated with CHD were found to reside proximal to a recombination peak while those associated with T2D were located distal to that peak.76 Consideration of recent results provide a clearer picture of this shared signal. Figure 1 displays associations of SNPs in this region with CHD and T2D, using summary data from large-scale GWAS.41, 77 This reveals two linked clusters of SNPs for CHD, both proximal to the recombination hotspot (clusters 1 and 2 in Figure 1A). For T2D, two independent signals are found on either side of the recombination hotspot. One of the T2D signals is highly linked with CHD cluster 2 (Figure 1D). Thus, while the strongest signal for T2D in this locus appears to be independent of the signals for CHD, there exists a second group of shared SNPs for T2D and CHD as well. One study closely examining this region concluded that the block tagged by rs4977574 (cluster 1) was primarily associated with atherosclerotic plaque deposition in the coronary arteries while cluster 2 is mainly associated with myocardial infarction (plaque rupture and thrombosis).78 That cluster 2 SNPs also affect T2D suggests a mechanism whereby some chromosome 9q21 SNPs predispose to atherosclerotic plaque formation while the diabetogenic SNPs in the region increase the risk of rupture and thrombosis of these plaques, potentially via hyperglycemia or other features specific to the diabetic state (e.g., hyperinsulinemia, increased inflammation). However, given that only a single study78 documented differential effects on plaque deposition versus plaque rupture, the above must be considered hypothesis generating until confirmed by future studies. While the CHD region overlaps the CDKN2A and CDKN2B genes, the long non-coding RNA CDKN2B-AS1 (also known as antisense non-coding RNA in the INK4 locus, ANRIL) is thought to be the effector at this locus. ANRIL has many different splice isoforms as well as circular RNA forms. Though not fully established, the balance of evidence suggests that CHD promoting genotype at 9p21 increases expression of linear ANRIL, which has pro-proatherogenic (pro-proliferative, anti-apoptotic) effects via altered expression of target genes, and decreases expresion of circular ANRIL, which has antiatherogenic effects via altered ribosomal RNA processing.79 In whole blood, peripheral blood monocytes, and atherosclerotic plaque tissue, linear ANRIL was positively associated with severity of atherosclerosis, while circular ANRIL was inversely correlated.80, 81 The target genes regulated by ANRIL, possibly including CDKN2A and/or CDKN2B, remain to be established. Given that this locus contains the strongest current signal for CHD (rs4977574, OR 1.3, p=8.8x10−223),77 further clarification of the underlying molecular mechanisms will be needed to enable clinical use of these findings, though efforts to incorporate 9p21 genotype to predict clinical outcomes are already underway.82 Whether ANRIL plays a role in the diabetogenic aspects of 9p21 also warrants investigation.
Figure 1.
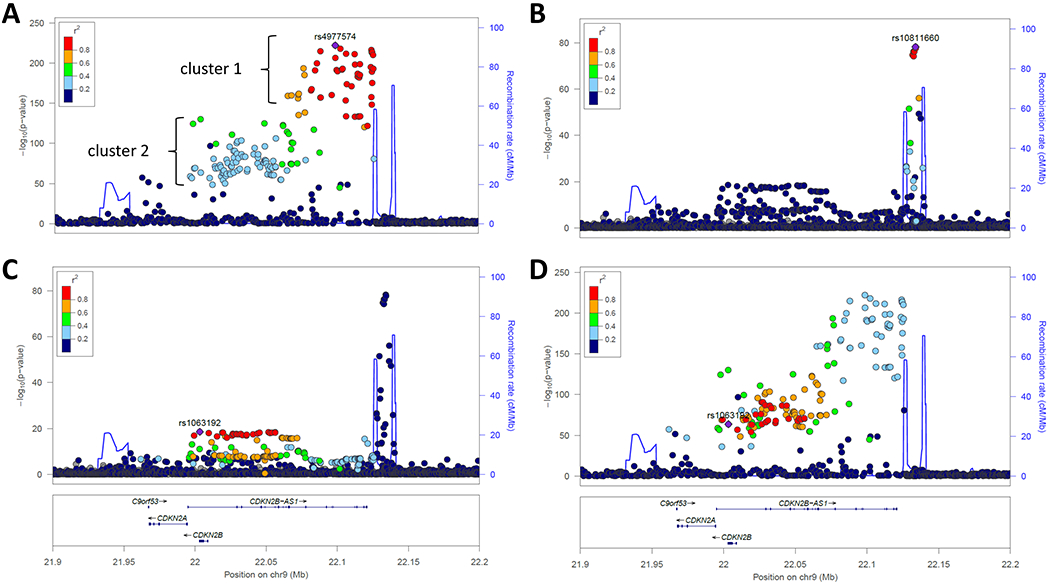
Regional association plot of the chromosome 9p21 locus for CHD and T2D. In these plots, P values are plotted as -log10 values as a function of genomic position. Each dot represents one SNP, with the index variant represented as a purple diamond. Color of other SNPs indicates their LD with the index SNP, per the scale on the top left. Recombination rates are represented by the blue line and genes in the region are shown at bottom. Plots were generated using publicly available summary statistics for CHD77 and T2D.41 A. Association with CHD, where the index SNP rs4977574 is the top known signal. Note that the CHD association lies proximal to a recombination hotspot; B. Association with T2D, index SNP rs10811660 is the top signal, in a region delimited by two recombination hotspots; C. Association with T2D at a second locus in the region, indexed by rs1063192, and independent of the first T2D signal; D. Association with CHD, indexed to the T2D SNP rs1063192. The cluster of SNPs constituting the second T2D signal is in LD with the CHD signal.
IRS1 (Insulin Receptor Substrate 1)
Another shared locus is the IRS1 locus at chromosome 2p36.3, where several GWAS have identified a group of SNPs in linkage disequilibrium (LD) that are associated with T2D, increased fasting insulin, increased triglycerides, lower HDL cholesterol, and/or increased visceral-to-subcutaneous fat ratio.83–87 While the review by Dauriz and Meigs76 listed six SNPs comprising this cluster, the GWAS Catalog88 now lists over 20 such SNPs, with additional associations with increased systolic and diastolic blood pressure (e.g., rs7590201), decreased body fat percentage (e.g., rs2943652), decreased body mass index (e.g., rs2176040), decreased waist circumference and waist-hip-ratio (e.g., rs2176040 and rs1515108), increased lean mass (e.g., rs2943656), and decreased adiponectin (e.g., rs1515110) (with respect to alleles corresponding to increased risk of T2D). In addition, recent GWAS have now associated at least two SNPs (rs2943634, rs2972146)77, 89 from this tightly linked cluster with CHD at genome-wide significance levels, with the diabetogenic allele increasing CHD risk. Though SNPs in this cluster are over 500 kb from IRS1, data from the Genotype-Tissue Expression (GTEx) Project reveal that these SNPs are eQTLs (expression quantitative trait loci) for IRS1 in subcutaneous and visceral adipose tissue, with the alleles associated with increased T2D and CHD associating with decreased IRS1 expression. Experimental evidence suggests that the region of these SNPs may be home to enhancers that affect IRS1 expression.90 Alternatively, the close proximity of these SNPs with non-coding RNA LOC646736 and microRNA MIR5702 raise the possibility that these might regulate IRS1 expression. Furthermore, the diabetogenic allele of rs2943641 (a representative of this cluster) was associated with oral glucose tolerance-based measures of insulin resistance as well as with reduced basal levels of IRS1 protein and decreased insulin induction of IRS1-associated phosphatidylinositol-3-OH kinase activity in human skeletal muscle biopsies.91 Given that IRS1 is the immediate second messenger of the insulin receptor, as well as the several metabolic syndrome-related traits associated with these SNPs, a likely mechanistic explanation is that insulin resistance may be a link between T2D and CHD. The lack of any association of SNPs in this loci with glycemic parameters (e.g., fasting glucose, hemoglobin A1c (HbA1c)) suggests, for this locus and possibly other loci, that insulin resistance and/or hyperinsulinemia, rather than hyperglycemia, are pathogenic in CHD in diabetes. The absense of association of the IRS1 locus SNP cluster with measures of structural atherosclerosis (e.g., coronary artery or abdominal aortic calcium, internal and common carotid intima-media thickness)92 suggests, similar to the chromosome 9p21 locus, that these diabetogenic SNPs may play a greater role in plaque destabilization than plaque formation.
Searches for Common Loci
In 2015, Jansen et al.93 examined 44 SNPs robustly associated with T2D in GWAS for their association with CHD in the largest dataset at the time, consisting of 22,233 cases with CAD and 64,762 controls from the Coronary Artery Disease Genome-wide Replication and Meta-analysis (CARDIoGRAM) Consortium.94, 95 Of the 44 T2D SNPs, 10 SNPs were associated with CAD (P<0.05), with the diabetogenic allele associated with increased odds of CAD, which greatly exceeded the number expected by chance. Indeed, 29 of the 44 SNPs exhibited a positive odds ratio for association with CAD, also greater than expected by chance. After study-wide Bonferroni correction, only rs2943651 remained individually statistically significantly associated with CAD (p=3.4x10−5); this is one of the SNPs in the IRS1 locus described above. This study went on to construct genetic risk scores based on the 44 T2D SNPs; these scores were significantly associated with CAD.93 Formal Mendelian randomization analyses provide an even more compelling story (reviewed below).
In contrast to the enrichment of CHD association seen among T2D SNPs, analyses in the opposite direction (i.e., assessing association of robust GWAS-discovered loci for CHD with T2D) have been much less dramatic. A study of 62 CHD loci examined their association with multiple cardiometabolic traits.96 After correction for multiple testing, the largest number of associations were seen with lipid traits and blood pressure traits. The only association with T2D (OR 1.12, p=2.7x10−5) was that of rs3130683, which lies within C2, which codes for complement C2 protein. There is no genome-wide signal for this SNP in the latest T2D GWAS.41 These findings suggest that recognized CHD genetic susceptibility loci act largely through lipids and blood pressure, with little action through T2D. Coupled with the frequent CHD association seen for T2D SNPs, it appears that T2D may cause CHD but CHD does not cause T2D, as detailed further below.
In 2017, Zhao et al. conducted a large GWAS for T2D with additional steps to elucidate shared genetics with CHD.97 Their GWAS increased the number of T2D SNPs at the time to 106, of which 17 were at least nominally associated with CHD (P<0.01), more than expected by chance. The only instance where the SNP associations with T2D and CHD both met genome-wide significance was for rs7578326 at the IRS1 locus, providing the first such evidence for the IRS1 group of SNPs discussed above. They also examined shared genetic risk more broadly across the genome; 76% of all SNPs associated with T2D at P<5 x 10−8 and 82% of all SNPs nominally associated with T2D (5 x 10−8 < P < 0.05) exhibited directionally consistent effects with CHD. In contrast, there was no such enrichment for CHD SNPs in terms of association with T2D. To further explore the shared genetics of T2D and CHD, Zhao et al. conducted a bivariate GWAS on the joint distribution of T2D and CHD, which identified 19 loci, including a novel signal at rs825476 near CCDC92 that is also an eQTL for this gene. They further refined these results with formal colocalization analysis (requiring colocalization probability >0.5) to identify the T2D-CHD associations due to a single underlying SNP, yielding 8 signals, 7 with consistent effects on T2D and CHD, two of which are coding variants (Table 1). At the apolipoprotein E (APOE) locus, the diabetogenic allele was also associated with decreased LDL-C (see possible explanation below in Therapeutic Implications). The IRS1 SNP did not meet the colocalization criterion. Additional loci of interest that did not meet this criterion but that had directionally consistent effects on T2D include CDKN2A/2B (discussed above), and FTO and MC4R (adiposity loci, see discussion below).
Table 1.
SNPs that are associated with both T2D and CHD with genome-wide significance on bivariate scan
| SNP | Nearest genes | Effect allele/other allele | CHD: OR | CHD: P value | T2D: OR | T2D: P value | Bivariate P value | Colocalization probability |
|---|---|---|---|---|---|---|---|---|
| rs7903146 | TCF7L2 | T/C | 1.04 (1.02-1.05) | 2.87E-05 | 1.35 (1.33-1.38) | 1.33E-219 | 2.64E-212 | 0.95 |
| rs1169288 (I27L) | HNF1A | C/A | 1.04 (1.03-1.06) | 3.86E-07 | 1.06 (1.04-1.08) | 9.25E-10 | 1.98E-12 | 0.69 |
| rs7202877 | CTRB1, CTRB2 | C/T | 1.06 (1.04-1.09) | 2.90E-06 | 1.06 (1.03-1.08) | 3.92E-09 | 6.33E-11 | 0.97 |
| rs2306374 | MRAS | C/T | 1.06 (1.04-1.08) | 2.35E-08 | 1.05 (1.02-1.07) | 6.51E-04 | 9.76E-09 | 0.55 |
| rs11556924 (R342H) | ZC3HC1 | C/T | 1.08 (1.06-1.10) | 3.28E-20 | 1.03 (1.01-1.05) | 4.94E-04 | 1.38E-19 | 0.63 |
| rs7985179 | GPC5, MIR17HG | T/A | 1.05 (1.02-1.08) | 6.37E-04 | 1.07 (1.05-1.10) | 3.72E-09 | 1.54E-09 | 0.54 |
| rs825476 | CCDC92 | T/C | 1.03 (1.02-1.05) | 2.96E-07 | 1.04 (1.03-1.06) | 2.21E-06 | 2.71E-09 | 0.98 |
| rs4420638 | APOE, APOC1 | G/A | 0.89 (0.85-0.93) | 1.76E-06 | 1.08 (1.05-1.11) | 8.82E-08 | 2.59E-13 | 0.99 |
Data are from Zhao et al.97 All loci have r2>0.7 between T2D and CHD associations and colocalization probability >0.5. Individual associations have P<1x10−3.
Triglyceride Metabolism Loci
A series of shared loci are centered on triglyceride metabolism. Lipoprotein lipase (LPL) is the enzyme on endothelial cells that hydrolyzes triglycerides, yielding monoacylglycerol and free fatty acids, making them available for tissue uptake. Thus, LPL acts to decrease triglyceride levels. GWAS studies have established LPL as a locus for triglycerides and HDL-C, two lipid fractions that are inversely correlated. Impressive P values (3 x 10−293 for rs79407615 and HDL-C and 1x10−300 for rs1569209 and triglycerides) have been recorded among a group of SNPs in LD.98 As high triglycerides and low HDL-C are hallmarks of the metabolic syndrome, it is not surprising that this locus has also been associated with the metabolic syndrome as well.99 In 2018, GWAS meta-analyses for CHD77 and T2D41 identified SNPs in LPL as associated with T2D at the genome-wide significance level, top SNPs being rs17091891 and rs10096633, respectively (Figure 2). These SNPs are highly linked (r2=0.97), thus representing a shared signal. A study of exomic variation identified rs328 in LPL as associated with T2D, with the common allele conferring risk.100 SNP rs328 is in LD with rs17091891 and rs10096633 (r2=0.75). SNP rs328 is a nonsense variant (S447X) that truncates the last two amino acids from the enzyme. A link between rs328 and CHD was noted in candidate gene work over 20 years ago.101 We and others have found that the Ter allele of rs328 confers gain of function in LPL.102, 103 This is consistent with GWAS findings from multiple race/ethnicities that the Ter allele is associated with decreased triglycerides and increased HDL-C and reduced risk of T2D.100, 104–106 Large-scale exome chip work confirmed the consistent directional effect of LPL variants on CHD and T2D.107 This work prompted studies of factors that regulate LPL. Angiopoietin-like 4 (ANGPTL4) inhibits LPL. ANGPTL4 was sequenced and inactivating variants were tested for association with lipid levels and CAD; the missense variant (rs116843064, E40K) was associated with lower triglycerides, higher HDL-C, and less CAD (OR 0.81, 95% CI 0.70-0.92 in lysine allele carriers), consistent with inactivation leading to higher LPL activity.108 GWAS have found consistent associations with triglycerides, HDL-C, and CAD.77, 98 Furthermore, the lysine allele was recently found to protect against T2D (OR 0.89, 95% CI 0.85-0.92) and associate with lower fasting glucose and greater insulin sensitivity index from the oral glucose tolerance test.109 All these results on LPL and ANGPTL4 are more broadly consistent with the genetic epidemiologic evidence that variants affecting triglycerides are risk factors for CHD.110
Figure 2.
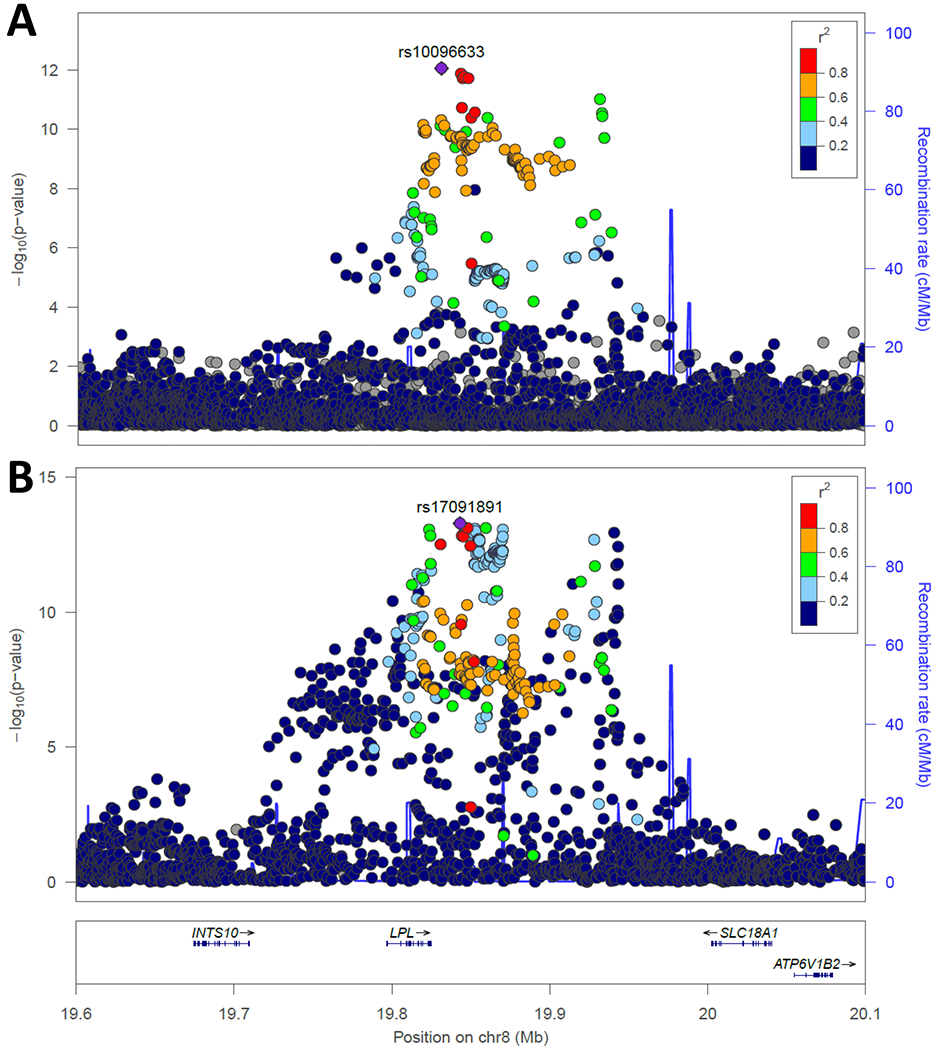
Regional association plot of the LPL locus for T2D and CHD. In these plots, P values are plotted as -log10 values as a function of genomic position. Each dot represents one SNP, with the index variant represented as a purple diamond. Color of other SNPs indicates their LD with the index SNP, per the scale on the top right. Recombination rates are represented by the blue line and genes in the region are shown at bottom. Plots were generated using publicly available summary statistics for CHD77 and T2D.41 A. Association with T2D, indexed to rs10096633. B. Association with CHD indexed to rs17091891. The two index SNPs are highly linked (r2=0.97 in Europeans).
The diverse forms of shared genetic susceptibility for T2D and CHD are illustrated in Figure 3.
Figure 3.
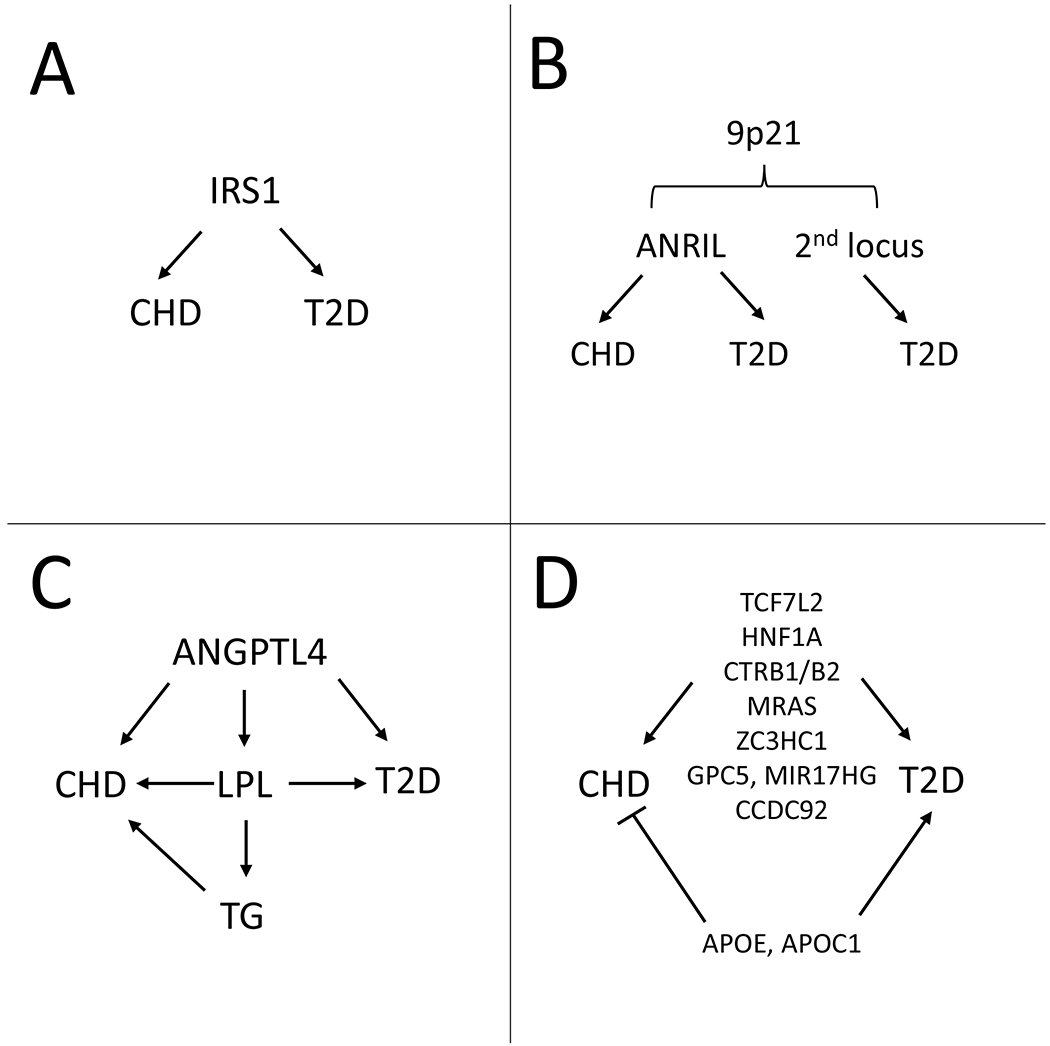
Different types of shared loci for CHD and T2D. A. IRS1 locus where the same variants are associated with CHD and T2D. B. More complex structure of chromosome 9p21 locus, which houses overlapping signals for CHD and T2D as well as an independent signal for T2D (see also Figure 1). C. Loci involved in triglyceride metabolism. D. Multi-locus colocalization approach97 that revealed seven signals with consistent direction of effect and one locus with opposite effects on CHD and T2D.
HETEROGENEITY WITHIN T2D AND THE IMPACT ON CHD
Those who see patients with T2D readily recognize different patterns of patients, such as lean apparently insulin sensitive patients versus the more common obese, insulin resistant patients. Race/ethnic differences appear to play a role in this heterogeneity.1 Phenotype-based studies have suggested different subtypes of diabetes; however, given that phenotype can evolve over time, it has been suggested that genetic-based classification could have greater utility over the lifespan, including before diabetes develops.111
One study of 94 independent T2D SNPs assembled their association from GWAS with 47 cardiometabolic and anthropometric traits and performed Bayesian nonnegative matrix factorization cluster analysis (a “soft” cluster analysis that allows SNPs to fall into more than one cluster, allowing for pleiotropic effects of T2D SNPs).112 This identified five clusters, each with distinct tissue-specific enhancer enrichment. Two clusters were related to reduced insulin secretion, one also with high proinsulin levels (“beta-cell” cluster) and one with low proinsulin levels (“proinsulin” cluster). The remaining clusters appear to have effects on insulin resistance, including an “obesity” cluster (high BMI), a “lipodystrophy-like” cluster (low BMI and HDL-C and higher triglycerides), and a “liver/lipid” cluster (low triglycerides). Pertinent to the current review, these clusters were associated with distinct clinical outcomes from GWAS. The beta-cell and lipodystrophy clusters were associated with CAD, whereas only the beta-cell cluster was associated with ischemic, large vessel, and small vessel stroke. No cluster was associated with cardioembolic stoke. The lipodystrophy-like cluster was also associated with elevated systolic and diastolic blood pressure. The proinsulin, obesity, and liver/lipid clusters were benign in terms of cardiovascular outcomes. If these clusters prove robust in additional analyses, they may pave the way to methods to prioritize which patients with T2D require the most intensive cardiovascular risk factor reduction.
The lipodystrophy-like cluster harkens to earlier data that used GWAS to suggest the existence of a normal-weight but “metabolically obese” phenotype.113 This study performed hierarchical cluster analysis on the association of 19 SNPs for fasting insulin with 8 traits (HDL-C, triglycerides, BMI, visceral-to-subcutaneous adipose tissue ratio, CT-measured hepatic steatosis, alanine aminotransferase (ALT), adiponectin, and sex hormone binding globulin). A cluster of 11 SNPs were identified that were characterized by higher triglycerides, lower HDL-C, greater hepatic steatosis and central adiposity but lower BMI. A genetic risk score of these 11 variants was associated with higher risk of T2D and CAD and elevated systolic and diastolic blood pressure, but not with carotid IMT or carotid plaque. These results suggest a lipodystrophy-like phenotype that increases risk of T2D and CHD. Another study using a different approach mined the GWAS literature for SNPs associated with elevated fasting insulin, lower HDL-C and higher triglyceride levels, finding 53 loci.114 These loci were found to play a role in rare, severe insulin resistance (association with familial partial lipodystrophy type 1) as well as common, mild states of insulin resistance (association with oral glucose tolerance and euglycemic clamp-derived measures of reduced insulin sensitivity). Genetic risk scores based on these SNPs were associated with reduced peripheral fat and reduced ability to expand peripheral fat over time; these loci exhibited expression enrichment in adipose tissue. By impairing peripheral and subcutaneous adipose storage capacity, these SNPs may lead to visceral and hepatic fat deposition, ultimately increasing risk of insulin resistance and diabetes. Genetic risk scores based on these SNPs were also associated with CHD, consistent with observations above.112, 113
We catalogued and classified over 450 independent SNPs for T2D, representing the state of the field at the time.42 Based on association with physiologic measures or hierarchical cluster analyses, the T2D SNPs were categorized as acting through beta-cell dysfunction, insulin resistance, lipodystrophy, or obesity/lipid pathways. Genetic risk scores based on the beta-cell and insulin resistance categories displayed the expected associations with oral glucose tolerance test-derived decreased insulin secretion and euglycemic clamp-derived reduced insulin sensitivity (two of the major endophenotypes leading to T2D), respectively; in contrast, the genetic risk score based on all T2D SNPs did not associate with these endophenotypes. One explanation of these results is that in any given individual, a limited number of genetically-driven metabolic derangements contribute to the development of T2D, which is consistent with the emerging concept of subtypes of T2D.111, 112, 115 Indeed, the recent soft clustering analysis found that the genetic profile of ~30% of individuals analyzed fell predominantly into one subclass.112 Studies are ongoing to examine the association of the clusters with the natural history of T2D, including microvascular and macrovascular complications. Future studies focusing on the component loci within each classification may shed light not only on the pathogenesis of T2D but also the differential cardiovascular risk associated with each subtype of T2D. Elucidation of key driver genes within each subtype could allow development of specific agents to prevent and/or better treat T2D and CHD in people based on their genetic profile obtained before the development of disease. Examination of the relationships between genes underlying each subtype may identify mechanistic pathways from normal glucose tolerance to T2D to CHD.
MENDELIAN RANDOMIZATION (MR): DOES T2D CAUSE CHD?
While the shared genetic basis discussed above suggests common origins, it raises the question of whether T2D causes CHD. Epidemiologic studies suggest this is the case, but causality cannot be deduced from observational studies. Randomized trials of intensive glucose lowering have not answered this question, as results have been mixed. However, we can turn to genetics to help answer this critially important question.
Overview of MR
The causal effect of a risk factor on an outcome can be assessed using Mendelian randomization (MR). The risk factor is represented by genetic variants (referred to in this context as instrument variables) for that factor, which avoids confounding and reverse causation in relation to the outcome, issues that plague observational studies. If a risk factor is truly causal for an outcome, then genetic variants for the risk factor will be associated with the outcome. Formal MR analysis assesses the instrument variable associations with risk factor and outcome to yield causal estimates. Instrument variables can be based on a single variant or multiple variants, the latter increasing power but also the chance of pleiotropy (see below). Random assortment of alleles during gametogenesis simulates a clinical trial where cases and controls differ only in the risk factor, in this case the genetic variants. A key advantage over clinical trials is that the genetic exposure can occur over long time periods (even lifelong), unlike clinical trials where the exposure is introduced late and only for months to years. Thus, lifelong exposure to even very subtle alteration in risk factor phenotype (due to different genetic variants) may have a significant impact on cardiometabolic diseases that develop later in life. MR may be useful in predicting whether drugs targeting certain risk factors will be effective in preventing outcomes, if the drug effect can be represented by genetic instruments (e.g., variants that lower LDL-C mimic statins). MR can also be used to predict drug adverse effects (see statins and T2D below). MR is based on three core assumptions: 1. Instrument variables are strongly associated with the risk factor of interest, 2. Variants are not associated with confounders of the risk factor and the outcome. 3. Variants are associated with disease through that risk factor and not through other mediators (Figure 4). Thus, MR may not be valid if the genetic instrument exhibits pleiotropy, whereby the variant (or variants in high LD) are associated with other traits. Modern MR methods such as MR Egger and MR PRESSO can assess whether substantial pleiotropy is present.116 Population stratification, where the race/ethnic composition of the cases and controls are different, may also interfere with MR, as race/ethnic-specific allelic differences may spuriously be associated with disease. In GWAS datasets, population stratification can be detected and corrected statistically. In early MR studies, the instrument to risk factor and instrument to outcome estimates were generated in a single cohort. In two-sample MR, these estimates are derived from separate, non-overlapping cohorts. The latter technique has allowed MR to be conducted using summary data from large GWAS consortia. Another advantage of two-sample MR is that it is less susceptible to weak instrument bias; in two-sample MR, this bias is towards the null whereas in one-sample MR, this bias is in the direction of the observational association.117 Multiple MR algorithms exist, with the workhorse being the sensitive inverse-variance weighted (IVW) method. Many studies also include additional methods, typically MR Egger and weighted median as sensitivity analyses to bolster results seen with IVW. Below, in reports that utilized multiple methods, we report results derived from IVW.
Figure 4.
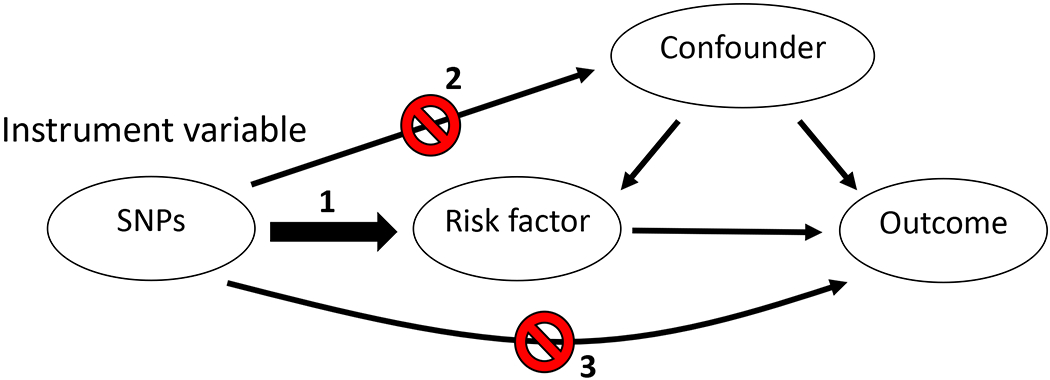
Illustrations of the core assumptions of Mendelian randomization. 1. The instrument variables must be strongly associated with the risk factor of interest, 2. Variants must not be associated with confounders of the risk factor and the outcome. 3. Variants are associated with disease through that risk factor and not through other mediators.
MR Studies of T2D as a Cause of CHD
Numerous MR studies examining T2D or CHD as outcomes have been published. These have been reviewed extensively and will be touched on briefly here.118–120 Obesity (as BMI) has been the most extensively studied risk factor, with strong evidence that it is causal for both T2D and CHD. One study went further to determine that the BMI to CHD relationship is mediated in part through systolic blood pressure, LDL-C, and remnant cholesterol.121 Another study concluded that the causality of BMI for CHD is mediated in large part by triglycerides and glycemia (HbA1c),122 while yet another concluded that adiposity’s effect on CHD is independent of blood pressure, glycemia, or T2D.123 Central fat distribution (as waist-hip ratio adjusted for BMI) also appears to cause T2D and CHD.124, 125 Effects of lipid fractions vary, with HDL-C not causing T2D or CHD (or possibly protecting against T2D126), triglycerides causing CHD but not T2D, and LDL-C causing CHD but protecting against T2D. Below, we will describe in detail MR studies wherein T2D and its component quantitative traits were examined as risk factors for CHD and cerebrovascular disease.
In 2015, SNPs for T2D were leveraged in MR analyses examining whether T2D may be causal for CHD. This was performed in two studies127, 128 that used summary data from the Diabetes Genetics Replication and Meta-Analysis Consortium (DIAGRAMv3)84 and CARDIoGRAM plus the Coronary Artery Disease Genetics (C4D) consortium (now known as CARDIoGRAMplusC4D).129 Using 59 variants associated with T2D, Ross et al. calculated the causal effect of T2D on CHD as an odds ratio of 1.63 (95% CI 1.23-2.07), which remained significant after adjusting for LDL-C, HDL-C, total cholesterol, triglycerides and BMI.128 Ahmad et al. took a strict approach to avoid pleiotropy, excluding any T2D variants that were also associated with BMI, systolic or diastolic blood pressure, LDL-C, or triglycerides, resulting in 26 SNPs for the MR analysis, which supported a causal effect of T2D on CHD (Table 2).127 Zhao et al. also conducted Mendelian randomization and found that an instrument based on 16 SNPs exclusively associated with T2D was significantly associated with CHD (Table 2).97
Table 2.
Summary of Mendelian randomization studies examining T2D and related traits as causal for CHD or MI
| Risk factor | Outcome | Year | Authors | Cohorts | Number of SNPs | OR for CHD | 95% CI | Increment |
|---|---|---|---|---|---|---|---|---|
| T2D | CHD | 2015 | Ross et al. | DIAGRAMv3, CARDIoGRAMplusC4D (n=344,248) | 59 | 1.63 | 1.23-2.07 | Not specified |
| T2D | CHD | 2015 | Ross et al. | DIAGRAMv3, CARDIoGRAMplusC4D (n=344,248) | 26 (BC) | 1.83 | 1.19-2.62 | Not specified |
| T2D | CHD | 2015 | Ross et al. | DIAGRAMv3, CARDIoGRAMplusC4D (n=344,248) | 11 (IR) | 2.35 | 1.46-3.53 | Not specified |
| T2D | CHD | 2015 | Ahmad et al. | DIAGRAMv3, CARDIoGRAMplusC4D (n=344,248) | 26 | 1.11 | 1.05-1.17 | Unit increase in odds of T2D |
| T2D | CHD | 2015 | Ahmad et al. | DIAGRAMv3, CARDIoGRAMplusC4D (n=344,248) | 8 (BC) | 1.07 | 1.01-1.14 | Unit increase in odds of T2D |
| T2D | CHD | 2019 | Gan et al. | CKB (n=159,528 Chinese) | 48 | 1.12 | 1.02-1.23 | 1 unit log-odds of T2D |
| T2D | CHD | 2019 | Gan et al. | DIAGRAMv4, CARDIoGRAMplusC4D (n=343,513) | 86 | 1.11 | 1.05-1.17 | 1 unit log-odds of T2D |
| T2D | CHD | 2019 | Gan et al. | CKB (n=159,528 Chinese) | 24 (BC) | 1.13 | 1.02-1.25 | 1 unit log-odds of T2D |
| T2D | CHD | 2019 | Gan et al. | CKB (n=159,528 Chinese) | 6 (IR) | 1.43 | 0.88-2.31 | 1 unit log-odds of T2D |
| T2D | CHD | 2017 | Zhao et al. | Several cohorts (n=526,043) | 16 (T2D only) | 1.26 | 1.16-1.37 | Not specified |
| Nonfasting glucose | CHD | 2012 | Benn et al. | CIHDS (n=80,522) | 5 | 1.25 | 1.03-1.52 | 1 mmol/L |
| Nonfasting glucose | MI | 2012 | Benn et al. | CIHDS (n=80,522) | 5 | 1.69 | 1.28-2.23 | 1 mmol/L |
| HbA1c | CHD | 2015 | Ross et al. | MAGIC, CARDIoGRAMplusC4D (n=240,795) | 9 | 1.53 | 1.14-2.05 | 1% |
| HbA1c | CAD | 2018 | Au Yeung et al. | MAGIC, UKBB (n=515,693) | 14 | 2.24 | 1.55-2.35 | 1% |
| HbA1c | CAD | 2018 | Au Yeung et al. | MAGIC, CARDIoGRAMPlusC4D (n=307,960) | 14 | 1.27 | 0.90-1.78 | 1% |
| HbA1c | CHD | 2019 | Leong et al. | MAGIC, CARDIoGRAMPlusC4D, UKBB (n=819,131) | 50 | 1.61 | 1.40-1.84 | 1% |
| HbA1c | CHD | 2019 | Leong et al. | MAGIC, CARDIoGRAMPlusC4D, UKBB (n=819,131) | 16 (Gly) | 2.23 | 1.73-2.89 | 1% |
| HbA1c | CHD | 2019 | Leong et al. | MAGIC, CARDIoGRAMPlusC4D, UKBB (n=819,131) | 20 (Ery) | 1.30 | 1.08-1.57 | 1% |
| FG | CHD | 2015 | Ross et al. | MAGIC, CARDIoGRAMplusC4D (n=327,437) | 30 | 1.18 | 0.97-1.42 | 1 mmol/L |
| FG | CHD | 2015 | Ahmad et al. | MAGIC, CARDIoGRAMplusC4D (n=327,437) | 24 | 1.15 | 1.00-1.32 | 1 mmol/L |
| FG | CAD | 2017 | Merino et al. | MAGIC, CARDIoGRAMplusC4D, FHS (n=332,550) | 12 | 1.43 | 1.14-1.79 | 1 mmol/L |
| FG | CAD | 2017 | Merino et al. | MAGIC, CARDIoGRAMplusC4D, FHS (n=332,550) | 5 | 1.33 | 1.02-1.73 | 1 mmol/L |
| FI | CHD | 2016 | Tikkanen et al. | Finnish cohort (n=26,554) | 20 | 1.06 | 1.02-1.10 | SD |
| FI | CHD | 2017 | Zhan et al. | MAGIC, CARDIoGRAM (n=195,552) | 12 | 2.64 | 1.55-4.48 | Not specified |
| FI | MI | 2019 | Zhao et al. | MAGIC, UKBB (n=500,567) | 7 | 2.87 | 1.30-6.33 | 1 pmol/l |
| FI | MI | 2019 | Zhao et al. | MAGIC, UKBB (n=500,567) (men only) | 7 | 4.27 | 1.60-11.3 | 1 pmol/l |
| FI | MI | 2019 | Zhao et al. | MAGIC, CARDIoGRAMplusC4D (n=292,862) | 7 | 1.90 | 1.04-3.49 | 1 pmol/l |
| FI | MI | 2019 | Zhao et al. | MAGIC, UKBB + CARDIoGRAMplusC4D (n=684,872) | 7 | 2.21 | 1.37-3.58 | 1 pmol/l |
All cohorts are European or majority European unless otherwise indicated.
BC, SNPs implicated in beta-cell dysfunction; CARDIoGRAMplusC4D, Coronary Artery Disease Genome-wide Replication and Meta-Analysis (CARDIoGRAM) Consortium plus the Coronary Artery Disease Genetics (C4D) Consortium; CIHDS, Copenhagen Ischemic Heart Disease Study; CKB, China Kadoorie Biobank; DIAGRAM, Diabetes Genetics Replication and Meta-Analysis; Ery, SNPs that affect HbA1c via erythrocytic pathways; FHS, Framingham Heart Study; FI, fasting insulin; Gly, SNPs that affect HbA1c via glycemic pathways; IR, SNPs implicated in insulin resistance; MAGIC, Meta-Analyses of Glucose and Insulin-Related Traits Consortium; MI, myocardial infarction; NINDS-SiGN, National Institute of Neurologic Disorders and Stroke-Stroke Genetics Network; SD, standard deviation in genetic risk score; REACTION, Risk Evaluation of Cancers in Chinese Diabetic Individuals: a Longitudinal Study; UKBB, UK Biobank
Among ~160,000 individuals in the China Kadoorie Biobank (CKB), 48 T2D SNPs were used in MR analysis supporting causality of T2D for CHD and ischemic stroke (mainly non-lacunar stroke as numbers were small for lacunar stroke) but not intracerebral hemorrhage (Tables 2 and 3).69 The genetically predicted outcomes were similar to the observational associations in CKB. This study also conducted MR for similar outcomes using summary statistics from DIAGRAM, CARDIoGRAMplusC4D, and the International Stroke Genetics Consortium (ISGC) (Tables 2 and 3), notably finding no evidence of heterogeneity in results between Chinese and Europeans. A study in Europeans found causal evidence for T2D but not fasting glucose or fasting insulin with lacunar (small vessel) stroke; none of these were causal for intracerebral hemorrhage, and large vessel stroke was not evaluated.130 Another study found causal evidence of T2D with all ischemic stroke, large vessel stroke, and small vessel stroke, and of fasting glucose with large vessel stroke (Table 3); the latter two findings were not considered robust given sensitivity analysis and multiple testing correction, respectively.131 In the latter study, fasting insulin had no causal evidence for any of the stroke types, and T2D and fasting glucose had no causal evidence for cardioembolic stroke. These studies generally support T2D as causal for large and small vessel ischemic stroke but not intracerebral hemorrhage or cardioembolic stroke.
Table 3.
Summary of Mendelian randomization studies examining T2D and related traits as causal for cerebrovascular disease
| Risk factor | Outcome | Year | Authors | Population | Number of SNPs | OR for outcome | 95% CI | Increment |
|---|---|---|---|---|---|---|---|---|
| T2D | Ischemic stroke | 2019 | Gan et al. | CKB (n=159,528 Chinese) | 48 | 1.08 | 1.02-1.14 | 1 unit log-odds of T2D |
| T2D | Intracerebral hemorrhage | 2019 | Gan et al. | CKB (n=159,528 Chinese) | 48 | 1.01 | 0.94-1.10 | 1 unit log-odds of T2D |
| T2D | Ischemic stroke | 2019 | Gan et al. | DIAGRAMv4, ISGC (n=233,661) | 86 | 1.13 | 1.05-1.21 | 1 unit log-odds of T2D |
| T2D | Intracerebral hemorrhage | 2019 | Gan et al. | DIAGRAMv4, ISGC (n=162,234) | 86 | 1.12 | 0.94-1.34 | 1 unit log-odds of T2D |
| T2D | Ischemic stroke | 2017 | Larsson et al. | DIAGRAM, METASTROKE, NINDS-SiGN (n=205,593) |
49 |
1.12 | 1.07-1.17 | 1 unit log-odds of T2D |
| T2D | Large vessel stroke | 2017 | Larsson et al. | DIAGRAM, METASTROKE, NINDS-SiGN (n=205,593) | 49 | 1.28 | 1.16-1.40 | 1 unit log-odds of T2D |
| T2D | Small vessel stroke | 2017 | Larsson et al. | DIAGRAM, METASTROKE, NINDS-SiGN (n=205,593) | 49 | 1.21 | 1.10-1.33 | 1 unit log-odds of T2D |
| T2D | Intracerebral hemorrhage | 2018 | Liu et al. | DIAGRAMv4, UKBB, 2 studies (n=169,657) | 84 | 1.07 | 0.95-1.20 | Not specified |
| T2D | Lacunar stroke | 2018 | Liu et al. | DIAGRAMv4, 3 studies (n=188,696) | 84 | 1.15 | 1.04-1.28 | Not specified |
| FI | Ischemic stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=164,329) | 18 | 1.03 | 0.78-1.37 | 0.6 pmol/L |
| FI | Large vessel stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=164,329) | 18 | 1.42 | 0.80-2.50 | 0.6 pmol/L |
| FI | Small vessel stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=164,329) | 18 | 1.15 | 0.65-2.02 | 0.6 pmol/L |
| FI | Lacunar stroke | 2018 | Liu et al. | MAGIC, 3 studies (n=138,045) | 18 | 1.52 | 0.45-5.08 | Not specified |
| FG | Ischemic stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=188,782) | 36 | 1.12 | 0.98-1.28 | 0.65 mmol/L |
| FG | Large vessel stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=188,782) | 36 | 1.42 | 1.08-1.85 | 0.65 mmol/L |
| FG | Small vessel stroke | 2017 | Larsson et al. | MAGIC, METASTROKE, NINDS-SiGN (n=188,782) | 36 | 1.09 | 0.83-1.43 | 0.65 mmol/L |
| FG | Lacunar stroke | 2018 | Liu et al. | MAGIC, 3 studies (n=162,498) | 36 | 1.10 | 0.63-1.90 | Not specified |
All cohorts are European or majority European unless otherwise indicated.
BC, SNPs implicated in beta-cell dysfunction; CARDIoGRAMplusC4D, Coronary Artery Disease Genome-wide Replication and Meta-Analysis (CARDIoGRAM) Consortium plus the Coronary Artery Disease Genetics (C4D) Consortium; CIHDS, Copenhagen Ischemic Heart Disease Study; CKB, China Kadoorie Biobank; DIAGRAM, Diabetes Genetics Replication and Meta-Analysis; Ery, SNPs that affect HbA1c via erythrocytic pathways; FHS, Framingham Heart Study; FI, fasting insulin; Gly, SNPs that affect HbA1c via glycemic pathways; IR, SNPs implicated in insulin resistance; ISGC, International Stroke Genetics Consortium; MAGIC, Meta-Analyses of Glucose and Insulin-Related Traits Consortium; MI, myocardial infarction; NINDS-SiGN, National Institute of Neurologic Disorders and Stroke-Stroke Genetics Network; SD, standard deviation in genetic risk score; REACTION, Risk Evaluation of Cancers in Chinese Diabetic Individuals: a Longitudinal Study; UKBB, UK Biobank
What Components of T2D Cause CHD?
The compelling MR evidence that T2D causes CHD raises the question of what feature(s) of diabetes may be responsible. Molecular biologic and animal model studies have suggested several potential mechanisms, including inflammation, hyperthrombotic state, endothelial dysfunction, oxidative stress, and advanced glycation end products, among others. Epidemiologic studies have associated fasting glucose, even in the non-diabetic range, with incident CHD.132 Similar observations have been made with markers of insulin resistance.48 Currently, we can look to formal MR studies addressing hyperglycemia, hyperinsulinemia, insulin resistance, and insulin secretion as potential mediators of the effect of T2D on CHD.
Hyperglycemia
In a study of 80,522 Danish individuals, five SNPs for fasting glucose (FG) were also associated with nonfasting glucose.133 These were used in MR analyses that found causal effects for nonfasting glucose on CHD and MI (Table 2). To avoid pleiotropy, the authors selected the five SNPs because they were not associated with other risk factors. However, three (GCK rs4607517, ADCY5 rs1170867, DGKB rs2191349) of the SNPs have been found to be associated with T2D.134 In addition, two SNPs (GCK rs4607517, G6PC2 rs560887) are also associated with increased birth weight,135 leaving only one SNP (ADRA2A rs10885122) free of pleiotropic effects. Thus, this study is inconclusive regarding whether nonfasting glucose is causative for CHD.
Ross et al. conducted MR using 9 SNPs for HbA1c (HbA1c) and 30 SNPs for FG.128 HbA1c (OR 1.53 (95% CI 1.14-2.05) per 1% increase) but not FG (OR 1.18 (95% CI 0.97-1.42) per 1 mmol/L increase) was associated with increased odds of CHD; after adjustment for lipid fractions and BMI, the effect of HbA1c lost significance (OR 1.66, 95% CI 0.44-6.35). The authors noted that their study was underpowered regarding FG. Using the same datasets but excluding SNPs with pleiotropic effects, Ahmad found supportive evidence for FG SNPs as increasing CHD risk (Table 2).127
To create an instrument variable that purely represented FG, Merino et al. assembled 12 FG SNPs that had no evidence of association with T2D or other risk factors such as lipids, obesity, or blood pressure.136 Using these 12 SNPs, a causal effect of FG on CHD was suggested (Table 2). SNP rs560887 (G6PC2) was the strongest contributor to the MR instrument; this SNP and another (PDX1 rs11619319) are associated with increased birth weight.135 Given that low birthweight has been causally linked to CHD,137 inclusion of these SNPs would not result in false positive results, as the FG increasing alleles are associated with increased birth weight.135, 138, 139 After verifying that the 12-SNP instrument was not associated with T2D, they then removed 7 SNPs that displayed even nominal (P<0.05) evidence for association with lipid fractions, blood pressure, or obesity; the resulting 5 SNPs (including PDX1 rs11619319 and G6PC2 rs560887) still supported causality of FG for diabetes (Table 2).
Two recent MR studies confirmed a causal effect of HbA1c on CHD (Table 2).140, 141 Both used the same datasets,142, 143 but approached the analysis differently. SNPs associated with HbA1c have been classified as acting via glycemic pathways or erythrocytic pathways.142 Au Yeung et al. estimated HbA1c effects on CHD separately in UK Biobank (UKBB) and CARDIoGRAMplusC4D (Table 2), excluding not only SNPs associated with potential confounders but also the erythrocytic SNPs.141 Leong et al. relied on MR Egger to exclude pleiotropy and analyzed all HbA1c SNPs as well as glycemic and erythrocytic SNPs separately, combining UKBB and CARDIoGRAMplusC4D via meta-analysis.140 That the erythrocytic SNP instrument was associated with CHD suggests that glycosylated hemoglobin may influence CHD through mechanisms not only related to hyperglycemia, but also potentially independent of hyperglycemia, such as reduced hemoglobin and increased LDL-C.140
Hyperinsulinemia
Tikkanen et al. conducted MR using summary-level data from GWAS consortia as well as individual-data analyses.144 This study uniquely first used summary-level data to simultaneously model multiple risk factors for CHD, including LDL-C, HDL-C, triglycerides, BMI, WHR, fasting insulin, fasting glucose, systolic and diastolic pressure (257 SNPs in total). In this multivariate model, the significant predictors of CHD were LDL-C, triglycerides, fasting insulin and fasting glucose. Notably, when insulin was removed from the model, BMI and WHR became significant, suggesting adiposity may affect CHD in part via insulinemia. In their individual level analysis, of the same nine risk factors, BMI, WHR, and fasting insulin were significantly associated with CHD both individually (Table 2) and in a multivariate model. While this difference from the summary-level may be due to sample size, it suggests insulin may also act through non-adiposity mechanisms on CHD. To this point, a subsequent network MR study placed fasting insulin (out of 10 evaluated potential mediators) on the causal pathway between shortened telomeres (a marker of cellular aging) and CHD (Table 2), suggesting that fasting insulin may account for 18% of the total effect of telomere length on CHD.145
After carefully excluding any SNPs with evidence of pleiotropic effects, investigators conducted MR analyses in the UKBB,146 using various combinations of SNPs associated with fasting insulin and fasting insulin adjusted for BMI in MAGIC,83 several of which were also associated with direct physiologic measures of insulin resistance.147 Genetically predicted fasting insulin was associated with increased risk of MI overall; when stratified by sex, the effect was significant only in the men (Table 2). A similar pattern was seen for angina. The fasting insulin instruments were also associated with LDL-C, systolic blood pressure, apolipoprotein B, and reticulocyte count, raising these as possible mediators of the effect on CHD.
Insulin resistance and insulin secretion
Ross et al. examined instruments based on 26 T2D SNPs implicated in beta-cell dysfunction and 11 T2D SNPs associated with insulin resistance based on their review of the literature; both of these were similarly associated with increased CHD (beta-cell: OR 1.83, 95% CI 1.19-2.62; insulin resistance: OR 2.35, 95% CI 1.46-3.53).128 Gan et al. also examined subsets of T2D SNPs thought to act through beta-cell dysfunction and insulin resistance; the effects on CHD were statistically similar, though the insulin resistance risk score was insignificant, likely due to the low number of SNPs (Table 2).69 Another study found that beta-cell dysfunction SNPs appear to increase CHD risk.127
Lessons Learned from MR Studies
As reviewed above, a wealth of studies have shed light on how T2D and related traits drive CHD and ischemic stroke (Figure 5). Results are compelling when several studies arrive at consistent conclusions; however, a caveat is that many studies utilized summary data from the same large genetics consortia, sometimes arriving at different conclusions, in part when different sets of SNPs are used or when pleiotropy is handled differently (e.g., statistically or by removing pleiotropic variants) or due to different sample sizes depending on when the summary data were accessed (e.g., DIAGRAMv3 or DIAGRAMv4). We note that while MR studies provide strong clues on causality, they do not elucidate detailed molecular or physiologic mechanisms.
Figure 5.
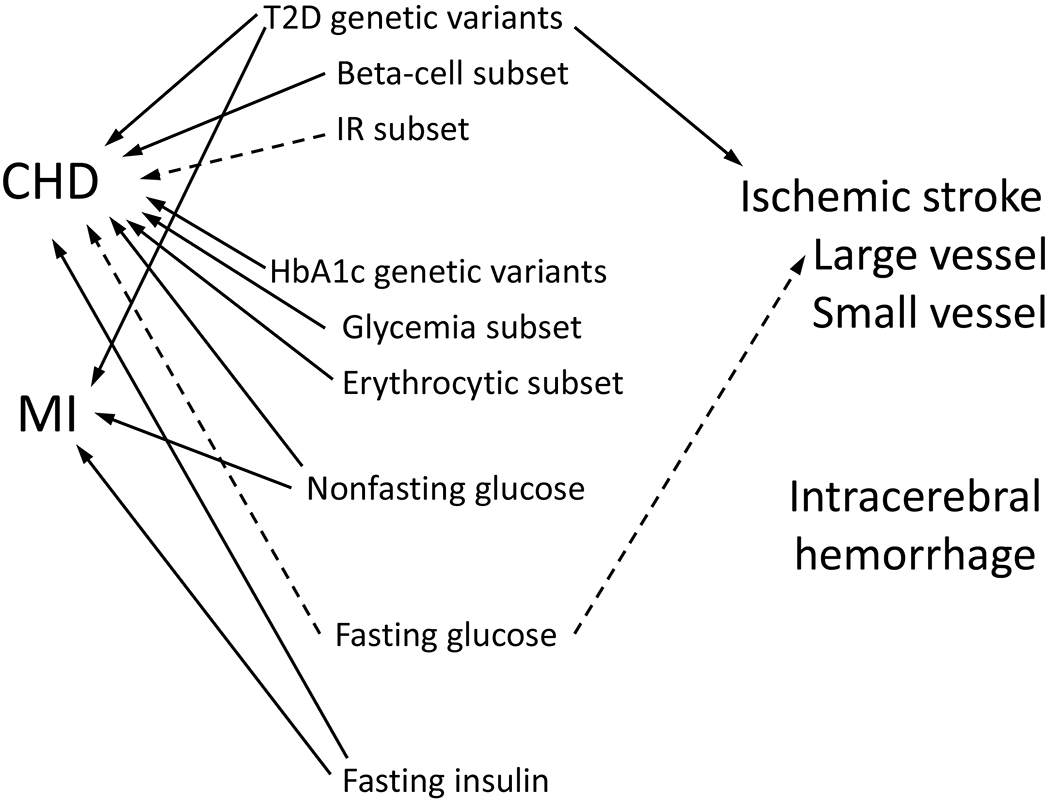
Summary of Mendelian randomization literature examining T2D and related traits as risk factors for CVD. Solid lines represent compelling evidence for causality. Dotted lines represent mixed results.
Given the tight relationships (including genetic correlation68) between adiposity, lipids, T2D, and CHD, another challenge is disentangling the effects of T2D from those of adiposity (BMI, WHR) and lipid fractions on the risk of CHD. Formal mediation analyses are needed to dissect the causal effect of T2D on CHD. This will be a challenging task, evidenced by the fact that such analyses have yielded conflicting results regarding the causal effect of BMI on CHD, as noted above.121–123 Given the well-recognized differences in risk of T2D and CHD between women and men, the paucity of studies genetically interrogating these differences is notable. A sensitivity analysis in an MR study examining effects of WHR adjusted for BMI on cardiometabolic traits found that while a risk score composed of SNPs associated with WHR in women but not men exhibited greater magnitudes of association with T2D and CHD in women than men, the differences were not statistically significant, possibly due to low power.125 Further studies examining the genetic relationships between T2D and CHD in the context of sex differences are needed.
The field still needs instrument variables strongly associated with direct measures of insulin resistance and insulin secretion (e.g., by euglycemic clamp or FSIGT). To date, fasting insulin has been interpreted as a marker of insulin resistance, yet fasting insulin reflects insulin resistance, insulin clearance, adiposity, and insulin secretion; in fact, it represents the former three in roughly similar proportions.148 Use of subsets of T2D SNPs implicated in insulin resistance or beta-cell function is useful, but conflated with T2D itself. Given the recent expansion of the number of independent T2D SNPs to 403,41 the field has not yet had the time to fully exploit this range of SNPs in MR studies.
We note that MR studies where fasting glucose is the risk factor have had mixed results with CHD as the outcome, whereas results are stronger when T2D or HbA1c are the instrumented risk factors. This may reflect that total glycemia, which includes postprandial hyperglycemia, may be more important to CHD risk than simply fasting glucose. Consistent with this notion, some epidemiologic studies found that postprandial glucose may be a stronger predictor of CVD events than fasting glucose.149, 150 Experimental evidence has suggested that postprandial hyperglycemia may be especially damaging to the vasculature.151
Given that the MR studies suggest that T2D and glucose may be causal for CHD and stroke, how do we reconcile this with the largely negative results of randomized trials of intensive glucose lowering that were designed specifically with cardiovascular disease as the outcome, including Action to Control Cardiovascular Risk in Diabetes (ACCORD),152 Action in Diabetes and Vascular Disease: Preterax and Diamicron - MR Controlled Evaluation (ADVANCE)153, and the Veterans Affairs Diabetes Trial (VADT)?154 The median durations of these trials were 3.5 years, 5 years, and 5.6 years, respectively, whereas MR represents lifelong exposure to risk. Nevertheless, meta-analyses including these trials and others found that intensive glucose lowering reduces non-fatal MI by 10-16% but does not reduce stroke or cardiovacular mortality.155–159 Interestingly, and perhaps more consistent with the exposure timeframe of MR, long-term follow-up after the trials ended did in several instances reveal that intensive glycemic control eventually resulted in lower rates of major cardiovasclar events, for example at 10 years of follow-up in the VADT160 (though not seen at 15 years161). A similar later emergence of benefit on cardiovascular events was seen during the post-trial observational studies of the United Kingdom Prospective Diabetes Study (UKPDS) and the Diabetes Control and Complications Trial (DCCT).162, 163
THERAPEUTIC IMPLICATIONS OF GENETIC FINDINGS
Lipid Lowering Agents and T2D
While CHD and T2D share many underying risk factors, especially obesity, their relationship with LDL-C is dramatically different. Low LDL-C appears to be a pathway promoting T2D. This emerged when statin treatment was found to confer an increased risk of new onset T2D, especially in those with pre-existing impaired glucose tolerance.164 This observation led to MR studies that found that genetic variants that lower LDL-C in the 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and proprotein convertase subtilisin/kexin type 9 (PCSK9) loci were associated with increased risk of T2D (Table 4).165, 166 Another study examined variants at genes coding multiple targets of LDL-C lowering agents (HMGCR, NPC1L1 (target of ezetimibe), PCSK9, ABGC5/G8, LDLR) and found that while the effect of a genetically predicted 1 mmol/L lowering of LDL-C at each locus was statistically similar regarding CHD (OR 0.54 to 0.61), the effects on T2D were heterogeneous (non-significant to OR 2.42) (Table 4).167 MR studies also found that genetically mediated higher LDL-C appears to be protective against T2D (Table 4)126, 168, 169 and that LDL-C is inversely related with age of onset of T2D.170 This is supported by an electronic medical records study that found that people with LDL-C ≤ 60 mg/dl have doubled prevalence of T2D, which remained significant in sensitivity analyses stratifying by sex and weight but not race (effect seen in European but not African ancestry).171 Additional genetic evidence is the observation that diabetes is less prevelent in those with familial hypercholesterolemia.172 This may explain the association of the APOE SNP with lower LDL-C and increased odds of T2D referred to earlier.97 Adiposity might be the link between lower LDL-C and T2D. LDL-C lowering variants in HMGCR and PSCK9 were also associated with higher body weight, especially centrally distributed weight.92, 93 In aggregate, weight increased slightly in statin trials.166 Whether subjects on PCSK9 inhibitors gain weight has not been reported.173 Unlike statins, clinical trials of PCSK9 inhibitors have not been associated with increased incident diabetes; a meta-analysis of PCSK9 inhibitor trials involving 26,123 subjects found no effect on incident diabetes, fasting glucose, or HbA1c.174 In contrast, the meta-analysis that documented increased diabetes risk with statins involved 91,140 patients.164 It has been suggested that sample sizes of PCSK9 inhibitor and ezetimibe trials have been insufficient to observe an increased risk of diabetes. Another suggested explanation for the discrepancy between genetic evidence and trial evidence is that because PCSK9 inhibitors mainly target circulating PCSK9, they may have little impact on local PCSK9 (e.g., in the beta-cell) where alteration may influence development of diabetes.175 It is also possible that other (not related to LDL-C lowering) effects of PCSK9 inhibition may counteract the effect on glycemia.
Table 4.
Mendelian randomization studies of relevance to lipid lowering therapy
| Risk factor | Outcome | Year | Authors | Cohorts | Number of SNPs | OR for T2D | 95% CI | Increment |
|---|---|---|---|---|---|---|---|---|
| NPC1L1 | T2D | 2016 | Lotta et al. | EPIC-Interact, DIAGRAM, UKBB (n=321,044) | 2 | 2.42 | 1.70-3.43 | −1 mmol/L in LDL-C |
| HMGCR | T2D | 2016 | Lotta et al. | EPIC-Interact, DIAGRAM, UKBB, 11 studies (n=376,217) | 3 | 1.39 | 1.12-1.73 | −1 mmol/L in LDL-C |
| PCSK9 | T2D | 2016 | Lotta et al. | EPIC-Interact, DIAGRAM, UKBB (n=321,044) | 1 | 1.19 | 1.02-1.38 | −1 mmol/L in LDL-C |
| ABCG5/G8 | T2D | 2016 | Lotta et al. | EPIC-Interact, DIAGRAM, UKBB (n=321,044) | 1 | 1.15 | 0.89-1.48 | −1 mmol/L in LDL-C |
| LDLR | T2D | 2016 | Lotta et al. | EPIC-Interact, DIAGRAM, UKBB (n=321,044) | 1 | 1.13 | 1.00-1.29 | −1 mmol/L in LDL-C |
| HMGCR | T2D | 2015 | Swerdlow et al. | 43 studies (n=223,463) | 1 | 1.02 | 1.00-1.05 | −0.06 mmol/L in LDL-C |
| PCSK9 | T2D | 2017 | Schmidt et al. | GLGC, DIAGRAM, UKBB, 50 studies (n=568,448) | 4 |
1.29 | 1.11-1.50 | −1 mmol/L in LDL-C |
| LDL-C | T2D | 2016 | White et al. | GLGC, DIAGRAMv3 (n=338,398) | 130 | 0.79 | 0.71-0.88 | +38 mg/dl in LDL-C |
| LDL-C | T2D | 2015 | Fall et al. | GLGC, DIAGRAMv3 (n=338,398) | 139 | 0.81 | 0.75-0.89 | +40 mg/dl in LDL-C |
| LDL-C | T2D | 2016 | Tragante et al. | GLCG, DIAGRAMv3 (n=245,275) | 197 | 0.86 | 0.81-0.91 | +1 SD in LDL-C |
All cohorts are European or majority European.
DIAGRAM, Diabetes Genetics Replication and Meta-Analysis; EPIC-Interact, European Prospective Investigation into Cancer and Nutrition-Interact Study; GLGC, Global Lipids Genetics Consortium; UKBB, UK Biobank
One study used genetics to prioritize existing and potential lipid lowering drugs that reduce CHD risk without adverse glycemic consequences.169 SNPs with associated with LDL-C at genome-wide significance (in the Global Lipids Genetics Consortium (GLGC) GWAS) and at nominal significance (P<0.05) with CHD (in CARDIoGRAMplusC4D) were collected and assessed for association with a glycemic burden composite consisting of T2D, fasting glucose, fasting insulin, and fasting proinsulin (from DIAGRAM and MAGIC); loci were then selected whose genes coded drug targets. In contrast to MR findings167 (but consistent with clincal experience), the PCSK9 and NPC1L1 loci were associated with reduced LDL-C and CHD risk but not with the glycemic composite. APOB and LPA loci also exhibited associations with reduced LDL-C and CHD risk without adverse glycemic effects. These have been recently targeted by a new form of pharmaceutical, antisense oligonucleotides (ASO).176 An ASO against APOB, mipomersen, has been approved by the FDA as an adjunct to first-line therapies in patients with homogygous familial hypercholesterolemia. ASOs targeting LPA have entered phase I trials, and have been found to lower Lp(a) levels.177 The HMGCR and SLC22A3 loci were also associated with reduced LDL-C and CHD; whereas HMGCR was also associated with an increase in the glycemic composite (consistent with the statin experience), SLC22A3 was associated with a decrease in the glycemic composite.169 SLC22A3 codes for the organic cation transporter OCT3, a widely expressed transporter that has been found to modulate metformin pharmacokinetics and pharmacodynamics.178 Different coding variants in SLC22A3 have been found to increase or decrease cellular metformin uptake.179 In the UKPDS, overweight patients receiving intensive glucose-lowering with metformin experienced decreased MI, sudden death, angina, stroke and peripheral vascular disease compared to conventional therapy; compared to insulin- or sulfonylurea-based intenstive control regimens, metformin also decreased all-cause mortality and stroke.180
Sulfonylureas and CHD
Sulfonylureas are one of the oldest drug classes for the treatment of T2D. For many years, their cardiovascular safety has been questioned. Almost 50 years ago, the University Group Diabetes Program reported an increased risk of CAD with use of sulfonylureas.181 As this result was not confirmed in subsequent studies, it was not considered in the choice of T2D treatment for many years; it was thought by many that this was a problem only with first generation sulfonylureas. However, a series of recent studies revived this issue, leading many clinicians to prescribe sulfonylureas much less frequently. Among 4,902 diabetic women without CVD at baseline in the Nurses’ Health Study, longer duration of sulfonylurea use was associated with increased risk of CHD but not of stroke.182 On the other hand, a randomized trial primarily designed to examine glycemic durability of rosiglitazone compared to metformin or glyburide found in secondary analysis that the risk of cardiovascular events was lowest with the sulfonylurea.183 Meta-analyses yielded conflicting results regarding the cardiovascular risk of sulfonylureas, suggesting increased,184 decreased,185 or neutral risk.186 Given the conflicting observational and trial evidence, Emdin et al. approached the question using genetics, specifically a variant (rs757110) for T2D that codes for an amino acid change (p.A1369S) in the gene (ABCC8) for one of the subunits (ATP binding cassette subfamily C member 8) of the sulfonylurea receptor.187 This variant is in high LD with GWAS signals for T2D (r2=0.92 with rs5215). The serine allele promotes closure of the channel in vitro, mimicking the action of sulfonylureas to promote insulin secretion.188 In a meta-analysis of UKBB with large-scale GWAS, the serine allele was associated with a lower odds of T2D (OR 0.93, 95% CI 0.91-0.95) and CHD (OR 0.98, 95% CI 0.96-0.99). Consistent with the known effects of sulfonylureas, the serine allele was also associated with higher BMI, higher fasting insulin and lower 2 hour glucose, but also with reduced WHR. Though not perfectly simulating sulfonylurea action (typically started late in life and the variant was not associated with HbA1c), this study provides some reassurance regarding the cardiovascular safety of sulfonylureas. Given that many studies suggesting harm compared sulfonylureas to metformin, one explanation is that both are cardioprotective, but metformin moreso, making it appear that sulfonylureas are harmful. Indeed, in observational trials where metformin was not the comparator, sulfonylureas had the lowest predicted risk.189 Alternatively, patients who are prescribed sulfonylureas rather than metformin may have more advanced T2D, biasing CHD associations. Recent trials finding similar rates of CV outcomes for the dipeptidyl peptidase-4 inhibitor linagliptin, the sulfonylurea glimepiride, and placebo are reassuring.190,191
GLP-1 Agonists and CHD
Unlike the uncertainty with sulfonylureas, recent randomized clinical trials have firmly established specific glucagon like peptide-1 (GLP-1) agonists as cardioprotective agents. This has been demonstrated for liraglutide, semaglutide, albuglutide, and dulaglutide, but not exenatide or lixisenatide.192–197 Given this picture, it is noteworthy that GLP-1 agonists are based on two distinct molecular structures. Liraglutide, dulaglutide, and albiglutide are covalently modified analogs of human GLP-1 while exenatide and lixisenatide are based on the Gila monster exendin-4, which possesses a nine amino acid C-terminal extension that is absent from human GLP-1 and increases affinity for binding to GLP-1 receptors.198 Perhaps these structural differences explain the different CV outcomes. It has also been suggested that the CV outcome trial results are related to the glycemic efficacy of the doses tested. Nevertheless, a meta-analysis combining positive and negative trials found that compared with placebo, GLP-1 agonist treatment yielded a 10% relative risk reduction in the CVD primary outcome (CV mortality, non-fatal MI, non-fatal stroke; HR 0.9, 95% CI 0.82-0.99, p=0.033); cardiovascular mortality (HR 0.87, 95% CI 0.79-0.96, p=0.007) and all-cause mortality (HR 0.88, 95% CI 0.81-0.95, p=0.002) were also reduced, with low-to-moderate between-trial heterogeneity, suggesting the benefit may be a class effect.199 The latter notion is supported by genetic studies. In the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium, we assessed association of exonic variants with fasting glucose (~60,000 non-diabetic individuals) and fasting insulin (~48,000 non-diabetic individuals) from 23 cohorts. We found a variant coding variant (rs10305492, A316T) in the gene for the GLP-1 receptor (GLP1R) associated with fasting glucose, with the threonine allele associated with lower fasting glucose.200 Additional examination of this variant found that the threonine allele was associated with reduced T2D (OR 0.86, 95% CI 0.76-0.96), reduced insulin secretion (insulinogenic index from oral glucose tolerance testing) but increased 2 hour glucose. We hypothesized that the threonine allele confers a gain-of-function that consitutively activates the GLP-1 receptor, causing beta-cells to secrete insulin at a lower ambient glucose level, thereby maintaining a lower fasting glucose, which in turn causes down-regulation of the receptors over time, causing incretin resistance and a higher 2 hour glucose after an oral carbohydrate load. A subsequent analysis examined coding variation in six genes for drug targets for obesity or T2D, including GLP1R, for association with disease outcomes to predict cardiovascular safety of those drugs.201 This analysis found that the threonine allele at rs10305492 was associated with reduced CHD (OR 0.93, 95% CI 0.87-0.98), along with the previously noted association with lower fasting glucose and protection against diabetes. This is elegant example of genetic findings paralleling drug effects, in this case the glucose-lowering and cardioprotective effects of GLP-1 agonists, which act on the GLP-1 receptor.
CONCLUSIONS
There have been dramatic advances in our understanding the genetic basis of T2D and of other cardiovascular risk factors. The data reviewed herein indicate that not only is T2D an epidemiologic risk factor for CHD, but that it is a genetic risk factor for CHD as well. Elucidating this relationship has identified shared genes between T2D and CHD with noteworthy examples being the 9p21 locus, the IRS1 locus, and the LPL and ANGPTL4 genes involved in triglyceride metabolism. Formal Mendelian randomization analyses are beginning to clarify the relationship between T2D, its underpinnings and related traits (fasting glucose, fasting insulin, HbA1c, insulin resistance, and insulin secretion), and the various pathophysiologic pathways of CHD development. Finally, genetic studies helped to clarify whether CHD therapies (e.g., lipid lowering) are diabetogenic, whether diabetes therapies increase CAD risk (e.g., sulfonylureas), and whether certain diabetes therapies (GLP-1 agonists) are cardioprotective. The increased understanding of the shared genetics between T2D and CHD will provide tools to subdivide diabetes patients into those who will need interventions to prevent CHD and help identify the therapeutic agents that will be most effective in such patients. That is, with continued success in identifying the genes and the variants in those genes that predispose to T2D and CHD, we are starting to subdivide T2D loci into various pathophysiologic subsets. It is likely that these different subsets will confer different risks for CHD, and that the elucidation of the responsible genes will provide clues to the mechanisms of CHD risk. These concepts lead to the following priorities for future research: (a) conduct large-scale genetic epidemiologic investigations of CHD occurring in patients with T2D; (b) test defined subsets of the T2D loci as risk factors for CHD; (c) examine the molecular mechanisms of the loci that link T2D subsets to CHD; and (d) genotype participants of large-scale therapeutic clinical trials of T2D and of CHD to examine the effect of these diabetogenic loci. The ultimate goal is improved therapy and even prevention in those at risk for T2D and CHD.
Acknowledgments
Sources of funding
This work was supported in part by National Institutes of Health grants from the National Institute of Diabetes and Digestive and Kidney Disease (P30-DK063491), and UL1TR001881 (University of California Los Angeles Clinical and Translational Science Institute Grant from the National Center for Advancing Translational Sciences). M.O.G. was supported by the Eris M. Field Chair in Diabetes Research.
Non-standard Abbreviations and Acronyms
- ACCORD
Action to Control Cardiovascular Risk in Diabetes
- ADVANCE
Action in Diabetes and Vascular Disease: Preterax and Diamicron - MR Controlled Evaluation
- ANGPTL4
angiopoietin like 4
- ANRIL
antisense non-coding RNA in the INK4 locus
- APOE
apolipoprotein E
- ASO
antisense oligonucleotide
- C4D
Coronary Artery Disease Genetics
- CARDIoGRAM
Coronary Artery Disease Genome-wide Replication and Meta-analysis
- CHARGE
Cohorts for Heart and Aging Research in Genomic Epidemiology
- CHD
coronary heart disease
- CIHDS
Copenhagen Ischemic Heart Disease Study
- CKB
China Kadoorie Biobank
- CV
cardiovascular
- CVD
cardiovascular disease
- DCCT
Diabetes Control and Complications Trial
- DIAGRAM
Diabetes Genetics Replication and Meta-Analysis
- DIAMANTE
DIAbetes, Meta-ANalysis Trans-Ethnic
- EPIC
Interact, European Prospective Investigation into Cancer and Nutrition-Interact Study
- eQTL
expression quantitative trait locus
- FG
fasting glucose
- FHS
Framingham Heart Study
- FI
fasting insulin
- FSIGT
frequently-sampled intravenous glucose tolerance test
- GLGC
Global Lipids Genetics Consortium
- GLP-1
glucagon like peptide-1
- GLP1R
glucagon like peptide-1 receptor
- GWAS
genome-wide association study
- HbA1c
hemoglobin A1c
- HMGCR
3-hydroxy-3-methylglutaryl-CoA reductase
- IMT
intima-media thickness
- IRS1
insulin receptor substrate 1
- ISGC
International Stroke Genetics Consortium
- LD
linkage disequilibrium
- LPL
lipoprotein lipase
- MAGIC
Meta-Analyses of Glucose and Insulin-Related Traits Consortium
- MI
myocardial infarction
- MR
Mendelian randomization
- NINDS-SiGN
National Institute of Neurologic Disorders and Stroke-Stroke Genetics Network
- PCSK9
proprotein convertase subtilisin/kexin type 9
- REACTION
Risk Evaluation of Cancers in Chinese Diabetic Individuals: a Longitudinal Study
- SNP
single nucleotide polymorphism
- T2D
type 2 diabetes
- UKBB
United Kingdom Biobank
- UKPDS
United Kingdom Prospective Diabetes Study
- VADT
Veterans Affairs Diabetes Trial
- WHR
waist hip ratio
Footnotes
Disclosures
The authors have no conflicts of interest.
References
- 1.Staimez LR, Deepa M, Ali MK, Mohan V, Hanson RL, Narayan KMV. Tale of two Indians: Heterogeneity in type 2 diabetes pathophysiology. Diabetes Metab Res Rev. 2019:e3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787 [DOI] [PubMed] [Google Scholar]
- 3.Magliano DJ, Islam RM, Barr ELM, Gregg EW, Pavkov ME, Harding JL, Tabesh M, Koye DN, Shaw JE. Trends in incidence of total or type 2 diabetes: systematic review. Bmj. 2019;366:l5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayan KM, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF. Lifetime risk for diabetes mellitus in the United States. Jama. 2003;290:1884–1890 [DOI] [PubMed] [Google Scholar]
- 5.Perry JR, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care. 2008;11:371–377 [DOI] [PubMed] [Google Scholar]
- 6.Preis SR, Hwang SJ, Coady S, Pencina MJ, D’Agostino RB, Sr., Savage PJ, Levy D, Fox CS. Trends in all-cause and cardiovascular disease mortality among women and men with and without diabetes mellitus in the Framingham Heart Study, 1950 to 2005. Circulation. 2009;119:1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinman JC, Donahue RP, Harris MI, Finucane FF, Madans JH, Brock DB. Mortality among diabetics in a national sample. Am J Epidemiol. 1988;128:389–401 [DOI] [PubMed] [Google Scholar]
- 8.Rao Kondapally Seshasai S, Kaptoge S, Thompson A, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia. 1981;20:87–93 [DOI] [PubMed] [Google Scholar]
- 10.Avery AR, Duncan GE. Heritability of type 2 diabetes in the Washington State Twin Registry. Twin Res Hum Genet. 2019;22:95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willemsen G, Ward KJ, Bell CG, et al. The concordance and heritability of type 2 diabetes in 34,166 twin pairs from international twin registers: the Discordant Twin (DISCOTWIN) Consortium. Twin Res Hum Genet. 2015;18:762–771 [DOI] [PubMed] [Google Scholar]
- 12.Almgren P, Lehtovirta M, Isomaa B, Sarelin L, Taskinen MR, Lyssenko V, Tuomi T, Groop L. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia. 2011;54:2811–2819 [DOI] [PubMed] [Google Scholar]
- 13.Wallace TM, Matthews DR. The assessment of insulin resistance in man. Diabet Med. 2002;19:527–534 [DOI] [PubMed] [Google Scholar]
- 14.Lillioja S, Mott DM, Zawadzki JK, Young AA, Abbott WG, Knowler WC, Bennett PH, Moll P, Bogardus C. In vivo insulin action is familial characteristic in nondiabetic Pima Indians. Diabetes. 1987;36:1329–1335 [DOI] [PubMed] [Google Scholar]
- 15.Goodarzi MO, Taylor KD, Guo X, Quinones MJ, Cui J, Li X, Hang T, Yang H, Holmes E, Hsueh WA, Olefsky J, Rotter JI. Variation in the gene for muscle-specific AMP deaminase is associated with insulin clearance, a highly heritable trait. Diabetes. 2005;54:1222–1227 [DOI] [PubMed] [Google Scholar]
- 16.Lehtovirta M, Kaprio J, Forsblom C, Eriksson J, Tuomilehto J, Groop L. Insulin sensitivity and insulin secretion in monozygotic and dizygotic twins. Diabetologia. 2000;43:285–293 [DOI] [PubMed] [Google Scholar]
- 17.Xiang AH, Azen SP, Raffel LJ, Tan S, Cheng LS, Diaz J, Toscano E, Henderson PC, Hodis HN, Hsueh WA, Rotter JI, Buchanan TA. Evidence for joint genetic control of insulin sensitivity and systolic blood pressure in hispanic families with a hypertensive proband. Circulation. 2001;103:78–83 [DOI] [PubMed] [Google Scholar]
- 18.Poulsen P, Levin K, Petersen I, Christensen K, Beck-Nielsen H, Vaag A. Heritability of insulin secretion, peripheral and hepatic insulin action, and intracellular glucose partitioning in young and old Danish twins. Diabetes. 2005;54:275–283 [DOI] [PubMed] [Google Scholar]
- 19.Beaty TH, Fajans SS. Estimating genetic and non-genetic components of variance for fasting glucose levels in pedigrees ascertained through non-insulin dependent diabetes. Ann Hum Genet. 1982;46:355–362 [DOI] [PubMed] [Google Scholar]
- 20.Hong Y, Weisnagel SJ, Rice T, Sun G, Mandel SA, Gu C, Rankinen T, Gagnon J, Leon AS, Skinner JS, Wilmore JH, Bergman RN, Bouchard C, Rao DC. Familial resemblance for glucose and insulin metabolism indices derived from an intravenous glucose tolerance test in Blacks and Whites of the HERITAGE Family Study. Clin Genet. 2001;60:22–30 [DOI] [PubMed] [Google Scholar]
- 21.Watanabe RM, Valle T, Hauser ER, Ghosh S, Eriksson J, Kohtamaki K, Ehnholm C, Tuomilehto J, Collins FS, Bergman RN, Boehnke M. Familiality of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Finland-United States Investigation of NIDDM Genetics (FUSION) Study investigators. Hum Hered. 1999;49:159–168 [DOI] [PubMed] [Google Scholar]
- 22.Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance--a population-based twin study. Diabetologia. 1999;42:139–145 [DOI] [PubMed] [Google Scholar]
- 23.Schousboe K, Visscher PM, Henriksen JE, Hopper JL, Sorensen TI, Kyvik KO. Twin study of genetic and environmental influences on glucose tolerance and indices of insulin sensitivity and secretion. Diabetologia. 2003;46:1276–1283 [DOI] [PubMed] [Google Scholar]
- 24.Hanson RL, Imperatore G, Narayan KM, Roumain J, Fagot-Campagna A, Pettitt DJ, Bennett PH, Knowler WC. Family and genetic studies of indices of insulin sensitivity and insulin secretion in Pima Indians. Diabetes Metab Res Rev. 2001;17:296–303 [DOI] [PubMed] [Google Scholar]
- 25.Lindsten J, Cerasi E, Luft R, Morton N, Ryman N. Significance of genetic factors for the plasma insulin response to glucose in healthy subjects. Clin Genet. 1976;10:125–134 [DOI] [PubMed] [Google Scholar]
- 26.Lehtovirta M, Kaprio J, Groop L, Trombetta M, Bonadonna RC. Heritability of model-derived parameters of beta cell secretion during intravenous and oral glucose tolerance tests: a study of twins. Diabetologia. 2005;48:1604–1613 [DOI] [PubMed] [Google Scholar]
- 27.Elbein SC, Hasstedt SJ, Wegner K, Kahn SE. Heritability of pancreatic beta-cell function among nondiabetic members of Caucasian familial type 2 diabetic kindreds. J Clin Endocrinol Metab. 1999;84:1398–1403 [DOI] [PubMed] [Google Scholar]
- 28.Colilla S, Cox NJ, Ehrmann DA. Heritability of insulin secretion and insulin action in women with polycystic ovary syndrome and their first degree relatives. J Clin Endocrinol Metab. 2001;86:2027–2031 [DOI] [PubMed] [Google Scholar]
- 29.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80 [DOI] [PubMed] [Google Scholar]
- 30.Franks PW, Rolandsson O, Debenham SL, Fawcett KA, Payne F, Dina C, Froguel P, Mohlke KL, Willer C, Olsson T, Wareham NJ, Hallmans G, Barroso I, Sandhu MS. Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia. 2008;51:458–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandhu MS, Weedon MN, Fawcett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parikh H, Groop L. Candidate genes for type 2 diabetes. Rev Endocr Metab Disord. 2004;5:151–176 [DOI] [PubMed] [Google Scholar]
- 33.Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323 [DOI] [PubMed] [Google Scholar]
- 34.Guo X, Rotter JI. Genome-wide association studies. Jama. 2019;322:1705–1706 [DOI] [PubMed] [Google Scholar]
- 35.Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336 [DOI] [PubMed] [Google Scholar]
- 36.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885 [DOI] [PubMed] [Google Scholar]
- 38.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–775 [DOI] [PubMed] [Google Scholar]
- 39.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris AP. Progress in defining the genetic contribution to type 2 diabetes susceptibility. Curr Opin Genet Dev. 2018;50:41–51 [DOI] [PubMed] [Google Scholar]
- 41.Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodarzi MO, Palmer ND, Cui J, Guo X, Chen YI, Taylor KD, Raffel LJ, Wagenknecht LE, Buchanan TA, Hsueh WA, Rotter JI. Classification of type 2 diabetes genetic variants and a novel genetic risk score association with insulin clearance. J Clin Endocrinol Metab. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodarzi MO. Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018;6:223–236 [DOI] [PubMed] [Google Scholar]
- 44.Fraser GE, Strahan TM, Sabate J, Beeson WL, Kissinger D. Effects of traditional coronary risk factors on rates of incident coronary events in a low-risk population. The Adventist Health Study. Circulation. 1992;86:406–413 [DOI] [PubMed] [Google Scholar]
- 45.Folsom AR, Szklo M, Stevens J, Liao F, Smith R, Eckfeldt JH. A prospective study of coronary heart disease in relation to fasting insulin, glucose, and diabetes. The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 1997;20:935–942 [DOI] [PubMed] [Google Scholar]
- 46.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234 [DOI] [PubMed] [Google Scholar]
- 47.Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, Salonen JT. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. Jama. 2002;288:2709–2716 [DOI] [PubMed] [Google Scholar]
- 48.Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care. 2002;25:1177–1184 [DOI] [PubMed] [Google Scholar]
- 49.Pyorala M, Miettinen H, Laakso M, Pyorala K. Hyperinsulinemia predicts coronary heart disease risk in healthy middle-aged men: the 22-year follow-up results of the Helsinki Policemen Study. Circulation. 1998;98:398–404 [DOI] [PubMed] [Google Scholar]
- 50.Despres JP, Lamarche B, Mauriege P, Cantin B, Dagenais GR, Moorjani S, Lupien PJ. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med. 1996;334:952–957 [DOI] [PubMed] [Google Scholar]
- 51.Pyorala M, Miettinen H, Laakso M, Pyorala K. Plasma insulin and all-cause, cardiovascular, and noncardiovascular mortality: the 22-year follow-up results of the Helsinki Policemen Study. Diabetes Care. 2000;23:1097–1102 [DOI] [PubMed] [Google Scholar]
- 52.Rutter MK, Meigs JB, Sullivan LM, D’Agostino RB Sr., Wilson PW. Insulin resistance, the metabolic syndrome, and incident cardiovascular events in the Framingham Offspring Study. Diabetes. 2005;54:3252–3257 [DOI] [PubMed] [Google Scholar]
- 53.Jeppesen J, Hansen TW, Rasmussen S, Ibsen H, Torp-Pedersen C, Madsbad S. Insulin resistance, the metabolic syndrome, and risk of incident cardiovascular disease: a population-based study. J Am Coll Cardiol. 2007;49:2112–2119 [DOI] [PubMed] [Google Scholar]
- 54.Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Meigs JB, Bonadonna RC, Muggeo M. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes Care. 2007;30:318–324 [DOI] [PubMed] [Google Scholar]
- 55.Ruige JB, Assendelft WJ, Dekker JM, Kostense PJ, Heine RJ, Bouter LM. Insulin and risk of cardiovascular disease: a meta-analysis. Circulation. 1998;97:996–1001 [DOI] [PubMed] [Google Scholar]
- 56.Pyorala M, Miettinen H, Laakso M, Pyorala K. Hyperinsulinemia and the risk of stroke in healthy middle-aged men: the 22-year follow-up results of the Helsinki Policemen Study. Stroke. 1998;29:1860–1866 [DOI] [PubMed] [Google Scholar]
- 57.Andreozzi F, Gastaldelli A, Mannino GC, Sciacqua A, Succurro E, Arturi F, Folli F, Perticone F. Increased carotid intima-media thickness in the physiologic range is associated with impaired postprandial glucose metabolism, insulin resistance and beta cell dysfunction. Atherosclerosis. 2013;229:277–281 [DOI] [PubMed] [Google Scholar]
- 58.Zavaroni I, Ardigo D, Zuccarelli A, Pacetti E, Piatti PM, Monti L, Valtuena S, Massironi P, Rossi PC, Reaven GM. Insulin resistance/compensatory hyperinsulinemia predict carotid intimal medial thickness in patients with essential hypertension. Nutr Metab Cardiovasc Dis. 2006;16:22–27 [DOI] [PubMed] [Google Scholar]
- 59.Izzo R, de Simone G, Trimarco V, Gerdts E, Giudice R, Vaccaro O, De Luca N, Trimarco B. Hypertensive target organ damage predicts incident diabetes mellitus. Eur Heart J. 2013;34:3419–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doerr R, Hoffmann U, Otter W, et al. Oral glucose tolerance test and HbA1c for diagnosis of diabetes in patients undergoing coronary angiography: the Silent Diabetes Study. Diabetologia. 2011;54:2923–2930 [DOI] [PubMed] [Google Scholar]
- 61.Stern MP. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes. 1995;44:369–374 [DOI] [PubMed] [Google Scholar]
- 62.Pannacciulli N, De Pergola G, Ciccone M, Rizzon P, Giorgino F, Giorgino R. Effect of family history of type 2 diabetes on the intima-media thickness of the common carotid artery in normal-weight, overweight, and obese glucose-tolerant young adults. Diabetes Care. 2003;26:1230–1234 [DOI] [PubMed] [Google Scholar]
- 63.Kao WH, Hsueh WC, Rainwater DL, O’Leary DH, Imumorin IG, Stern MP, Mitchell BD. Family history of type 2 diabetes is associated with increased carotid artery intimal-medial thickness in Mexican Americans. Diabetes Care. 2005;28:1882–1889 [DOI] [PubMed] [Google Scholar]
- 64.Dash DK, Choudhury AK, Singh M, Mangaraj S, Mohanty BK, Baliarsinha AK. Effect of parental history of diabetes on markers of inflammation, insulin resistance and atherosclerosis in first degree relatives of patients with type 2 diabetes mellitus. Diabetes Metab Syndr. 2018;12:285–289 [DOI] [PubMed] [Google Scholar]
- 65.Makimattila S, Ylitalo K, Schlenzka A, Taskinen MR, Summanen P, Syvanne M, Yki-Jarvinen H. Family histories of Type II diabetes and hypertension predict intima-media thickness in patients with Type I diabetes. Diabetologia. 2002;45:711–718 [DOI] [PubMed] [Google Scholar]
- 66.de Andrade Junior CR, Silva EL, da Matta Mde F, Castier MB, Rosa ML, Gomes MB. Influence of a family history of type 2 diabetes, demographic and clinical data on carotid intima-media thickness in patients with type 1 diabetes: a cross-sectional study. Cardiovasc Diabetol. 2014;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Afarideh M, Noshad S, Ghajar A, Aryan Z, Khajeh E, Hosseini Shirvani S, Bonnet F, Esteghamati A. Family history of diabetes and the risk of coronary heart disease in people with or without type 2 diabetes. Diabetes Metab. 2017;43:180–183 [DOI] [PubMed] [Google Scholar]
- 68.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Duncan L, Perry JR, Patterson N, Robinson EB, Daly MJ, Price AL, Neale BM. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gan W, Bragg F, Walters RG, et al. Genetic predisposition to type 2 diabetes and risk of subclinical atherosclerosis and cardiovascular diseases among 160,000 chinese adults. Diabetes. 2019;68:2155–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493 [DOI] [PubMed] [Google Scholar]
- 74.Broadbent HM, Peden JF, Lorkowski S, et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17:806–814 [DOI] [PubMed] [Google Scholar]
- 75.Gori F, Specchia C, Pietri S, Crociati L, Barlera S, Franciosi M, Nicolucci A, Signorini S, Brambilla P, Franzosi MG. Common genetic variants on chromosome 9p21 are associated with myocardial infarction and type 2 diabetes in an Italian population. BMC Med Genet. 2010;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dauriz M, Meigs JB. Current insights into the joint genetic basis of type 2 diabetes and coronary heart disease. Curr Cardiovasc Risk Rep. 2014;8:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122:433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan M, Dandona S, McPherson R, Allayee H, Hazen SL, Wells GA, Roberts R, Stewart AF. Two chromosome 9p21 haplotype blocks distinguish between coronary artery disease and myocardial infarction risk. Circ Cardiovasc Genet. 2013;6:372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holdt LM, Teupser D. Long noncoding RNA ANRIL: lnc-ing genetic variation at the chromosome 9p21 locus to molecular mechanisms of atherosclerosis. Front Cardiovasc Med. 2018;5:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holdt LM, Stahringer A, Sass K, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, Buerki C, McLean BW, Cook RC, Parker JS, McPherson R. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–1677 [DOI] [PubMed] [Google Scholar]
- 82.Nikulina S, Artyukhov I, Shesternya P, Gavrilyuk O, Maksimov V, Voyevoda M, Brusentsov D. Clinical application of chromosome 9p21.3 genotyping in patients with coronary artery disease. Exp Ther Med. 2019;18:3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scott RA, Lagou V, Welch RP, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manning AK, Hivert MF, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kilpelainen TO, Zillikens MC, Stancakova A, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klarin D, Zhu QM, Emdin CA, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rung J, Cauchi S, Albrechtsen A, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41:1110–1115 [DOI] [PubMed] [Google Scholar]
- 92.Lim S, Hong J, Liu CT, Hivert MF, White CC, Murabito JM, O’Donnell CJ, Dupuis J, Florez JC, Meigs JB. Common variants in and near IRS1 and subclinical cardiovascular disease in the Framingham Heart Study. Atherosclerosis. 2013;229:149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jansen H, Loley C, Lieb W, et al. Genetic variants primarily associated with type 2 diabetes are related to coronary artery disease risk. Atherosclerosis. 2015;241:419–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Preuss M, Konig IR, Thompson JR, et al. Design of the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet. 2010;3:475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Webb TR, Erdmann J, Stirrups KE, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao W, Rasheed A, Tikkanen E, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Klarin D, Damrauer SM, Cho K, et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet. 2018;50:1514–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kraja AT, Vaidya D, Pankow JS, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011;60:1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet. 2018;50:559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Humphries SE, Nicaud V, Margalef J, Tiret L, Talmud PJ. Lipoprotein lipase gene variation is associated with a paternal history of premature coronary artery disease and fasting and postprandial plasma triglycerides: the European Atherosclerosis Research Study (EARS). Arterioscler Thromb Vasc Biol. 1998;18:526–534 [DOI] [PubMed] [Google Scholar]
- 102.Ranganathan G, Unal R, Pokrovskaya ID, Tripathi P, Rotter JI, Goodarzi MO, Kern PA. The lipoprotein lipase (LPL) S447X gain of function variant involves increased mRNA translation. Atherosclerosis. 2012;221:143–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hayne CK, Lafferty MJ, Eglinger BJ, Kane JP, Neher SB. Biochemical analysis of the lipoprotein lipase truncation variant, LPL(S447X), reveals increased lipoprotein uptake. Biochemistry. 2017;56:525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kathiresan S, Melander O, Guiducci C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tan A, Sun J, Xia N, et al. A genome-wide association and gene-environment interaction study for serum triglycerides levels in a healthy Chinese male population. Hum Mol Genet. 2012;21:1658–1664 [DOI] [PubMed] [Google Scholar]
- 106.Wojcik GL, Graff M, Nishimura KK, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu DJ, Peloso GM, Yu H, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49:1758–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gusarova V, O’Dushlaine C, Teslovich TM, et al. Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun. 2018;9:2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Udler MS. Type 2 diabetes: multiple genes, multiple diseases. Curr Diab Rep. 2019;19:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Udler MS, Kim J, von Grotthuss M, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 2018;15:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yaghootkar H, Scott RA, White CC, et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes. 2014;63:4369–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ahlqvist E, Storm P, Karajamaki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6:361–369 [DOI] [PubMed] [Google Scholar]
- 116.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Swerdlow DI. Mendelian randomization and type 2 diabetes. Cardiovasc Drugs Ther. 2016;30:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Frayling TM, Stoneman CE. Mendelian randomisation in type 2 diabetes and coronary artery disease. Curr Opin Genet Dev. 2018;50:111–120 [DOI] [PubMed] [Google Scholar]
- 120.Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J. 2014;35:1917–1924 [DOI] [PubMed] [Google Scholar]
- 121.Varbo A, Benn M, Smith GD, Timpson NJ, Tybjaerg-Hansen A, Nordestgaard BG. Remnant cholesterol, low-density lipoprotein cholesterol, and blood pressure as mediators from obesity to ischemic heart disease. Circ Res. 2015;116:665–673 [DOI] [PubMed] [Google Scholar]
- 122.Xu L, Borges MC, Hemani G, Lawlor DA. The role of glycaemic and lipid risk factors in mediating the effect of BMI on coronary heart disease: a two-step, two-sample Mendelian randomisation study. Diabetologia. 2017;60:2210–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lv WQ, Zhang X, Fan K, Xia X, Zhang Q, Liu HM, Jiang BY, Zhang WD, Deng HW. Genetically driven adiposity traits increase the risk of coronary artery disease independent of blood pressure, dyslipidaemia, glycaemic traits. Eur J Hum Genet. 2018;26:1547–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dale CE, Fatemifar G, Palmer TM, et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a Mendelian randomization analysis. Circulation. 2017;135:2373–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, Kathiresan S. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. Jama. 2017;317:626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.White J, Swerdlow DI, Preiss D, Fairhurst-Hunter Z, Keating BJ, Asselbergs FW, Sattar N, Humphries SE, Hingorani AD, Holmes MV. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1:692–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ahmad OS, Morris JA, Mujammami M, Forgetta V, Leong A, Li R, Turgeon M, Greenwood CM, Thanassoulis G, Meigs JB, Sladek R, Richards JB. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6:7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ross S, Gerstein HC, Eikelboom J, Anand SS, Yusuf S, Pare G. Mendelian randomization analysis supports the causal role of dysglycaemia and diabetes in the risk of coronary artery disease. Eur Heart J. 2015;36:1454–1462 [DOI] [PubMed] [Google Scholar]
- 129.Deloukas P, Kanoni S, Willenborg C, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu J, Rutten-Jacobs L, Liu M, Markus HS, Traylor M. Causal impact of type 2 diabetes mellitus on cerebral small vessel disease: a Mendelian randomization analysis. Stroke. 2018;49:1325–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Larsson SC, Scott RA, Traylor M, Langenberg CC, Hindy G, Melander O, Orho-Melander M, Seshadri S, Wareham NJ, Markus HS. Type 2 diabetes, glucose, insulin, BMI, and ischemic stroke subtypes: Mendelian randomization study. Neurology. 2017;89:454–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Benn M, Tybjaerg-Hansen A, McCarthy MI, Jensen GB, Grande P, Nordestgaard BG. Nonfasting glucose, ischemic heart disease, and myocardial infarction: a Mendelian randomization study. J Am Coll Cardiol. 2012;59:2356–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Warrington NM, Beaumont RN, Horikoshi M, et al. Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet. 2019;51:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Merino J, Leong A, Posner DC, Porneala B, Masana L, Dupuis J, Florez JC. Genetically driven hyperglycemia increases risk of coronary artery disease separately from type 2 diabetes. Diabetes Care. 2017;40:687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zeng P, Zhou X. Causal association between birth weight and adult diseases: evidence from a Mendelian randomization analysis. Front Genet. 2019;10:618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Horikoshi M, Beaumont RN, Day FR, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538:248–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Freathy RM, Mook-Kanamori DO, Sovio U, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat Genet. 2010;42:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Leong A, Chen J, Wheeler E, Hivert MF, Liu CT, Merino J, Dupuis J, Tai ES, Rotter JI, Florez JC, Barroso I, Meigs JB. Mendelian randomization analysis of hemoglobin A1c as a risk Factor for coronary artery disease. Diabetes Care. 2019;42:1202–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Au Yeung SL, Luo S, Schooling CM. The Impact of glycated hemoglobin (HbA1c) on cardiovascular disease risk: a Mendelian randomization study using UK Biobank. Diabetes Care. 2018;41:1991–1997 [DOI] [PubMed] [Google Scholar]
- 142.Wheeler E, Leong A, Liu CT, et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLoS Med. 2017;14:e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tikkanen E, Pirinen M, Sarin AP, Havulinna AS, Mannisto S, Saltevo J, Lokki ML, Sinisalo J, Lundqvist A, Jula A, Salomaa V, Ripatti S. Genetic support for the causal role of insulin in coronary heart disease. Diabetologia. 2016;59:2369–2377 [DOI] [PubMed] [Google Scholar]
- 145.Zhan Y, Karlsson IK, Karlsson R, Tillander A, Reynolds CA, Pedersen NL, Hagg S. Exploring the causal pathway from telomere length to coronary heart disease: a network Mendelian randomization study. Circ Res. 2017;121:214–219 [DOI] [PubMed] [Google Scholar]
- 146.Zhao JV, Luo S, Schooling CM. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun Biol. 2019;2:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scott RA, Fall T, Pasko D, et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes. 2014;63:4378–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goodarzi MO, Cui J, Chen YD, Hsueh WA, Guo X, Rotter JI. Fasting insulin reflects heterogeneous physiological processes: role of insulin clearance. Am J Physiol Endocrinol Metab. 2011;301:E402–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shahim B, De Bacquer D, De Backer G, Gyberg V, Kotseva K, Mellbin L, Schnell O, Tuomilehto J, Wood D, Ryden L. The prognostic value of fasting plasma glucose, two-hour postload glucose, and HbA1c in patients with coronary artery disease: a report from EUROASPIRE IV: a survey from the European Society of Cardiology. Diabetes Care. 2017;40:1233–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cavalot F, Petrelli A, Traversa M, Bonomo K, Fiora E, Conti M, Anfossi G, Costa G, Trovati M. Postprandial blood glucose is a stronger predictor of cardiovascular events than fasting blood glucose in type 2 diabetes mellitus, particularly in women: lessons from the San Luigi Gonzaga Diabetes Study. J Clin Endocrinol Metab. 2006;91:813–819 [DOI] [PubMed] [Google Scholar]
- 151.Kawano H, Motoyama T, Hirashima O, Hirai N, Miyao Y, Sakamoto T, Kugiyama K, Ogawa H, Yasue H. Hyperglycemia rapidly suppresses flow-mediated endothelium-dependent vasodilation of brachial artery. J Am Coll Cardiol. 1999;34:146–154 [DOI] [PubMed] [Google Scholar]
- 152.Gerstein HC, Miller ME, Byington RP, Goff DC, Jr., Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr., Probstfield JL, Simons-Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572 [DOI] [PubMed] [Google Scholar]
- 154.Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139 [DOI] [PubMed] [Google Scholar]
- 155.Kelly TN, Bazzano LA, Fonseca VA, Thethi TK, Reynolds K, He J. Systematic review: glucose control and cardiovascular disease in type 2 diabetes. Ann Intern Med. 2009;151:394–403 [DOI] [PubMed] [Google Scholar]
- 156.Ray KK, Seshasai SR, Wijesuriya S, Sivakumaran R, Nethercott S, Preiss D, Erqou S, Sattar N. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet. 2009;373:1765–1772 [DOI] [PubMed] [Google Scholar]
- 157.Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, Lafont S, Bergeonneau C, Kassai B, Erpeldinger S, Wright JM, Gueyffier F, Cornu C. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. Bmj. 2011;343:d4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seidu S, Achana FA, Gray LJ, Davies MJ, Khunti K. Effects of glucose-lowering and multifactorial interventions on cardiovascular and mortality outcomes: a meta-analysis of randomized control trials. Diabet Med. 2016;33:280–289 [DOI] [PubMed] [Google Scholar]
- 159.Hemmingsen B, Lund SS, Gluud C, Vaag A, Almdal T, Hemmingsen C, Wetterslev J. Intensive glycaemic control for patients with type 2 diabetes: systematic review with meta-analysis and trial sequential analysis of randomised clinical trials. Bmj. 2011;343:d6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hayward RA, Reaven PD, Wiitala WL, Bahn GD, Reda DJ, Ge L, McCarren M, Duckworth WC, Emanuele NV. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;372:2197–2206 [DOI] [PubMed] [Google Scholar]
- 161.Reaven PD, Emanuele NV, Wiitala WL, Bahn GD, Reda DJ, McCarren M, Duckworth WC, Hayward RA. Intensive glucose control in patients with type 2 diabetes - 15-year follow-up. N Engl J Med. 2019;380:2215–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589 [DOI] [PubMed] [Google Scholar]
- 164.Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742 [DOI] [PubMed] [Google Scholar]
- 165.Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Swerdlow DI, Preiss D, Kuchenbaecker KB, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385:351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lotta LA, Sharp SJ, Burgess S, et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: a meta-analysis. Jama. 2016;316:1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fall T, Xie W, Poon W, Yaghootkar H, Magi R, Knowles JW, Lyssenko V, Weedon M, Frayling TM, Ingelsson E. Using genetic variants to assess the relationship between circulating lipids and type 2 diabetes. Diabetes. 2015;64:2676–2684 [DOI] [PubMed] [Google Scholar]
- 169.Tragante V, Asselbergs FW, Swerdlow DI, Palmer TM, Moore JH, de Bakker PIW, Keating BJ, Holmes MV. Harnessing publicly available genetic data to prioritize lipid modifying therapeutic targets for prevention of coronary heart disease based on dysglycemic risk. Hum Genet. 2016;135:453–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.He L, Culminskaya I, Loika Y, Arbeev KG, Bagley O, Duan M, Yashin AI, Kulminski AM. Causal effects of cardiovascular risk factors on onset of major age-related diseases: A time-to-event Mendelian randomization study. Exp Gerontol. 2018;107:74–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Feng Q, Wei WQ, Chung CP, Levinson RT, Sundermann AC, Mosley JD, Bastarache L, Ferguson JF, Cox NJ, Roden DM, Denny JC, Linton MF, Edwards DRV, Stein CM. Relationship between very low low-density lipoprotein cholesterol concentrations not due to statin therapy and risk of type 2 diabetes: A US-based cross-sectional observational study using electronic health records. PLoS Med. 2018;15:e1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. Jama. 2015;313:1029–1036 [DOI] [PubMed] [Google Scholar]
- 173.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722 [DOI] [PubMed] [Google Scholar]
- 174.Cao YX, Liu HH, Dong QT, Li S, Li JJ. Effect of proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibodies on new-onset diabetes mellitus and glucose metabolism: A systematic review and meta-analysis. Diabetes Obes Metab. 2018;20:1391–1398 [DOI] [PubMed] [Google Scholar]
- 175.Vohnout B, Lisicanova J, Havranova A. PCSK9 inhibitors and diabetes mellitus. Vnitr Lek. 2019;64:1186–1189 [PubMed] [Google Scholar]
- 176.Lee RG, Crosby J, Baker BF, Graham MJ, Crooke RM. Antisense technology: an emerging platform for cardiovascular disease therapeutics. J Cardiovasc Transl Res. 2013;6:969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57:340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen EC, Liang X, Yee SW, Geier EG, Stocker SL, Chen L, Giacomini KM. Targeted disruption of organic cation transporter 3 attenuates the pharmacologic response to metformin. Mol Pharmacol. 2015;88:75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen L, Pawlikowski B, Schlessinger A, More SS, Stryke D, Johns SJ, Portman MA, Chen E, Ferrin TE, Sali A, Giacomini KM. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet Genomics. 2010;20:687–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865 [PubMed] [Google Scholar]
- 181.Goldner MG, Knatterud GL, Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. 3. Clinical implications of UGDP results. Jama. 1971;218:1400–1410 [PubMed] [Google Scholar]
- 182.Li Y, Hu Y, Ley SH, Rajpathak S, Hu FB. Sulfonylurea use and incident cardiovascular disease among patients with type 2 diabetes: prospective cohort study among women. Diabetes Care. 2014;37:3106–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O’Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443 [DOI] [PubMed] [Google Scholar]
- 184.Phung OJ, Schwartzman E, Allen RW, Engel SS, Rajpathak SN. Sulphonylureas and risk of cardiovascular disease: systematic review and meta-analysis. Diabet Med. 2013;30:1160–1171 [DOI] [PubMed] [Google Scholar]
- 185.Hemmingsen B, Schroll JB, Wetterslev J, Gluud C, Vaag A, Sonne DP, Lundstrom LH, Almdal T. Sulfonylurea versus metformin monotherapy in patients with type 2 diabetes: a Cochrane systematic review and meta-analysis of randomized clinical trials and trial sequential analysis. CMAJ Open. 2014;2:E162–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Varvaki Rados D, Catani Pinto L, Reck Remonti L, Bauermann Leitao C, Gross JL. The association between sulfonylurea use and all-cause and cardiovascular mortality: a meta-analysis with trial sequential analysis of randomized clinical trials. PLoS Med. 2016;13:e1001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Emdin CA, Klarin D, Natarajan P, Florez JC, Kathiresan S, Khera AV. Genetic variation at the sulfonylurea receptor, type 2 diabetes, and coronary heart disease. Diabetes. 2017;66:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fatehi M, Raja M, Carter C, Soliman D, Holt A, Light PE. The ATP-sensitive K(+) channel ABCC8 S1369A type 2 diabetes risk variant increases MgATPase activity. Diabetes. 2012;61:241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Azoulay L, Suissa S. Sulfonylureas and the risks of cardiovascular events and death: a methodological meta-regression analysis of the observational studies. Diabetes Care. 2017;40:706–714 [DOI] [PubMed] [Google Scholar]
- 190.Rosenstock J, Kahn SE, Johansen OE, et al. Effect of linagliptin vs glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes: the CAROLINA randomized clinical trial. Jama. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rosenstock J, Perkovic V, Johansen OE, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. Jama. 2019;321:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hernandez AF, Green JB, Janmohamed S, D’Agostino RB Sr., Granger CB, Jones NP, Leiter LA, Rosenberg AE, Sigmon KN, Somerville MC, Thorpe KM, McMurray JJV, Del Prato S. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519–1529 [DOI] [PubMed] [Google Scholar]
- 193.Holman RR, Bethel MA, Mentz RJ, et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2017;377:1228–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247–2257 [DOI] [PubMed] [Google Scholar]
- 195.Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121–130 [DOI] [PubMed] [Google Scholar]
- 196.Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–1844 [DOI] [PubMed] [Google Scholar]
- 197.Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Taylor SI. GLP-1 receptor agonists: differentiation within the class. Lancet Diabetes Endocrinol. 2018;6:83–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bethel MA, Patel RA, Merrill P, et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol. 2018;6:105–113 [DOI] [PubMed] [Google Scholar]
- 200.Wessel J, Chu AY, Willems SM, et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun. 2015;6:5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Scott RA, Freitag DF, Li L, et al. A genomic approach to therapeutic target validation identifies a glucose-lowering GLP1R variant protective for coronary heart disease. Sci Transl Med. 2016;8:341ra376. [DOI] [PMC free article] [PubMed] [Google Scholar]