

Abstract
The hydrogen isotopes deuterium (D) and tritium (T) have become essential tools of chemistry, biology, and medicine.1 Beyond their widespread use in spectroscopy, mass spectrometry, and mechanistic and pharmacokinetic studies, there has been considerable interest in incorporating deuterium into drug molecules.1 The deuterium kinetic isotope effect (DKIE), which compares the rate of a chemical reaction for a compound to its deuterated counterpart, can be dramatic.1–3 The strategic replacement of hydrogen with deuterium can affect both the rate of metabolism and distribution of metabolites for a compound,4 improving the efficacy and safety of the drug. Deutetrabenazine, a promising treatment for Huntington’s disease,5 recently became the first deuterated drug to win FDA-approval.The pharmacokinetics of a deuterated compound depend on the location(s) of D. While methods currently exist for deuterium incorporation at both early and late stages of a drug’s synthesis,6–7 these processes are often unselective and the stereoisotopic purity can be difficult to measure.7–8 Here, we describe the preparation of stereoselectively deuterated building blocks for pharmaceutical research. As a proof of concept, we demonstrate a four-step conversion of benzene to cyclohexene with varying degrees of D incorporation, as bound to a tungsten complex. Using different combinations of deuterated and proteated acid and hydride reagents, the deuterated positions can be precisely controlled on the cyclohexene ring. In total, 52 unique stereoisotopomers of cyclohexene are available, in the form of ten different isotopologues. This concept can be extended to prepare discrete stereoisotopomers of functionalized cyclohexenes. Such systematic methods for the preparation of pharmacologically active compounds as discrete stereoisotopomers could improve pharmacological and toxicological properties of drugs and provide new mechanistic information related to their distribution and metabolism in the body.
Typically, hydrogenation of benzene using D2 gas leads to isotopologue mixtures of cyclohexane.10–12 However, Taube et al. demonstrated that the complex [Os(NH3)5(η2-benzene)]2+ could be deuterated to form a single stereoisotopomer of [Os(NH3)5(η2-cyclohexene-d4)]2+ using D2 and a Pd/C catalyst.13 We posited that benzene bound in this manner could also be converted to cyclohexene using four well-defined additions of two protons and two hydrides, passing through an η2-1,3-cyclohexadiene intermediate (Fig. 1). If these reactions could be performed regio- and stereoselectively, one could access a diverse set of isotopologues and even stereoisotopomers of cyclohexene using various combinations of proteated and deuterated reagents.
Fig. 1. Methods for the deuteration of benzene:

a), Prior methods for the selective deuteration of benzene can lead to over-reduction and a mixture of isotopologues. b) The current approach provides access to cyclohexene isotopologues and stereoisotopomers, c) the dearomatized benzene complex WTp(NO)(PMe3)(η2-benzene).
The dearomatization agent {WTp(NO)(PMe3)} is considerably more activating than its osmium predecessor.9 Strong π-backbonding renders arene and diene complexes of this system highly nucleophilic, and resistant to substitution.9 Furthermore, this system displays significant electronic asymmetry, and the benzene complex WTp(NO)(PMe3)(η2-benzene) (1) can be prepared on a multi-gram scale,14 and in enantioenriched form.15 Treatment of an acetone-d6 solution of 1 with diphenylammonium triflate (DPhAT, pKa ~ 0) at −30 °C affords its clean conversion to the η2-benzenium complex [WTp(PMe3)(NO)(η2-C6H7)](OTf) (2; Fig. 2). Using chilled diethyl ether as a precipitating solvent, 2 can be isolated from dichloromethane in 86% yield (1.9 g). As an acetonitrile solution, the η2-benzenium complex 2 is moderately stable at room temperature but soon decomposes (t1/2 ~ 6 min). At 0°C, however, 2 exists in equilibrium with its diastereomer 3 in a 10:1 ratio (Fig. 2) and persists for three hours without significant decomposition. The major isomer (2) is formed with the metal binding two internal carbons of the five-carbon π-system, and with the newly formed sp3 carbon distal to the PMe3 ligand. The minor isomer (3) is bound at a terminus of the π-system with the sp3 carbon proximal to the phosphine. Proton NMR data and DFT calculations (Supplementary materials; Fig S1–S3) of these η2-benzenium complexes (2, 3) suggest that they are similar in structure to complexes of the form [WTp(NO)(PMe3)(η2-allyl)]+,16 where the allyl ligand is tightly bound to the metal through only two carbons. A third carbon, weakly associated to the metal, resembles a carbocation, and is indicated as such in figures herein (Fig.2). Combining cold solutions of 2 and tetrabutylammonium borohydride generates WTp(PMe3)(NO)(η2-1,3-cyclohexadiene) exclusively (4). Despite the coexistence of the allyl conformer 3 in solution, the WTp(PMe3)(NO)(η2-1,4-cyclohexadiene) complex (8) is undetected (Fig. 2) in the reaction mixture.16 The η2-diene complex 4 was then treated with either DPhAT or HOTf/MeOH acids to generate the η2-allyl complex (6).16 When 6 was subjected to base, it deprotonated to form 5, a stereoisomer of 4,16 in which the uncoordinated double bond is now distal to the PMe3.16 Combining the allyl complex 6 with a hydride source produced the desired η2-cyclohexene complex 7 (67%). Crystals suitable for X-ray structure determinations were grown for complexes of cyclohexadiene 4, allyl complex 6, and cyclohexene 7, and a rendering of these structures, along with key NOE interactions are provided in supplementary material (Fig. S4). Overlapping signals in the 1H NMR spectrum of cyclohexene complex 7 precluded unambiguous stereochemical assignments of some of the ring proton signals.
Fig. 2. Formation of tungsten-bound cyclohexene from benzene:

a)The sequential reduction of benzene to cyclohexene bound to tungsten (by addition of 1H or 2H). 35 confirmed by 13C-NMR and rotational spectroscopy, 9 confirmed by quantitative NOE, b) Crystal structure and relevant NOE interactions (red arrows) for methylated cyclohexene complex 9 (Ph2NH2+ as OTf salt).
By methylating the nitrosyl ligand of 7 (CH3OTf) to generate [WTp(NOMe)(PMe3)(η2-C6H10)]OTf, (9),17 the chemical shifts of the cyclohexene ring separated to the point that each proton could be assigned with high confidence (SI sections G and H). An X-ray structure determination of 9 provided conclusive evidence for methylation of the nitrosyl oxygen (Fig. 2), analogous to earlier literature reports.18 Strong NOE interactions between the ring endo protons and the methylated nitrosyl ligand further facilitated these assignments and quantitative NOE experiments were carried out that support the stereochemical assignments of all diastereotopic protons on the cyclohexene ring (SI, section H).
Deuterium studies
With all hydrogen resonances for the methylated η2-cyclohexene complex 9 fully assigned, we investigated the regio- and stereochemical fidelity of the reaction sequence (Fig. 3). When the η2-benzenium complex 11 is prepared from 1 using [MeOD2+]OTf, a loss of signal intensity is observed, corresponding to the methylene endo proton. This indicates that protonation of the η2-benzene occurs syn to the metal (red, Fig. 3). A complementary experiment was next performed starting with the fully deuterated benzene complex, 17, in which MeOH+ was used as the acid source. In this case, protonation led to a single broad proton resonance for the deuterated η2-benzenium complex 18. This proton signal is ~ 0.03 ppm upfield from its proteo counterpart, consistent with a primary H/D isotopic shift.19 The endo-selective protonation of the benzene ligand in 1 is in stark contrast to the addition of carbon and heteroatom electrophiles, which have been observed to add anti to η2-arene and η2-diene ligands of tungsten complexes.9 When η2-benzenium complexes 11 and 18 were treated with NaBD4 or NaBH4, respectively, the complementary cyclohexadiene complexes 12 and 19 were formed (Fig. 3). A comparison of NOESY data for all three isotopologues of the cyclohexadiene complex (4, 12, 19) confirms that the proton delivered from the borohydride reagent is anti to the metal (Fig. 3). The cyclohexadiene complexes 12 and 19 were then taken forward to their π-allyl analogs 13, 15, and 20 (Fig. 3). In contrast to protonation of the η2-benzene ligand of 1, the acidic hydrogen was delivered predominantly anti to the metal (Fig.3).
Fig. 3. Synthesis of isotopologues and stereoisotopomers of the cyclohexene complex 7.
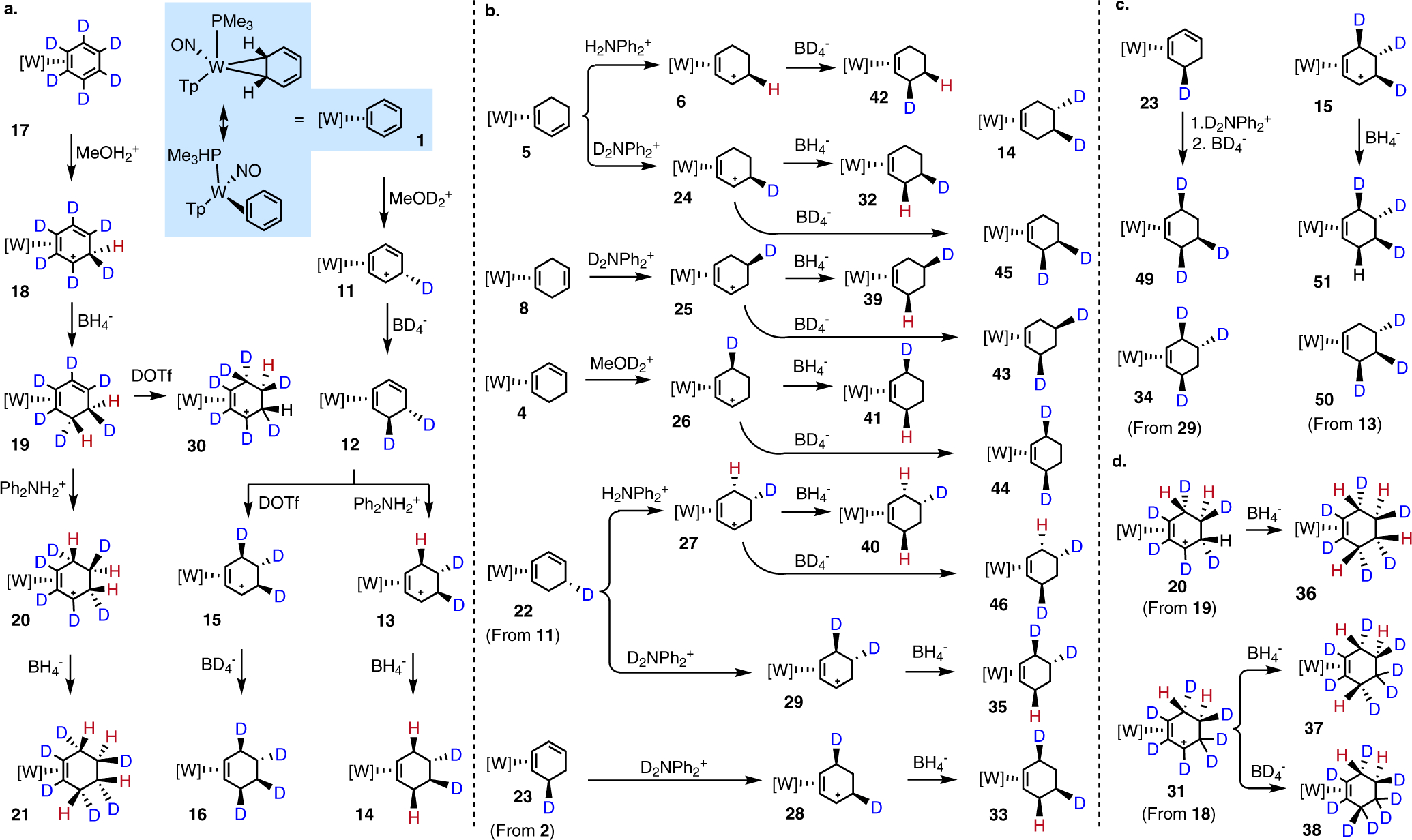
a) detailed syntheses of d2, d4, and d6 isotopologues b) synthesis of d1 and d2 isotopologues c) synthesis of d3 isotopologues d) synthesis of d6, d7, and d8 isotopologues.
The resulting η2-allyl complexes (13, 15, 20) underwent a conformational change (“allyl shift”) such that the second proton added becomes H6exo (conversion of 4 to 6, Fig. 2), while the first proton added is now H5endo. For allyl complexes 13 and 20, full stereoselective protonation was achieved. However, with the preparation of 15 or 26 we experienced difficulties in achieving full deuterium incorporation, owing to an unusually large DKIE (kH/kD ~37 at −30 °C for the deuteration of 12 or 4). This DKIE was determined for 4 as the average value from three separate experiments in which 26 was generated from acidic solutions with differing H/D ratios (SI, section K). This DKIE could be decreased by raising the temperature to 22 °C, however such action compromised the stereofidelity of the resulting deuterated product (15), with endo deuteration of the η2-diene 12 now competing with exo deuteration. Consequently, stereoselective deuterium incorporation at the H6exo position of cyclohexene (i.e., 16, 33-35, 41, 44, 49, 51; Fig. 3) could not be achieved above ~75–80%. A similar outcome was observed when we tried to convert the d6-isotopologue, diene 19 to allyl 30. Finally, as before, treatment of 13, 15, or 20 with a hydride or deuteride source confirmed that the corresponding η2-cyclohexene products (14, 16, 21) are formed by nucleophilic addition anti to the metal (Fig. 3). Similar to the 1,3-diene complex 4, its isomer 5 undergoes exo protonation to form the allyl complex 24. Remarkably, treatment of the 1,4-cyclohexadiene complex (8) with D+ (D2NPh2+ in MeOD) also undergoes direct exo protonation (Fig. 3), this time providing allyl 25. The direct exogenous protonation of the unconjugated C=C bond in 8 appears to result in a carbocation that DFT calculations reveal can be stabilized by the participation of the nitrosyl ligand. A subsequent [1,2]-hydride shift results in the formation of the allyl complex 25 (Fig. S5). Unambiguous assignment of the deuterated hydrogen atom in 25 comes from its conversion to 9-d1 (via 39; Fig. 3). In order to demonstrate regio- and stereocontrol of deuterium incorporation, additional deuterated isotopomers of the allyl complex were prepared from the monodeuterated dienes 22 and 23, and from the benzene-d6-derived allyls 30 and 31(Fig. 3). The allyl complexes 24-31 were then combined with deuteride or hydride to form 18 additional cyclohexene complexes 32-46, 49-51. In principle, one can selectively make 10 different isotopologues of the cyclohexene complex using the procedures outlined above (d0-d4; d6-d10), eight of which (7, 16, 32-38) are reported herein.
Levels of isotopic purity for the cyclohexene ligand isotopologues were determined by recording HRMS data for the corresponding complexes as their methylated adducts (Fig. 2. 9-dn), in order to create a suitable cation for ESI mass analysis. Using the isotope envelope of 9-d0 as a reference (Fig. S6), the isotopic purity of 7, 16, and 32-38 (as converted to 9-dn) was estimated to be >90%, with the exception of 16 (79%), for which the high DKIE of the second protonation prevented complete deuteration at the H6exo position (vide supra). Finally, as a demonstration of how the {WTp(NO)(PMe3)} system precisely governs both the stereochemistry and regiochemistry of protonation and hydride addition, a series of five monodeuterated (32, 39-42), seven dideuterated (14, 33, 35, 43-46), and four trideuterated (34, 49-51) isotopomers of the cyclohexene complex were prepared from these methods (Fig. 3).
Oxidation of the tungsten complex 7 with DDQ releases the free cyclohexene (Fig. 2; 10). Such action on 32, 42, 45, and 46 confirmed the expected regiochemistry of these d1 and d2 isotopomers of cyclohexene via 13C NMR. Introduction of a single deuterium in 3-deuterocyclohexene or 4-deuterocyclohexene allows one to distinguish all six of the carbons in the 13C NMR spectrum, owing to isotopic shifting of the now asymmetric cyclohexene carbons (Fig. S7). Alternatively, solvent-free heating of various isotopologues of the methylated complex 9 effected the release of the cyclohexene ligand for analysis by MRR spectroscopy (SI, section L).20 These experiments determined that 1) over-deuteration is exceedingly low (< 2%). 2) the stereoselectivity is excellent when assessed by observation of undesired cis/trans isomers, which in the worst case is 22:1 and in other cases it is 40:1 or higher. 3) The dominant stereoisotopomers in all cases are those predicted by 1H NMR data. As a final check of stereochemical assignments, the locations of the deuterium atoms were confirmed for complex 45 by neutron diffraction (SI section I; TOPAZ at ONRL).
Mechanistic considerations
The reaction of 1 with D+ to form 11 results in deuterium incorporation exclusively endo to the metal, but this does not definitively show which carbon is initially protonated (Fig. S8). Given that the endo proton of the benzene ligand in 1 completely preempts protonation from an exogenous acid (exo), we propose that the protonation must be concerted - that C-H bond formation is intramolecular and simultaneous with electronic changes at the metal - which could lower the activation barrier for this process relative to protonation by an external acid. Such a mechanism could occur via a hydride intermediate, but this seems sterically untenable. Rather, we propose a mechanism (SI section M) in which the nitrosyl ligand first is protonated to form a hydroxylimido ligand analogous to that reported by Legzdins et al.21 This action is followed by a concerted proton transfer in which a gamma carbon of the benzene is protonated simultaneously with release of electron density back into the tungsten through the NO group. The role of nitrosyl ligands in intramolecular proton transfer has been previously documented.22 In contrast, the stereochemistry and kinetics of η2-diene protonation (e.g., 4, Fig. 2) indicates the hydrogen is delivered exogenously, anti to the tungsten (Fig 1). We speculate that while endo protonation may still be accessible for these 1,3-cyclohexadiene complexes, the less-delocalized diene ligand is most likely more basic than its η2-benzene predecessor, and its direct exo protonation apparently preempts the purported endo mechanism at −30 °C.
Transition metal promoted protonation of benzene was observed in the η4-benzene complexes Cr(CO)3(η4-benzene)− and Mn(CO)3(η4-benzene)− by Cooper et al,23–24 and was proposed to occur via hydride intermediates.23–24 More recently, Chirik et al have explored the molybdenum-catalyzed reduction of benzene and cyclohexadiene, with D2 (g), which resulted in mixtures of isotopologues of cyclohexane.12 However, reduction of cyclohexene with D2 produced a single cis isotopomer of 1,2-dideuterocyclohexane using the molybdenum catalyst.
The high stereoselectivity enabled by the tungsten system provides unprecedented control over the preparation of specific isotopologues and isotopomers of cyclohexene, starting from either benzene complex 1 or its deuterated analog 17, and utilizing either proteated or deuterated sources of acids and hydrides (Table S1). As illustration, consider the d2 isotopologue of the cyclohexene complex, 7-d2. Given that the {WTp(NO)(PMe3)} system is available in enantioenriched form,15 one has access to 14 different isotopomers (individual enantiomers of 14, 33, 35, 43-46; Table S2). The cyclohexene-d2 ligand of these complexes, once removed from the metal by oxidative decomplexation, would be available as 11 individual isotopomers: both enantiomers of cis-3,4-, trans-3,4-, cis-3,5-, trans-3,5-, trans-4,5-, and the meso compound cis-3,6-dideuterocycloohexene. Similarly, 11 distinct isotopomers of cyclohexene-d8 should be available from this methodology starting from benzene-d6. Regarding cyclohexene complex 7-d3 and 7-d7, 8 isotopomers of each would be available, and all 16 of these complexes would yield a unique, chiral cyclohexene (8 cyclohexene-d3, and 8 cyclohexene-d7). All totaled, (Table S2) the methodology outlined herein could provide access to 52 unique isotopomers of cyclohexene, as derived from benzene and benzene-d6. For reference, the total number of isotopomers for cyclohexene is 528.
The ability of {WTp(NO)(PMe3)}to be optically resolved on a practical scale and to retain its stereochemical configuration, even when undergoing ligand displacement,15 also makes it a valuable tool for determining the isotopic pattern of cyclohexene H/D isotopomers produced by other methods.8 Consider for example a scenario in which an unknown isotopomer of cyclohexene-d1 is combined with the resolved form of benzene complex (R)-1 in solution and allowed to undergo ligand exchange. Even though the two faces of the cyclohexene ring will bind to tungsten with equal probability, the 1H NMR spectrum will be unique for each of the five possible isotopomers (SI section C; Fig. S11). A similar approach could be taken for any cyclic alkene (e.g., dehydropiperidines, pyrrolines, cyclopentenes) for which an 1H NMR spectrum of a fully proteated species can be fully assigned (vide supra).
Deuterated building blocks for medchem
The development of deutetrabenazine, is considered by many as a prelude to a new generation of medicines and therapies that incorporate deuterium into the active pharmaceutical ingredient.5 Given that each stereoisotopomer of a biologically active substance will have its own unique pharmacokinetic profile, the ability to stereoselectively deuterate cyclohexene or other medchem building blocks could enable the development of new probes, fragment libraries, and leads for medicinal chemists, as well as providing a new tool for organic and organometallic mechanistic studies. Cyclohexene can be readily converted into perhydroindoles,25 perhydroisoquinolines,26 and azepines.27 However, the inability to chemically differentiate the two alkene carbons or the enantioface of the deuterated cyclohexene limits its potential. But by replacing the benzene ligand in Fig. 2 with a substituted benzene, or utilizing a non-hydrogenic nucleophile in the conversion of 6 to 7 (Fig. 2), one can envision a series of 3-substituted cyclohexenes with highly defined isotopic patterns. As proof of concept, we prepared the α,α,α-trifluorotoluene complex WTp(NO)(PMe3)(η2-CF3Ph),28 which can be elaborated into a 3-(trifluoromethyl)cyclohexene complex (47) analogous to the cyclohexene complex 7 (Fig. S14). Liberation of the cyclohexene from {WTp(NO)(PMe3)} can be accomplished by a one-electron oxidant such as DDQ, Fe(III), or NOPF6 in yields ranging from 70–75%.28 Oxidation of 47 would generate a cyclohexene that has been previously shown to undergo diastereoselective epoxidation, and would therefore be an attractive building block for medicinal chemistry.29 Repeating the synthesis of 47 with deuteride in the final step yields the cis-6-deutero-3-(trifluoromethyl)cyclohexene complex 52 in 95% yield. Various other isotopologues of 47 and 52 were also prepared (47, 52, 53, 54), and the reaction pattern was found to be similar to that observed for benzene. Prepared compounds are summarized in Fig. 4, with synthetic details provided in SI section B. Notably, in the syntheses of 47, 52, 53, 54, protonation at the carbon bearing the CF3 group ultimately occurs endo to the metal, allowing the CF3 group to assume an exo stereochemistry. However, if the purported diene intermediate is protonated under kinetic control, exo protonation forces the CF3 group endo, and the result after a second hydride reduction is the cyclohexene complex 55. Exploiting this reactivity feature, we were able to prepare other isotopologues of 55 with inversion of the stereocenter bearing the -CF3 substituent (Fig. 4, 56, 57; Fig. S14).
Fig. 4. Examples of functionalized cyclohexene isotopomer complexes.

a) elaboration (speculated) of functionalized cyclohexenes; reference 29 (Murray, R. W.; Singh, M.; Williams, B. L.; Moncrieff, H. M.,Diastereoselectivity in the Epoxidation of Substituted Cyclohexenes by Dimethyldioxirane J. Org Chem. 1996, 61, 1830–1841) describes a single step reaction with DMDO (70%); reference 30 (Leiris, S.; Lucas, M.; Dupuy d’Angeac, A.; Morère, A., Synthesis and biological evaluation of cyclic nitrogen mustards based on carnitine framework. European Journal of Medicinal Chemistry 2010, 45, 4140–4148) describes a 9-step synthesis that includes: i) HCl/H2O. ii) isobutylene. iii) mCPBA. iv) NaN3. v) acetyl chloride. vi) H2/C. vii) ethylene oxide. viii) TsCl. ix) TFA. b) See SI section B for full synthetic details of 47, 48, 52-63.
As further demonstration of the ability of this methodology to selectively prepare isotopomers of functionalized cyclohexenes, we have prepared the tungsten complex of cis,trans-3-cyano-4,5-dideuterocyclohexene (58) by the addition of cyanide to the allyl intermediate 13 (57%; dr > 98%; Fig 4.). Other d1-isotopolouges were also prepared (Fig. S15) and the stereochemistry could again be controlled with the sequence of nucleophiles. For example, 58, 59 and 60 could be prepared by generating the appropriate isotopologue of the tungsten-allyl complex and then treating with NaCN (Fig. S16). Conversely, treating the benzenium 2 with NaCN leads to a cyano-substituted cyclohexadiene that can be subsequently combined with acid and hydride source to generate other cyclohexene isotopomers (61–63; SI section B). 3-cyanocyclohexene (proteo form) has been previously used as a precursor to cytotoxic mustards that are of interest in cancer research.30 Allyl-substituted cyclohexenes theoretically exist as 1024 different H/D isotopomers (512 for each enantiomer). Using the tungsten dearomatization methodology, the CF3- and CN-substituted cyclohexenes are accessible as 64 and 60 unique isotopomers respectively. We further note that a full range of both carbon and nitrogen nucleophiles has now been demonstrated to add to tungsten benzenium and allyl tungsten complexes,31 which demonstrates the broad scope of compounds that can now be prepared as various deuteroisotopomers.
Data Availability
All data is available in the main text or the supplementary materials, including NMR spectra, experimental details, crystallographic information, DFT calculations, Rotational spectroscopy, and HRMS data. CCDC 1885723–1885725 and 1972890 contains the supplementary crystallographic data for this paper (4, 7, 9 [X-ray] and 45 [neutron]). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures
Supplementary Material
Acknowledgements:
The authors acknowledge the assistance of Dr. Earl Ashcraft in collecting HRMS data.
Funding: National Institutes of Health (1R01GM132205-01); University of Virginia. Single crystal neutron diffraction experiment performed on TOPAZ used resources at the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory, under Contract No. DE-AC05-00OR22725 with UT-Battelle, LLC.
Footnotes
Online content:
Full synthetic procedures, spectra, compound characterizations, DFT calculations, and supplementary figures and tables.
The authors declare no competing interests.
References
- 1.Gant TG, Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem 2014, 57, 3595–3611. [DOI] [PubMed] [Google Scholar]
- 2.Thibblin A; Ahlberg P, Reaction Branching and Extreme Kinetic Isotope Effects in the Study of Reaction Mechanisms. Chemical Society Reviews 1989, 18, 209–224. [Google Scholar]
- 3.Thibblin A, Unusually Large Kinetic Deuterium Isotope Effects on Oxidation Reactions. 1. The Mechanism of Hydroxide-Catalysed Permanganate Oxidation of PhCD(CF3)OH and PhCD(CH3)OH in Water. Journal of Physical Organic Chemistry 1995, 8, 186–190. [Google Scholar]
- 4.Nelson SD; Trager WF, The Use of Deuterium Isotope Effects to Probe the Active Site Properties, Mechanism, of Cyctochrome P450-Catalyzed Reactions, and Mechanisms of Metabolically Dependent Toxicity. Drug Metabolism and Disposition 2003, 31, 1481–1497. [DOI] [PubMed] [Google Scholar]
- 5.Dean M; Sung VW, Review of Deutetrabenazine: a Novel Treatment for Chorea Asociated with Huntington’s Disease. Drug design, development and therapy 2018, 12, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC, Photoredox-catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358 (6367), 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pony Yu R; Hesk D; Rivera N; Pelczer I; Chirik PJ, Iron-catalysed Tritiation of Pharmaceuticals. Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin JE; Kiemle DJ; Kostikov AP, Quantitative Analyses of Stereoisomeric 3,4-d2-Cyclohexenes in the Presence of 3,6-d2-Cyclohexenes. J. Org Chem 2009, 74, 3866–3874. [DOI] [PubMed] [Google Scholar]
- 9.Liebov BK; Harman WD, Group 6 Dihapto-Coordinate Dearomatization Agents for Organic Synthesis. Chem. Rev 2017, 117, 13721–13755. [DOI] [PubMed] [Google Scholar]
- 10.Eisen MS; Marks TJ, Supported organoactinide complexes as heterogeneous catalysts. A Kinetic and Mechanistic Study of Facile Arene Hydrogenation. J. Am. Chem. Soc 1992, 114, 10358–68. [Google Scholar]
- 11.Jones RA; Seeberger MH, Synthesis of polymer-supported transition metal catalysts via phosphido linkages: heterogeneous catalysts for the hydrogenation of aromatic compounds under mild conditions. J. Chem. Soc., Chem. Commun 1985, 373–4. [Google Scholar]
- 12.Joannou MV; Bezdek MJ; Chirik PJ, Pyridine(diimine) Molybdenum-Catalyzed Hydrogenation of Arenes and Hindered Olefins: Insights into Precatalyst Activation and Deactivation Pathways. ACS Catalysis 2018, 8, 5276–5285. [Google Scholar]
- 13.Harman WD; Taube H, The selective hydrogenation of benzene to cyclohexene on pentaammineosmium(II). J. Am. Chem. Soc 1988, 110, 7906–7907. [Google Scholar]
- 14.Welch KD; Harrison DP; Lis EC; Liu W; Salomon RJ; Harman WD; Myers WH, Large-Scale Syntheses of Several Synthons to the Dearomatization Agent {TpW(NO)(PMe3)} and Convenient Spectroscopic Tools for Product Analysis. Organometallics 2007, 26, 2791–2794. [Google Scholar]
- 15.Lankenau AW; Iovan DA; Pienkos JA; Salomon RJ; Wang S; Harrison DP; Myers WH; Harman WD, Enantioenrichment of a Tungsten Dearomatization Agent Utilizing Chiral Acids. J. Am. Chem. Soc 2015, 137,10, 3649–3655. [DOI] [PubMed] [Google Scholar]
- 16.Harrison DP; Nichols-Nielander AC; Zottig VE; Strausberg L; Salomon RJ; Trindle CO; Sabat M; Gunnoe TB; Iovan DA; Myers WH; Harman WD, Hyperdistorted Tungsten Allyl Complexes and Their Stereoselective Deprotonation to Form Dihapto-Coordinated Dienes. Organometallics 2011, 30, 2587–2597. [Google Scholar]
- 17.Lis EC; Delafuente DA; Lin Y; Mocella CJ; Todd MA; Liu W; Sabat M; Myers WH; Harman WD, The Uncommon Reactivity of Dihapto-Coordinated Nitrile, Ketone, and Alkene Ligands When Bound to a Powerful π-Base. Organometallics 2006, 25, 5051–5058. [Google Scholar]
- 18.Arashiba K; Matsukawa S; Kuwata S; Tanabe Y; Iwasaki M; Ishii Y, Electrophilic O-Methylation of a Terminal Nitrosyl Ligand Attained by an Early–Late Heterobimetallic Effect. Organometallics 2006, 25, 560–562. [Google Scholar]
- 19.Jamison CJ Isotope Effects on Chemical Shifts and Coupling Constants 2007, eMagRes. doi:10.1002/9780470034590.emrstm0251 [Google Scholar]
- 19.Pérez C; Lobsiger S; Seifert NA; Zaleski DP; Temelso B; Shields GC; Kisiel Z; Pate BH, Broadband Fourier transform rotational spectroscopy for structure determination: The water heptamer. Chemical Physics Letters 2013, 571, 1–15. [Google Scholar]
- 20.Sharp WB; Legzdins P; Patrick BO, O-Protonation of a Terminal Nitrosyl Group To Form an η1-Hydroxylimido Ligand. J. Am. Chem. Soc 2001, 123, 8143–8144. [DOI] [PubMed] [Google Scholar]
- 21.Llamazares A; Schmalle HW; Berke H, Ligand-Assisted Heterolytic Activation of Hydrogen and Silanes Mediated by Nitrosyl Rhenium Complexes. Organometallics 2001, 20, 5277–5288. [Google Scholar]
- 22.Leong VS; Cooper NJ, Electrophilic activation of benzene in [Cr(.eta.4-C6H6)(CO)3]2. J. Am. Chem. Soc 1988, 110, 2644–2646. [Google Scholar]
- 23.Thompson RL; Lee S; Rheingold AL; Cooper NJ, Reductive activation of the coordinated benzene in manganese complex [Mn(.eta.6-C6H6)(CO)3]+: synthesis and characterization of the .eta.4-naphthalene complex PPN[Mn(.eta.4-C10H8)(CO)3]. Organometallics 1991, 10, 1657–1659. [Google Scholar]
- 24.Ungureanu I; Klotz P; Mann A, Phenylaziridine as a Masked 1,3 Dipole in Reactions with Nonactivated Alkenes. 2000, 39, 4615–4617. [PubMed] [Google Scholar]
- 25.Sarkar N; Banerjee A; Nelson SG, [4 + 2] Cycloadditions of N-Alkenyl Iminium Ions: Structurally Complex Heterocycles from a Three-Component Diels–Alder Reaction Sequence. J. Am. Chem. Soc 2008, 130, 9222–9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shim SC; Doh CH; Kim TJ; Lee HK; Kim KD, A new and convenient synthesis of N-substituted perhydroazepines from adipaldehyde and primary amines with tetracarbonylhydridoferrate, HFe(CO)4-, as a selective reducing agent. J. Heterocycl. Chem 1988, 25, 1383–5. [Google Scholar]
- 27.Wilson KB; Myers JT; Nedzbala HS; Combee LA; Sabat M; Harman WD, Sequential Tandem Addition to a Tungsten–Trifluorotoluene Complex: A Versatile Method for the Preparation of Highly Functionalized Trifluoromethylated Cyclohexenes. J. Am. Chem. Soc 2017, 139, 11401–11412. [DOI] [PubMed] [Google Scholar]
- 28.Murray RW; Singh M; Williams BL; Moncrieff HM, Diastereoselectivity in the Epoxidation of Substituted Cyclohexenes by Dimethyldioxirane J. Org Chem 1996, 61, 1830–1841. [DOI] [PubMed] [Google Scholar]
- 29.Leiris S; Lucas M; Dupuy d’Angeac A; Morère A, Synthesis and biological evaluation of cyclic nitrogen mustards based on carnitine framework. European Journal of Medicinal Chemistry 2010, 45, 4140–4148. [DOI] [PubMed] [Google Scholar]
- 30.Wilson KB; Smith JA; Nedzbala HS; Pert EK; Dakermanji SJ; Dickie DA; Harman WD, Highly Functionalized Cyclohexenes Derived from Benzene: Sequential Tandem Addition Reactions Promoted by Tungsten. J. Org Chem 2019, 84, 6094–6116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials, including NMR spectra, experimental details, crystallographic information, DFT calculations, Rotational spectroscopy, and HRMS data. CCDC 1885723–1885725 and 1972890 contains the supplementary crystallographic data for this paper (4, 7, 9 [X-ray] and 45 [neutron]). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures