

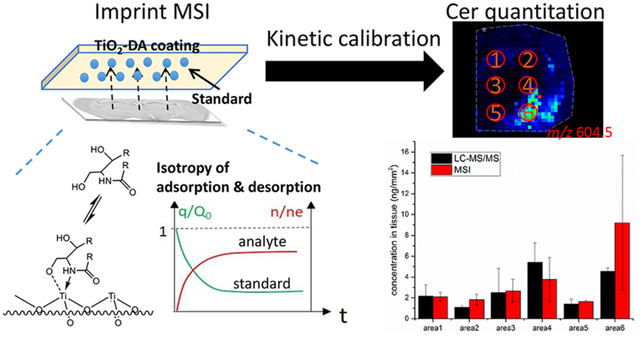
Abstract
Quantitative mass spectrometry imaging (MSI) is an effective technique for determining the spatial distribution of molecules in a variety of sample types; however, the quality of the ion signals is related to the chemical and morphological properties of both the same matrix and targeted analyte(s). Issues may arise with the incorporation of standards into the sample at repeatable, well-defined concentrations, as well as with the extraction and incorporation of endogenous analytes versus standards from tissue into the matrix. To address these concerns, we combine imprint MSI (iMSI) with kinetic calibration, and use it to quantify lipids in rat brain tissue samples. Briefly, tissues were imprinted on slides coated with a dopamine-modified TiO2 monolith pretreated with analyte standards, resulting in the adsorption of endogenous analytes onto the coating and desorption of standards into the tissue. The incorporation of standards into the tissue enabled quantification of the measured analytes using kinetic calibration. Moreover, matrix effects were reduced, and the intensities of analyte standard signals became more uniform. The symmetry of the adsorption of endogenous ceramides and the desorption of ceramide standards suggests that the content of adsorbed endogenous ceramide can be determined by measuring the content of desorbed ceramide standard. Using kinetic calibration, endogenous ceramide concentrations were calculated for a range of dry and wet tissue imprinting conditions and compared with quantitative MSI using a standard spiking approach. We validated quantitative iMSI using liquid chromatography tandem mass spectrometry (LC-MS/MS) and found that the concentrations determined using iMSI compared with LC-MS/MS were in the range of 70 to 200% over the concentration range of endogenous ceramides; the correlation coefficient between iMSI and LC-MS/MS was over 0.9 (Pearson’s r), while the recoveries via traditional standard spiking were between 200% and 5000% depending on the brain region and sample preparation conditions.
Graphical Abstract
INTRODUCTION
Mass spectrometry imaging (MSI)1 is a sensitive and multiplexed approach for the spatiochemical characterization of a wide range of analytes. The field encompasses a variety of methods based on different analyte desorption (sputtering)/ionization techniques, including secondary ion mass spectrometry imaging (MSI), laser desorption/ionization (LDI) MSI, matrix-assisted laser desorption/ionization (MALDI) MSI, and desorption electrospray ionization (DESI) MSI.2–6 Of these, MALDI MSI enables analysis across the widest molecular mass range7–10 at micrometer-scale spatial resolution, with detection limits comparable to other MS-based approaches,11–12 and is well suited for the detection and localization of small metabolites,8,10 lipids,13 and peptides and proteins14 across a range of sample types, including brain tissues. However, matrix interference in the low molecular mass range is an issue for MALDI MS. Accordingly, surface assisted laser desorption/ionization (SALDI) and other related methods utilizing metal oxides, silicon, gold, platinum, silver nanoparticles, and carbon nanotubes to assist analyte desorption/ionization have been developed.15–17
Because quantitative and semi-quantitative measurements of analytes in tissues aids our understanding of biological processes, quantitative MSI is an area of continued method development.18–23 Although MALDI- or SALDI-MSI often has sufficient spatial resolution and sensitivity for studying the distribution of molecules within a diverse range of samples, using the approach for quantitation can be challenging. Among the factors limiting the quantitative abilities of MALDI- or SALDI-MSI is the dependence of ion signal quality on the tissue’s physicochemical and morphological properties. For example, endogenous targeted compounds may be located within different chemical environments, such as the cytoplasm, organelles, and cellular membranes. Approaches using analyte separations, such as liquid chromatography (LC)–mass spectrometry (MS), reduce sample matrix effects by separating matrix and analyte molecules extracted from tissues. However, for MALDI- or SALDI-MSI, the tissue slice should retain its in vivo state to allow for determination of the endogenous spatial distribution of compounds. Therefore, approaches for ion signal calibration and normalization tailored to each specific microenvironment are needed.
A variety of analytical methods that incorporate standard-based calibration approaches, including LC-tandem MS (MS/MS) and autoradiography, are effectively used to correlate an analyte’s signal to its quantity.24 A range of standard addition methods have been developed for MALDI MSI, including deposition of standards at specific tissue locations.19 Standards can also been microspotted onto tissue to avoid uneven distribution during the longer evaporation times for large droplets and to realize pixel-to-pixel calibration,20 or spiked into tissue homogenates to create a mimetic model sliced together with targeted samples.21
Significant advantages have been reported using standard spiked tissue homogenates in quantitative MALDI MSI. However, endogenous analytes, which are often incorporated into membranes and organelles, may exhibit different mass transfer kinetics compared to spiked standards during their extraction and incorporation into MALDI matrix crystals, or adsorption by LDI-assisting nanomaterial surfaces. As one example, standards applied in the extracellular environment of the tissue are easily accessible to the MALDI matrix and may generate larger signals relative to the endogenous analytes located within the cells, including those incorporated into the cell membranes. Thus, the application of standards should be done in a manner that ensures that the mass transfer kinetics of both standards and endogenous analytes are similar.
One interesting imaging approach is imprint MSI (iMSI), which combines imprinting (blotting) approaches with various MSI methods, including MALDI,25–26 SALDI,16,27 and DESI.28 The imprinting process can selectively transfer/adsorb the analytes from a complex sample surface into a flat imprinting material surface, reduce matrix effects, and increase selectivity to detect targeted analytes. It has been used for a variety of sample types, including animals, plants, and bacterial cultures.16,25,27 By simplifying the biological samples during imprinting, quantitation may improve. However, because analyte adsorption efficiencies are dependent on the matrix used, local micro-environment, and analyte properties, quantitation using iMSI is not common. Since the imprinting process is similar to solid phase extraction (SPE) and solid phase micro-extraction (SPME), which are widely used sample preparation methods for purification and preconcentration steps,29–30 the quantitation methods used in SPME may ameliorate the issues of quantitative iMSI.31 For example, desorption of the standards preloaded onto the SPME fiber during its conditioning helps to calibrate signals produced by the analytes extracted from the sample according to the isotropy of analyte adsorption and desorption in the SPME fiber. With this approach, known as kinetic calibration or the in-fiber standardization technique,32–33 there is no need to spike the standard into samples before analyte extraction. The extraction kinetics of the sample’s endogenous compounds onto a solid phase is standardized using information on the desorption kinetics of the standards. Kinetic calibration has already been successfully used for in vivo SPME in which the standard cannot easily be spiked into samples.33–35 We speculated that if imprinting could be introduced into MSI and kinetic calibration used, the loading of the standards into tissue as a separate sample preparation step for quantitative MSI could be avoided.
Accordingly, we developed a quantitative method utilizing kinetic calibration and iMSI. More specifically, a dopamine-modified TiO2 (TiO2-DA) monolith, developed by our group,17 was applied onto indium tin oxide (ITO) slides and used as the solid phase for adsorption of analytes, including endogenous lipids extracted from tissue. As a proof of concept, we performed MSI of rat brain tissue imprinted on a TiO2-DA monolith and found that many lipids, including cholesterol, ceramides, phosphatidylethanolamines (PEs) and phosphatidylcholines (PCs), can be detected using this approach. To demonstrate the methodology, rat brain tissue slices were imprinted onto a TiO2-DA coating preloaded with ceramide standard. Ceramides, which are functionally important signaling lipids, are located in different rat brain regions and quantified using kinetic calibration. The endogenous ceramide signal adsorbed onto the coating was calibrated by comparison with the difference in signal intensity of preloaded ceramide standard before and after desorption from the coating. The imprint method was compared with the traditional standard spiking method, and the enhanced ability to compensate for the variation of extraction conditions of the new method was demonstrated (ratios of quantitation results in different conditions are near 1). Additionally, the results agreed with those obtained using quantitative LC-MS/MS according to the relative recoveries of iMSI results compared with LC-MS/MS results (72%–200%) and the correlation coefficient (0.93) of the results between these methods.
EXPERIMENTAL SECTION
Reagents and Materials
Concentrated nitric acid and phosphoric acid (analytical grade) were purchased from Fisher Scientific (Pittsburgh, PA). Acetonitrile (ACN), ethanol, and water (LC/MS grade) were from Fisher Scientific. The matrix, 2,5-dihydroxybenzoic acid (DHB), was purchased from Bruker (Billerica, MA). Titanium(IV) n-butoxide, dopamine hydrochloride (purity >98%), phospholipid mixture (1mg/mL, HPLC grade), cholesterol (purity >99%), Cer d18:1/6:0 (purity >98%), and galactocerebrosides mixture (1mg/mL, HPLC grade) were obtained from MilliporeSigma (St. Louis, MO).
Preparation of the TiO2-DA Monolithic Coating
TiO2 nanoparticles were prepared using hydrolysis of titanium(IV) n-butoxide in an ethanol-water solution under acidic condition, as previously described.17 Briefly, 3.4 mL of titanium(IV) n-butoxide and 1.6 mL of ethanol were mixed by vortex for 1 min, forming a precursor solution. Next, a solution containing 5 mL of ethanol with 0.1 M nitric acid and 1% HPLC grade water was added dropwise into the vigorously stirred precursor solution, which was cooled in an ice/water bath. TiO2 structures were formed in the solution (solution I) during stirring in an ice/water bath for 3 h; 250 μL of solution I was diluted in 5 mL of 5% water-ethanol solution with 0.005 M dopamine hydrochloride, and then incubated for 1 h (solution II).
To prepare the TiO2-DA monolith coating, solution II containing TiO2-DA was applied onto ITO glass slides (Delta Technologies, Loveland, CO) using a 0.2 mm nozzle airbrush (Paasche Airbrush Company, Chicago, IL) with an airbrush nozzle-to-target distance of ~50 cm and nitrogen gas pressure set at 35 psi for 5 min. The push-button on the airbrush was pushed to the middle level during spraying. The final density of the TiO2-DA layer was ~400 μg/cm2, which was determined by comparing the weight of the slide before and after coating. The TiO2-DA monolithic layers were washed in the following order: ethanol, 50% ethanol-water, and water.
Loading of Standards
In most of the experiments, arrays of 0.5-μL droplets containing 10 μg/mL of standards were manually deposited onto the TiO2-DA monolith. The droplets were dried in a N2 chamber. In experiments evaluating sample matrix effects on MSI results, standards were deposited with a microspotting device (CHIP-1000, Shimadzu Biotech, Kyoto, Japan) forming ~500-μm diameter dried spots on tissue sections, tissue imprints on a TiO2-DA monolith, and a bare TiO2-DA monolith. A total of 5 nL of solution containing 10 μg/mL of Cer d18:1/6:0 in ACN was applied to each spot. Five droplets of approximately 100 pL were deposited on each spot per cycle. A total of 10 iterations were thus necessary to obtain the final volume.
Brain Tissue Collection, Sectioning and Imprinting
Tissues were harvested from four male Sprague-Dawley rats (ENVIGO, Indianapolis, IN), 1–3 months old, maintained on a 12-h light/dark cycle and fed normal chow ad libitum. Euthanasia by decapitation was performed in compliance with local and federal regulations and according to animal use protocols approved by the Illinois Institutional Animal Care and Use Committee.
In all cases, the brains were surgically dissected, frozen in liquid nitrogen, and stored at −80 °C until use. Coronal brain sections, 12-μm thick, were cut through the cerebrum region using a Leica CM 3050 S cryostat (Leica Microsystems, Bannockburn, IL) at −19 °C and thaw-mounted onto ITO glass slides, as detailed below.
For SALDI MSI, tissue sections were placed onto ITO glass slides. Solution II was applied using the 0.2 mm nozzle airbrush as described in the section above, “Preparation of the TiO2-DA Monolithic Coating”, and quickly dried at ambient conditions. After application of solution II, additional ACN was applied using the airbrush for 2 min with an airbrush nozzle-to-target distance of ~50 cm and the nitrogen gas pressure set at 35 psi. To compare the calibration results under different sample preparation conditions, the gas pressures and spraying times of solution II and ACN were varied to provide wetter or dryer conditions (see the Supporting Information, Method S1 for details).
For iMSI, tissue sections were mounted onto TiO2-DA monolith-coated ITO glass slides, which were preloaded with standards. After thaw-mounting, the tissues were sprayed with ACN for 5 min using a 0.2 mm nozzle airbrush with a ~50 cm nozzle-to-target distance, with the nozzle nitrogen gas pressure set at 35 psi. The push-button on the airbrush was pushed to the middle level during the spraying. To determine the time curves of desorption/adsorption, different spraying times (1 min, 3 min, 5 min, 7 min, and 15 min) were evaluated. For comparison of the calibration results under different sample preparation conditions, gas pressures and spraying times were varied to provide wetter or dryer conditions for imprinting (see Method S1 for details). This step was followed by removal of tissue slices by rinsing the entire sample with water, followed by drying the TiO2-DA monolith-coated ITO glass slide in an N2 chamber at ~25 °C.
TiO2-DA Monolith-Assisted LDI-MSI Analyses
TiO2-DA monolith-assisted LDI-time-of-flight (TOF)/TOF MSI (for both SALDI and iMSI) was performed using an ultrafleXtreme II mass spectrometer (Bruker) equipped with a solid-state UV Smartbeam II laser. The laser was set to the “ultra” setting with a ~100-μm diameter footprint and a 100 μm raster step. Each color-coded pixel in the resulting MS images represents the relative intensity of an ion signal. Each single mass spectrum was acquired as a sum of 1000 laser shots fired at 1000 Hz frequency. MS spectra were acquired in the m/z range of 20–3000. A mixture of DHB, bradykinin, and angiotensin II was deposited near to the brain tissue section for mass spectra recalibration, needed in cases of molecular mass errors related to sample surface topography. MSI data acquisition, processing, and visualization were performed with flexImaging software (Version 3.0, Bruker). The MALDI TOF/TOF mass analyzer was calibrated with the Peptide Calibration Standard II set (Bruker) containing bradykinin, angiotensin II, angiotensin I, substance P, bombesin, ACTH clip 1–17, ACTH clip 18–39, renin substrate, and somatostatin 28.
LC-MS/MS
The LC-MS/MS details, including sample preparation, instrumentation and conditions for analyte detection, quantitation, and characterization are described in the Supporting Information, Method S2.
Data Processing
Details on data processing including the kinetic calibration methods are in the Supporting Information Method S3.
RESULTS AND DISCUSSION
TiO2-DA Monolith-Assisted iMSI
Previously, we reported that TiO2-DA was useful for assisting LDI MS to detect a number of lower molecular weight metabolites and lipids with high sensitivity and low background.17 In this work, a TiO2-DA monolith was used for brain tissue imprinting as well as for kinetic calibration. During sample preparation utilizing TiO2-DA-based analyte retention and kinetic calibration, ITO glass slides were coated with TiO2-DA monolith, followed by deposition of standards, tissue blotting (also called imprinting), and tissue removal by rinsing (Figure 1). iMSI enabled detection of the phospholipids and other Lewis basic lipids. As expected, because imprinting simplifies the analyte environment, the effects of the matrix were decreased and the sensitivity of detection of targeted analytes, including lipids, was increased.
Figure 1.
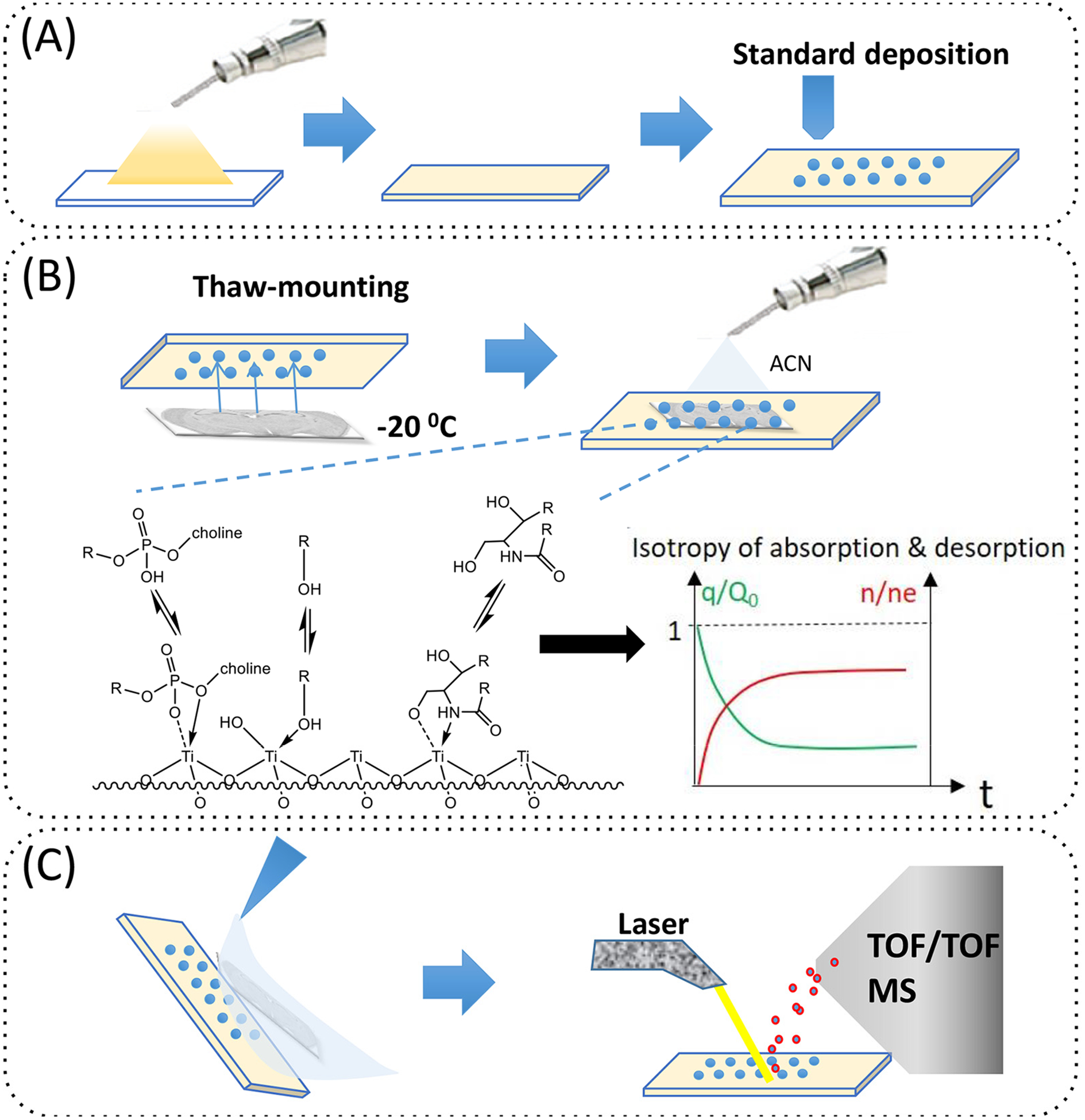
Schematic illustration of the sample preparation and analysis process in quantitative iMSI utilizing TiO2-DA monolith and kinetic calibration. (A) Preparation of the TiO2-DA monolithic coating of ITO glass slides with either manual or robotic deposition of standards. (B) Imprinting of the brain tissue slice onto TiO2-DA monolith and its wetting using ACN. Graphical illustrations of the affinity of metabolites onto the TiO2-DA monolithic coating and kinetic calibration fundamentals based on isotropy of adsorption and desorption of analytes onto/from SPE surfaces (two lower panels). (C) Brain tissue removal and remaining imprint conditioning using several brief rinses with different solvents followed by iMSI.
Although there are a number of advantages to the imprinting method, its reproducibility and quantitative aspects are yet to be established because the ability to move material from the tissue to the surface to be imaged via MSI depends on tissue properties. In order to account for such differences, we applied a kinetic calibration approach. The isotropic desorption of standards from the TiO2-DA monolith into imprinted tissue was used to calibrate the amounts of endogenous compounds adsorbed from the tissue into the monolith. The assumption we verified is that both processes are impacted by tissue properties in a similar way. The relative intensities of many signals were different between direct tissue SALDI MSI and iMSI; however, in both cases, the same major peaks were observed. Importantly, iMSI produced higher relative peak intensities for putative PC and PE signals in the mass range of 700–900 Da. These results corroborate our previous observation of the high affinity for TiO2-DA to phospholipids. Conversely, in the lower mass range (m/z 200–700), most peaks were 5 to 10 times less intense in the case of iMSI compared to direct tissue-SALDI MSI. As we demonstrated previously using the same animal model, most peaks in the m/z 200–700 range represent fatty acids (m/z 200–400), cholesterol (m/z 400–500), ceramides, and diacylglycerol (m/z 500–700).17 Observed differences in peak intensities may be explained by the lower affinity of the TiO2-DA material to common Lewis bases (such as the carboxyl group of fatty acids, hydroxyl group of cholesterol, and amino group of ceramides) than to bi-/tri-dentate ligands (such as the phosphate group of phospholipids).36–37 Thus, under the binding competition with phospholipids, the adsorption efficiencies of small molecules such as fatty acids are expected to be lower. Despite the multistep sample treatment used in iMSI, the analyte signal spatial distributions for detected compounds are similar to those observed with direct tissue SALDI MSI; e.g., ceramide and PE ion spatial distributions are similar in images produced by both approaches (Figure 2). Additionally, iMSI produces better contrast in the molecular images, suggesting that localization of compounds during sample preparation for iMSI is preserved.
Figure 2.
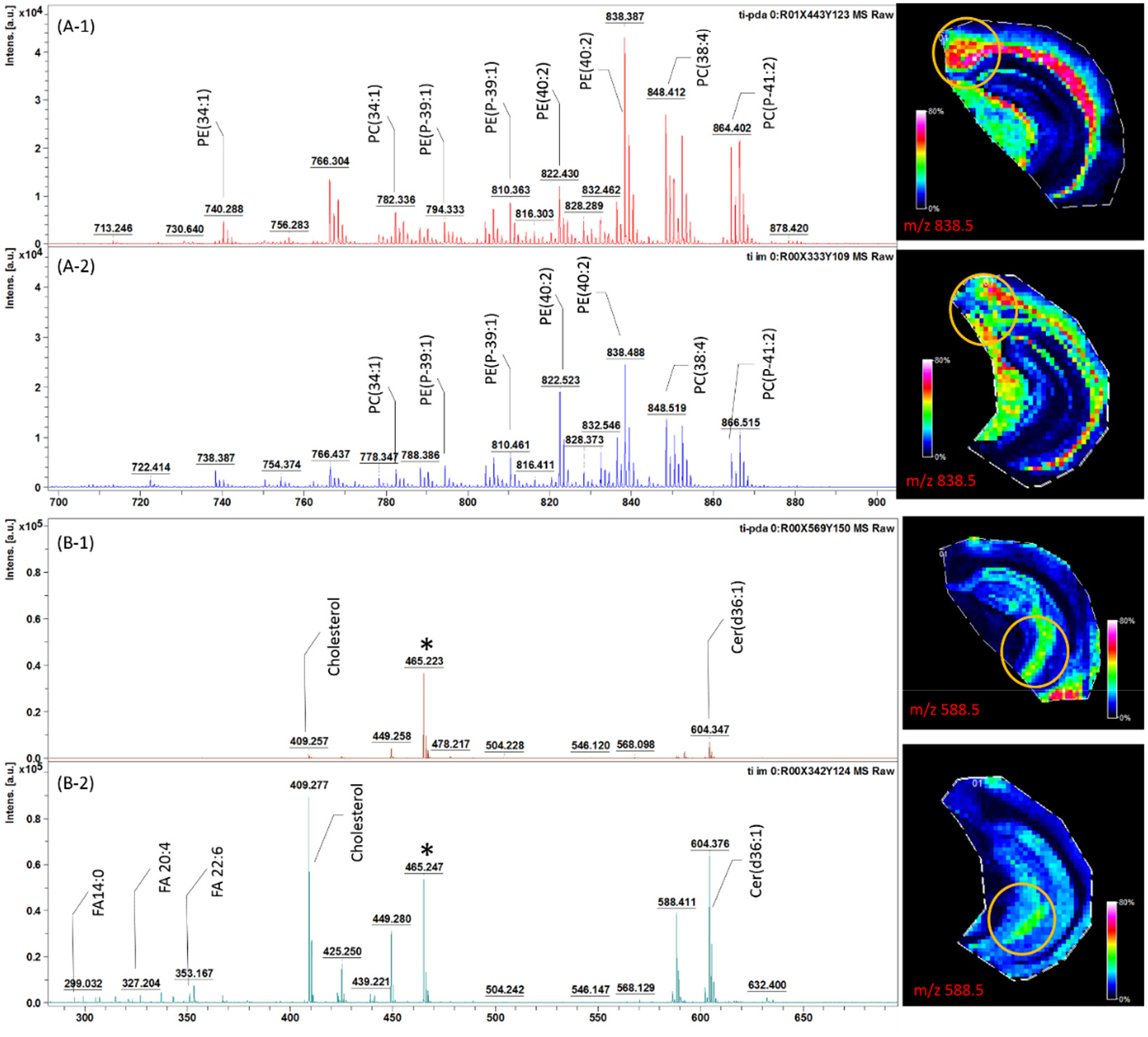
Representative mass spectra of the brain areas outlined by yellow circles in the ion images depicted on the right with (A-1, B-1) iMSI and (A-2, B-2) direct tissue SALDI MSI in the higher m/z range (A, m/z 200–700) and lower m/z range (B, m/z 700–900). Ion images on the right are of A, m/z 838.5 and B, m/z 588.5 signal distributions. Adjacent sections of the same mouse brain are used in these measurements.
We found that the pH influences the extraction and detection of fatty acids with iMSI; as expected, lower pHs facilitated the extraction/retention of fatty acids, and higher pHs decreased the extraction/retention efficiency of procedures for fatty acids.38 However, lower pH was associated with difficulties of tissue section removal from the TiO2-DA monolith, perhaps caused by the denaturation and protonation of the proteins, increasing their affinity to adhere to the surface.
Comparison of Chemical Matrix Effects on Results of iMSI and Direct Tissue-SALDI MSI Measurements
Matrix effects resulting from these two chemical imaging methods were determined by comparing signals acquired from arrays of ~500-μm diameter microspots containing ceramide analyte standard on: 1) brain sections mounted onto ITO glass slides (n=2) (SALDI MSI); 2) brain tissue imprints (n=2) (iMSI); 3) bare ITO glass slides (n=2) (SALDI MSI); 4) TiO2-DA monolith (n=2). Ratios of the signal areas observed in standard microspots illustrate matrix effects on the outcomes of iMSI and direct tissue SALDI MSI measurements (Table 1 and Figure S1). iMSI demonstrated matrix effects between 5% to 90% for both the Na and K adducts of ceramide. In contrast, direct tissue SALDI MSI exhibited the lowest matrix effects for the Na adduct of the ceramide peak (11%) and highest for its K adduct signal (1330%). Differences in the chemical complexity of tissue, tissue imprint, and blank areas, including inorganic salt composition and their amounts, contributed to the observed matrix effects.
Table 1.
Matrix effects on the results of direct tissue SALDI MSI and iMSI detection of different lipid standards in different brain regions.
| Approach | Region | [CEr+Na]+ (m/z 588.5) | [CEr+K]+ (m/z 604.5) | [PE+Na]+ (m/z 750.6) | [PE+K]+ (m/z 766.6) | [CB+Na]+ (m/z 850.6) | [CB+K]+ (m/z 866.6) |
|---|---|---|---|---|---|---|---|
| Direct tissue SALDI MSI | CTX | 18± 8 | 1330±670 | 0.34±0.08 | 53±25 | 85±47 | 1230±750 |
| CC | 7±3 | 320± 140 | 17±5 | 455±50 | 326±50 | 6490±1300 | |
| CA1 | 11±8 | 475±367 | 44 ±14 | 212±121 | 264±183 | 4560±3700 | |
| CA3 | 46±17 | 890±370 | 1.3 ±0. 3 | 0.11± 0.03 | 0.10±0.03 | 28±6 | |
| iMSI | CTX | 76±32 | 67±30 | 7±3 | 10±3 | 33±8 | 11±3 |
| CC | 84±11 | 43±26 | 12±6 | 16±6 | 124±25 | 12±6 | |
| CA1 | 89±18 | 29±52 | 21±16 | 27±6 | 51±6 | 34±12 | |
| CA3 | 32±33 | 5±3 | 10±5 | 16±3 | 46±17 | 20±7 |
CTX, cortex; CC, corpus callosum; CA1, region I of hippocampus proper; CA3, region III of hippocampus proper; Cer, ceramide; PE, phosphatidylethanolamine; CB, cerebroside. Mean ± standard deviations for all points/pixels were assessed. Matrix effects determined as ratios of lipid standard signal areas determined by measurements of arrays of microspots deposited onto (1) tissue slices and clean ITO glass surfaces (SALDI MSI) (n=2 animals); (2) tissue imprints and clean ITO glass slides coated with TiO2-DA monolith (iMSI) (n=2 animals).
In the case of PEs, matrix effects were typically lower than 20% and were similar in different brain areas (Table 1). In contrast, the matrix effects observed in direct tissue SALDI MSI measurements had large variability between different brain regions, affecting the final molecular images of PEs. Matrix effects on cerebroside signals were similar to those observed for PE signals.
These results suggest that iMSI not only exhibits overall lower matrix effects, but also allows chemical imaging with lower brain region-dependent variations caused by matrix effects. This increases the quality and accuracy of the analyses.
Kinetic Calibration and Measurement Variability Compensation in Quantitative iMSI Analysis
Isotropy of adsorption and desorption of ceramides in kinetic calibration for quantitative iMSI measurements.
Standard deposition onto a sample for calibration is a common approach for both the relative and absolute analyte quantitation with chemical imaging.19,39 However, the accuracy and precision of quantitative measurements is reduced by chemical heterogeneities within the sample and the typical inability to uniformly add a known series of standards at well-defined concentrations across the entire tissue sample. In addition, spotted standard and endogenous analytes may have different mass transfer kinetics, so they are not extracted into the MALDI matrix or onto SALDI nanomaterials with similar efficiency. Because kinetic calibration has been successfully applied to the SPE field,32–33 we applied it here to increase accuracy and precision. The kinetic calibration is based on the analyte’s isotropy of adsorption and desorption. As described in the Supporting Information, Method S4, the adsorption time curve of an analyte from the sample matrix into the SPE material is symmetric with the desorption time curve of the standard from SPE coating into tissue matrix.33 Thus, when applying the standard on SPE material instead of the sample, the amount of standard remaining in the coating after desorption (Q), the initial amount of analyte standard loaded on the coating (q0), and the amount of adsorbed analyte at that time (n) can be used to calculate the amount of analyte adsorbed by the SPE material at equilibrium (ne). ne is related to the concentration of analytes in the sample (Cs) if the adsorption equilibrium parameters of the analytes on the surface are known. The following equation
| (Eq. 1) |
allows us to determine ns, the amount of analyte in the sample, where Vs is the sample volume, K is the adsorption equilibrium constant (in cm2 mol−1), A is the surface area of the fiber (in cm2), and S is the concentration of unoccupied sites on the surface of the sorbent (in mol cm−2). K × A × S is determined as the detected analyte contents in samples and on the TiO2 coating at equilibrium.33 According to Eq. 1, if the amount of standard remaining in the coating (Q) and the initial amount of standard (q0) are known, we can calibrate the amount of adsorbed analyte on the coating to obtain the original content of analyte in the sample (ns).33
To develop our quantitative iMSI approach using kinetic calibration, ceramide (Cer d18:1/18:0) was chosen as the targeted analyte. It is present at relatively low concentrations in brain tissue and can be detected with TiO2-DA monolith-assisted LDI.17 Cer d18:1/6:0, which is not present in mouse brain tissue but has similar detection properties as endogenous Cer d18:1/18:0, was used as the standard. To control the adsorption/desorption time for kinetic investigation, the solvent application time was varied by spraying solvent (ACN) with different volumes at a constant flowrate using an airbrush system. Results of the iMSI measurements demonstrated that the adsorption curve of endogenous Cer d18:1/18:0 is symmetric with the desorption curve of standard Cer d18:1/6:0 (Figure 3). However, a longer analyte extraction time (when ACN application time exceeded 5 min) resulted in increases of the sum of n/ne and Q/q0.
Figure 3.

Isotropy of the adsorption of endogenous ceramide (Cer d18:1/18:0) by TiO2-DA monolith and ceramide standard (Cer d18:1/6:0) desorption from the TiO2-DA monolith in relation to applied ACN solvent volume. ACN was applied onto the sample at different times (1 min, 3 min, 5 min, 7min and 15 min) at a constant spraying flowrate (~1mL/min) using an airbrush. The sample area covered by spray was larger than the area occupied by brain tissue. The experimental time allowed for adsorption and desorption increases with ACN volume. Means and standard deviations are shown.
An assumption of our model is the unlimited sample volume compared to the solid phase coating. Thus, the limited volume of a brain tissue section may cause deviations in the outcomes of the analyses of our finite sample compared to our model for longer duration of analyte desorptions. An equilibrium concentration of standard retained on SPE material would be reached instead of its full desorption. The isotropy between desorption and adsorption is maintained at experimental conditions described there when the ACN application time is kept below 5 min.
K× A×S /Vs can be calculated using the desorption curve of the standard. In the described experiment, K× A×S /Vs is 1.32 for Cer d18:1/6:0. This parameter was used in the calibration process via Eq. 1.
Comparison of kinetic calibration and standard addition-based spiking calibration.
To evaluate the ability of kinetic calibration with iMSI to produce robust data for multiple brain regions, we prepared a set of adjacent brain section samples using different analyte extraction conditions. In all cases, an airbrush was used to apply the ACN solvent onto brain tissue placed above the TiO2-DA monolith preloaded with standard for iMSI, or to coat the standard-spotted brain tissue with TiO2-DA for SALDI MSI. The ACN spray-covered area of the ITO glass slide is much larger than the surface occupied by tissue, and so the droplet sizes sprayed on the tissue were relatively uniform. To control the droplet size, the pressure of the nebulizing gas (N2) was adjusted to control the nebulizing gas-solvent ratio at the airbrush spray tip. As a result, differences in the applied solvent droplet size and solvent application time led to dryer or wetter analyte extraction conditions. Under dryer spray conditions, a higher gas pressure was used with a lower spraying time of solvent; under wetter conditions, a lower gas pressure was used with a higher spraying time of solvent (see Method S1 for details).
For standard addition calibration, the standard solutions were deposited onto the tissues and dried, followed by coating of the samples with the TiO2-DA solution under both dryer and wetter conditions (Figure S2A). The standard signal acquired from areas containing ceramide standard were used directly to calibrate the signal of endogenous ceramide for calculation of their absolute concentrations. For kinetic calibration, the steps described above were performed for analyte extraction and standard desorption under both dryer and wetter conditions, and the tissue was rinsed off. The TiO2-DA coating with tissue imprint and the remaining ceramide standard after desorption was used for iMSI (FigureS2B). To determine whether the ceramide standard signal was affected by the analyte desorption step, another set of tissues from the same animals was imprinted onto TiO2-DA without preloading standard (in the latter case, the standards were added to the slide after the tissue was rinsed off). In all cases, manual deposition of the standards at six locations on the imprint was performed. These samples allowed us to determine the signals from ceramide standard without its desorption from the TiO2-DA monolith (Figure S2B). The signal intensity of the standard remaining on the TiO2-DA coating, with and without desorption, was used to calibrate the signal of endogenous ceramide according to Eq. S1 to determine its absolute concentration.
Since the adjacent brain sections were used (presumably with very similar ceramide levels), the calculated absolute concentrations under different preparation conditions should be similar and their ratios close to 1. However, with standard addition calibration method and SALDI MSI, much larger values were calculated under the wetter condition (Figure 4A). From Figure S2A, it appears that the endogenous ceramide signal intensities had distinct mass transfer kinetics under these conditions than our selected ceramide standard; as always, proper choice of an internal standard is important.
Figure 4.

Quantitative results of MSI of the same tissue under different sample preparation conditions with (A) standard addition calibration and (B) kinetic calibration. The details of wetter and dryer conditions for the two calibrations are described in the Supporting Information, Method S1.The compared brain regions are numbered 1–6: the hippocampus (1), cortex/fiber tracts (2), thalamus (3), cauduputamen/fiber tracts (4), hypothalamus (5), and cortical subplate (6); the corresponding images are shown in Figure S2. Means with standard deviations, as well as ratios of mean quantitation results in the dryer and wetter conditions (r), are shown.
While neither approach was ideal, Figure 4 shows that kinetic calibration leads to more uniform quantitative iMSI measurements, even under an extreme range of dryer and wetter sample preparation conditions; their ratios of absolute concentrations under these two conditions ranged between 0.66–4.24) (Figure 4B) compared to the standard addition calibration method and SALDI MSI (ratios of quantitation results in two conditions ranging between 3.04–34.3) (Figure 4A). This result suggests that the variation in the desorbed amount of standard from TiO2-DA monolith can be used to compensate for changes in the adsorbed amount of endogenous ceramides under different conditions.
Validation of the Method with LC-MS/MS
We evaluated the accuracy of the iMSI measurements utilizing kinetic calibration using tissue samples of the same brain areas assayed with the “gold standard” LC-MS/MS approach with external standard calibration.40 In these experiments, six cylindrical tissue punches with 1 mm in diameter were collected from frozen tissue blocks at specified brain locations (shown as red circles in Figure 5). Lipids were extracted from the samples using the Folch’s extraction procedure,41–42 extracts dried, and then analytes reconstituted in ACN solvent for loading onto the HPLC system. Data acquired from all examined brain areas, except Area #1 for rat 2 and 3, demonstrated similar quantitative results using iMSI and LC-MS/MS (relative recoveries of iMSI results comparing with LC - MS/MS results are 72%–200%), and the correlation coefficients for quantitative results obtained with the two methods is 0.9 (Figure S3). However, by comparing Figure 4A with the results of rat #1 in Figure 5, the absolute quantitation results with standard addition calibration are much higher than the results of LC - MS/MS, resulting in much higher relative recoveries in the range of 200%–5000%.
Figure 5.

Comparison of iMSI with kinetic calibration and LC-MS/MS with external calibration. Histograms illustrate the quantitation results of iMSI with kinetic calibration (red bars) and LC-MS/MS with external calibration (black bars) measurements of samples collected from six regions of interest (red circles in the inset images; the regions shown are the hippocampus (1), cortex/fiber tracts (2), thalamus (3), cauduputamen/fiber tracts (4), hypothalamus (5), and cortical subplate (6)). RR indicates relative recovery or accuracy of iMSI results, calculated by dividing the quantitation results with iMSI with the LC-MS/MS results. Inset: molecular images from 3 different rats of Cer d18:1/18:0 signal distributions in 6 rat brain sections collected from each brain. Means and standard deviations are shown. Only one technical replicate was performed for the iMSI with kinetic calibration for rat #3.
An interesting question is why Area #1 for rat 2 and 3 had discrepancies between the two approaches. This area covered one of the more spatially heterogeneous brain regions,43 and includes the CA1 and dentate gyrus (DG) cell layers of the hippocampus. Because of this, it may be that slight differences in punches and ROIs could have caused the issues. In addition, we often observe cracks in the dried tissues separating these brain regions from the cortex when drying tissue sections on ITO glass slides. Both effects can be mitigated using high spatial resolution MSI (as opposed to tissue punches) and indicate the need for optimum sample preparation approaches. Moreover, laser caption microdissection44–45 could be used to sample smaller tissue areas at a higher spatial precision, alleviating issues with slight differences in tissue punch locations for the LC-MS measurements. Overall, the quantitative results between MSI and LC-MS are comparable (relative recoveries 72%–200%, r=0.93) (Figure 5), with LC-MS achieving slightly higher precision.
CONCLUSIONS
Many MSI studies have moved from analyte identification to comparing levels of analytes between brain regions. Given the different composition of tissues, even within closely located brain regions, approaches that can deal with chemical heterogeneity are needed. Different sample preparation techniques have been developed to obtain well-defined concentrations of standards on the sample surface, allowing calibration in quantitative analysis. However, differences in the mass transfer rates between endogenous analytes and spiked standards in tissues makes quantitation in chemically and structurally complex biological samples difficult. In this work, a TiO2-DA monolith with pre-loaded standards and imprinted brain tissue with kinetic calibration enabled MSI with robust semi-quantitative results. Experiments investigating tissue imprinting on TiO2-DA monolith revealed that molecules exhibiting Lewis base properties, such as phospholipids, ceramides and cholesterol, could be desorbed from brain tissue without obvious spatial delocalization. After imprinting, matrix effects were reduced, making measurements repeatable and less variable across distinct tissues. We find that quantitative iMSI utilizing kinetic calibration allows one to compensate for differences in local mass transfer for endogenous analytes. Comparison of absolute quantitation with iMSI with kinetic calibration and LC-MS/MS of analytes located in same brain regions produced similar quantitation results for the two distinct approaches (correlation coefficient >0.9, ratio between results 0.72–2.0), while the quantitation results of traditional standard addition calibration differed much more when compared to LC-MS/MS (ratios = 2–50).
Supplementary Material
ACKNOWLEDGMENTS
Funding was provided by Abbott Nutrition through the Center for Nutrition, Learning, and Memory, University of Illinois, Urbana–Champaign, and the National Institute on Drug Abuse by Award No. P30 DA018310. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Supporting Information
Supporting Methods S1–S5 detailing sample preparations for wetter/dryer conditions; LC-MS/MS, data processing and kinetic analyses, mass transfer kinetics, and protein analysis; and Figures S1–S3 (as noted in the text).
The authors declare no competing financial interest.
References
- (1).Caprioli RM; Farmer TB; Gile J, Molecular Imaging of Biological Samples: Localization of Peptides and Proteins Using MALDI-TOF MS. Anal. Chem 1997, 69, 4751–4760. [DOI] [PubMed] [Google Scholar]
- (2).Schwartz SA; Caprioli RM, Imaging Mass Spectrometry: Viewing the Future In Mass Spectrometry Imaging: Principles and Protocols, Rubakhin SS; Sweedler JV, Eds. 2010; Vol. 656, pp 3–19. [DOI] [PubMed] [Google Scholar]
- (3).Monroe EB; Annangudi SP; Hatcher NG; Gutstein HB; Rubakhin SS; Sweedler JV, SIMS and MALDI MS imaging of the spinal cord. Proteomics 2008, 8, 3746–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Buchberger AR; DeLaney K; Johnson J; Li L, Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem 2018, 90, 240–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dufresne M; Thomas A; Breault-Turcot J; Masson J-F; Chaurand P, Silver-Assisted Laser Desorption Ionization For High Spatial Resolution Imaging Mass Spectrometry of Olefins from Thin Tissue Sections. Anal. Chem 2013, 85, 3318–3324. [DOI] [PubMed] [Google Scholar]
- (6).Wiseman JM; Ifa DR; Song Q; Cooks RG, Tissue Imaging at Atmospheric Pressure Using Desorption Electrospray Ionization (DESI) Mass Spectrometry. Angew. Chem. Int. Ed 2006, 45, 7188–7192. [DOI] [PubMed] [Google Scholar]
- (7).Chaurand P; Norris JL; Cornett DS; Mobley JA; Caprioli RM, New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J. Proteome Res 2006, 5, 2889–2900. [DOI] [PubMed] [Google Scholar]
- (8).Garrett TJ; Prieto-Conaway MC; Kovtoun V; Bui H; Izgarian N; Stafford G; Yost RA, Imaging of small molecules in tissue sections with a new intermediate-pressure MALDI linear ion trap mass spectrometer. Int. J. Mass spectrom 2007, 260, 166–176. [Google Scholar]
- (9).Aksenov AA; Bier ME, The analysis of polystyrene and polystyrene aggregates into the mega Dalton mass range by cryodetection MALDI TOF MS. J. Am. Soc. Mass Spectrom 2008, 19, 219–230. [DOI] [PubMed] [Google Scholar]
- (10).Liu HL; Dai JY; Zhou JH; Huang HY; Chen F; Liu ZL, A hybrid ionic liquid-matrix material, TiO2-Si-NH3+ CHC−, as a novel matrix for the analysis of small molecules by MALDI-TOF MS. Int. J. Mass spectrom 2015, 376, 85–89. [Google Scholar]
- (11).Cornett DS; Reyzer ML; Chaurand P; Caprioli RM, MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods 2007, 4, 828–833. [DOI] [PubMed] [Google Scholar]
- (12).McDonnell LA; Heeren RMA, Imaging mass spectrometry. Mass Spectrom. Rev 2007, 26, 606–643. [DOI] [PubMed] [Google Scholar]
- (13).Berry KAZ; Hankin JA; Barkley RM; Spraggins JM; Caprioli RM; Murphy RC, MALDI Imaging of Lipid Biochemistry in Tissues by Mass Spectrometry. Chem. Rev 2011, 111, 6491–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Goodwin RJA; Pennington SR; Pitt AR, Protein and peptides in pictures: Imaging with MALDI mass spectrometry. Proteomics 2008, 8, 3785–3800. [DOI] [PubMed] [Google Scholar]
- (15).Law KP; Larkin JR, Recent advances in SALDI-MS techniques and their chemical and bioanalytical applications. Anal. Bioanal. Chem 2011, 399, 2597–2622. [DOI] [PubMed] [Google Scholar]
- (16).Vidova V; Novak P; Strohalm M; Pol J; Havlicek V; Volny M, Laser Desorption-Ionization of Lipid Transfers: Tissue Mass Spectrometry Imaging without MALDI Matrix. Anal. Chem 2010, 82, 4994–4997. [DOI] [PubMed] [Google Scholar]
- (17).Wu Q; Chu JL; Rubakhin SS; Gillette MU; Sweedler JV, Dopamine-modified TiO2 monolith-assisted LDI MS imaging for simultaneous localization of small metabolites and lipids in mouse brain tissue with enhanced detection selectivity and sensitivity. Chemical Science 2017, 8, 3926–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ellis SR; Bruinen AL; Heeren RMA, A critical evaluation of the current state-of-the-art in quantitative imaging mass spectrometry. Anal. Bioanal. Chem 2014, 406, 1275–1289. [DOI] [PubMed] [Google Scholar]
- (19).Pirman DA; Reich RF; Kiss A; Heeren RMA; Yost RA, Quantitative MALDI Tandem Mass Spectrometric Imaging of Cocaine from Brain Tissue with a Deuterated Internal Standard. Anal. Chem 2013, 85, 1081–1089. [DOI] [PubMed] [Google Scholar]
- (20).Chumbley CW; Reyzer ML; Allen JL; Marriner GA; Via LE; Barry CE; Caprioli RM, Absolute Quantitative MALDI Imaging Mass Spectrometry: A Case of Rifampicin in Liver Tissues. Anal. Chem 2016, 88, 2392–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Groseclose MR; Castellino S, A Mimetic Tissue Model for the Quantification of Drug Distributions by MALDI Imaging Mass Spectrometry. Anal. Chem 2013, 85, 10099–10106. [DOI] [PubMed] [Google Scholar]
- (22).Barry JA; Ait-Belkacem R; Hardesty WM; Benakli L; Andonian C; Licea-Perez H; Stauber J; Castellino S, Multicenter Validation Study of Quantitative Imaging Mass Spectrometry. Anal. Chem 2019, 91, 6266–6274. [DOI] [PubMed] [Google Scholar]
- (23).Song X; He J; Pang X; Zhang J; Sun C; Huang L; Li C; Zang Q; Li X; Luo Z; Zhang R; Xie P; Liu X; Li Y; Chen X; Abliz Z, Virtual Calibration Quantitative Mass Spectrometry Imaging for Accurately Mapping Analytes across Heterogenous Biotissue. Anal. Chem 2019, 91, 2838–2846. [DOI] [PubMed] [Google Scholar]
- (24).Frick DA; Giesen C; Hemmerle T; Bodenmiller B; Guenther D, An internal standardisation strategy for quantitative immunoassay tissue imaging using laser ablation inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom 2015, 30, 254–259. [Google Scholar]
- (25).Mullen AK; Clench MR; Crosland S; Sharples KR, Determination of agrochemical compounds in soya plants by imaging matrix-assisted laser desorption/ionisation mass spectrometry. Rapid Commun. Mass Spectrom 2005, 19, 2507–2516. [DOI] [PubMed] [Google Scholar]
- (26).Si T; Li B; Comi TJ; Wu Y; Hu P; Wu Y; Min Y; Mitchell DA; Zhao H; Sweedler JV, Profiling of Microbial Colonies for High-Throughput Engineering of Multistep Enzymatic Reactions via Optically Guided Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. J. Am. Chem. Soc 2017, 139, 12466–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).de Oliveira DN; Ferreira MS; Catharino RR, Rapid and Simultaneous In Situ Assessment of Aflatoxins and Stilbenes Using Silica Plate Imprinting Mass Spectrometry Imaging. PLoS One 2014, 9, e90901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hemalatha RG; Ganayee MA; Pradeep T, Electrospun Nanofiber Mats as “Smart Surfaces” for Desorption Electrospray Ionization Mass Spectrometry (DESI MS)-Based Analysis and Imprint Imaging. Anal. Chem 2016, 88, 5710–5717. [DOI] [PubMed] [Google Scholar]
- (29).Ouyang G; Vuckovic D; Pawliszyn J, Nondestructive Sampling of Living Systems Using in Vivo Solid-Phase Microextraction. Chem. Rev 2011, 111, 2784–2814. [DOI] [PubMed] [Google Scholar]
- (30).Tian R; Zhang H; Ye M; Jiang X; Hu L; Li X; Bao X; Zou H, Selective Extraction of Peptides from Human Plasma by Highly Ordered Mesoporous Silica Particles for Peptidome Analysis. Angew. Chem. Int. Ed 2007, 46, 962–965. [DOI] [PubMed] [Google Scholar]
- (31).Ai J, Solid Phase Microextraction for Quantitative Analysis in Nonequilibrium Situations. Anal. Chem 1997, 69, 1230–1236. [Google Scholar]
- (32).Chen Y; O’Reilly J; Wang Y; Pawliszyn J, Standards in the extraction phase, a new approach to calibration of microextraction processes. Analyst 2004, 129, 702–703. [DOI] [PubMed] [Google Scholar]
- (33).Zhou SN; Zhang X; Ouyang G; Es-Haghi A; Pawliszyn J, On-fiber standardization technique for solid-coated solid-phase microextraction. Anal. Chem 2007, 79, 1221–1230. [Google Scholar]
- (34).Zhang X; Es-haghi A; Musteata FM; Ouyang G; Pawliszyn J, Quantitative in vivo microsampling for pharmacokinetic studies based on an integrated solid-phase microextraction system. Anal. Chem 2007, 79, 4507–4513. [DOI] [PubMed] [Google Scholar]
- (35).Musteata FM; Musteata ML; Pawliszyn J, Fast in vivo microextraction: A new tool for clinical analysis. Clin. Chem 2006, 52, 708–715. [DOI] [PubMed] [Google Scholar]
- (36).Kweon HK; Håkansson K, Selective Zirconium Dioxide-Based Enrichment of Phosphorylated Peptides for Mass Spectrometric Analysis. Anal. Chem 2006, 78, 1743–1749. [DOI] [PubMed] [Google Scholar]
- (37).Guerrero G; Mutin PH; Vioux A, Anchoring of Phosphonate and Phosphinate Coupling Molecules on Titania Particles. Chem. Mater 2001, 13, 4367–4373. [Google Scholar]
- (38).Tanl K; Sumizawa T; Watanabe M; Tachibana M; Koizumi H; Kiba T, Evaluation of titania as an ion-exchanger and as a ligand-exchanger in HPLC. Chromatographia 2002, 55, 33–37. [Google Scholar]
- (39).Shrivas K; Hayasaka T; Sugiura Y; Setou M, Method for Simultaneous Imaging of Endogenous Low Molecular Weight Metabolites in Mouse Brain Using TiO2 Nanoparticles in Nanoparticle-Assisted Laser Desorption/Ionization-Imaging Mass Spectrometry. Anal. Chem 2011, 83, 7283–7289. [DOI] [PubMed] [Google Scholar]
- (40).Salamatipour A; Blair IA; Mesaros C, Targeted Lipid Biomarker Quantitation Using Liquid Chromatography-Mass Spectrometry (LC-MS) In Targeted Biomarker Quantitation by LC-MS, Weng N; Jian W, Eds. 2017; p 273–287. [Google Scholar]
- (41).Folch J; Ascoli I; Lees M; Meath JA; LeBaron N, Preparation of lipide extracts from brain tissue. J. Biol. Chem 1951, 191, 833–841. [PubMed] [Google Scholar]
- (42).Folch J; Lees M; Sloane Stanley GH, A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem 1957, 226, 497–509. [PubMed] [Google Scholar]
- (43).Hawrylycz M; Ng L; Feng D; Sunkin S; Szafer A; Dang C, The Allen Brain Atlas. 2014; p 1111–1126. [Google Scholar]
- (44).Knittelfelder O; Traikov S; Vvedenskaya O; Schuhmann A; Segeletz S; Shevchenko A; Shevchenko A, Shotgun Lipidomics Combined with Laser Capture Microdissection: A Tool To Analyze Histological Zones in Cryosections of Tissues. Anal. Chem 2018, 90, 9868–9878. [DOI] [PubMed] [Google Scholar]
- (45).Bastrup J; Birkelund S; Asuni AA; Volbracht C; Stensballe A, Dual strategy for reduced signal-suppression effects in matrix-assisted laser desorption/ionization mass spectrometry imaging. Rapid Commun. Mass Spectrom 2019, 33, 1711–1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.