

Abstract
Alcohol exposure during fetal and early postnatal development can lead to an increased incidence of later life adult-onset diseases. Examples include central nervous system dysfunction, depression, anxiety, hyperactivity, and an inability to deal with stressful situations, increased infection and cancer. Direct effects of alcohol leading to developmental abnormalities often involve epigenetic modifications of genes that regulate cellular functions. Epigenetic marks carried over from the parents are known to undergo molecular programming events that happen early in embryonic development by a wave of DNA demethylation, which leaves the embryo with a fresh genomic composition. The proopiomelanocortin (Pomc) gene controls neuroendocrine-immune functions and is imprinted by fetal alcohol exposure. Recently, this gene has been shown to be hypermethylated through three generations. Additionally, the alcohol epigenetic marks on the Pomc gene are maintained in the male but not in the female germline during this transgenerational transmission. These data suggest that the male-specific chromosome might be involved in transmitting alcohol epigenetic marks through multiple generations.
Keywords: DNA methylation, fetal alcohol, male germline, proopiomelanocortin, stress hormone, transgenerational epigenetic
INTRODUCTION
Epigenetic state is a term used to define chemical modifications that occur within a genome without changing the DNA sequence. Epigenetic changes often occur during development where they help maintain the specification of cells during mitotic cell division. These changes can also be meiotically heritable, passing from one generation to the next. Thus, they are referred to as transgenerational epigenetic events, which is the focus here. The transgenerational inheritance of epigenetic changes in the genome provides an additional molecular mechanism, along with induction of genetic mutations, for the germline transmission of environmentally induced phenotypic change (Jirtle & Skinner 2007). In humans, a number of studies have recently identified diseases that result from disruption to the epigenetic state that is transmitted across generations. One of these diseases is Prader–Willi syndrome, which is characterized by decreased mental capacity and obesity. The syndrome is often caused by aberrant methylation, and appears to be the result of an allele that has passed through the male germline without clearing of the silent epigenetic state previously established in the grandmother (Buiting et al. 2003). Evidence for multi-generational transfer of alcohol-related disease is beginning to appear. It has been shown that maternal grandmothers who had alcohol-related medical problems have grandchildren with higher rates of fetal alcohol spectrum disorders (FASD) than those from control grandmothers (Kvigne et al. 2008). Major depressive disorders that often co-occur with alcohol use disorders in the grandparental generation are known to increase the risk to grandchildren (Olino et al. 2008). These findings suggest that inheritance of alcohol-related disorders occurs for multiple generations in humans. In this review, I will present evidence from animal studies to show alcohol-induced epigenetic changes and associated pathologies pass through the male germline without clearing of the silent epigenetic state previously established in the great grandparent, therefore providing support for transgenerational inheritance.
TRANSGENERATIONAL EPIGENETIC EFFECTS
First, it will be important to differentiate the transgenerational epigenetic effects from parental and grandparental effects. In a recent review, Dr. Grossniklaus and colleagues have argued that while some biologists consider all outcomes observed in both parents and offspring to be transgenerational, an epigenetic basis of environmentally induced effects can be inferred only if they last over multiple generations (Grossniklaus et al. 2013). Furthermore, some additional aspects need to be considered. For example, parents can influence their offspring not only by providing their DNA but also by contributing bioactive molecules in the egg and sperm cytoplasm, by providing nutrients and hormonal information during embryo-genesis and by taking care of offspring after birth. Many of these parental (and sometimes grandparental) effects will not have an epigenetic basis and will not involve modification in the germline (Grossniklaus et al. 2013). It is suggested that an epigenetic basis of transgenerational environmental effects can be considered only if they last over three generations for maternal exposures and two generations for paternal exposures (Jirtle & Skinner 2007). The reasoning behind this assumption is that an environmental exposure of an F0 generation gestating female directly exposes both the F1 generation fetuses and the germ cells present in those fetuses that will generate the F2 generation. The subsequent F3 generation would be the first generation that would not have been directly exposed to the environmental factor. Therefore, effects on the F1 and F2 generations can be due to direct exposure and so should be considered parental effects (Skinner, Manikkam & Guerrero-Bosagna 2010). In contrast, a transgenerational effect following exposure of an F0 generation gestating female is defined as an effect seen in the F3 or later generations (Jirtle & Skinner 2007; Skinner 2008). In the case of the male, an environmental exposure of an F0 generation male directly exposes his germ cells which will generate F1 generation fetuses. Therefore, transgenerational effects following exposure of an F0 male can be inferred only if they last in F2 generation fetuses. Transgenerational phenomena by definition do not involve direct exposure but involve epigenetic changes induced in the germline (Guerrero-Bosagna et al. 2010; Thornburg et al. 2010). We will name these two routes of transmission as germline-dependent and germline-independent inheritance, to distinguish between the transgenerational effects and the parental effects of environmentally induced epigenetic variations (summarized in Fig. 1).
Figure 1.
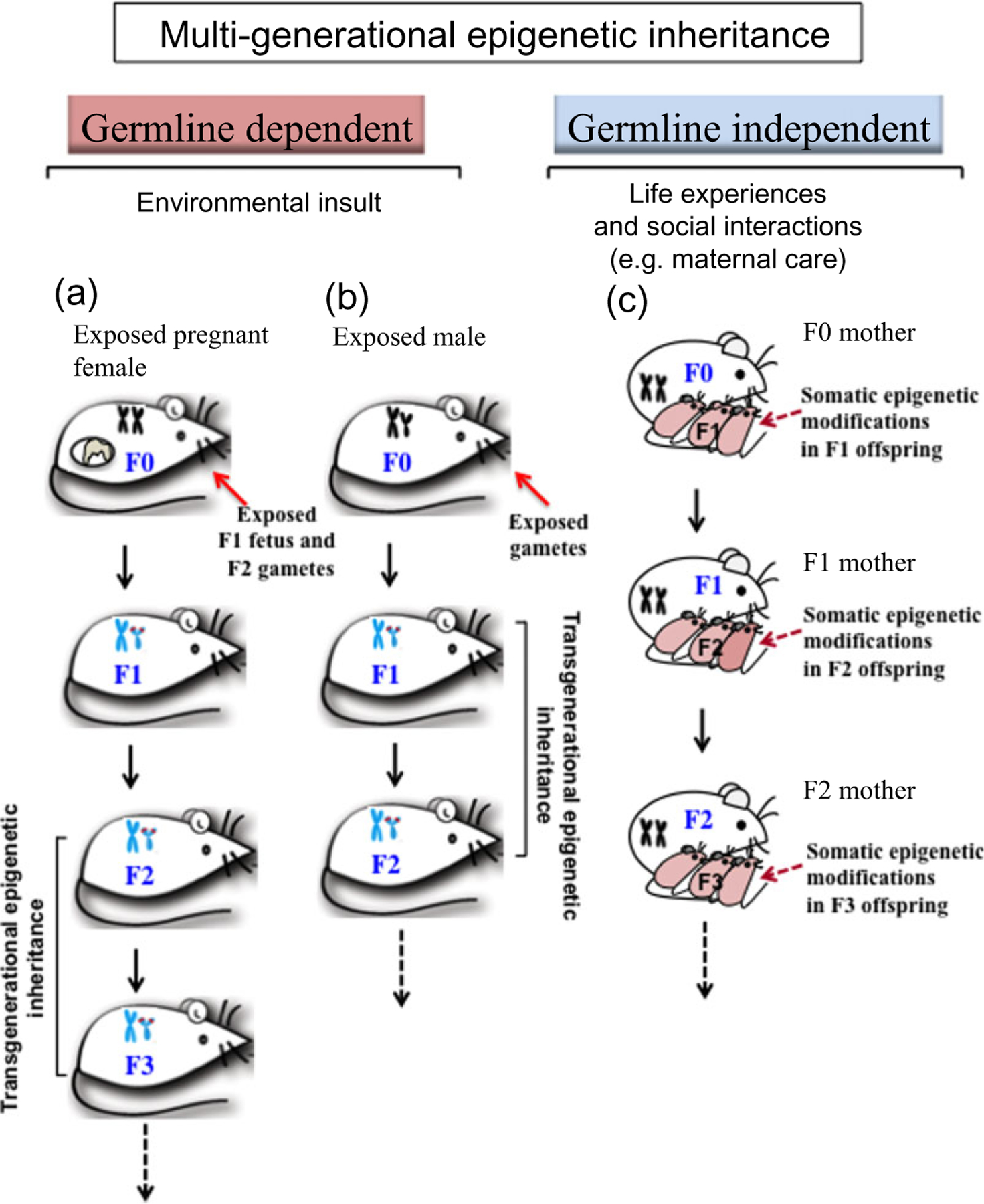
Proposed model of transgenerational epigenetic inheritance.The transmission of epigenetic marks across multiple generations is proposed to follow two paths: (1) germline dependent and (2) germline independent. In the germline-dependent inheritance, two mechanisms have been suggested. (a) Exposure of a gestating mother (F0) to an environmental stressor directly exposes both the F1 generation fetuses and the germ cells present in those fetuses that will generate the F2 generation.The subsequent F3 generation would be the first generation that would not have been directly exposed to the environmental factor. Hence, the transgenerational effect in this case is only observed at the F3 generation. (b) In the case of an F0 male, exposure to an environmental factor directly exposes his germ cells which will generate F1 generation fetuses. Therefore, transgenerational effects are seen at the F2 generation. (c) Germline-independent inheritance is acquired through social and behavioral interactions and through parental and grandparental effects. For example, low maternal licking and grooming of pups during the early postnatal period led to somatic epigenetic modifications that were associated with stress intolerance and were maintained in the adult female offspring (F1 mother) which in turn perpetuated the phenotype of low licking and grooming to the next generation of mothers (F2). Adapted from Rachdaoui & Sarkar (2014)
A. Germline-independent epigenetic inheritance
This type of inheritance is acquired through social and behavioral interactions and through parental and grandparental effects. For example, good maternal care through grooming and licking was associated with a decrease in DNA methylation at the nerve growth factor-inducible protein-A transcription factor (TF) binding site in the promoter region of the glucocorticoid receptor in the hippocampus of offspring rats (Weaver 2007; Weaver et al. 2007). This effect was associated with stress resilience and was passed on to the next generation. This seems to be, at least in part, socially transmitted, because pups that were given less maternal care gave less maternal care themselves as parent. Furthermore, cross-fostering reversed the effect. In addition, early postnatal cross-fostering of these pups with low licking and grooming mothers reversed these effects, which suggests that the transmission of these epigenetic changes is germline independent (Weaver 2009; McGowan et al. 2011).
B. Germline-dependent epigenetic transmission
This type of transgenerational transmission of epigenetic marks requires the action of environmental factors during germline differentiation, a critical narrow window during which the germline undergoes epigenetic reprogramming (Jirtle & Skinner 2007; Skinner et al. 2010). The most sensitive developmental period to environmental perturbations is the fetal gonadal sex determination (Anway et al. 2005; Skinner et al. 2010). After fertilization, the primordial germ cells undergo an erasure of DNA methylation to produce pluripotent embryonic stem cells. At the blastula stage of embryonic development, during gonadal sex determination, the DNA is remethylated in a sex-specific manner to generate male or female germlines (Reik, Dean & Walter 2001; Morgan et al. 2005). It was suggested that some germline imprinted genes or epimutations escape this DNA demethylation process, allowing transmission of permanent epigenetic changes across generations (Skinner et al. 2010). For example, exposures to an anti-androgen endocrine disruptor vinclozolin have been shown to modulate the epigenome (as reflected by alterations in DNA methylation patterns of several genes) in adult rat males of the F1 generation, and this was followed by DNA methylation disruption in the spermatozoa of the F2 and F3 generations (Anway, Leathers & Skinner 2006a; Anway et al. 2006b). In addition, many male offspring phenotypes observed following initial F0 vinclozolin exposures persisted to the F4 generation. These phenotypes include excess tumors and disrupted kidney development (Anway et al. 2006a,b), increased anxiety traits and decreased reproductive behaviors (Crews et al. 2007). These data suggest that an altered germline epigenome induced by environmental insults can promote the transmission of abnormal states of cell and tissue differentiation and behavioral deficits for many generations.
KEY ELEMENTS OF TRANSGENERATIONAL EFFECTS
A. Epigenetic processes
The mechanisms by which environmental exposures cause transgenerational effects are not well understood. The process involving epigenetic modifications is believed to be an important component that involves chemical modifications to the DNA rather than mutations of the DNA sequence itself. Epigenetic modifications can be achieved in various ways including the addition of methyl groups to DNA, known as DNA methylation, or chemical modifications (e.g. methylation, acetylation, phosphorylation, ubiquitination, ADP-ribosylation and sumoylation) to the histone proteins that surround the DNA, known as histone modifications, and/or interference of gene transcription by small non-coding RNAs (ncRNAs) (Kugel & Goodrich 2012).
The process of DNA methylation involves the transfer of a methyl group onto the C5 position of the cytosine to form 5-methylcytosine (5mC). Methylation is a covalent modification of the DNA that is catalyzed by the activity of DNA methyltransferases (DNMTs) that transfer a methyl group from S-adenyl methionine (SAM) to the fifth carbon of a cytosine residue. The majority of DNA methylation occurs on cytosines next to a guanine nucleotide, also known as CpG sites. CpG islands (CGIs) are stretches of DNA roughly about 1000 base pairs long containing strings of CpG dinucleotides and often are not methylated (Bird et al. 1985). The majority of gene promoters in mammals (70 percent) show the presence of CGIs in or near them (Saxonov, Berg & Brutlag 2006). Methylation of CGIs is often regulated during development to control gene expression (Doerfler 1983). DNA methylation regulates gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of TS(s) to DNA (Moore, Le & Fan 2013). It is well recognized that DNA methylation, in concert with other regulators, is a major epigenetic factor controlling gene activities.
DNA demethylation is also an important epigenetic component for gene transcription and epigenetic programming. Recent studies have shown that 5mC can be oxidized to 5-hydroxymethylcytosine (5hmC). Tet proteins were recently identified as 5mC hydrolases that catalyze the conversion of 5mC of DNA to 5hmC, raising the possibility that DNA demethylation may be a Tet-mediated process (Tahiliani et al. 2009). It is also suggested that demethylation may be a process during which 5mC is converted into 5hmC without going through demethylation (Wu & Zhang 2009). Biological roles of 5hmC are not clear. Ficz et al. (2011) showed that the presence of 5hmC in promoter regions was associated with high levels of transcription, suggesting a role on long- and short-term regulation of gene expression. 5hmC is not recognized by DNMTs, which will prevent maintenance methylation during DNA replication, resulting in passive DNA demethylation. Current evidence suggests that both ‘passive’ and ‘active’ mechanisms may contribute to the DNA demethylation of the parental genome and participate in epigenetic programming and reprogramming during development (Cantone & Fisher 2013).
Histone modifications are another major biological process by which epigenetic regulation of gene expression occurs. Histones are highly basic proteins that help in the packaging of DNA into nucleosomes, the binding blocks of chromatin. These proteins are subject to epigenetic changes to the histone proteins that surround the DNA, known as histone modifications in response to internal or external factors including alcohol. Histone 3 (H3) and histone 4 (H4) are commonly studied, as they have tails that can be covalently modified by methylation or acetylation. The histone tail undergoes malleable posttranslational modification and allows the chromatin to exist in a relaxed state (euchromatin) that is conductive for gene activation or in a compacted state (heterochromatin) that is conductive for gene repression. Heterochromatin is associated with hypoacetylation (deacetylated state) for H3 and H4, and di- or trimethylation of the ninth lysine residue on H3 (H3K9me2 or H3K9me3). The open state, euchromatin, is associated with acetylated H3 and H4, and di- or trimethylation of the fourth lysine residue of H3 (H3K4me2 or H3K4me3) (Arney & Fisher 2004). Thus, differential histone modifications produce dual effects on gene transcription, such as gene activation or repression.
Recent evidence has demonstrated involvement of a number of small ncRNAs (~20–30 nucleotides), such as short interfering RNAs (siRNAs), microRNAs (miRNAs) and PIWI-interacting RNAs (piRNAs) (Aalto & Pasquinelli 2012), in epigenetic silencing of specific genes and in the protection of the genome against viruses and transposons (Moazed 2009; Luteijn & Ketting 2013).
B. Epigenetic marks
The DNA methylation/demethylation and histone modifications are passed on to the daughter cells when a cell divides. They are referred to as epigenetic changes because the level of regulation is above (‘epi’) the direct genetic modifications of the DNA (Hemberger, Dean & Reik 2009). Scientists often use the term ‘marks’ or ‘tags’ to define environmental factors that induce chemical modifications of DNA and/or histones. Epigenetic marks carried over from the parents are generally cleaned off during molecular programming events that happen during embryonic development. After fertilization, a wave of DNA demethylation leaves the embryo with a fresh genomic composition with the exception of certain imprinted genes which remain methylated. Later, cells in the developing embryo are remethylated as they develop into the somatic cells that make up different organs and tissues in the body (Shi & Wu 2009). Germ cells, meanwhile, undergo their own wave of demethylation and remethylation programming events, which are specific to the sex of the developing embryo. However, it is now becoming increasingly clear that epigenetic marks in germ cells are more stable and persistent than initially assumed (Bohacek & Mansuy 2013). A recent genome-wide DNA methylation study revealed that a substantial number of genes retain parental DNA methylation in promoter regions after fertilization, which is an important requirement for the transgenerational transmission of DNA methylation (Borgel et al. 2010). Similarly, not all histones in sperm are replaced by protamines and those that are maintained likely keep their posttranslational modifications and can be transferred to the oocyte (Hammoud et al. 2009; Brykczynska et al. 2010). In addition to DNA methylation and histone modifications, new data also point to a role for ncRNAs in the transfer of transgenerational information through the germline (Johnson et al. 2011). A number of studies provided evidence to show that a subset of cytosine methylation patterns in sperm is known to be heritable (Rakyan & Whitelaw 2003; Rakyan et al. 2003; Waterland & Jirtle 2003; Cropley et al. 2006; Chong et al. 2007). Additionally, RNA molecules packaged in sperm have shown to affect offspring phenotype (Rassoulzadegan et al. 2006; Wagner et al. 2008; Carone et al. 2010). Furthermore, since sperms are largely devoid of histone proteins, chromatin structure has been proposed to carry epigenetic information (Chong et al. 2007; Arpanahi et al. 2009; Brykczynska et al. 2010; Carone et al. 2010).
A role of site-specific non-CpG methylation has also been proposed in transgenerational inheritance. While a majority of the methylated residues are in the symmetrical (meC)pG:Gp(meC) configuration, a smaller but significant fraction is found in the CpA, CpT and CpC asymmetric (non-CpG) dinucleotides. Nishino et al. (2011) found existence of a unique DNA methylation at the CCTGG site in the Sry promoter regions and that this methylation occurred reciprocally to CpG methylation in the developmental process. However, this non-CpG methylation is transient and unstable on the Y chromo-some. Additionally, Grandjean et al. (2007) presented data to show that paternal inheritance of epigenetic information in the form of CpG methylation patterns directs the establishment of site-specific non-CpG methylation. Hence, further studies are needed to establish the role of non-CpG methylation in transgenerational inheritance.
A number of studies have recently reported the presence of extensive DNA methylation in gene bodies downstream of transcription start sites (TSS) (intragenic methylation) (Hellman & Chess 2007; Ball et al. 2009; Kulis et al. 2013). It was noticed that in human cells most of the DNA methylation peaks are found in gene bodies rather than in promoters. Although incompletely understood, intragenic DNA methylation exerts multiple potential functions such as reducing gene expression by regulating transcriptional elongation efficiency (Lorincz et al. 2004), determining alternative polyA site choice (Wood et al. 2008) and tissue-specific selection of alternative promoters (Maunakea et al. 2010). However, the functional role of gene body in sex-specific methylation imprinting is not well studied and remains to be elucidated.
Most available studies on transgenerational epigenetic inheritance have focused on understanding the role of maternal transmission of environmentally induced epigenetic modifications; studies on the effects of paternal epigenetic alterations on offspring are still scarce. In a recent study by Petropoulos, Matthews & Szyf (2014), it was reported that the administration of the synthetic glucocorticoid, dexamethasone, to adult male mice led to alterations of the DNA methylation patterns in the mature sperm cells of these animals which was associated with alterations in the expression and DNA methylation of nuclear steroid receptors (i.e. mineralocorticoid receptor, estrogen alpha receptor and glucocorticoid receptor) in somatic tissues such as hippocampus and kidney of offspring.
In our own work, we attempted to answer the question of whether alcohol-related disorders are transgenerationally inherited through multiple generations. We tested this hypothesis by using an animal model of fetal alcohol exposure. I will discuss the data from these studies in the next section of this review.
ALCOHOL EPIGENETIC MARKS ON PROOPIOMELANOCORTIN (Pomc) GENE AND THE STRESS AXIS
The question of whether the inheritance of alcohol-related disorders occurs for multiple generations has recently been tested using an animal model of fetal alcohol exposure. Drinking alcohol during pregnancy produces children with FASD. These children often show behavioral and physiological changes such as depression, anxiety, hyperactivity and an inability to deal with stressful situations (Schneider et al. 2002; Kelly et al. 2009; Kelley et al. 2010), as well as increased neonatal infection and childhood leukemia (Gauthier et al. 2005; Latino-Martel et al. 2010). Rodents exposed to alcohol during fetal life also show pathologies including anxiety, stress hyperresponsiveness, immune problems and increased susceptibility to carcinogenesis (Sarkar et al. 2007; Boyadjieva et al. 2009; Hellemans et al. 2010; Polanco et al. 2010; Murugan et al. 2013).
The hypothalamic β-endorphin is implicated in the behavioral as well as the biological responses associated with stress stimuli. The well-studied effect of β-endorphin is its ability to modulate pain, but the peptide has also been implicated in the central regulation of hypothalamic corticotrophin-releasing hormone (CRH) (Plotsky 1986). Indeed, β-endorphin has been shown to play an important role in the negative feedback loop controlling the stress response. In response to stress, the secretion of CRH and catecholamines stimulates the secretion of β-endorphin from the hypothalamus that participates in the negative feedback loop involving adrenocorticotropic hormone (ACTH) from the pituitary gland and glucocorticoids from the adrenal gland. Central β-endorphin binds to δ and μ opioid receptors and modulates the autonomic nervous system via neurons within the paraventricular nucleus. β-endorphin produced from pituitary POMC that circulates in the periphery is primarily regulated by CRH and argi-nine vasopressin and has less impact on autonomic nervous system function (reviewed in Wynne & Sarkar 2013).
POMC neurons in the arcuate nucleus of the hypothalamus play a critical function in regulation of the HPA (hypothalamic-pituitary-adrenal) axis as well as reward pathways, and the immune system, through the neuropeptides melanocortin, ACTH and β-endorphin derived from the POMC precursor polypeptide (Chen et al. 2006). POMC neurons were found to be impaired in both fetal alcohol-exposed (FAE) rats and postnatally ethanol-exposed rats (Sarkar et al. 2007; Boyadjieva et al. 2009). Glucocorticoids show levels that are low in the alcohol-exposed fetus, whereas they are elevated in the FAE neonate and FAE adult rats (Hellemans et al. 2010). Recent experiments to replace β-endorphin/POMC-producing cells in FAE rats demonstrated an improvement in stress and immune response in the animals, demonstrating a role for POMC in FASD (Boyadjieva et al. 2009).
Using an animal model of fetal alcohol exposure (Chen et al. 2006), we determined whether alcohol exposure during the developmental period incites epigenetic marks leading to permanent alteration of Pomc gene expression in the hypothalamus. Furthermore, we tested whether fetal alcohol produces transgenerational effects on the Pomc gene. The results of this study are recently published (Chen et al. 2006; Govorko et al. 2012; Bekdash, Zhang & Sarkar 2013). Alcohol feeding via a liquid diet to pregnant dams produced a significant deficit in Pomc neuronal functions in the hypothalamus, including lower Pomc gene expression and its protein product β-endorphin production, of both male and female offspring during the adult period. Alcohol fetal programming that causes this lower expression of Pomc gene appears to involve DNA methylation. This is because FAE animals showed increased methylation of several CpG dinucleotides in the proximal part of the Pomc gene promoter region. CpG methylation in the promoter region of a gene most often correlates with silencing of its promoter activity (Deaton & Bird 2011). A group of CpG dinucleotides in the promoter region or around the TSS of a gene are usually unmethylated. Abnormal methylation of the promoter CGIs in response to external or internal factors has been associated with gene silencing. We found that one of the hypermethylated CpGs at position −62 upstream of Pomc gene TSS coincided with the CCAAT box which is a binding site for TFs essential for transcriptional activation. Another methylated CpG we found in FAE rats at position −216 contained an 11 bp sequence which is highly conserved in vertebrates and is important for Pomc gene expression in target tissues (Bumaschny et al. 2007), suggesting that the hypermethylation of this site by FAE could be one of the causes of the decrease in Pomc gene expression.
The mechanism by which FAE programs the POMC cells to hypermethylate the Pomc gene promoter has also been studied (Govorko et al. 2012; Bekdash et al. 2013). It has been shown that administrations of Trichostatin A (TSA), the inhibitor of histone deacetylases (HDACs), or 5′-Azacytidine (5′-Aza), the inhibitor of DNMTs, during the neonatal period normalized fetal alcohol-induced Pomc gene hypermethylation in the adult stage, suggesting roles for both histone modification and DNA methylation machineries in fetal alcohol programming of POMC neurons. Measurements of the changes in protein and gene levels of histone-modifying proteins and DNMT levels in POMC neurons provided plausible mechanisms by which alcohol programs histone modification and DNA methylation to increase Pomc gene methylation and expression. It was observed that FAE decreased the level of histone-modifying enzymes that methylate H3K4 and its associated gene Set7/9, and acetylate H3K9 and its associated gene CREB-binding protein (CBP). Additionally, FAE increased the level of HDAC2, which is known to suppress H3K9 acetylation in the brain (Pandey et al. 2008). H3K4 methylation and H3K9 acetylation are known to activate gene expression. On the other hand, FAE increased the levels of histone-modifying enzymes that methylate H3K9 and its associated genes G9a and Setdb1. These are repressive marks for gene activation. Hence, increased H3K9 methylation/deacetylation and decreased H3K4 methylation might be the key modifications of the histone tail surrounding the POMC DNA in FAE animals (Fig. 2).
Figure 2.
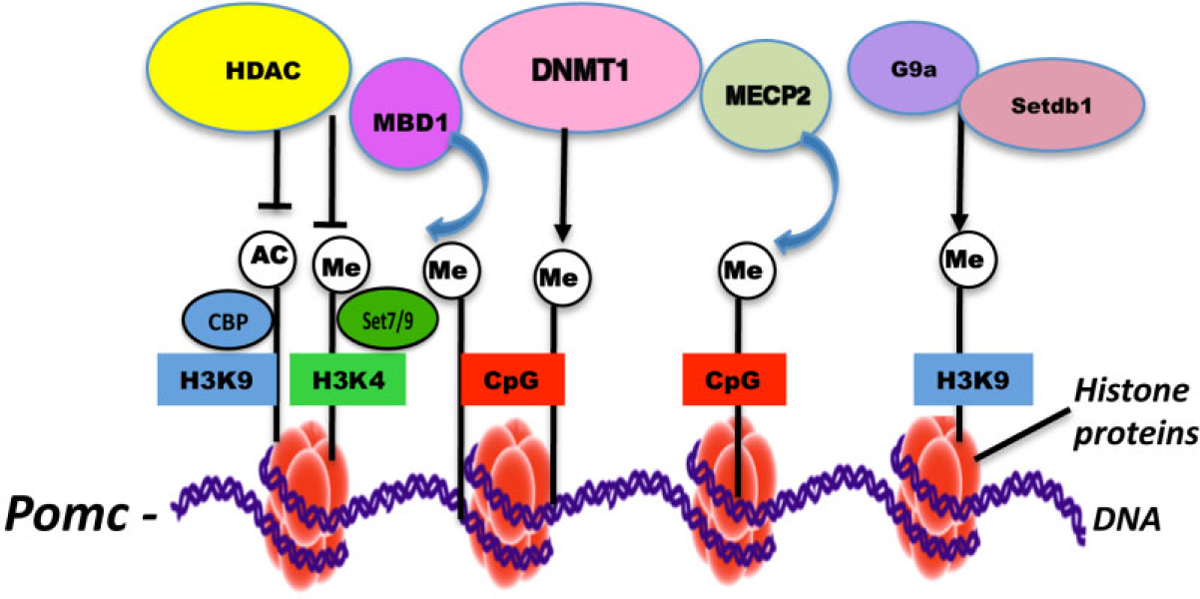
Fetal alcohol marks on epigenetic machinery that may repress POMC gene expression. It was observed that fetal alcohol exposure decreases the level of histone-modifying enzymes that methylate H3 lysine 4 (H3K4) and its associated gene Set7/9 and acetylate H3 lysine 9 (H3K9) and its associated gene CREB-binding protein (CBP) expression. Fetal alcohol exposure also increases the levels of histone-modifying enzymes that methylate H3K9 and its associated genes G9a and Setdb1 expression. Increased levels of DNA methyltransferases1 (DNMT1), methyl-CpG-binding protein (MeCP2) and methyl-binding proteins1 (MBD1) were also observed in POMC cells in fetal alcohol exposure offspring. It is postulated that histone modifications such as H3 lysine 9 acetylation and H3K4 methylation create a signal that regulates POMC gene transcription. Histone deacetylases (HDACs) remove acetyl groups from H3 lysine residues making way for methylation. HDACs also activate DNA methyltransferases (DNMT) and methyl-C-binding proteins (MBD1 and MeCP2) that aid in spreading the silencing signal
Ethanol exposure during the human third trimester equivalent has been shown to alter histone acetylation in the developing rat cerebellum (Guo et al. 2011), enhanced activity of G9a (lysine dimethyltransferase) and increased levels of histone H3 lysine 9 (H3K9me2) and 27 (H3K27) dimethylation in developing mouse hippocampus (Subbanna et al. 2013). Using mice neural stem cell population in vitro, Veazey et al. (2013) also have shown ethanol-induced reductions in H3K27me3 and H3K4me3 at promoters of genes involved in neural precursor cell identity and differentiation. Global transcriptome analysis also showed corresponding changes in gene expression in the brain in a maternal voluntary consumption model of FASD (Kleiber et al. 2014). These results suggest FAE affects histone modification globally rather than in POMC-specific cells to cause HPA-associated phenotypes relevant to FASD.
It has been proposed that DNMTs could only access the DNA that is wrapped around nucleosomal histones with H3K9 methylation signal (Vaissière, Sawan & Herceg 2008). Ethanol has been shown to interfere with one-carbon metabolism, the primary methyl donor in the DNMT pathway (Halsted et al. 2002; Fowler et al. 2012). This is accomplished in part by reducing folate availability. Folate is converted in a stepwise process to methionine, which is then converted to the active methyl donor SAM. Ethanol can also reduce SAM levels by reducing the activity of methionine synthase (Barak, Beckenhauer & Tuma 1996). Reductions in SAM impair the ability of DNMTs to maintain DNA methylation. DNA methylation involves covalent modification of the cytosine residue in CpG dinucleotides in the promoter region to ‘lock in’ the silent state of a gene and is catalyzed by the activity of DNMTs (Deaton & Bird 2011). DNMTs include DNMT1, DNMT2, DNMT3a, DNMT3b and DNMT3L. DNA methylation studies in mice have shown that adult mice prenatally exposed to ethanol have alterations in methylation-sensitive genes (Kaminen-Ahola et al. 2010) and show broad alterations when examined at the whole-genome scale, including within imprinted regions (Laufer et al. 2013). Alcohol exposures also have shown to alter DNA methylation profiles in mouse embryos at early neurulation (Liu et al. 2009). Hence, we tested whether significant changes in DNMT activities have also occurred in POMC cells of FAE animals. We found that the protein and transcript levels of DNMT1 are increased in POMC cells of FAE offspring. In enkaryotes, this enzyme utilizes SAM to methylate carbon C5 of cytosine (5-mC) that is located next to a guanine (G) in CpG dinucleotides in the promoter. It has been shown that there is ‘cross-talk’ between the DNA methylation machinery and the histone modifiers (Bird 2001). Methylated DNA most often recruits several methyl-binding proteins (MBDs) to promote gene repression. Increased levels of methyl-CpG-binding protein (MeCP2) and MBD1 were also observed in POMC cells of FAE offspring. H3K9 methylation itself recruits MBDs or MeCP2 and other chromatin-modifying factors to the promoter of a specific gene and leads to gene repression (Sarraf & Stancheva 2004; Vaissière et al. 2008; Guibert & Weber 2013). In summary, it could be hypothesized that FAE epigenetic marks involve methylation of Pomc gene via suppression of H3 lysine 9 acetylation, activation of HDACs (e.g. HDAC1) and various methylation-promoting genes (G9a, Setdb1). DNMT 1 and MBD1/MeCP2 assist in spreading the silencing signal (Fig. 2).
Fetal alcohol exposures also induced some endophenotypes of the Pomc gene expression defect, including elevated basal and immune stimulus (lipopolysaccharide)-activated ACTH and corticosterone levels in the plasma of both male and female offspring (Govorko et al. 2012). Noticeably, the suppression of histone deacetylation and DNA methylation by pharmacological agents normalized Pomc gene expression and POMC neuronal functional abnormalities (elevated corticosterone and ACTH responses to a stress challenge). These data suggest that FAE epigenetic marks on the Pomc gene might lead to the abnormal production of stress hormones and hyperstress response for a prolonged period of time in the offspring.
TRANSGENERATIONAL EPIGENETIC MODIFICATIONS CAUSED BY MATERNAL ALCOHOL CONSUMPTION CAN PROCEED THROUGH THE MALE GERMLINE
To determine whether the effect of fetal alcohol exposure on the Pomc gene is transmitted transgenerationally, we compared the POMC gene methylation and expression changes as well as the endophenotypes related to POMC gene expression defects in F1–F3 offspring of male germline and female germline (see Fig. 3). F1, F2 and F3 male progenies of male germline but not female germline demonstrated a significant increase in Pomc gene methylation and a decrease in expression levels. However, female progeny showed the fetal alcohol-induced changes in Pomc gene methylation and expression only in the F1 generation but not in F2 and F3 generation irrespective of their germline differences (Govorko et al. 2012). Similar transgenerational transmission of fetal alcohol effects was also observed on the stress hyperresponse (lipopolysaccharide-induced ACTH and corticosterone levels), which is considered one of the endophenotypes of POMC defects (Boyadjieva et al. 2009; Sarkar et al. 2012; Sarkar & Zhang 2013). Furthermore, the Pomc gene methylation defect was observed in sperm in F1 through F3 generations of male rats derived from the male germline. These results provide the first direct evidence that fetal alcohol effects on Pomc gene hypermethylation and stress axes abnormalities persist throughout adulthood and perpetuate into subsequent generations through the male germline.
Figure 3.
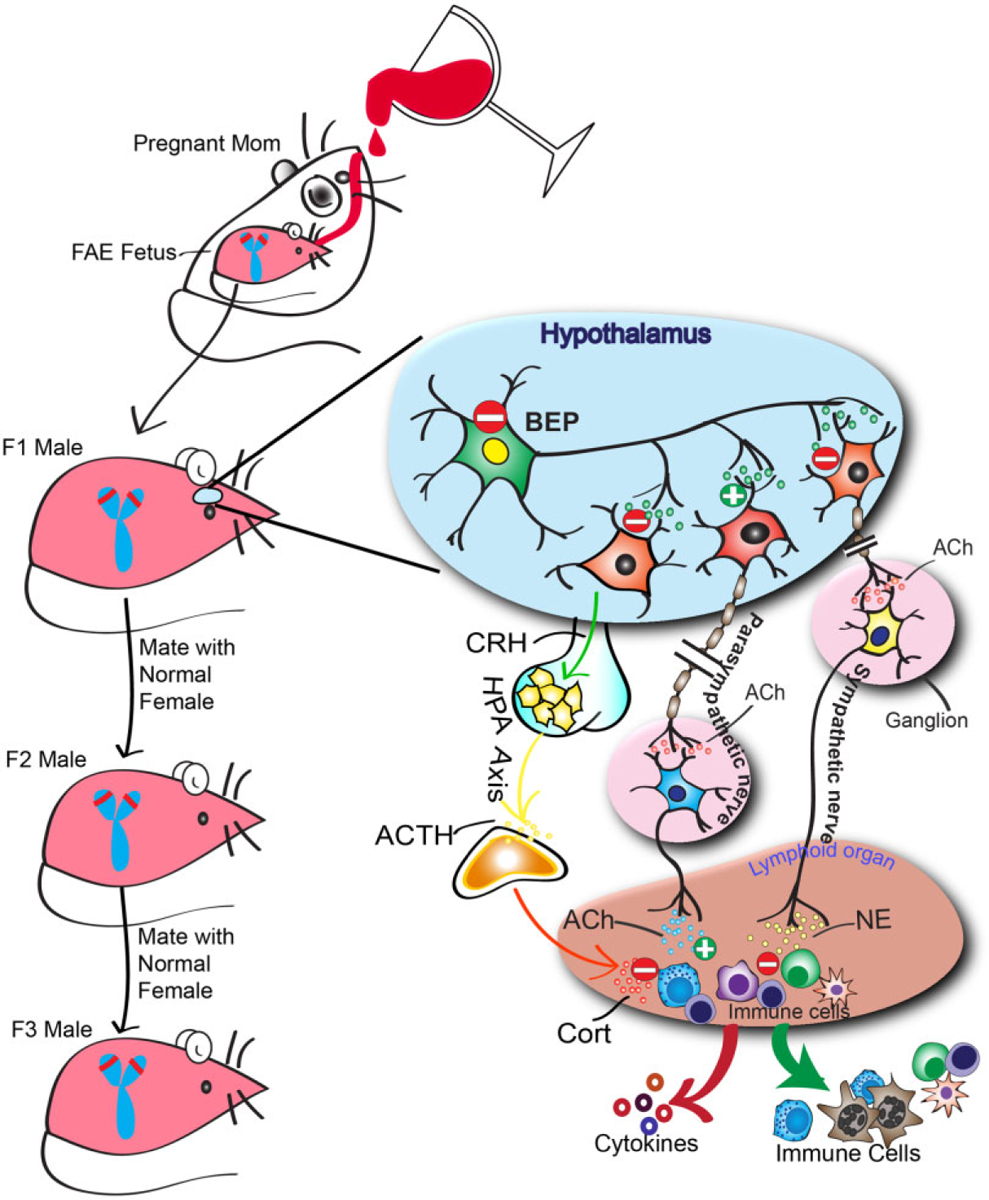
Transgenerational inheritance of fetal alcohol marks on the stress axis. Alcohol feeding in the mother produces epigenetic marks on the proopiomelanocortin (Pomc) gene of the offspring that leads to reduced (indicated by ‘-’) production of POMC-derived peptide β-endorphin (BEP), hyperstress response and compromised immune functions. Effects may include the suppression of the BEP stimulatory action (indicated by ‘+’) on the parasympathetic nervous system (which releases acetylcholine,Ach) and inhibitory action (indicated by ‘−’) on sympathetic neurons (which release norepinephrine, NE), thereby altering the autonomic neuronal control of innate immune cell functions and anti-inflammatory cytokine production.The hypothalamic-pituitary-adrenal axis that produces corticotrophin-releasing hormone (CRH) from the hypothalamus, adrenocorticotropic hormone (ACTH) from the pituitary and subsequent stress hormones released from the adrenal gland (corticosterone, Cort) may also be suppressed.As fetal alcohol exposure causes heritable sex-linked changes in Pomc expression in male rats, the possibility is raised that a sex-linked decrease in Pomc expression caused by fetal alcohol exposure (indicated by the red line on the short p arms of the Y chromosome) creates the transgenerational pattern observed
There are a number of studies in humans that provide indirect support of the heritability of alcohol-related disorders in the literature. In a recent study of Native American women who abused alcohol, F2 generation offspring (i.e. the grandchildren) of an alcohol-abusing woman have a higher tendency to show fetal alcohol syndrome than those who do F2 progeny of control women (Kvigne et al. 2008). There is also evidence that hypomethylation occurs in the sperm of alcoholic men (Ouko et al. 2009). Transmission of the effects of alcohol through the paternal line has precedents in the literature for induction of symptoms like those found in FASD. These include mental impairment, cardiac defects, low birth weight and hyperactivity, compared with controls, as assessed in human epidemiological studies and backed by animal studies (Abel 2004). This supports the findings of Govorko and colleagues that factors that impact POMC and subsequently affect the HPA axis and FASD can be transmitted by males through the germline.
HYPOTHETICAL MECHANISM OF TRANSGENERATIONAL TRANSMISSION OF THE ALCOHOL EPIGENETIC MARKS
As discussed earlier, transgenerational epigenetic marks on genes are less common as most epigenetic signatures are typically lost during gametogenesis. However, as discussed earlier, genome-wide DNA methylation studies revealed a number of genes retain parental DNA methylation in promoter regions after fertilization, which is an important factor for the transgenerational transmission of DNA methylation (Borgel et al. 2010; Bohacek & Mansuy 2013). The data presented here suggest that the Pomc gene is one of the genes that can retain alcohol-induced modification of the parental DNA promoter. Why is the Pomc gene resistant to developmental clearance? Prior to fertilization, the male gamete carries the fathers’ germline epigenetic signature. During development, protamines that are necessary for stabilization and dense condensation of the male DNA in spermatozoa replace most histones in late spermatogenesis. During gamete fusion, protamines are exchanged with female histones, typically acetylated histones, and the male DNA is demethylated (Fulka et al. 2004). This process leads to clearance of most of the environmentally induced epigenetic marks on the maternally or paternally derived DNA methylation. However, euchromatic regions including imprinted regions and CGIs may escape demethylation (Li, Kirschmann & Wallrath 2002). In the hypothalamus, POMC is regulated in part by the non-pairing region of the Y chromosome (YNPAR) (Botbol et al. 2011). Additionally, transgenerational changes in the expression of the Pomc gene in rats were detected in sperm (Botbol et al. 2011). As fetal alcohol exposure causes heritable sex-linked changes in Pomc expression in male rats, the possibility is raised that a sex-linked decrease in Pomc expression is due in part to epigenetic changes in the Y chromosome (YNPAR) caused by fetal alcohol exposure, creating the transgenerational pattern observed. Current studies are underway to test this hypothesis.
CONCLUSIONS AND FUTURE DIRECTIONS
Recent studies with various environmental toxins have shown that a genetic or developmental defect in the offspring of a mother exposed to a toxin during gestation may appear in later generations. Substances that have the ability to induce a transgenerational effect cause stable chromosomal alterations or an epigenetic phenomenon such as DNA methylation (Rakyan & Whitelaw 2003). I have summarized evidence to indicate that alcohol intake during pregnancy produces epigenetic marks on the Pomc gene, which involves increases in DNA methylation levels of the proximal part of the Pomc gene promoter with the concomitant decrease in Pomc gene expression levels and stress hyperresponse. Furthermore, this Pomc gene methylation defect was observed in germ cells. More importantly, these fetal alcohol-induced long-lasting changes at the molecular, cellular and organismal levels impacted subsequent generations via the male germline. The epigenetic mechanisms described in this review may provide a route through which developmental plasticity in one generation can be transmitted across multiple generations. Presently, it is not apparent how the epigenetic marks carried over from the parents bypass the demethylation process during molecular programming events that happen during embryonic development. One possibility is that the non-pairing euchromatic region of the Y chromosome (YNPAR) bypasses developmental molecular programming and therefore participates in the male-specific transgenerational transfer of the epigenetic mark (Li et al. 2002). Additional studies are needed for understanding the mechanism by which alcohol’s epigenetic marks are transmitted across generations in a sex-specific manner. It should also be emphasized that the lack of genome-wide studies employing ChIP-seq and RNA-seq is a limitation in the field and should be prioritized in future research. Genome-wide analyses may reveal the significance of POMC in FASD, particularly whether epigenetic changes in POMC play a major or a minor role in generating the endophenotypes observed in FASD. Furthermore, whether POMC and other players in the HPA axis interplay in a network to affect the neuroendocrine-immune axis functions.
Acknowledgements
The author acknowledges the contribution of Changqing Zhang for drawing the cartoon (Fig. 3). This study was supported by NIH R21 grant (AA16695) and R37 award (AA08757).
Footnotes
Disclosure/Conflict of Interest
The author reports no financial interests or potential conflicts of interest.
References
- Aalto AP, Pasquinelli AE (2012) Small non-coding RNAs mount a silent revolution in gene expression. Curr Opin Cell Biol 24:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel E (2004) Paternal contribution to fetal alcohol syndrome. Addict Biol 9:127–133. [DOI] [PubMed] [Google Scholar]
- Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308:1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Leathers C, Skinner MK (2006a) Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 12:5515–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Memon MA, Uzumcu M, Skinner MK (2006b) Transgenerational effect of the endocrine disruptor vinclozolin on male spermatogenesis. J Androl 6:868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arney KL, Fisher AG (2004) Epigenetic aspects of differentiation. J Cell Sci 117 (Pt 19):4355–4363. [DOI] [PubMed] [Google Scholar]
- Arpanahi A, Brinkworth M, Iles D, Krawetz SA, Paradowska A, Platts AE, Saida M, Steger K, Tedder P, Miller D (2009) Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res 19:1338–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM (2009) Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak AJ, Beckenhauer HC, Tuma DJ (1996) Betaine effects on hepatic methionine metabolism elicited by short-term ethanol feeding. Alcohol 13:483–486. [DOI] [PubMed] [Google Scholar]
- Bekdash RA, Zhang C, Sarkar DK (2013) Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res 37:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A (2001) Molecular biology. Methylation talk between histones and DNA. Science 294:2113–2115. [DOI] [PubMed] [Google Scholar]
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D (1985) A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40:91–99. [DOI] [PubMed] [Google Scholar]
- Bohacek J, Mansuy IM (2013) Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 38:220–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forné T, Weber M (2010) Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 42:1093–1100. [DOI] [PubMed] [Google Scholar]
- Botbol M, Roubertoux PL, Carlier M, Trabado S, Brailly-Tabard S, Perez-Diaz F, Bonnot O, Bronsard G, Tordjman S (2011) Modulation of brain β-endorphin concentration by the specific part of the Y chromosome in mice. PLoS ONE 6:e16704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, Ortigüela M, Arjona A, Cheng X, Sarkar DK (2009) Beta-endorphin neuronal cell transplant reduces corticotropin releasing hormone hyperresponse to lipopolysaccharide and eliminates natural killer cell functional deficiencies in fetal alcohol exposed rats. Alcohol Clin Exp Res 33:931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brykczynska U, Hisano M, Erkek S, Ramos L, Oakeley EJ, Roloff TC, Beisel C, Schübeler D, Stadler MB, Peters AH (2010) Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol 17:679–687. [DOI] [PubMed] [Google Scholar]
- Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B (2003) Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet 72:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumaschny VF, de Souza FS, López Leal RA, Santangelo AM, Baetscher M, Levi DH, Low MJ, Rubinstein M (2007) Transcriptional regulation of pituitary POMC is conserved at the vertebrate extremes despite great promoter sequence divergence. Mol Endocrinol 21:2738–2749. [DOI] [PubMed] [Google Scholar]
- Cantone I, Fisher AG (2013) Epigenetic programming and reprogramming during development. Nat Struct Mol Biol 20:282–289. [DOI] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, Meissner A, Weng Z, Hofmann HA, Friedman N, Rando OJ (2010) Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Kuhn P, Advis JP, Sarkar DK (2006) Prenatal ethanol exposure alters the expression of period genes governing the circadian function of beta-endorphin neurons in the hypothalamus. J Neurochem 97:1026–1033. [DOI] [PubMed] [Google Scholar]
- Chong S, Vickaryous N, Ashe A, Zamudio N, Youngson N, Hemley S, Stopka T, Skoultchi A, Matthews J, Scott HS, de Kretser D, O’Bryan M, Blewitt M, Whitelaw E (2007) Modifiers of epigenetic reprogramming show paternal effects in the mouse. Nat Genet 39:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews D, Gore AC, Hsu TS, Dangleben NL, Spinetta M, Schallert T, Anway MD, Skinner MK (2007) Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci USA 14:5942–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cropley JE, Suter CM, Beckman KB, Martin DI (2006) Germ-line epigenetic modification of the murine A vy allele by nutritional supplementation. Proc Natl Acad Sci USA 103:17308–17312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler W (1983) DNA methylation and gene activity. Annu Rev Biochem 52:93–124. [DOI] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473:398–402. [DOI] [PubMed] [Google Scholar]
- Fowler AK, Hewetson A, Agrawal RG, Dagda M, Dagda R, Moaddel R, Balbo S, Sanghvi M, Chen Y, Hogue RJ, Bergeson SE, Henderson GI, Kruman II (2012) Alcohol-induced one-carbon metabolism impairment promotes dysfunction of DNA base excision repair in adult brain. J Biol Chem 287:43533–43542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulka H, Mrazek M, Tepla O, Fulka J Jr. (2004) DNA methylation pattern in human zygotes and developing embryos. Reproduction 128:703–708. [DOI] [PubMed] [Google Scholar]
- Gauthier TW, Drews-Botsch C, Falek A, Coles C, Brown LA (2005) Maternal alcohol abuse and neonatal infection. Alcohol Clin Exp Res 29:1035–1043. [DOI] [PubMed] [Google Scholar]
- Govorko D, Bekdash RA, Zhang C, Sarkar DK (2012) Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry 72:378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean V, Yaman R, Cuzin F, Rassoulzadegan M (2007) Inheritance of an epigenetic mark: the CpG DNA methyltransferase 1 is required for de novo establishment of a complex pattern of non-CpG methylation. PLoS ONE 2:e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossniklaus U, Kelly WG, Ferguson-Smith AC, Pembrey M, Lindquist S (2013) Transgenerational epigenetic inheritance: how important is it? Nat Rev Genet 14:228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Settles M, Lucker BJ, Skinner MK (2010) Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE 5:e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibert S, Weber M (2013) Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol 104:47–83. [DOI] [PubMed] [Google Scholar]
- Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF, Xao X (2011) Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 6:e19351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halsted CH, Villanueva JA, Devlin AM, Niemela O, Parkkila S, Garrow TA, Wallock LM, Shigenaga MK, Melnyk S, James SJ (2002) Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci USA 99:10072–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR (2009) Distinctive chromatin in human sperm packages genes for embryo development. Nature 460:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KG, Sliwowska JH, Verma P, Weinberg J (2010) Prenatal alcohol exposure: fetal programming and later life vulnerability to stress, depression and anxiety disorders. Neurosci Biobehav Rev 34:791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman A, Chess A (2007) Gene body-specific methylation on the active X chromosome. Science 315:1141–1143. [DOI] [PubMed] [Google Scholar]
- Hemberger M, Dean W, Reik W (2009) Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol 10:526–537. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK (2007) Environmental epigenomics and disease susceptibility. Nat Rev Genet 8:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GD, Lalancette C, Linnemann AK, Leduc F, Boissonneault G, Krawetz SA (2011) The sperm nucleus: chromatin, RNA, and the nuclear matrix. Reproduction 141:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S (2010) Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 6:e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley ML, Braitman A, Henson JM, Schroeder V, Ladage J, Gumienny L (2010) Relationships among depressive mood symptoms and parent and peer relations in collegiate children of alcoholics. Am J Orthopsychiatry 80:204–212. [DOI] [PubMed] [Google Scholar]
- Nishino K, Hattori N, Sato S, Arai Y, Tanaka S, Nagy A, Shiota K (2011) Non-CpG methylation occurs in the regulatory region of the Sry gene. J Reprod Dev 57:586–593. [DOI] [PubMed] [Google Scholar]
- Olino TM, Pettit JW, Klein DN, Allen NB, Seeley JR, Lewinsohn PM (2008) Influence of parental and grandparental major depressive disorder on behavior problems in early childhood: a three-generation study. J Am Acad Child Adolesc Psychiatry 47:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouko LA, Shantikumar K, Knezovich J, Haycock P, Schnugh DJ, Ramsay M (2009) Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: implications for fetal alcohol spectrum disorders. Alcohol Clin Exp Res 33:1615–1627. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A (2008) Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28:3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S, Matthews SG, Szyf M (2014) Adult glucocorticoid exposure leads to transcriptional and DNA methylation changes in nuclear steroid receptors in the hippocampus and kidney of mouse male offspring. Biol Reprod 90:43. [DOI] [PubMed] [Google Scholar]
- Plotsky PM (1986) Opioid inhibition of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation of rats. Regul Pept 16:235–242. [DOI] [PubMed] [Google Scholar]
- Polanco TA, Crismale-Gann C, Reuhl KR, Sarkar DK, Cohick WS (2010) Fetal alcohol exposure increases mammary tumor susceptibility and alters tumor phenotype in rats. Alcohol Clin Exp Res 34:1879–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachdaoui N, Sarkar DK (2014) Transgenerational epigenetics and brain disorders. Int Rev Neurobiol 115:51–73. [DOI] [PubMed] [Google Scholar]
- Rakyan V, Whitelaw E (2003) Transgenerational epigenetic inheritance. Curr Biol 13:R6. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, Whitelaw E (2003) Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci USA 100:2538–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F (2006) RNA-mediated non-Mendelian inheritance of an epigenetic change in the mouse. Nature 441:469–474. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293:1089–1093. [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Zhang C (2013) Beta-endorphin neuron regulates stress response and innate immunity to prevent breast cancer growth and progression. Vitam Horm 93:263–276. [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Kuhn P, Marano J, Chen C, Boyadjieva N (2007) Alcohol exposure during the developmental period induces beta-endorphin neuronal death and causes alteration in the opioid control of stress axis function. Endocrinology 148:2828–2834. [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Murugan S, Zhang C, Boyadjieva N (2012) Regulation of cancer progression by β-endorphin neuron. Cancer Res 72:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Stancheva I (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15:595–605. [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL (2006) A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA 103:1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider ML, Moore CF, Kraemer GW, Roberts AD, DeJesus OT (2002) The impact of prenatal stress, fetal alcohol exposure, or both on development: perspectives from a primate model. Psychoneuroendocrinology 27:285–298. [DOI] [PubMed] [Google Scholar]
- Shi L, Wu J (2009) Epigenetic regulation in mammalian preimplantation embryo development. Reprod Biol Endocrinol 7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK (2008) What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol 25:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK, Manikkam M, Guerrero-Bosagna C (2010) Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab 21:214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Umapathy NS, Saito M, Mohan PS, Kumar A, Nixon RA, Verin AD, Psychoyos D, Basavarajappa BS (2013) G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol Dis 54:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg KL, Shannon J, Thuillier P, Turker MS (2010) In utero life and epigenetic predisposition for disease. Adv Genet 71:57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaissière T, Sawan C, Herceg Z (2008) Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 659:40–48. [DOI] [PubMed] [Google Scholar]
- Veazey KJ, Carnahan MN, Muller D, Miranda RC, Golding MC (2013) Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol Clin Exp Res 37:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KD, Wagner N, Ghanbarian H, Grandjean V, Gounon P, Cuzin F, Rassoulzadegan M (2008) RNA induction and inheritance of epigenetic cardiac hypertrophy in the mouse. Dev Cell 14:962–969. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23:5293–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC (2007) Epigenetic programming by maternal behavior and pharmacological intervention. Nature versus nurture: let’s call the whole thing off. Epigenetics 2:22–28. [DOI] [PubMed] [Google Scholar]
- Weaver IC (2009) Epigenetic effects of glucocorticoids. Semin Fetal Neonatal Med 14:143–150. [DOI] [PubMed] [Google Scholar]
- Weaver IC, D’Alessio AC, Brown SE, Hellstrom IC, Dymov S, Sharma S, Szyf M, Meaney MJ (2007) The transcription factor nerve growth factor-inducible protein A mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci 27:1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Schulz R, Woodfine K, Koltowska K, Beechey CV, Peters J, Bourc’his D, Oakey RJ (2008) Regulation of alternative polyadenylation by genomic imprinting. Genes Dev 22:1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Zhang Y (2009) Minireview: role of protein methylation and demethylation in nuclear hormone signaling. Mol Endocrinol 23:1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne O, Sarkar DK (2013) Stress and neuroendocrine-immune interaction: a therapeutic role for β-endorphin In: Kusnecov A, Anisman H, eds. Handbook of Psychoneuroimmunology, pp. 198–211. Oxford, UK.: Wiley Blackwell. [Google Scholar]