

Abstract
The striking and complex phenotype of Cockayne syndrome (CS) patients combines progeria-like features with developmental deficits. Since the establishment of the in vitro culture of skin fibroblasts derived from patients with CS in the 1970s, significant progress has been made in the understanding of the genetic alterations associated with the disease and their impact on molecular, cellular, and organismal functions. In this review, we provide a historic perspective on the research into CS by revisiting seminal papers in this field. We highlighted the great contributions of several researchers in the last decades, ranging from the cloning and characterization of CS genes to the molecular dissection of their roles in DNA repair, transcription, redox processes and metabolism control. We also provide a detailed description of all pathological mutations in genes ERCC6 and ERCC8 reported to date and their impact on CS-related proteins. Finally, we review the contributions (and limitations) of many genetic animal models to the study of CS and how cutting-edge technologies, such as cell reprogramming and state-of-the-art genome editing, are helping us to address unanswered questions.
Keywords: Cockayne syndrome, transcription-coupled nucleotide excision repair, neurodegeneration, progeroid syndrome, DNA repair
The epidemiology of Cockayne Syndrome (CS) and the discovery of the CS genes
Edward Alfred Cockayne first described CS in 1936. He diagnosed it in two young siblings (born to healthy parents) that displayed a set of very similar characteristics that included skin photosensitivity, short stature, prominent superior maxillae, disproportionally large hands and feet, sunken eyes with retinal atrophy, hearing impairment, below-average intelligence, a severely limited speaking ability, and muscle contraction, conditions that would later become characteristic of CS patients (Cockayne, 1936, 1946; Laugel, 2013). Subsequent reports also confirmed that patients with CS feature complex and heterogeneous neuropathology that includes calcification of the basal ganglia, cerebellar atrophy, loss of Purkinje and granular cells, hyperchromatic macroglial cells, microcephaly, and patchy demyelination (Guardiola et al., 1999; Karam et al., 2000; Weidenheim et al., 2009; Wilson et al., 2015; Karikkineth et al., 2017).
CS is an autosomal recessive disorder with a prevalence of ~2.7 per million births in Western Europe and in Japan (Kleijer et al., 2008; Kubota et al., 2015). The phenotype of the patients can range from mild to very severe and is subdivided into three types. The classical type (type I) corresponds to the moderate phenotype in which life expectancy is 16 years. In type II (the most severe and with the earliest onset), life expectancy is 5 years, whereas in the third type (mild and atypical), the phenotype manifests itself later in life, with life expectancy above 30 years. In all cases, pneumonia/respiratory ailments are the most common causes of death (Natale, 2010).
In vitro culture culture of skin fibroblasts derived from patients with CS in the 1970s was the first step toward the development of experimental models of the disease. CS fibroblasts are characterized by extreme sensitivity to ultraviolet light (UV) despite a normal ability to excise pyrimidine-dimers from the genome (Schmickel et al., 1977; Andrews et al., 1978). In fact, CS cells display a marked defect in the recovery of RNA synthesis after UV irradiation (Mayne and Lehmann, 1982) owing to a failure in the repair of transcriptionally active genes (Venema et al., 1990; van Hoffen et al., 1993). By evaluation of post-UV RNA synthesis recovery in multinucleated cells obtained by the fusion of cells from different CS donors, three complementation groups (A, B, and C) were initially identified (Tanaka et al., 1981; Lehmann, 1982). Group C identified by Lehmann corresponded to a patient that had combined features of CS and xeroderma pigmentosum (XP). Patients that fall in this category (termed XP/CS) manifest, in addition to CS features, the classical XP characteristics (skin pigmentation and extremely high skin cancer predisposition) and harbor mutations in the genes XPB, XPD, XPG, or (more recently identified) XPF or ERCC1 (Weeda et al., 1990; Kashiyama et al., 2013; Lehmann et al., 2014; Moriel-Carretero et al., 2015).
In the 1990s, the genes corresponding to groups A and B were cloned, characterized, and termed CSA and CSB, respectively. CSB was originally termed ERCC6 (excision repair cross-complementation group 6) because it was found to complement the nucleotide excision repair (NER) defect of the Chinese Hamster Ovary mutant cell line UV61, a representative of complementation group 6 of rodent cell lines defective in excision repair (Troelstra et al., 1990). Two years later, Hoeijmaker’s group demonstrated that the expression of this gene could reverse UV sensitivity and rescue post-UV RNA synthesis in a cell line (CS1AN) assigned to CS group B but not in group A cells (Troelstra et al., 1992). Another two years later, using episomal vectors to drive the expression of a cDNA library, Friedberg’s group was able to identify the gene capable of reversing the UV sensitivity of CS cells from group A (but not B) and to reactivate the expression of a UV-inactivated reporter gene (Henning et al., 1995). They discovered that the CSA protein, encoded by ERCC8 (Itoh et al., 1996), can interact with the CSB protein. Both genes play a critical role in the transcription-coupled nucleotide excision repair (TC-NER) of damaged DNA, which is described below in more detail.
CSA and CSB in TC-NER
Cells evolved complex and refined mechanisms to prevent genome instability in response to the presence of exogenously and endogenously generated DNA lesions. One of these mechanisms, conserved from bacteria to humans, is the NER pathway (Schärer, 2013; Marteijn et al., 2014; Gregersen and Svejstrup, 2018). This system drives the repair of bulky distorting DNA lesions (such as those induced by UV and by some redox processes) in four sequential steps: i) detection of a lesion, ii) excision of a DNA single-strand fragment containing the lesion, iii) DNA synthesis by a polymerase to fill the gap, and iv) nick sealing by a ligase (Costa et al., 2003; Reardon and Sancar, 2005; Menck and Munford, 2014). In bacteria, three proteins (UvrA, UvrB, and UvrC) are critical for the detection and excision of the lesion (Seeberg and Strike, 1976 ; Sancar and Rupp, 1983), whereas in humans, more than 30 proteins acting in an orchestrated manner are required for these steps, as reviewed elsewhere in detail (Menck and Munford, 2014). NER is subdivided into two sub-pathways: global genome repair (GG-NER) and transcription-coupled repair (TC-NER). They differ in how DNA lesions are detected, although the excision and DNA re-synthesis steps are shared by the two pathways (Menck and Munford, 2014). In humans, GG-NER is initiated by the XPC protein (Sugasawa et al., 1998), which is constantly scanning the whole genome for the presence of helix-distorting lesions (Hoogstraten et al., 2008), and the detection of UV products is facilitated by XPE/DDB2 (Cleaver et al., 2009). In TC-NER, as shown in Figure 1, the triggering event is the arrest of RNA polymerase II (RNA pol II) owing to the presence of a lesion in the actively transcribed strand of a gene (Bohr et al., 1985; Hanawalt and Spivak, 2008). It is in this pathway that the CSA and CSB proteins play a critical part. Upon RNA pol II blockade, the binding of CSB to RNA pol II is stabilized (Tantin et al., 1997; van den Boom et al., 2004; Fousteri et al., 2006) and CSB then wraps DNA around itself, altering its conformation and recruiting histone acetyltransferase p300 and core NER factors (such as RPA, XPG, and TFIIH) to RNA pol II arrest sites (Fousteri et al., 2006), as presented in Figure 1 (upper panel). CSB also recruits an E3-ubiquitin ligase complex, highlighted in green, composed of CSA (which contains WD motifs involved in protein-protein interactions), DDB1, Cullin 4A, and ROC1/Rbx1 proteins (Groisman et al., 2003; Fousteri et al., 2006). Although CSA is not necessary for the recruitment of NER factors, it is required for recruiting HMGN1, XAB2, and TFIIS to RNA pol II arrest sites (Fousteri et al., 2006). XAB2 is an XPA-interacting protein (Nakatsu et al., 2000) and might act as a scaffolding factor for protein assembly during TC-NER (Fousteri and Mullenders, 2008), while the nucleosome-binding protein, HMGN1, was suggested to promote chromatin changes that allow for the incision step (Fousteri and Mullenders, 2008).
Figure 1. Physical blockage of RNA polymerase II facilitates CSB binding to it. The following is responsible for recruiting p300, CSA and the other NER factors. CSA, together with CUL4, RBX1 and DDB1 are constantly ubiquitinating CSB, however UVSSA-USP7 complex are constantly removing ubiquitin tags from it. CSA also recruits HMGN1, which together with p300 unwinds chromatin upstream RNApol II, allowing it to backtrack and expose the lesion site to NER factors. TFIIS stimulates RNA cleavage by RNApol II during this process. NER factors unwinds DNA around the lesion. While RPA protects ssDNA from degradation, XPG and XPF-ERCC1 endonucleases cleave the strand containing the lesion. DNA polymerase and ligase then fill up the gap. CSB degradation is necessary to RNA synthesis recovery.

The fates of the nascent transcript and that of the stalled RNA pol II are still debated. One hypothesis is that transcript cleavage may occur in an elongation factor for RNA pol II (ELL)-dependent manner (Gregersen and Svejstrup, 2018). As for the stalled RNA pol II — which occupies a space that ranges from 25 nucleotides downstream of the lesion to 10 nucleotides upstream (Spivak and Ganesan, 2014), thus impairing the assembly of NER factors — it may undergo reverse translocation/backtracking (Donahue et al., 1994; Fousteri et al., 2006) or be targeted for proteasomal degradation by ubiquitination (Harreman et al., 2009), as shown in Figure 1 (lower panel).
Upon lesion resolution, the CSA-E3 ubiquitin ligase complex performs a critical function in the recovery of transcription by targeting CSB for proteasomal degradation (Groisman et al., 2006). To avoid premature degradation of CSB, UVSSA, which binds firmly to stalled RNA pol II, recruits USP7, an enzyme that promotes deubiquitination of CSB (Schwertman et al., 2012), highlighted in blue in Figure 1. Therefore, once the DNA lesion is removed, USP7-mediated deubiquitination of CSB ceases, and CSB is finally targeted for degradation.
In the absence of the CSA or CSB protein, the arrest of RNA pol II persists, an event that leads to p53 activation and cell death, thus explaining the extreme sensitivity of CS cells to UV damage (Ljungman and Zhang, 1996). Besides its participation in TC-NER, proteins CSA and CSB play several other important roles. In the following sections, we provide detailed descriptions of the structures of these proteins, their functions outside of TC-NER, and a comprehensive review of pathological mutations, their consequences for protein function, and their association with patients’ clinical characteristics.
The structure and functions of proteins CSA and CSB, or how pathological mutations are (not) associated with clinical phenotypes
CSB is a 168 kDa protein composed of 1493 amino acid residues and is encoded by the ERCC6 gene located in chromosomal region 10q11 (Troelstra et al., 1992, 1993). It belongs to the SWI2/SNF2 family of helicases, and just as all the proteins in this family, it does not have the capacity to open the DNA double helix (Selby and Sancar, 1997a). In contrast, the SWI2/SNF2 proteins temporarily modify DNA conformation via ATP hydrolysis, thereby altering the DNA contact with histones and nucleosome positioning. Therefore, SWI2/SNF2 proteins are considered chromatin remodelers (Lusser and Kadonaga, 2003; Beerens et al., 2005). In addition to its known classic function in TC-NER, CSB takes part in the regulation of transcription and assists with nuclear and mitochondrial base excision repair (BER). It has been reported that CSB interacts with proteins XPB, XPD, XPG, TFIIH, RNA pol I and II, and glycosylases (Tantin, 1998; Bradsher et al., 2002; Tuo et al., 2002; Sarker et al., 2005; Stevnsner et al., 2008; Kamenisch et al., 2010), although some of these interactions were described only once and need additional confirmation of their biological relevance.
The CSB structure mainly includes the following domains: an acidic domain, SNF2/ATPase region, ubiquitin-binding domain, and a nuclear localization signal (Liu et al., 2015). The acidic domain comprising amino acid residues 356 to 394 is located in the N-terminal portion, which is mostly negatively charged (Troelstra et al., 1992). In other proteins, this domain facilitates protein-protein interactions, especially those of nuclear and DNA-binding proteins, such as transcriptional activators and chromatin remodelers (Melcher, 2000; Carpenter et al., 2005; Wu et al., 2017). Given that CSB is classified as a chromatin remodeler, it has been hypothesized that its acidic domain facilitate this activity, but the possible underlying mechanisms have not yet been fully elucidated (Brosh et al., 1999).
Mutations in the acidic domain of CSB in UV61 cells do not compromise the ability to repair lesions caused by UV, 4-QNO (4-Nitroquinoline 1-oxide), and NA-AAF (N-acetoxy-2-acetylaminofluorene), or cell viability after exposure to these agents (Brosh et al., 1999; Sunesen et al., 2000). These data indicate that the integrity of this domain is not essential for this protein’s function in TC-NER. Similar results were obtained by Lake et al. (2010) with UV irradiation of CS1AN-SV cells expressing CSB protein lacking the first 454 amino acid residues in the N-terminal portion, demonstrating that the absence of this region does not compromise the ability of the protein to associate with chromatin but instead makes such associations much more frequent even without UV exposure. It was also observed that this deletion increases the ATPase activity of CSB, indicating that the N-terminal portion acts as a negative regulator of its association with chromatin via ATP hydrolysis (Lake et al. (2010)). CSB protein structure and homozygous and heterozygous pathological alterations are illustrated in Figure 2 A and B, whereas Table S1 lists all ERCC6 mutations reported in the literature.
Figure 2. Representation of CSB protein and domains. Acidic region (A), nuclear localization signal (N), helicase motifs (I, Ia -VI) and ubiquitin binding domain (UBD). (A) Homozygous mutations are indicated: frameshifts and nonsense mutations are indicated above the protein, while deletions and missense mutations are indicated below the protein. (B) Represents heterozygous mutations.
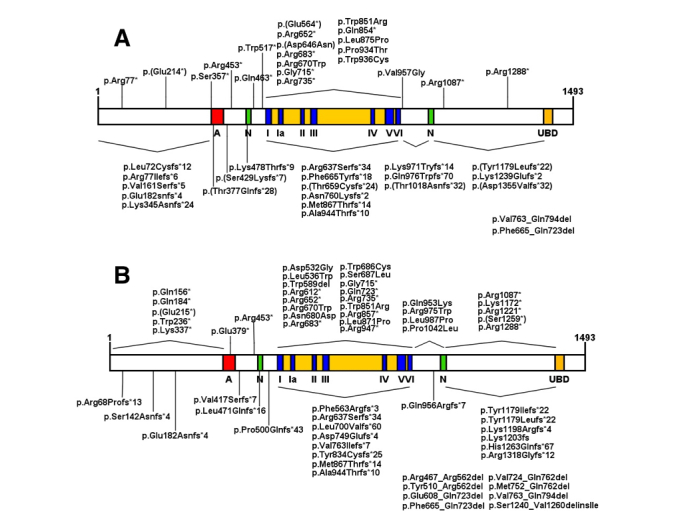
The nuclear localization signal is found within regions 466–481 and 1038–1055 (amino acid positions) (Lange et al., 2007). In a recent work, Iyama et al. (2018) identified through computational analysis the existence of a third region of nuclear localization signal, in addition to the three nucleolar localization signals that cooperate for the distribution of the protein between the nucleus and nucleolus.
Among these regions there is also the SNF2/ATPase domain, which is highly conserved in the SWI2/SNF2 family (Pazin and Kadonaga, 1997). This domain extends from amino acid residue 510 to residue 960 and contains seven ATPase motifs: I, Ia, II, II, IV, V, and VI (Troelstra et al., 1992), essential for the functioning of the protein (Brosh et al., 1999; Selzer et al., 2002). The function of the ATPase region is the most relevant for the activity of CSB, because this function provides energy for its association with (and remodeling of) chromatin by altering the positioning of nucleosomes (Citterio et al., 2000). Through this activity, CSB enables the repair of DNA lesions by promoting the access of other proteins, such as CSA and NER factors, to the site of stalled RNA pol II (Stadler and Richly, 2017). During the transcription process, CSB alters in situ chromatin conformation, favoring the binding of transcription factors (Lake et al., 2014).
To understand and characterize the functional importance of each ATPase motif, several cell lines carrying mutations in different and highly conserved regions in these motifs have been created. In general, amino acid substitutions in these regions decrease cell survival, RNA synthesis recovery, and DNA repair after exposure to UV, as well as increase the sensitivity to 4-NQO (Brosh et al., 1999; Muftuoglu et al., 2002; Selzer et al., 2002). Notably, mutations in domains I and II, named “Walker A” and “Walker B,” respectively, can completely inactivate the ATPase activity (Citterio et al., 1998; Christiansen et al., 2003). Mutations in motifs V and VI also compromise the ATPase activity, although to a lesser extent (Christiansen et al., 2003). Different motifs can also contribute in different ways to other activities carried out by CSB. Tuo et al. (2001) demonstrated that cells mutated in motifs V and VI are more sensitive to γ-radiation than wildtype cells, and DNA lesions such as 7,8-dyhydro-2’-deo-xyguanosine (8-oxoGua) accumulate in CSB-null and VI mutant-CSB cells after exposure to γ-radiation, indicating a possible relation between CSB and the BER pathway (Tuo et al., 2002a).
The ubiquitin-binding domain (UBD) is located in the C-terminal region of the CSB protein (amino acid residues 1400–1428). UBD-CSB-deficient cells have a phenotype similar to that of cells that do not express the CSB protein at all. Although the TC-NER complex is fully assembled around the lesion and RNA pol II in these cells, the repair does not proceed because of the inability of CSB to leave the lesion site (Anindya et al., 2010). The replacement of the CSB UBD by another UBD, such as UBA2 of Rad23, an otherwise unrelated Saccharomyces cerevisiae DNA repair gene, also enables CSB dissociation from the lesion region and progression of the repair process, thereby demonstrating the need for CSB ubiquitination for the correct functioning of the protein in this TC-NER (Anindya et al., 2010). Cells lacking UBD in the CSB protein are sensitive to oxidatively induced DNA damage (Ranes et al., 2016), suggesting that this domain is important for the repair of this kind of lesion. The conserved amino acid lysine at position 911 was recently found to be a ubiquitination site that is also required for this function, but is dispensable for TC-NER (Ranes et al., 2016).
By constructing several CSB mutants with different deletions in the C-terminal region, Sin et al. (2016) found that the integrity of the amino acid sequence in this region is important for this sumoylation of this protein and association with chromatin. Aside from this region, a functional UBD domain is necessary for RNA Pol II interaction and CSA recruitment to the nucleus (Sin et al., (2016)). In addition, Groisman (2006) demonstrated that the degradation of CSB depends on the action of an E3-ubiquitin ligase complex that contains CSA.
CSA is a 44 kDa protein 396 amino acid residues long and is encoded by the ERCC8 gene located in chromosomal region 5q12.1 (Henning et al., 1995). It belongs to the WD-repeat family because it contains 7 WD40 domains that are repeated in its structure. These domains consist of approximately 40 amino acid residues that start with a conserved glycine and histidine sequence and terminate in tryptophan and aspartic acid (WD), a seven-bladed propeller structure with its N terminus attached to DDB1 via a helix-loop-helix motif (Fischeret al., 2011). Proteins with the WD40 domain characteristically interact with other proteins and are typically known for their ability to form protein complexes (Xu and Min, 2011). Although they do not have a catalytic activity, they are involved in a variety of cellular functions, such as the regulation of transcription and chromatin conformation, apoptosis, signal transduction, and cell cycle control, among others (Xu and Min, 2011). CSA protein structure and homozygous/heterozygous pathological alterations are illustrated in Figure 3A and B. Table S2 shows all the ERCC8 mutations reported in the literature.
Figure 3. Representation of CSA protein and its seven WD-repeat domains. (A) Homozygous mutations are indicated: frameshifts and nonsense mutations are indicated above the protein, while deletions and missense mutations are indicated below the protein. (B) Represents heterozygous mutations.
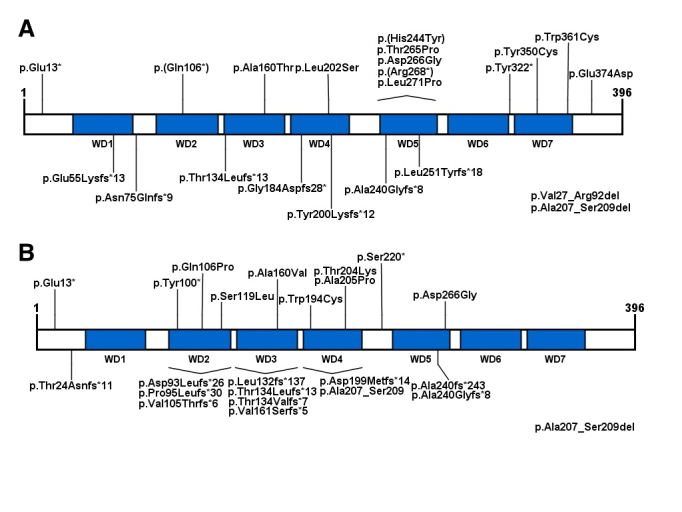
It is known that CSA is part of the E3 ubiquitin ligase complex, along with DDB1, RBX1, and CUL4A (Groisman et al., 2003; Fischer et al., 2011), responsible for the ubiquitination and degradation of TC-NER proteins when the repair is finalized (Groisman et al., 2006). In addition, CSA interacts and complexes with several proteins involved in transcription, ribosomal biogenesis, and TC-NER (reviewed by Aamann et al., 2014; Koch et al., 2014). Nonetheless, the mechanisms via which these interactions occur, and which CSA regions are involved, are not yet completely understood.
The three-dimensional structure and conserved amino acid residues in specific regions of the protein are of great importance for the regulatory activity and the interactions. Mutations in these regions that lead to the alteration of one of these factors may inevitably impair these functions (Muftuoglu et al., 2002:; Christiansen et al., 2003).
To identify a possible relation between the mutations and the phenotype of patients with CS, we mapped all the mutations in the ERCC8 and ERCC6 genes and their impact on proteins CSA and CSB, respectively, that is already reported in the literature. In total, we found 102 mutations in ERCC6 (50 homozygous and 52 heterozygous) and 37 mutations in ERCC8 (23 homozygous and 14 heterozygous). These numbers indicate that 70% of all the cases of the disease are caused by CSB mutations and 30% by CSA mutations. Analyzing only homozygous mutations, we observed the lack of an obvious correlation between the type of mutation or the affected region with the severity of CS (Tables S1 and S2).
Nevertheless, as discussed by Laugel (2013), the large prevalence of type I CS clinical classification (moderate phenotype) is noted in CSA patients, with most of the mutations located in the WD domains. Regarding CSB, mutations are predominant among the domains (such as domains III and IV, for example) and are mostly nonsense mutations and frameshifts, with lower prevalence of missense mutations and deletions. Unlike CSA cases, type II CS clinical manifestation is predominant among CSB patients (Laugel, 2013; Calmelset al., 2018).
The mutations that give rise to truncated CS proteins and the phenotype of the patients are also non-correlating variables. Proteins with mutations in the amino acid end sequences, such as R1087X, all have the integral ATPase domains but still lead to the manifestation of type II CS (more severe phenotype). On the other hand, the most striking example is the total absence of functional CSB resulting from the R77X mutation, as reported by Horibata et al. (2004), not leading to CS manifestation but instead causing UV-sensitive syndrome (UVSS) (Spivak, 2005). Another interesting observation is that the same mutation may cause different forms of CS in different patients. Other examples of one specific mutation resulting in different phenotypes exist in the literature. For example, Colella et al. (2000) described two siblings with the R735X mutation in CSB that do not cause CS but instead induce De Sanctis-Cacchione syndrome (a variant form of XP), whereas Mallery et al. (1998) described an association of the same mutation with type I of CS. In addition, Jaakkola et al. (2010) discovered the R1288X mutation (in CSB), which causes a severe neurological disorder known as cerebro-ocular-facial-skeletal syndrome (COFS) in several members of the same family, while the same mutation was also associated with type II CS manifestation (Laugel et al., 2009). This unexpected difference in phenotypes suggests that the genetic background may be a key factor that also needs to be taken into account in studies on the development, diversification of forms, and manifestations of the disease.
CS as a transcription syndrome
A deficiency in TC-NER and the consequent inability to recover RNA synthesis upon DNA damage in actively transcribed strands of genes could contribute to the neurological CS phenotype. Nonetheless, patients with UVSS (which can be caused by mutations in genes CSA, CSB, or UVSSA) lack TC-NER and are sun sensitive but do not manifest any of the neurological symptoms of CS (Itoh et al., 1996; Horibata et al., 2004; Spivac, 2005; Nardo et al., 2009; Zhang et al., 2012; Brooks, 2013).
In 1997, both the Hoeijmakers and Sancar groups discovered that CSB associates with RNA pol II, suggesting that CSB is implicated in transcription (Selby and Sancar, 1997a,b; van Gool et al., 1997). In fact, CSB was shown to increase the rate of transcription elongation up to threefold in an in vitro assay that reconstituted the transcription system (Selby and Sancar, 1997b). Similar observations were made by Balajee et al. (1997). These results prompted the authors to suggest that CS may be mainly a transcription, rather than a DNA repair related syndrome.
Five years later, Bradsher et al. (2002) stated that CSB was found in the nucleolus in a complex containing RNA pol I, which regulates ribosomal RNA (rRNA) transcription. The authors noticed that a CSB mutant cell line showed 8- to 10-fold lower rates of rRNA synthesis than did wild-type cells, confirming a critical role of CSB not only in mRNA synthesis (by RNA pol II) but also in rRNA synthesis (by RNA pol I). Of note, the CSA protein turned out to be associated with RNA pol I in the nucleolus, and a knockdown of CSA reduced rRNA synthesis (Koch et al., 2014). This malfunction in RNA pol I transcription has also been linked to endoplasmic-reticulum stress, leading to an unfolded protein response and the loss of proteostasis, which may be linked to the CS phenotype (Alupei et al., 2018).
In agreement with the idea that CS is a transcription syndrome, Newman et al. (2006) found that CSB-null fibroblasts feature a gene dysregulation pattern similar to that induced by HDAC inhibitors. Wang et al. (2014) reported dysregulation of several genes (linked to neurons) in CSB-mutant and CSA-mutant fibroblasts and in post-mortem brain tissue of patients. The authors also noticed that in vitro transdifferentiation of fibroblasts into neurons and neuroblast differentiation are impaired in CSB-deficient cells. These results strongly indicate that CSB is critical for neuronal differentiation and maintenance, and that gene expression defects might underlie the neurodegenerative and the neurodevelopmental defects observed in patients.
By taking advantage of cell reprogramming, Andrade et al. (2012) for the first time reprogrammed CSB-mutated primary fibroblasts into induced-pluripotent stem cells. Aside from making it possible to obtain a cell type that can be used to model CS development in vitro, the authors noticed dysregulation of hundreds of targets (including p53 and TXNIP) in these cells (Andrade et al., 2012). More recently, by combining cell reprogramming with neuron differentiation protocols, Vessoni et al. (2016) were, for the first time, able to obtain live neurons from patients’ skin fibroblasts. By RNA sequencing, the authors were able to find that pathological mutations in the ERCC6 gene changed the expression of almost 5000 transcripts in neurons of CSB-deficient patients compared to unaffected controls. Pathways related to axonogenesis, the action potential of neurons, neurotransmission, as well as transcripts related to the growth hormone–IGF-1 pathway were found to be dysregulated in the CSB-deficient neurons. Collectively, these results confirm that CSB deficiency heavily impacts the transcriptional process of the cell types relevant for the disease, even in the absence of exogenous DNA-damaging agents. Such extensive transcriptional dysregulation may underlie the complex and heterogeneous CS phenotype.
Nonetheless, as mentioned before, some mutations in genes XPB, XPD, XPF, XPG, or ERCC1, which participate in NER (Menck and Munford, 2014), may result in a combined phenotype of CS and XP (Lehmann 1982, 2014; Weeda et al., 1990; Moriel-Carretero et al., 2015). How can we explain the CS phenotype in all these cases? A likely answer to this question may depend on a multiprotein complex, TFIIH, which is indispensable for NER and for transcription. TFIIH consists of two functional subcomplexes (Core and CAK) that participate in initiation, promoter escape, and early elongation (Compe and Egly, 2012). The core subcomplex consists of seven proteins, including 3’ to 5’ ATP-dependent helicase XPB, and XPD, a 5’ to 3’ ATP-dependent helicase that binds to the core and CAK complexes together and facilitates optimal transcription (Tirode et al., 1999; Egly and Coin, 2011). In this sense, two mutations in the XPB gene (associated with the XP/CS phenotype) reduce transcriptional activity in a reconstituted transcription assay in vitro (Coin et al., 1999). Moreover, XPD or XPB mutations associated with CS were found to disrupt the interactions among CSB, TFIIH, and RNA pol I (Bradsher et al., 2002). The XPG nuclease was also found to associate with and stabilize TFIIH, and mutations in the XPG gene related to an XP-G/CS phenotype abrogate the XPG–TFIIH interaction (Ito et al., 2007; Lehmann et al., 2014; Narita et al., 2015). The recently described XP/CS patients with mutations in XPF or ERCC1 (Kashiyama et al., 2013) pose a challenge to the “CS as a transcription syndrome” point of view, because XPF/ERCC1 is implicated in NER and not in basal transcription. Although XPF is recruited to the promoter of inducible genes (to facilitate chromatin modification for transcription) in the absence of exogenous DNA damage, other NER factors are recruited as well, including XPC, in which mutations do not result in neurological abnormalities (Le May et al., 2010). XPF mutations that cause the XP/CS phenotype were recently found to cause persistent recruitment of NER proteins to DNA damage sites, which may induce the stalling of RNA and DNA polymerases, thereby interfering with the transcription and replication processes (Sabatella et al., 2018). Nevertheless, endogenous levels of DNA lesions that are substrates for NER have been reported to accumulate in mammalian cells and tissues to the levels that would not be consistent with the notion of defective TC-NER as a cause of the neurological symptoms of CS (Brooks, 2013). Therefore, more studies are needed to clarify the mechanism behind XPF/ERCC1 mutations and the development of the CS phenotype in these patients (Kashiyama et al., 2013).
CS and redox processes
The manifestation of CS occurs only when some NER proteins are mutated, while the complete deactivation of this pathway, via mutations in XPA, leads to the development of XP. Consequently, the roles of proteins CSA and CSB in addition to those known classic functions come into question. In addition, the main symptoms of patients with CS, e.g., neurological aberrations, cannot be explained only by the inefficient repair of UV damage because neurons are not exposed to this type of radiation. Due to the systemic presence of reactive species and their ability to chemically and structurally modify biomolecules, especially DNA, these compounds have been investigated as possible contributing factors of CS.
Experiments with keratinocytes derived from patients with CS suggest that these cells contain high concentrations of reactive oxygen species (ROS) with redox balance alterations under baseline conditions, characteristics that are related to the senescence phenotype of these cells (Cordisco et al., 2018). The induction of redox processes by the exposure of mice and CSB-/- mouse embryonic fibroblasts to ionizing radiation or paraquat revealed high sensitivity to the toxic effects of these agents, while such high sensitivity is not observed in CSA-mutant mice (de Waard et al., 2003, 2004). The sensitivity to the redox processes was also observed in CSA-/- keratinocytes and fibroblasts treated with potassium bromide (D’Errico et al., 2007) and in CSB-/- cells exposed to MMS (methyl methansulfonate) and 5-hydroxymethyl-2’deoxyuridine (Wong et al., 2007).
Moreover, lipid peroxidation products such as HNE (4-hydroxynonenal) at high cellular concentrations perform direct modifications on the CSB protein, compromising its ATPase activity required for the DNA repair by TC-NER (Maddukuri et al., 2009). On the other hand, Boetefuer et al. (2018) demonstrated that this activity is not needed for CSB–chromatin association when CS1AN-sv cells are exposed to menadione. In that condition, these associations are loci-specific and are stimulated by the PARP1 protein, thereby indicating possible participation of CSB in the transcriptional regulation in response to oxidative stress (Boetefuer et al., 2018a). These findings are in agreement with previous results that point to the involvement of a CSB function in this process, thereby showing that CSB localization and interaction with transcriptional repressor CTCF in promoter regions are greater in cells under oxidative stress (Lake et al., 2016; Boetefuer et al., 2018b).
Although DNA oxidation products are typically repaired by the BER pathway, it has been demonstrated that BER and NER proteins not only show crosstalk, but also that some oxidatively generated lesions are substrates for the NER pathway (D’Errico et al., 2007; Berra et al., 2013). For example, 8-oxoGua, thymine glycol, malondialdehyde, and etheno adducts induce distorting modifications in the double helix and have the potential to block transcription (reviewed by Tornaletti, 2005; Chaim et al., 2017), with cyclopurines being DNA oxidation products that are repaired only by NER (Brooks et al., 2000). Numerous experiments have confirmed the importance of the CSA and CSB proteins for the repair of oxidized bases, by demonstrating that in the absence of CSA or CSB there is accumulation of 8-oxoGua in DNA (Dianov et al., 1999; Tuo et al., 2001; D’Errico et al., 2007; Aamann et al., 2014a; Cordisco et al., 2018). 8-OH-Ade, 5-hydroxycytosine, and cyclopurines are also lesions that are inefficiently repaired in cells harboring mutations in CS genes (Tuo et al., 2002b, 2003; D’Errico et al., 2013). Because of the inefficient repair in the cells of important tissues such as the brain, the accumulation of these lesions in DNA may be one of the factors that cause and aggravate the neurological symptoms of the disease, as seen in patients with neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, Huntington’s disease, and amyotrophic lateral sclerosis (Ayala-Peña, 2013; Coppedè et al., 2016; Abolhassani et al., 2017).
The mutational impact of oxidatively induced DNA damage in CS was investigated by Lodato et al. (2018). By means of single-cell whole-genome sequencing followed by genome-wide somatic single-nucleotide variant identification, they detected an increased number of mutations in the neurons of patients with CS as compared to the control. In addition, C > A variants, a signature for mutations induced by oxidatively induced DNA damage, are found in a higher frequency among patients with CS (Lodato et al., 2018).
It is noteworthy that in response to irradiation or hydrogen peroxide, CSA is translocated to the nuclear matrix by a CSB-dependent mechanism (Kamiuchi et al., 2002), but as discussed above, these two proteins perform different functions in chromatin remodeling and in recruitment of the factors associated with the repair and blocking of RNA pol II in vivo (Fousteri et al., 2006). This cooperation between CSA and CSB was not observed in response to treatment with alkylating agents and is independent of XPA and XPC, thus suggesting that TC-NER plays a key part in this cellular response mechanism (Kamiuchi et al., 2002).
Brain biopsies of CS and XPA patients yielded distinct results in response to the accumulation of oxidized bases in DNA and SOD expression, with only XPA-mutant patients showing upregulation of 8-oxoGua in the nucleus and alteration in SOD expression (Hayashi et al., 2005).
In addition to the participation of these proteins in the repair of oxidation-induced lesions through the NER pathway, there is also a contribution to the removal of DNA damage via direct and indirect activity in the BER pathway (Khobta and Epe, 2013). This contribution is mediated by the direct modulation through interactions of CSB with BER protein glycosylases, APE1, NEIL1 and NEIL2, and association with the OGG1 complex, which stimulates the incision activity of these proteins and drives the repair (Wong et al., 2007; Muftuoglu et al., 2009; Aamann et al., 2014b). Csbm/m/Ogg1-/- mice show high concentrations of 8-oxoGua in comparison with Ogg1-/- animals, thereby confirming the importance of the cooperation between these two proteins in the removal of these lesions (Osterod et al., 2002; Trapp et al., 2007). Pastoriza-Gallego et al. (2007) also demonstrated that the 8-oxoGua lesions exert different effects on gene expression depending upon the promoter and sequence context, and that both proteins, Csb and Ogg1, are required for full repair.
Recent evidence also indicates that CSB recruits the XRCC1 protein to single-strand break regions after oxidatively induced DNA damage for subsequent processing by BER (Menoni et al., 2018). In the indirect modulation of the BER pathway, CSB acts as an important factor in the expression of hOGG1, inducing its transcription. It has also been observed that in CSB-deficient cells, the amounts of OGG1 mRNA and protein are low (Dianov et al., 1999; Tuo et al., 2002a; Aamann et al., 2014b).
Some studies point to a possible implication of CSB in the repair of oxidized bases in genomic and mitochondrial DNA, to CSB migration to mitochondria, and modulation of p53 activity in response to a redox process (Stevnsner et al., 2008; Frontini and Proietti-De-Santis, 2009; Aamann et al., 2010).
The repair of oxidized bases also has a fundamental role for the mitochondria. These organelles produce ATP through the electron transport chain, a process that not only provides cells with energy, but also produces ROS as a by-product of respiration (reviewed by Kowaltowski et al., 2009). For this reason, mitochondria are also considered the primary source of ROS in cells, having a much more oxidizing environment within the mitochondrial matrix as compared with the cellular cytosol (Hu et al., 2008).
Because of proximity to the initial site of formation and constant exposure to ROS, mitochondrial DNA (mtDNA) is the main target of these molecules, and just as nuclear DNA, it is susceptible to oxidation in the DNA bases and undergoes other modifications in its structure (reviewed by Muftuoglu et al., 2014). Besides, the accumulation of lesions in mtDNA causes instability and compromises its functioning, which can lead to mutations and affect genes that are important for mitochondrial metabolism. In addition, various aging-related diseases, such as cardiovascular diseases, Parkinson’s disease, Alzheimer’s disease, and cancer are associated with significant amounts of lesions in mitochondria, thus being considered one of the causes of the pathologies related to mitochondrial dysfunction (Mecocci et al., 1994; Ide et al., 2001; Stichel et al., 2007; Wallace, 2012).
Mitochondria have some specific repair pathways, such as BER and mismatch repair (MMR). As in the repair of nuclear DNA, the CSA and CSB proteins are present and participate in this process, even though there is no mitochondrial NER (Kamenisch et al., 2010). It has been demonstrated that CSA and CSB, after induction of redox processes by the exposure to H2O2, UV, or menadione, are directed toward mitochondria and interact with OGG1 (Aamann et al., 2010; Kamenisch et al., 2010; reviewed by Prates Mori and de Souza-Pinto, 2018). In mitochondria, CSB may act as a modulator of the BER pathway, in association with the inner mitochondrial membrane for mtDNA lesion removal (Stuart et al., 2005; Aamann et al., 2010). In fact, CSBm/m mice accumulate 2.5-fold more 8-oxoGua in mtDNA than wild-type animals (Osenbroch et al., 2009). The mutation in CSBm/m mice is the same as that in the ERCC6 gene of the CS1AN patient, K337 → stop, which generates a truncated nonfunctional protein (Troelstra et al., 1992; van der Horst et al., 1997).
In addition to the influence on DNA repair, CSB is involved in gene expression by engaging in the elongation by RNA polymerase II (Selby and Sancar, 1997a). In in vitro transcription, through interactions with proteins of the mitochondrial nucleoid complex, CSB promotes POLRMT transcriptional elongation and allows it to access mtDNA by TFAM removal from the region to be transcribed (Berquist et al., 2012).
Regarding CS mitochondrial metabolism, Scheibye-Knudsen et al. (2012) reported a significant increase of this metabolism in both CSBm/m mice and CSB-deficient cells, and this phenomenon is also manifested in CSA- or CSB- deficient fibroblasts and CSA-/-/XPA-/- mice (Pascucci et al., 2012; Brace et al., 2016). This increase in energy metabolism is accompanied by a large production of ROS through electron leakage (Pascucci et al., 2012; Cleaver et al., 2014), and is the main source of DNA damage induction (Scheibye-Knudsen et al., 2012) and of changes in the membrane potential, excessive fragmentation, and mitochondrial fission (Pascucci et al., 2012, 2016).
Under normal cell conditions, organelles and defective proteins are eliminated by autophagy. On the other hand, dysfunctional or damaged mitochondria are subjected to a special form of autophagy, called mitophagy (Fivenson et al., 2017). This mechanism contributes to cell homeostasis, and its malfunction is present in several aging-related diseases (reviewed by Redmann et al., 2014). Human and murine CSB-mutant fibroblasts feature a dysfunction in this pathway owing to defects in the recruitment of P62, an essential player in mitophagy (Scheibye-Knudsen et al., 2012).
Thus, dysfunctional mitochondria, with large amounts of mtDNA lesions, can produce more ROS via their metabolism. Consequently, ROS accumulate in the cells, promoting apoptosis (Pinto and Moraes, 2015; van Houten et al., 2016), which might be associated with reduced amounts of subcutaneous fat in CSA-/- and CSBm/m mice (Kamenisch et al., 2010). Treatment with pharmacological activators of autophagy, such as rapamycin (Scheibye-Knudsen et al., 2012) and an HDAC inhibitor (Majora et al., 2018), attenuates the accumulation of damaged mitochondria in CSB-deficient cells and the loss of subcutaneous fat in CSBm/m mice.
Models for CS study
To better understand the mechanisms of CS progression, several animal models have been developed for research on the disease, e.g., mice, Caenorhabditis elegans, and zebrafish. In general, such models carry knockout mutations or mutations identical to those found in humans, enabling a more accurate analysis in different cells and tissues of an organism.
The first models generated to study CS were created on the basis of mice with the C57BL/6 background - CSBm/m mice (van der Horst et al., 1997), and CSA-/- mice (van der Horst et al., 2002). In CSA-/- cells, the interruption of the gene sequence in exon 2 of the Ercc8 gene results in protein absence (van der Horst et al., 2002). When exposed to UV, the fibroblasts of these animals manifest higher UV sensitivity, and they have an inability to resume RNA synthesis and defective TC-NER, these being the classic characteristic of CS (Lehmann, 1982; van der Horst et al., 1997, 2002).
The CSBm/m and CSA-/- models have similar phenotypic characteristics, although neither has the severe neurodegenerative phenotype and lifespan reduction seen in patients with CS. Despite differences between them, these mouse models have a few characteristics that resemble their human counterpart, such as the tendency toward reduced body weight via a decrease in the amounts of subcutaneous and visceral fat (Gorgels et al., 2007; Scheibye-Knudsen et al., 2012), mild neurological dysfunction with changes in myelin (Jaarsma et al., 2011), activation of glial cells in white-matter regions (Jaarsma et al., 2011), progressive deafness (Nagtegaal et al., 2015), photophobia and sensitivity to UV exposure (van der Horst et al., 1997, 2002). In contrast to what is manifested in humans, the mouse models are prone to skin and eye cancers when exposed to UV, a feature that can be explained by the inefficiency of murine GG-NER in dealing with UV-induced cyclobutane pyrimidine dimer-like lesions, which are preferentially repaired by TC-NER (van der Horst et al., 1997, 2002; de Boer and Hoeijmakers, 1999).
CSBm/m cells (from several tissues, mainly the brain and kidneys) also contain high levels of formamidopyrimidines and 8-oxoGua in DNA when compared to wild-type mice (Muftuoglu et al., 2009). In addition to nuclear DNA damage, mtDNA damage and mutational load in the mtDNA of CSA-/- and CSBm/m mice increased during aging and is related to subcutaneous fat loss, one of the main characteristics of CS, probably mediated by the apoptosis caused by mitochondrial dysfunction (Kamenisch et al., 2010) and by cell senescence, a feature commonly present in progeroid models (Carrero et al., 2016).
The combined deletion of other NER proteins such as XPA in these CS mice leads to a more severe phenotype. These animals manifest severe neurological symptoms, ataxia, compromised growth, low weight, a lack of motor coordination, kyphosis, and abnormal behavior and development with premature death within 20 days (Murai et al., 2001; van der Pluijm et al., 2007). Inactivation of GG-NER by an XPC knockout in CSBm/m mice leads to a similar phenotype, with dysmyelination, slow development, and low body weight: characteristics that resemble those of patients with CS (Laposa et al., 2007; van der Pluijm et al., 2007; Revet et al., 2012). CSA-/-/XPA-/- mice also show evidence of neurological problems, life expectancy reduced to ~1 month, an increase in cellular oxidative phosphorylation, and lipodystrophy (Brace et al., 2013, 2016).
The mechanism that underlies this aggravation of the CS phenotype by the double inactivation of CS and XP genes in mice has yet to be uncovered. Since CS and XP proteins act in the same DNA repair pathway, it is assumed that this severe CS phenotype in mice is due to the disruption of both NER-related and other functions of the CS proteins, such as transcription regulation (Brooks, 2013) and/or interactions with other DNA repair pathways (Murai et al., 2001). Inactivation of NER by an XPC or XPA gene knockout can result in the accumulation of transcription-blocking DNA lesions, and the loss of either CS protein may further interfere with transcription and further increase the DNA damage load, which can in turn have detrimental effects, such as cell death or early cell senescence, both of which have been observed in progeroid mice (Weeda et al., 1997; van der Pluijm et al., 2007; Brooks, 2013).
Combined inactivation of TC-NER and NER in neurons has also been reported to generate an age-related progressive neurodegenerative phenotype. CSBm/m mice featuring a neuron-specific conditional XPA knockout have a shorter lifespan, behavioral abnormalities, and brain atrophy. These characteristics are possibly related to a synergistic effect of the TC-NER and NER pathways or to other functions of the inactivated proteins, such as chromatin remodeling or other functions in transcription (Newman et al., 2006; Jaarsma et al., 2011; Wang et al., 2018). Cell type–specific conditional knockouts may also help to better understand the participation of other processes in the progression of the disease, along with their molecular mechanisms, e.g., the role of oligodendrocytes (Howng et al., 2010) or other glial cells (Raj et al., 2014) in CS dysmyelination and in the neurodegenerative phenotype.
Mutations in XPG or ERCC1 can yield mice with the characteristics similar to those of CS and other diseases on the CS spectrum, such as XP/CS and COFS (Jaarsma et al., 2013). These NER nuclease–deficient models have several neurodegenerative features, altered metabolism, and a reduced lifespan (Weeda et al., 1997; Barnhoorn et al., 2014). Notably, despite the lipodystrophy observed in all these animal models, dietary restriction and methionine restriction (which are metabolic interventions known to increase the lifespan of several species) reversed some of the neurodegeneration indicators of the CSA-/-/XPA-/- and ERCC1Δ/- models, with dietary restriction nearly doubling the lifespan of ERCC1Δ/- mice. This finding indicates that metabolic interventions are a possible therapy for NER-related progeroid diseases (Brace et al., 2016; Vermeij et al., 2016).
The current murine models for CS research do not fully reproduce the phenotype seen in humans because these animals manifest milder symptoms of the disease when only a single TC-NER protein is mutated. One of the possible explanations is the adaptation of NER to deal with DNA damage that would be repaired initially by CSA and CSB. This explains the severe symptoms when both TC-NER and NER are inactivated (van der Pluijm et al., 2007). In this case, it is important to consider the evolutionary distance that separates mice from humans. This distance entails differences in gene expression and metabolic and physiological profiles, among others (Seok et al., 2013; Lin et al., 2014). Such differences contribute to the following phenomenon: the phenotype resulting from the same mutation and the activity of the repair pathway are not identical between the two species. Table 1 summarizes the mouse models carrying mutations in CS genes and other single-NER-mutation models that recapitulate CS-like phenotypes.
Table 1. Reported neurological and metabolic characteristics of Cockayne Syndrome and related mouse models.
| Mouse model | Genetic background | Neurological abnormalities | Metabolic characteristics | Lifespan | Other reported features | References |
|---|---|---|---|---|---|---|
| Csbm/m | C57BL/6J | Mild, progressive neurodegeneration with no behavioral phenotype, mild astro and microgliosis, hearing loss associated with loss of neurosensorial cells, late retinal degeneration | Increased metabolism and mitochondrial numbers, Progressive lipodistrophy | > 2 years | Increased skin cancer predisposition after UV exposure | Van der Horst et al., 1997; Jaarsma et al., 2011 |
| Csa-/- | C57BL/6J | Mild, progressive neurodegeneration with no behavioral phenotype, mild astro and microgliosis, hearing loss associated with loss of neurosensorial cells | Increased metabolism | > 2 years | Increased skin cancer predisposition after UV exposure | van der Horst et al., 2002; Gorgels et al, 2007; Jaarsma et al., 2011; Brace et al., 2016 |
| Csbm/m/Xpa-/- | C57BL/6J; CBA/C57BL6/CD-1/129Ola hybrid | Profound early postnatal ataxia, abnormal cerebellar development and degeneration, loss of purkinjecells, progressive neurological dysfunction, motor coordination problems, early retinal degeneration | Severe post-natal growth deficiency, Severe lipodistrophy, decreased oxidative metabolism | 3 weeks | Suppression of the GH/IGF1 somatotroph axis increased antioxidant responses, hypoglycemia | Murai et al., 2001, van der Pluijm et al., 2007; Jaarsma et al., 2011 |
| Csb-/- / Xpac/- CamKIIα-Cre | C57BL/6J | Progressive neurological dysfunction, Purkinje cell death, neurodegeneration, motor coordination problems starting at 6 months | Weight reduction in adult animals | 90 weeks | - | Jaarsma et al., 2011 |
| Csbm/m/Xpc-/- | C57BL/6J, 129Ola/C57BL/6J hybrid | Depletion of purkinje cells, neurodegeneration in cerebellum, motor coordination problems, dysmielination | Severe post-natal growth deficiency, Severe lipodistrophy | 3 weeks | - | van der Pluijm et al., 2007; Laposa et al., 2007; Revet et al., 2012 |
| Csbm/m/Ogg1-/- | C57BL/6J | No neurological phenotype has been observed | Not described | > 2 years | Accumulation of oxidative stress related DNA damage and mutations in various tissues | Osterod et al., 2002; Pastoriza-Gallego et al., ?2007; Fusser et al., 2011 |
| Csa-/-/Xpa-/- | C57BL/6J | Early, progressive neurological dysfunction, dysmyelination, abnormal cerebellar development and degeneration, motor coordination problems, severe astro and microgliosis | Post-natal growth deficiency, increased metabolism, severe lipodistrophy, senescence of fat tissue, no fat tissue inflammation | 20 weeks | - | Brace et al., 2013; Brace et al., 2016 |
| Xpg-/- | C57BL/6J | Purkinje cell degeneration, cerebellum neurodegeneration | Severe post-natal growth deficiency, Severe lipodistrophy | 3 weeks | Abnormalities of the gastro-intestinal tract, early fibroblast senescence | Harada et al., 1999; Sun et al., 2001; Barnhoorn et al., 2014 |
| Xpg-/- | C57BL6/FVB F1 hybrid | Purkinje cell death, neurodegeneration, motor coordination problems axonal spheroids, severe astrogliosis | Post-natal growth deficiency, loss of subcutaneous fat | 18 weeks | Skeletal abnormalities, mild anisokaryosis, increased mean nuclear size of liver cells, supression of GH/IGF1 axis | Barnhoorn et al, 2014; Vermeij, 2016 |
| Ercc1-/- | Hybrid C57Bl/6/129 | Early, progressive neurological dysfunction, neurodegeneration and motor coordination problems | Severe post-natal growth deficiency, decreased oxidative metabolism, severe lipodistrophy, increase in fat tissue inflammation | 3 weeks | Liver nuclear abnormalities, p53 upregulation in liver and kidney, early fibroblast senescence | Mcwhir et al., 1993, Weeda et al., 1997 |
| Ercc1Δ/- | C57Bl6J/FVB hybrid | Early, progressive neurological dysfunction, myelin abnormalities, abnormal cerebellar development and degeneration, Motor coordination problems, severe astro and microgliosis | Post-natal growth deficiency, decreased oxidative metabolism, severe lipodistrophy, increase in fat tissue inflammation | 22-25 weeks | Early fibroblast and vascular tissue senescence, reduced bone density | Weeda et al., 1997; Nevedomskaya et al., 2010; de Waard et al., 2010; Durik et al., 2012; Vermeij et al., 2016; |
Different animal models of lower complexity have also been developed to study the role of CSA and CSB proteins, and are an interesting alternative for the research into protein functions. Among these models are Caenorhabditis elegans and zebrafish.
C. elegans has advantages, such as the ease of laboratory maintenance, of genetic manipulation, of tissue differentiation, rapid reproduction with several offspring, a generally fixed and genetically determined number of cells, and a short life cycle, allowing for the study of its development within short periods. It has been demonstrated that the NER pathway is well conserved in C. elegans, resembling the repair mechanisms of mammals (Meyer et al., 2007; reviewed by Lans and Vermeulen, 2011), and because they do not repair DNA by photoreactivation (Hartman et al., 1989). Rather, the lesions caused by UV are repaired exclusively by NER. CSA and CSB analogs, csa-1 (Babu et al., 2014) and csb-1 (CeCSB) (Lee et al., 2002), respectively, were found to be a part of this pathway. Animals mutated in csa-1 are hypersensitive to UV-B light exposure (Babu et al., 2014), and csb-1–deficient animal germ cells show apoptosis induction and morphological abnormalities after exposure to this agent (Lee et al., 2002). Moreover, a knockout of either csa-1 or csb-1 in C. elegans resulted in increased oxygen consumption and in changes in the transcription of genes related to mitochondrial ATP production, ubiquitin pathways, and transcriptional regulation (Scheibye-Knudsen et al., 2016).
In general, C. elegans is a good model for the investigation of the DNA damage response to UV via the NER pathway, because this species makes it possible to reproduce and examine the effects of irradiation or other DNA-damaging agents on an entire organism and throughout its developmental stages within a short period of time.
The zebrafish is a vertebrate model that is widely used in research on the effects of exposure to genotoxic agents, carcinogenesis processes, and mainly embryonic development (Spitsbergen and Kent, 2003; Titus et al., 2009). This animal has orthologous DNA repair genes in the pathways present in higher eukaryotes, e.g., BER, NER, MMR, Non-homologous end joining and Homologous recombination (Pei and Strauss, 2013). In NER, 44 genes are responsible for the functioning mechanisms of damage removal. Despite this observation, there are still few zebrafish studies where these characteristics are exploited from the perspective of DNA repair alone.
Zebrafish with the CSB depleted by morpholino oligonucleotides at the larval stage show an increased frequency of morphological abnormalities, which may recapitulate some of the congenital and developmental manifestations seen in patients with CS. Ionizing radiation can further increase morphological aberrations in CSB-depleted zebrafish, thereby pointing to an important role of CSB in the defense against oxidative DNA damage (Wei et al., 2015).
Due to the complexity of CS and the unique characteristics of patients with CS, which cannot be fully recapitulated in any animal model, a combination of human cell models with various animal models offers complimentary approaches to elucidate the various characteristics of this syndrome. In this sense, the use of somatic cell reprogramming coupled with genome editing allows investigators to obtain relevant and functional cell types (such as neurons) carrying patient-specific mutations. Moreover, this method offers an opportunity to investigate how the genetic background of different patients interacts with the one given pathological mutation. Such an approach may clarify why certain patients that share the same mutation in a CS gene have different phenotypes (Colella et al., 2000).
Conclusion
Almost 60 years of accumulated research in the NER field, especially regarding progeroid CS, provides extensive knowledge about the structure of proteins CSA and CSB and their participation in TC-NER and other mechanisms, such as transcription, repair, and mitochondrial functioning. For the advances made so far, human cellular models and animal models have been invaluable. They have revealed that the CS phenotype is likely a combination of altered gene transcription, metabolic adjustment, redox imbalance, and DNA repair defects, although the relative importance of each of these mechanisms for the disease is still largely unknown. Moreover, the full spectrum of the syndrome is not completely mirrored in animals, and this situation undoubtedly hinders further research. This difficulty is also associated with the impossibility of correlation between the mutations in ERCC6/ERCC8 genes and the phenotype of the patients, suggesting that the genetic background can heavily influence the manifestation of the symptoms.
Recently, the ability to generate pluripotent stem cells from patients with CS enabled for the first time the recapitulation of the full genetic signature of a patient in a cell type relevant for the disease, e.g., neurons. This observation expands and complements the previous CS models because it enables the investigation of metabolism, of the DNA damage response, and of gene expression in a patient’s cells. These data were previously impossible or very difficult to obtain. Still, one needs to keep in mind that CS is a systemic disease, and for this reason, investigating how dysfunction in one tissue/organ impacts others is critical. All these difficulties reveal how complex and diverse CS is and imply that a multimodel approach will therefore help to better recapitulate certain characteristics of CS. Moreover, although not entirely equivalent to normal aging, CS has clinical and cellular similarities to some aging-related diseases. Thus, improvements of (and new approaches to) CS models may have a broad impact on the study of these diseases as well.
Acknowledgments
We acknowledge the scholarship to CCCG from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES Brasil – Finance code 001), the scholarships to LLSN and GSK from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and CAPES. This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, grant APQ00828-14) and the Pró-Reitoria de Pesquisa da Universidade Federal de Ouro Preto. Last but not least, we thank Prof. Dr. Carlos Menck for critical reading and encouragement.
Supplementary material
The following online material is available for this article:
References
- Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wilson DM, Stevnsner T. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010;24 doi: 10.1096/fj.09-147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aamann MD, Hvitby C, Popuri V, Muftuoglu M, Lemminger L, Skeby CK, Keijzers G, Ahn B, Bjoras M, Bohr VA. Mech Ageing Dev. 2014;135 doi: 10.1016/j.mad.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aamann MD, Muftuoglu M, Bohr VA, Stevnsner T. Mech Ageing Dev. 2014;134 [Google Scholar]
- Abolhassani N, Leon J, Sheng Z, Oka S, Hamasaki H, Iwaki T, Nakabeppu Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech Ageing Dev. 2017;161 doi: 10.1016/j.mad.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Alupei MC, Maity P, Esser P, Krikki I, Tuorto F, Parlato R, Penzo M, Schelling A, Laugel V, Montanaro L. Loss of proteostasis is a pathomechanism in Cockayne syndrome. Cell Rep. 2018;23 doi: 10.1016/j.celrep.2018.04.041. [DOI] [PubMed] [Google Scholar]
- Andrade LN, Nathanson JL, Yeo GW, Menck CFM, Muotri AR. Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum Mol Genet. 2012;21 doi: 10.1093/hmg/dds211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AD, Barrett SF, Yoder FW, Robbins JH. Cockayne’s Syndrome fibroblasts have increased sensitivity to ultraviolet light but normal rates of unscheduled DNA synthesis. J Invest Dermatol. 1978;70 doi: 10.1111/1523-1747.ep12541383. [DOI] [PubMed] [Google Scholar]
- Anindya R, Mari PO, Kristensen U, Kool H, Giglia-Mari G, Mullenders LH, Fousteri M, Vermeulen W, Egly JM, Svejstrup JQ. A ubiquitin-binding domain in cockayne syndrome b required for transcription-coupled nucleotide excision repair. Mol Cell. 2010;38 doi: 10.1016/j.molcel.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Peña S. Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic Biol Med. 2013;62 doi: 10.1016/j.freeradbiomed.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu V, Hofmann K, Schumacher B. A C. elegans homolog of the Cockayne syndrome complementation group A gene. DNA Repair (Amst) 2014;24 doi: 10.1016/j.dnarep.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee AS, May A, Dianov GL, Friedberg EC, Bohr VA. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc Natl Acad Sci U S A. 1997;94 doi: 10.1073/pnas.94.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhoorn S, Uittenboogaard LM, Jaarsma D, Vermeij WP, Tresini M, Weymaere M, Menoni H, Brandt RM, de Waard MC, Botter SM. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG Deficiency. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Hoeijmakers JHJ, Kanaar R, Vermeulen W, Wyman C. The CSB protein actively wraps DNA. J Biol Chem. 2005;280 doi: 10.1074/jbc.M409147200. [DOI] [PubMed] [Google Scholar]
- Berquist BR, Canugovi C, Sykora P, Wilson DM, Bohr VA. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gks565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra CM, de Oliveira CS, Garcia CCM, Rocha CRR, Lerner LK, Lima LC, Baptista M, Menck CFM. Nucleotide excision repair activity on DNA damage induced by photoactivated methylene blue. Free Radic Biol Med. 2013;61 doi: 10.1016/j.freeradbiomed.2013.03.026. [DOI] [PubMed] [Google Scholar]
- Boetefuer EL, Lake RJ, Dreval K, Fan HY. promotes oxidative stress–induced association of Cockayne syndrome group B protein with chromatin. J Biol Chem. 2018;293 doi: 10.1074/jbc.RA118.004548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetefuer EL, Lake RJ, Fan HY. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40 doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- Brace LE, Vose SC, Vargas DF, Zhao S, Wang XP, Mitchell JR. Lifespan extension by dietary intervention in a mouse model of Cockayne syndrome uncouples early postnatal development from segmental progeria. Aging Cell. 2013;12 doi: 10.1111/acel.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradsher J, Auriol J, Proietti de, Iben S, Vonesch JL, Grummt I, Egly JM. CSB Is a Component of RNA Pol I Transcription. Mol Cell. 2002;10 doi: 10.1016/s1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- Brace LE, Vose SC, Stanya K, Gathungu RM, Marur VR, Longchamp A, Treviño-Villarreal H, Mejia P, Vargas D, Inouye K. Increased oxidative phosphorylation in response to acute and chronic DNA damage. Aging Mech Dis. 2016;2 doi: 10.1038/npjamd.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ. Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair (Amst) 2013;12 doi: 10.1016/j.dnarep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K. The oxidative DNA lesion 8,5’-(S)-Cyclo- 2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275 doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Balajee AS, Selzer RR, Sunesen M, Proietti De, Bohr VA. The ATPase domain but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair. Mol Biol Cell. 1999;10 doi: 10.1091/mbc.10.11.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmels N, Botta E, Jia N, Fawcett H, Nardo T, Nakazawa Y, Lanzafame M, Moriwaki S, Sugita K, Kubota M. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J Med Genet. 2018;55 doi: 10.1136/jmedgenet-2017-104877. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Memedula S, Plutz MJ, Belmont AS. Common effects of acidic activators on large-scale chromatin structure and transcription. Mol Cell Biol. 2005;25 doi: 10.1128/MCB.25.3.958-968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero D, Soria-Valles C, López-Otín C. Hallmarks of progeroid syndromes: Lessons from mice and reprogrammed cells. Dis Model Mech. 2016;9 doi: 10.1242/dmm.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaim IA, Gardner A, Wu J, Iyama T, Wilson DM, Samson LD. A novel role for transcription-coupled nucleotide excision repair for the in vivo repair of 3,N4-ethenocytosine. Nucleic Acids Res. 2017;45 doi: 10.1093/nar/gkx015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen M, Stevnsner T, Modin C, Martensen PM, Brosh RM, Bohr VA. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gkg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio E, Rademakers S, van der Horst GT, van Gool AJ, Hoeijmakers JH, Vermeulen W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J Biol Chem. 1998;273 doi: 10.1074/jbc.273.19.11844. [DOI] [PubMed] [Google Scholar]
- Citterio E, van den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JHJ, Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20 doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10 doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Brennan-Minnella AM, Swanson RA, Fong K, Chen J, Chou KM, Chen YW, Revet I, Bezrookove V. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci U S A. 2014;111 doi: 10.1073/pnas.1414135111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1936;11 doi: 10.1136/adc.11.61.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1946;11 [PMC free article] [PubMed] [Google Scholar]
- Coin F, Bergmann E, Tremeau-Bravard A, Egly J. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999;18 doi: 10.1093/emboj/18.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Nardo T, Botta E, Lehmann AR, Stefanini M. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-Cacchione variant of Xeroderma pigmentosum. Hum Mol Genet. 2000;9 doi: 10.1093/hmg/9.8.1171. [DOI] [PubMed] [Google Scholar]
- Compe E, Egly JM. TFIIH: When transcription meets DNA repair. Nat Rev Mol Cell Biol. 2012;13 doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- Coppedè F, Tannorella P, Stoccoro A, Chico L, Siciliano G, Bonuccelli U, Migliore L. Methylation analysis of DNA repair genes in Alzheimer’s disease. Mech Ageing Dev. 2016;161 doi: 10.1016/j.mad.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Cordisco S, Tinaburri L, Teson M, Orioli D, Cardin R, Degan P, Stefanini M, Zambruno G, Guerra L, Dellambra E. Cockayne syndrome type A protein protects primary human keratinocytes from senescence. J Invest Dermatol. 2018;139 doi: 10.1016/j.jid.2018.06.181. [DOI] [PubMed] [Google Scholar]
- Parlanti E, Teson M, Degan P, Lemma T, Calcagnile A, Iavarone I, Jaruga P, Ropolo M, Pedrini AM. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene. 2007;26 doi: 10.1038/sj.onc.1210232. [DOI] [PubMed] [Google Scholar]
- Pascucci B, Iorio E, van Houten B, Dogliotti E. The role of CSA and CSB protein in the oxidative stress response. Mech Ageing Dev. 2013;134 doi: 10.1016/j.mad.2013.03.006. [DOI] [PubMed] [Google Scholar]
- de Boer J, Hoeijmakers JH. Cancer from the outside, aging from the inside: Mouse models to study the consequences of defective nucleotide excision repair. Biochimie. 1999;81 doi: 10.1016/s0300-9084(99)80045-5. [DOI] [PubMed] [Google Scholar]
- de Waard H, de Wit J, Gorgels TGMF, van den Aardweg G, Andressoo JO, Vermeij M, Van Steeg H, Hoeijmakers JHJ, van der Horst GTJ. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair (Amst) 2003;2 doi: 10.1016/s1568-7864(02)00188-x. [DOI] [PubMed] [Google Scholar]
- de Waard MC, de Wit J, Andressoo JO, van Oostrom CTM, Riis B, Weimann A, Poulsen HE, Van Steeg H, Hoeijmakers JHJ, van der Horst GT. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol. 2004;24 doi: 10.1128/MCB.24.18.7941-7948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G, Bishoff C, Sunesen M, Bohr VA. Repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Nucleic Acids Res. 1999;27 doi: 10.1093/nar/27.5.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue BA, Yint S, Taylort JS, Reines D, Hanawalt PC. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci U S A. 1994;91 doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durik M, Kavousi M, van der Pluijm I, Isaacs A, Cheng C, Verdonk K, Loot AE, Oeseburg H, Bhaggoe UM, Leijten F. Nucleotide Excision DNA Repair is Associated with Age-Related Vascular Dysfunction. Circulation. 2013;126 doi: 10.1161/CIRCULATIONAHA.112.104380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egly JM, Coin F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10 doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147 doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- Fivenson EM, Lautrup S, Sun N, Sheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF. Mitophagy in neurodegeneration and aging. Neurochem Int. 2017;109 doi: 10.1016/j.neuint.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri M, Mullenders LHF. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008;18 doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- Fousteri M, Vermeulen W, van Zeeland AA, AA and, Mullenders LHF. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23 doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Frontini M, Proietti-De-Santis L. Cockayne syndrome B protein (CSB): Linking p53, HIF-1 and p300 to robustness, lifespan, cancer and cell fate decisions. Cell Cycle. 2009;8 doi: 10.4161/cc.8.5.7754. [DOI] [PubMed] [Google Scholar]
- Fusser M, Nesse GJ, Khobta A, Xia N, Li H, Klungland A, Epe B. Spontaneous mutagenesis in csbm/mogg1-/- mice is attenuated by dietary resveratrol. Carcinogenesis. 2011;32 doi: 10.1093/carcin/bgq196. [DOI] [PubMed] [Google Scholar]
- Gorgels TG, van der Pluijm I, Brandt RM, Garinis GA, Van Steeg H, van den Aardweg G, Jansen GH, Ruijter JM, Bergen AA, van Norren D. Retinal degeneration and ionizing radiation hypersensitivity in a mouse model for Cockayne syndrome. Mol Cell Biol. 2007;27 doi: 10.1128/MCB.01037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH, Svejstrup JQ. The cellular response to transcription-blocking DNA damage. Trends Biochem Sci. 2018;43 doi: 10.1016/j.tibs.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113 doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, Kisselev AF, Harel-Bellan A, Nakatani Y. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006;20 doi: 10.1101/gad.378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola A, Álvares-da-Silva CR, Grisolia J, Silbermann R. Síndrome de Cockayne. Arq Neuro-Psiquiatr. 1999;57 doi: 10.1590/s0004-282x1999000100021. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nature. 2008;9 doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Harreman M, Taschner M, Sigurdsson S, Anindya R, Reid J, Somesh B, Kong SE, Banks CAS, Conaway RC, Conaway JW. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc Natl Acad Sci U S A. 2009;106 doi: 10.1073/pnas.0907052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman PS, Hevelone J, Dwarakanath V, Mitchell DL. Excision repair of UV radiation-induced DNA damage in Caenorhabditis elegans. Genetics. 1989;122 doi: 10.1093/genetics/122.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R. Oxidative nucleotide damage and superoxide dismutase expression in the brains of Xeroderma pigmentosum group A and Cockayne syndrome. Brain Dev. 2005;27 doi: 10.1016/j.braindev.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, Schultz RA, Stefanini M, Lehmann AR, Mayne LV. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995;82 doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- Hoogstraten D, Bergink S, Ng JM, Verbiest VH, Luijsterburg MS, Geverts B, Raams A, Dinant C, Hoeijmakers JHJ, Vermeulen W. Versatile DNA damage detection by the global genome nucleotide excision repair protein XPC. J Cell Sci. 2008;121 doi: 10.1242/jcs.031708. [DOI] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101 doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howng SY, Avila RL, Emery B, Traka M, Lin W, Watkins T, Cook S, Bronson R, Davisson M, Barres BA. ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev. 2010;24 doi: 10.1101/gad.1864510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Dong L, Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem. 2008;283 doi: 10.1074/jbc.M803028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura KI, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88 doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/Patients with CS. Mol Cell. 2007;26 doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Itoh T, Shiomi T, Shiomi N, Harada Y, Wakasugi M, Matsunaga T, Nikaido O, Friedberg EC, Yamaizumi M. Rodent complementation group 8 (ERCC8) corresponds to Cockayne syndrome complementation group A. Mutat Res. 1996;362 doi: 10.1016/0921-8777(95)00046-1. [DOI] [PubMed] [Google Scholar]
- Iyama T, Okur MN, Golato T, McNeill DR, Lu H, Hamilton R, Raja A, Bohr VA, Wilson III. Regulation of the intranuclear distribution of the Cockayne syndrome proteins. Sci Rep. 2018;8 doi: 10.1038/s41598-018-36027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola E, Mustonen A, Olsen P, Miettinen S, Savuoja T, Raams A, Jaspers NG, Shao H, Wu BL, Ignatius J. ERCC6 founder mutation identified in Finnish patients with COFS syndrome. Clin Genet. 2010;78 doi: 10.1111/j.1399-0004.2010.01424.x. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, van der Pluijm I, de Waard MC, Haasdijk ED, Brandt R, Vermeij M, Rijksen Y, Maas A, Van Steeg H, Hoeijmakers JHJ. Age-related neuronal degeneration: Complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, van der Pluijm I, van der Horst GTJ, Hoeijmakers JH. Cockayne syndrome pathogenesis: Lessons from mouse models. Mech Ageing Dev. 2013;134 doi: 10.1016/j.mad.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Kamenisch Y, Fousteri M, Knoch J, von Thaler AK, Fehrenbacher B, Kato H, Becker T, Dollé ME, Kuiper R, Majora M. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med. 2010;207 doi: 10.1084/jem.20091834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiuchi S, Saijo M, Citterio E, de Jager M, Hoeijmakers JH, Tanaka K. Translocation of Cockayne syndrome group A protein to the nuclear matrix: Possible relevance to transcription-coupled DNA repair. Proc Natl Acad Sci U S A. 2002;99 doi: 10.1073/pnas.012473199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam SM, Costa JC, Jardim L, Pires RF, Lehmann AR, Giugliani R. Cockayne syndrome: Report of a Brazilian family with confirmation of impaired RNA synthesis after UV-irradiation. Genet Mol Biol. 2000;275 [Google Scholar]
- Karikkineth AC, Scheibye-Knudsen M, Fivenson E, Croteau DL, Bohr VA. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev. 2017;33 doi: 10.1016/j.arr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, Xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92 doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khobta A, Epe B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mech Ageing Dev. 2013;134 doi: 10.1016/j.mad.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, Jaspers NGJ, Sarasin A, Stefanini M, Lehmann AR. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7 doi: 10.1016/j.dnarep.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Koch S, Gonzalez OG, Assfalg R, Schelling A, Schäfer P, Scharffetter-Kochanek K, Iben S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle. 2014;13 doi: 10.4161/cc.29018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47 doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Kubota M, Ohta S, Ando A, Koyama A, Terashima H, Kashii H, Hoshino H, Sugita K, Hayashi M. Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. Pedriatrics Int. 2015;57 doi: 10.1111/ped.12635. [DOI] [PubMed] [Google Scholar]
- Lake RJ, Geyko A, Hemashettar G, Zhao Y, Fan HY. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell. 2010;37 doi: 10.1016/j.molcel.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake RJ, Boetefuer EL, Tsai PF, Jeong J, Choi I, Won KJ, Fan HY. The sequence-specific transcription factor c-jun targets Cockayne syndrome protein B to regulate transcription and chromatin structure. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake RJ, Boetefuer EL, Won KJ, Fan HY. The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkv1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: Definition, function, and interaction with importin α. J Biol Chem. 2007;282 doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lans H, Vermeulen W. Nucleotide excision repair in Caenorhabditis elegans. Mol Biol Int. 2011;2011 doi: 10.4061/2011/542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci U S A. 2007;104 doi: 10.1073/pnas.0610619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugel V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech Ageing Dev. 2013;134 doi: 10.1016/j.mad.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz AC, Durand M, Sauvanaud F, Kristensen U, Vincent MC, Pasquier L, Odent S, Gener B, Tobias ES. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat. 2009;31 doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- Le May N, Mota-Fernandes D, Vélez-Cruz R, Iltis I, Biard D, Egly JM. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38 doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Lee MH, Ahn B, Choi IS, Koo HS. The gene expression and deficiency phenotypes of Cockayne syndrome B protein in Caenorhabditis elegans. FEBS Lett. 2002;522 doi: 10.1016/s0014-5793(02)02880-6. [DOI] [PubMed] [Google Scholar]
- Lehmann AR. Three complementation groups in Cockayne syndrome. Mutat Res. 1982;106 doi: 10.1016/0027-5107(82)90115-4. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Schubert S, Schäfer A, Apel A, Laspe P, Schiller S, Ohlenbusch A, Gratchev A, Emmert S. An unusual mutation in the XPG gene leads to an internal in-frame deletion and a XP/CS complex phenotype. Br J Dermatol. 2014;171 doi: 10.1111/bjd.13035. [DOI] [PubMed] [Google Scholar]
- Lin S, Lin Y, Nery JR, Urich MA, Breschi A, Davis CA, Dobin A, Zaleski C, Beer MA, Chapman WC. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci U S A. 2014;111 doi: 10.1073/pnas.1413624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U, Zhao Q, Zheng Y, Zhao Y. Sequence analysis IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31 doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M, Zhang F. Blockage of RNA polymerase as a possible trigger for UV light-induced apoptosis. Oncogene. 1996;13 [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Lovelace LJ, Yandava CN. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359 doi: 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusser A, Kadonaga JT. Chromatin remodeling by ATP-dependent molecular machines. BioEssays. 2003;25 doi: 10.1002/bies.10359. [DOI] [PubMed] [Google Scholar]
- Maddukuri L, Speina E, Christiansen M, Dudzinska D, Zaim J, Obtulowicz T, Kabaczyk S, Komisarki M, Bukowy Z, Szczegielniak J. Cockayne syndrome group B protein is engaged in processing of DNA adducts of lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat Res. 2009;666 doi: 10.1016/j.mrfmmm.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majora M, Sondenheimer K, Knechten M, Uthe I, Esser C, Schiavi A, Ventura N, Krutmann J. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci Transl Med. 2018;10 doi: 10.1126/scitranslmed.aam7510. [DOI] [PubMed] [Google Scholar]
- Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, Stefanini M, Lehmann AR. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62 doi: 10.1086/301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15 doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- Mayne LV, Lehmann AR. Failure of RNA synthesis to recover after UV irradiation: An early defect in cells from individuals with Cockayne’s syndrome and Xeroderma pigmentosum. Cancer Res. 1982;42 [PubMed] [Google Scholar]
- McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and dia before weaning. Nat Genet. 1993;5 doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36 doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Melcher K. The strength of acidic activation domains correlates with their affinity for both transcriptional and non-transcriptional protiens. J Mol Biol. 2000;301 doi: 10.1006/jmbi.2000.4034. [DOI] [PubMed] [Google Scholar]
- Menck CFM, Munford V. DNA repair diseases: What do they tell us about cancer and aging? Genet Mol Biol. 2014;37 doi: 10.1590/s1415-47572014000200008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menoni H, Wienholz F, Theil AF, Janssens RC, Lans H, Campalans A, Radicella JP, Marteijn JA, Vermeulen W. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JN, Boyd WA, Azzam GA, Haugen AC, Freedman JH, van Houten B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol. 2007;8 doi: 10.1186/gb-2007-8-5-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriel-Carretero M, Herrera-Moyano E, Aguilera A. A unified model for the molecular basis of Xeroderma pigmentosum-Cockayne syndrome. Rare Dis. 2015;3 doi: 10.1080/21675511.2015.1079362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Selzer R, Tuo J, Brosh RM, Bohr VA. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene. 2002;283 doi: 10.1016/s0378-1119(01)00870-8. [DOI] [PubMed] [Google Scholar]
- Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J Biol Chem. 2009;284 doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Mori MP, de Souza-Pinto NC. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion. 2014;17 doi: 10.1016/j.mito.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Murai M, Enokido Y, Inamura N, Yoshino M, Nakatsu Y, van der Horst GT, Hoeijmakers JH, Tanaka K, Hatanaka H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc Natl Acad Sci U S A. 2001;98 doi: 10.1073/pnas.231329598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagtegaal AP, Rainey RN, van der Pluijm I, Brandt RM, van der Horst GT, Borst JG, Segil N. Cockayne syndrome group B (Csb) and group A (Csa) deficiencies predispose to hearing loss and cochlear hair cell degeneration in mice. J Neurosci. 2015;35 doi: 10.1523/JNEUROSCI.5063-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu Y, Asahina H, Citterio E, Rademakers S, Vermeulen W, Kamiuchi S, Yeo J, Khaw M, Saijo M, Kodo N. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and rranscription. J Biol Chem. 2000;275 doi: 10.1074/jbc.M004936200. [DOI] [PubMed] [Google Scholar]
- Nardo T, Oneda R, Spivak G, Vaz B, Mortier L, Thomas P, Orioli D, Laugel V, Stary A, Hanawalt PC. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci U S A. 2009;106 doi: 10.1073/pnas.0902113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T, Narita K, Takedachi A, Saijo M, Tanaka K. Regulation of transcription elongation by the XPG-TFIIH complex is implicated in Cockayne syndrome. Mol Cell. 2015;35 doi: 10.1128/MCB.01401-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale V. A comprehensive description of the severity groups in Cockayne Syndrome. Am J Med Genet Part A. 2010;155 doi: 10.1002/ajmg.a.33933. [DOI] [PubMed] [Google Scholar]
- Nevedomskaya E, Meissner A, Goraler S, De Waard M, Ridwan Y, Zondag G, Van Der Pluijm I, Deelder AM, Mayboroda OA. Metabolic profiling of accelerated aging ERCC1d/- mice. J Proteome Res. 2010;9 doi: 10.1021/pr100210k. [DOI] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103 doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenbroch PO, Auk-Emblem P, Halsne R, Strand J, Forstrøm RJ, van der Pluijm I, Eide L. Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J. 2009;276 doi: 10.1111/j.1742-4658.2009.07004.x. [DOI] [PubMed] [Google Scholar]
- Osterod M, Larsen E, Le Page, Hengstler JG, van der Horst GT, Boiteux S, Klungland A, Epe B. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002;21 doi: 10.1038/sj.onc.1206027. [DOI] [PubMed] [Google Scholar]
- Pascucci B, Lemma T, Iorio E, Giovannini S, Vaz B, Iavarone I, Calcagnile A, Narciso L, Degan P, Podo F. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell. 2012;11 doi: 10.1111/j.1474-9726.2012.00815.x. [DOI] [PubMed] [Google Scholar]
- Pascucci B, D’Errico M, Romagnoli A, De Nuccio C, Savino M, Pietraforte D, Lanzafame M, Calcagnile AS, Fortini P, Baccarini S. Overexpression of Parkin rescues the defective mitochondrial phenotype and the increased apoptosis of Cockayne Syndrome A cells. Oncotarget. 2016;8 doi: 10.18632/oncotarget.9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoriza-Gallego M, Armier J, Sarasin A. Transcription through 8-oxoguanine in DNA repair-proficient and Csb-/Ogg1- DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis. 2007;22 doi: 10.1093/mutage/gem024. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Kadonaga JT. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell. 1997;88 doi: 10.1016/s0092-8674(00)81918-2. [DOI] [PubMed] [Google Scholar]
- Pei DS, Strauss PR. Zebrafish as a model system to study DNA damage and repair. Mutat Res. 2013;744 doi: 10.1016/j.mrfmmm.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med. 2015;85 doi: 10.1016/j.freeradbiomed.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prates Mori M, de Souza-Pinto NC. Role of mitochondrial dysfunction in the pathophysiology of DNA repair disorders. Cell Biol Int. 2018;42 doi: 10.1002/cbin.10917. [DOI] [PubMed] [Google Scholar]
- Raj DD, Jaarsma D, Holtman IR, Olah M, Ferreira FM, Schaafsma W, Brouwer N, Meijer MM, de Waard MC, van der Pluijm I. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol Aging. 2014;35 doi: 10.1016/j.neurobiolaging.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Ranes M, Boeing S, Wang Y, Wienholz F, Menoni H, Walker J, Encheva V, Chakravarty P, Mari PO, Stewart A. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkw216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res. 2005;79 doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- Revet I, Feeney L, Tang A, Huang EJ, Cleaver JE. Dysmyelination not demyelination causes neurological symptoms in preweaned mice in a murine model of Cockayne syndrome. Proc Natl Acad Sci U S A. 2012;109 doi: 10.1073/pnas.1202621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatella M, Theil AF, Ribeiro-Silva C, Slyskova J, Thijssen K, Voskamp C, Lans H, Vermeulen W. Repair protein persistence at DNA lesions characterizes XPF defect with Cockayne syndrome features. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Rupp WD. A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell. 1983;33 doi: 10.1016/0092-8674(83)90354-9. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: Insights for transcription-coupled repair and Cockayne syndrome. Mol Cell. 2005;20 doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DM, Cooper M, Spencer R, de Cabo R. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. 2012;209 doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Tseng A, Jensen MB, Scheibye-Alsing K, Fang EF, Iyama T, Bharti SK, Marosi K, Froetscher L, Kassahun H. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc Natl Acad Sci U S A. 2016;113 doi: 10.1073/pnas.1610198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickel RD, Chu EH, Trosko JE, Chang CC. Cockayne syndrome: A cellular sensitivity to ultraviolet light. Pediatrics. 1977;60 [PubMed] [Google Scholar]
- Schwertman P, Lagarou A, Dekkers DHW, Raams A, van der Hoek A, Laffeber C, Hoeijmakers JH, Demmers JAA, Fousteri M, Vermeulen W. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44 doi: 10.1038/ng.2230. [DOI] [PubMed] [Google Scholar]
- Seeberg E, Strike P. Excision repair of ultraviolet-irradiated deoxyribonucleic acid in plasmolyzed cells of Escherichia coli. J Bacteriol. 1976;125 doi: 10.1128/jb.125.3.787-795.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Proc Natl Acad Sci U S A. 1997;94 [Google Scholar]
- Selby CP, Sancar A. J Biol Chem. 1997;272 [Google Scholar]
- Selzer RR, Nyaga S, Tuo J, May A, Muftuoglu M, Christiansen M, Citterio E, Brosh RM, Bohr VA. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 2002;30 doi: 10.1093/nar/30.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110 doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin Y, Tanaka K, Saijo M. The C-terminal region and SUMOylation of Cockayne syndrome group B protein play critical roles in transcription-coupled nucleotide excision repair. J Biol Chem. 2016;291 doi: 10.1074/jbc.M115.683235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitsbergen JM, Kent ML. The state of the art of the zebrafish model for toxicology and toxicologic pathology research - advantages and current limitations. Toxicol Pathol. 2003;31 doi: 10.1080/01926230390174959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak G. UV-sensitive syndrome. Mutat Res. 2005;577 doi: 10.1016/j.mrfmmm.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Spivak G, Ganesan AK. The complex choreography of transcription-coupled repair. Int J Mol Sci. 2014;19 doi: 10.1016/j.dnarep.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev. 2008;129 doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lübbert H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum Mol Genet. 2007;16 doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gki683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker APM, Hanaoka F, Bootsma D, Hoeijmakers JHJ. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2 doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- Sunesen M, Selzer RR, Brosh Jr. Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acids Res. 2000;28 doi: 10.1093/nar/28.16.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghdiri M, Dastsooz H, Fardaei M, Mohammadi S, Fard MAF, Faghihi MA. A novel mutation in ERCC8 gene causing Cockayne syndrome. Front Pediatr. 2017;5 doi: 10.3389/fped.2017.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Kawai K, Kumahara Y, Ikenaga M, Okada Y. Genetic complementation groups in Cockayne syndrome. Somatic Cell Genet. 1981;7 doi: 10.1007/BF01542989. [DOI] [PubMed] [Google Scholar]
- Tantin D. RNA polymerase II elongation complexes containing the Cockayne syndrome group B protein interact with a molecular complex containing the transcription factor IIH components xeroderma pigmentosum B and p62. J Biol Chem. 1998;273 doi: 10.1074/jbc.273.43.27794. [DOI] [PubMed] [Google Scholar]
- Tantin D, Kansal A, Carey M. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol Cell Biol. 1997;17 doi: 10.1128/mcb.17.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3 doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Titus TA, Yan YL, Wilson C, Starks AM, Frohnmayer JD, Canestro C, Rodriguez-Mari A, He X, Postlethwait JH. The Fanconi anemia/BRCA gene network in zebrafish: Embryonic expression and comparative genomics. Mutat Res. 2009;668 doi: 10.1016/j.mrfmmm.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S. Transcription arrest at DNA damage sites. Mutat Res. 2005;577 doi: 10.1016/j.mrfmmm.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Trapp C, Reite K, Klungland A, Epe B. Deficiency of the Cockayne syndrome B (CSB) gene aGG-NERavates the genomic instability caused by endogenous oxidative DNA base damage in mice. Oncogene. 2007;26 doi: 10.1038/sj.onc.1210167. [DOI] [PubMed] [Google Scholar]
- Troelstra C, Odijk LH, de Wit J, Westerveld A, Thompson LH, Bootsma D, Hoeijmakers JHJ. Molecular cloning of the human DNA excision repair. Mol Cell Biol. 1990;10 doi: 10.1128/mcb.10.11.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JHJ. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71 doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- Troelstra C, Hesen W, Bootsma D, Hoeijmakers JHJ. Structure and expression of the excision repair gene ERCC6, involved in the human disorder Cockayne’s syndrome group B. Nucleic Acids Res. 1993;21 doi: 10.1093/nar/21.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Müftüoglu M, Chen C, Jaruga P, Selzer RR, Brosh RM, Rodriguez H, Dizdaroglu M, Bohr VA. The Cockayne syndrome group b gene product is involved in general genome base excision repair of 8-Hydroxyguanine in DNA. J Biol Chem. 2001;276 doi: 10.1074/jbc.M107888200. [DOI] [PubMed] [Google Scholar]
- Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst) 2002;1 doi: 10.1016/s1568-7864(02)00116-7. [DOI] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Dizdaroglu M, Bohr VA. J Biol Chem. 2002;277 doi: 10.1074/jbc.M204814200. [DOI] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003;17 doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- van den Boom V, Citterio E, Hoogstraten D, Zotter A, Egly JM, van Cappellen WA, Hoeijmakers JHJ, Houtsmuller AB, Vermeulen W. DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J Cell Biol. 2004;166 doi: 10.1083/jcb.200401056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst GTJ, Van Steeg H, Berg RJW, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer pre-disposition. Cell. 1997;89 doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- van der Horst GTJ, Meira L, Gorgels TG, de Wit J, Velasco-Miguel S, Richardson JA, Kamp Y, Vreeswijk MPG, Smit B, Bootsma D. UVB radiation-induced cancer pre-disposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 2002;1 doi: 10.1016/s1568-7864(01)00010-6. [DOI] [PubMed] [Google Scholar]
- van der Pluijm I, Garinis GA, Brandt RMC, Gorgels TG, Wijnhoven SW, Diderich KEM, De Wit J, Mitchell JR, van Oostrom C, Beems R. Impaired genome maintenance suppresses the growth hormone-insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007;5 doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool AJ, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, Egly JM, Bootsma D, Hoeijmakers JH. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997;16 doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoffen A, Natarajan AT, Mayne LV, Zeeland AA, Mullendersl LHF, Venemal J. Deficient repair of the transcribed strand of active in Cockayne’s syndrome cells. Nucleic Acids Res. 1993;21 doi: 10.1093/nar/21.25.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houten B, Hunter SE, Meyer JN. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front Biosci. 2016;21 doi: 10.2741/4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Mullenders LH, Natarajan AT, van Zeeland AA, AA and, Mayne LV. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Natl Acad Sci U S A. 1990;87 doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij WP, Dollé MET, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, Wu H, Roks AJM, Botter SM, van der Eerden BC. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016;537 doi: 10.1038/nature19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessoni AT, Herai RH, Karpiak JV, Leal AMS, Trujillo CA, Quinet A, Lima LFA, Menck CFM, Muotri AR. Cockayne Syndrome-derived neurons display reduced synapse density and altered neural network synchrony. Hum Mol Genet. 2016;25 doi: 10.1093/hmg/ddw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12 doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu J, Chong J, Wang D. Structural basis of DNA lesion recognition for eukaryotic transcription-coupled nucleotide excision repair. DNA Repair (Amst) 2018;71 doi: 10.1016/j.dnarep.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chakravarty P, Ranes M, Kelly G, Brooks PJ, Neilan E, Stewart A, Schiavo G, Svejstrup JQ. Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc Natl Acad SciUSA. 2014;111 doi: 10.1073/pnas.1412569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, van Ham RCA, Vermeulen W, Bootsma D, van der Eb AJ, Hoeijmakers JHJ. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders Xeroderma pigmentosum and Cockayne’s syndrome. Cell. 1990;62 doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, Van Steeg H, Bootsma D, Hoeijmakers JHJ. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7 doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M. DNA damage during the G0 / G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc Natl Acad Sci U S A. 2015;112 doi: 10.1073/pnas.1507105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenheim KM, Dickson DW, Rapin I. Neuropathology of Cockayne syndrome: Evidence for impaired development, premature aging, and neurodegeneration. Mech Ageing Dev. 2009;130 doi: 10.1016/j.mad.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Wilson BT, Stark Z, Sutton RE, Danda S, Ekbote AV, Elsayed SM, Gibson L, Goodship JA, Jackson AP, Keng WT. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet Med. 2015;18 doi: 10.1038/gim.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35 doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wei W, Chen J, Cong R, Shi T, Li J, Wong J, Du JX. Acidic domains differentially read histone H3 lysine 4 methylation status and are widely present in chromatin-associated proteins. Sci China Life Sci. 2017;60 doi: 10.1007/s11427-016-0413-3. [DOI] [PubMed] [Google Scholar]
- Xu C, Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2 doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Chen L, Ye L, Fei L, Tang W, Tian Y. Identification of two missense mutations of ERCC6 in three chinese sisters with Cockayne syndrome by Whole Exome Sequencing. PloS One. 2014;9 doi: 10.1371/journal.pone.0113914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat Genet. 2012;44 doi: 10.1038/ng.2228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.