

Abstract
Coupling reactions of feedstock alkenes are promising, but few of these reactions are practiced industrially. Even though recent advances in the synthetic methodology have led to excellent regio- and enantioselectivies in the dimerization reactions between 1,3-dienes and acrylates, the efficiency as measured by the turnover numbers (TON) in the catalyst has remained modest. Through a combination of reaction progress kinetic analysis (RPKA) of a prototypical dimerization reaction, characterization of isolated low-valent cobalt catalyst precursors involved, several important details of the mechanism of this reaction have emerged. (i) The prototypical reaction has an induction period that requires at least two hours of stir time to generate the competent catalyst. (ii) Reduction of a Co(II) complex to a Co(I) complex, and subsequent generation of a cationic [Co(I)]+ species are responsible for this delay. (iii) Through RPKA using in situ IR spectroscopy, same excess experiments reveal inhibition by the product towards the end of the reaction and no catalyst deactivation is observed as long as diene is present in the medium. The low TON observed is most likely the result of the inherent instability of the putative cationic Co(I)-species that catalyzes the reaction. (iv) Different excess experiments suggest that the reaction is first order in the diene and zero order in the acrylate. (v) Catalyst loading experiments show that the catalyst is first order. The orders in the various regents were further confirmed by Variable Time Normalization Analysis (VTNA). (vi) A mechanism based on oxidative dimerization [via Co(I)/Co(III)-cycle] is proposed. Based on the results of this study, it is possible to increase the TON by a factor of 10 by conducting the reaction at an increased concentration of the starting materials, especially, the diene, which seems to stabilize the catalytic species.
Keywords: heterodimerization; 1,3-dienes; acrylates; cobalt catalysis; counterion effects; kinetics; mechanism
Graphical Abstract
INTRODUCTION
Development of highly selective and catalytic coupling reactions between feedstock materials can greatly impact the design, synthesis, and manufacture of molecules of interest from materials to medicines.1 While the most readily available precursors used in the chemical industry are alkenes, 1,3-dienes, and alkyl acrylates, large-scale applications of heterodimerizations between such compounds are rare.2 These precursors are most often used as monomers3 since current heterodimerization methods are hindered by the need for expensive catalysts and low turnover number (TON). The key challenge is the limited mechanistic understanding of the chemical driving forces that could be modified to achieve higher TON. Recent work on heterodimerizations between 1,3-dienes and ethylene or alkyl acrylates has examined catalytic systems that show very high regio- and stereoselectivity, albeit with limited TONs (rarely exceeding 200 for cobalt, for example4). It is hoped that the yield and TON for the overall processes could be improved through developing a thorough mechanistic understanding of these reactions. Elucidating the details of the kinetics would provide valuable insights into the reaction mechanism that could be used to improve catalyst performance (selectivity, TON, rates, and yield).
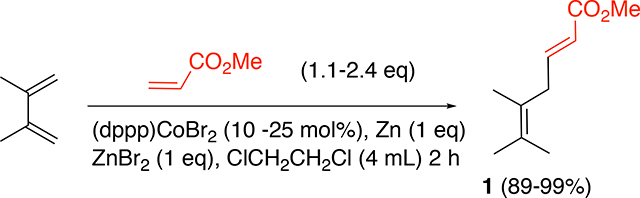
Heterodimerization has received considerable attention with notable examples including a Rh-catalyzed 1,3-diene-ethylene dimerization that has been extensively studied in connection with the synthesis of 1,4-hexadiene as a monomer for elastomers.5 A related reaction, the codimerization between 1,3-butadiene and methyl acrylate, which first appeared in the literature in 1964,6 has never been fully developed into a commercial scale process (Figure 1, Eq 1). The reported scope for various diene and acrylate precursors, and the selectivities observed for this dimerization reaction has been somewhat limited until recently.7–9,4 A notable early example of this reaction was the cobalt-catalyzed heterodimerization between 2,3-dimethyl-1,3-butadiene and t-butyl acrylate (Figure 1, Eq 2a) reported by Hilt in 2001.8 More recently, Hirano has reported the applications of Ru(0)-complexes for codimerization of 1,3-dienes and acrylates (Figure 1, Eq 2b).9 In 2017, one of our groups reported4 the first example of an enantioselective version of this reaction where chiral cationic cobalt(I)-bis-phosphine complexes of the type [(S,S)-BDPP]Co]+ X− [BDPP = 2,4-bis(diphenylphosphino)pentane; X = [3,5- (CF3)2C6H3]4B−), abbreviated as BArF−] was found to be an excellent catalyst for heterodimerization between a broad class of 1,3-dienes and various acrylates (Figure 1, Eq 3). During these studies, we also found that a minor modification of the Hilt procedure8b using [1,3-bis-(diphenylphosphino)propane)]CoBr2 is an attractive alternative for the simple co-dimerization of various 1,3-dienes and acrylates (Figure 1, Eq 4).10 Since these precursors are abundantly available and, the chemistry highly selective, such reactions provide inexpensive routes to value-added intermediates.
Figure 1.

Heterodimerization of 1,3-dienes and methyl acrylate
Since heterodimerization can be catalyzed using inexpensive cobalt catalysts, this overall system could be powerful if the challenges with low TON can be overcome. A careful evaluation of the various dimerization catalysts suggests that low-valent cobalt species generated in situ from the 1,n-bis-diarylphosphinoalkane-cobalt (II) salts hold the greatest potential for these and related reactions; yet, very little is known about the mechanism of this reaction. Principally, two mechanistic scenarios (Figure 2) have been advanced for olefin dimerization reactions:2a (1) one involving discrete metal-hydrides with no changes in the oxidation state of the metal throughout catalysis (most often invoked in nickel-catalyzed reactions),11 and (2) one involving an oxidative dimerization in which the metal undergoes redox changes, such as proposed in the dimerization reactions catalyzed by Co4,12 and Ru13 systems. Elucidating the key mechanistic steps has the potential to provide the necessary insights that will allow this reaction to be performed at high TON using inexpensive catalysts.
Figure 2.

Possible mechanisms for heterodimerization
While traditional methods that use high substrate concentrations to cause the system to behave as pseudo-first order could be used to investigate the reaction mechanism, it is increasingly recognized that the high substrate concentrations may not be relevant to the actual reaction mechanism. Indeed, insights related to the actual chemistry are best obtained using reactant concentrations that are relevant to actual process conditions since competitive binding of substrates may alter the observed reaction mechanism. Use of in situ IR spectroscopy as a sampling technique offers considerable advantage in determining the concentration as a function of time since there is no need to withdraw aliquots from the highly oxygen and moisture sensitive reactions in progress. To maximize results while minimizing experiments, Reaction Progress Kinetic Analysis (RPKA)14 and variable time normalization analysis (VTNA)15 were performed. RPKA and VTNA were aided by in situ IR spectroscopy, which enabled continuous monitoring of the reactant concentrations until the reaction proceeded to near completion.
In this work, we will study a prototypical diene-acrylate dimerization between methyl acrylate and 2,3-dimethylbutadiene (Eq 5) with the goal of providing further details on the mechanism of an important class of reactions exemplified by this transformation. Initial experiments showed an induction period during which the pre-catalyst added to the reaction mixture presumably transformed into the active cobalt(I) species. The nature of this species was clarified via isolation and UV-Visible spectroscopy of some of the relevant intermediates responsible for catalysis. Using a consistent stirring period, reproducible experiments were achieved that were subsequently used to investigate the reaction order of the different components in the reaction, including the acrylate, the diene, and the catalyst. These results were used to propose one potential reason that current investigations have only observed low TON. Through modifying reaction conditions, the TON was increased by an order of magnitude.
Catalyst Activation
The cobalt-catalyzed heterodimerization was investigated using a standard probe reaction of 2,3-dimethyl-1,3-butadiene and methyl acrylate under conditions that had been previously established (Eq 5).10 The procedure involves combining (dppp)CoBr2 (10–25 mol%), Zn (1 eq), and ZnBr2 (1 eq) under a nitrogen atmosphere in 2 mL of dichloroethane at 30 °C. After combining these components in a flask equipped with a ReactIR probe, the reaction was initiated by the addition of the substrates 2,3-dimethyl-1,2-butadiene (1 eq) and methyl acrylate (1.1–2.4 eq) in 2 mL DCE. Upon addition of the reactants, several new peaks are observed in the FT-IR. From background experiments (p. S17-S18), it was determined that the peak at 896 cm−1 (=CH2 out-of-plane wagging)16a and 1401 cm−1 (scissoring vibration of the terminal methylene16b) are associated with the diene and acrylate respectively. These peaks could be reliably used to quantify the amount of each substrate in the reaction mixture over the course of the reaction. Through extracting the intensities of these peaks over time, it was observed that the concentration of both substrates decreases over time, as shown for acrylate in Figure 3.
Figure 3.


(a) Acrylate concentration, measured by the intensity at 1401 cm−1, vs time for stir time investigations at 5 min, 1 hour, 2 hours, and 8 hours. 0.31 M acrylate, 0.15 M diene, and 0.15 M (dppp)Co(II)Br2 was used for each run. (b) Zoomed in region of graph (a) from 0 to 0.2 hours.
 |
(5) |
One important early discovery was that the reaction exhibited an induction period that was ascertained through using different catalyst activation times before the addition of the reactants (Figure 3). When using an initial activation time of five minutes, the concentration of the substrates initially decreased slowly followed by a faster reaction. This observation suggested that the actual catalyst was not fully formed initially for the reaction to proceed. Additionally, it was determined that a procedure using a five-minute stir time for activation resulted in data that was difficult to reproduce. This has been attributed to an incomplete activation of the catalyst (i.e., reduction of Co(II) to a Co(I) species and, possibly, subsequent formation of a cationic species (see later for details) likely associated with the use of insoluble reagents, including metallic Zn.17
To obtain reproducible kinetics, a series of experiments was performed in which the stir time was systematically varied by allowing the (dppp)CoBr2, Zn, and ZnBr2 to mix for different times prior to adding in diene and acrylate. In Figure 3, the results were depicted for the experiments performed using different stir times. In comparison to the five-minute stir time, using longer stir times resulted in more reproducible data. The data when using a stir time of one hour still had an initial period of slow conversion over time followed by an acceleration in reaction rate, suggesting the catalyst was still being activated. An expanded version of the concentration vs time is shown in Figure 3b, showing the initial time t = 0 to t = 25 minutes. This shows more clearly the initial slow pace of the reaction upon shorter activation times. It was found that a stir time of 2 hours resulted in a reproducible reaction profile that was consistent with first order kinetics. Therefore, two hour stir time was selected for all of the subsequent kinetic and mechanistic investigations described in this work.
Induction period and UV-Vis spectroscopy
An induction period suggests that (dppp)CoBr2 is a pre-catalyst that transforms into the active catalyst when using the reducing agent, Zn, and the activator, ZnBr2. The presence of an induction period prompted investigations of the possible cobalt species present in solution using UV-Vis spectroscopy (Figure 4). UV-Vis measurements were used to analyze different cobalt species, purported to be involved in the catalytic cycle, including (dppp)Co(II)Br2, ‘(dppp)Co(I)Br’, and ‘[(dppp)Co(I)]+’ [dppp = 1,3-Bis-(diphenyl phosphino) propane].18 The activation conditions used for catalysis were simulated by stirring (dppp)CoBr2, Zn, and ZnBr2 in DCE for two hours with rigorous exclusion of oxygen at the concentrations that the reaction are typically run (0.0155–0.0375 M of (dppp)CoBr2]. The excess Zn was filtered off and the supernatant solution was used for UV-Vis measurements. For the UV-Vis analysis, it was necessary to dilute the sample with DCE to avoid excessive absorption in the spectral region of interest.
Figure 4.

UV-Vis spectra of (dppp)Co(II)Br2 (Blue), (dppp)3Co2(I)Br2 (3, Green), and [(dppp)Co(I)]+ (Red) in dichloroethane. (dppp)Co(II)Br2 (Blue) and (dppp)3Co2(I)Br2 (3, Green) were obtained from pure crystallized materials of the named compound. [(dppp)Co(I)]+ (Red) was obtained by the following: the supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 that was stirred for two hours in a glovebox. The three plots are offset (y-axis) for clarity.
For recrystallized (dppp)Co(II)Br2, the UV-vis spectra had three distinct peaks at wavelengths above 400 nm, including peaks at 622, 662, and 735 nm (Figure 4). This spectrum was consistent with previous characterizations of Co(II) complexes like X2Co(PPh3)2 [e.g., 605 nm, 635 nm and 745 nm for Cl2Co(PPh3)2 in benzene19a or 591, 636 and 728 for (dppp)CoCl2 in CH2Cl219c (Table 1)]. Importantly, the Co(II) complex did not exhibit any peaks around 500 and 900 nm that would be characteristic of Co(I) compounds. Indeed, a related Co(I) species (Ph3P)3CoCl was reported to have absorptions at 745 and 940 nm in benzene (run in the presence of excess ligand to avoid decomposition) in addition to peaks at 605 nm and 635 nm ascribed to Co(II) species formed in situ by slow disproportionation of Co(I) species to Co(II) and metallic Co.19a, 19c Additionally, a bromo-derivative (Ph3P)3CoBr had peaks at 750 nm and 970 nm.19b
Table 1.
UV-Visible Spectra of Cobalt(I) and Cobalt(II) Complexesa
| Compound | (Ph3P)2CoCl2 | 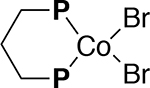 |
[Ph3P]3CoX | |
| λmax (nm) | (605, 635, 745)19a | (622,662,735)a | X =Cl (745, 970)19a,19c | X=Br (750, 970)19b |
| Compound | 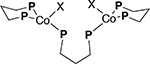 |
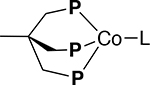 |
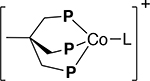 |
|
| Co(I): (dppp)3Co2X2 [2 and 3]a,b | Co(I) | [Co(I)]+ | ||
| λmax (nm) | 2 X = Cl (710, 910) |
3 X = Br (725, 935) 3 + NaBArF (500, 970) |
4 L = Cl (530, 826)19c | 5 L = η4-1,3-butadiene (460)21 |
UV spectra of (dppp)CoBr2, 2 and 3, this work.
P = Ph2P
We also prepared model Co(I) complexes 2 and 3 (Table 1) and fully characterized these compounds by X-ray crystallography.20 Their UV-Vis spectra were recorded in DCE, as shown in the Figure 5. The chloro-compound has the major absorptions are at 510 (sh), 710 (sh), and 910 nm, and the bromo-compound at 510 (sh), 725 (sh), and 935 nm (in addition to two broad absorptions around 600 nm that may be ascribed to unavoidable contaminant Co(II) species arising from the previously mentioned disproportionation). These absorptions are remarkably close to the authentic Co(I) complexes reported in the literature (see for example, of (Ph3P)3CoCl and (Ph3P)3CoBr, See Table 1.19a–b Upon storage or exposure to oxygen, characteristic peaks associated with in solution Co(I) or ‘[(dppp)Co(I)]+’ quickly disappears, as reported for other Co(I) complexes like (Ph3P)3CoBr (Figure 6).19b
Figure 5.

UV-Vis spectra of (dppp)3Co2Cl2 (2, black) and (dppp)3Co2Br2 (3, red). See Table 1 for structures.
Figure 6.

The supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 was mixed for two hours. After removing the excess Zn, the solution was allowed to sit at room temperature. The UV-Vis spectrum was recorded from 0 to 36 minutes. The plots shown are smoothed (binomial smoothing). The initial intensity (0 minutes) is shown red with the final intensity (36 minutes) is shown in blue, all intermediate intensities are gray.
Stirring a mixture of (dppp)CoBr2, Zn and ZnBr2 and following the UV absorptions of the supernatant (Figure 7) provided a clue about the induction period. Since these compounds (i.e., Co(I) and Co(II)) absorb strongly, these experiments were performed using lower concentrations of each component (p. S12) affecting the time for the catalyst to be activated. With time, the intensity of the characteristic peaks for Co(I) at 740 nm and the broad band above 900 nm progressively increased until it reaches a maximum at 240 min (Figure 7). Also, we observed a parallel behavior of a strong peak at 517 nm, which also reaches its maximum intensity at ca. 240 minutes. No further changes in the spectrum were noted after this period. We assigned the absorption at 517 nm to ‘[(dppp)Co(I)]+’.18 Further support for such a claim also came from NMR studies. While as expected 31P NMR of (dppp)CoBr2 in CDCl3 shows no signals, addition of excess Zn and mixing for 15 minutes gave two signals at δ −21.87 and 15.47 in an approximate ratio of 2:1 respectively. Further addition of ZnBr2 (2 eq with respect to Co(II)-complex) after 30 minutes only one peak at δ - 21.89 was observed. We believe this peak corresponds to the catalytically competent [LCo(I)]+ species. A similar species was produced when (dppp)CoBr2 is reduced with zinc in the presence of NaBArF. See Supporting Information (p. S51–54) for further experimental details and spectra.
Figure 7.

The supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 was mixed for different amounts of time. The peak at 517 nm stopped increasing at about four hours and thirty minutes.
Additional evidence for the intermediacy of such a species comes from the observation of identical peaks, when reduction of (dppp)CoBr2 was carried out with Zn in the presence NaBArF (Figure 8) or when an authentic Co(I) complex, which we have prepared, (dppp)3Co2Br2 (3), was treated with NaBArF (Figure 9). A comparable cationic species (5, Table 1) described in the literature had an absorption at 460 nm.21 The blue shift in this presumed metal-to-ligand charge transition in the complex (5, Table 1) compared to the corresponding complex ‘[(dppp)Co]+’ might be associated with the strain of the distorted tetrahedral geometry imposed by the tripodal ligand in 4.22
Figure 8.

Zn-Reduced dpppCo(II)Br2 activated with ZnBr2 (purple) and Zn-reduced dpppCo(II)Br2 activated with NaBArF (red) both stirred for 2 hours at the same concentrations.
Figure 9.

The UV-Vis spectrum of crystalline (dppp)3Co2(I)Br2 (3), with added NaBArF (black) compared to the UV spectrum of the crystalline [(dppp)2Co(I)]+ BArF− (7, red).
After the activation conditions had been examined experimentally through UV-Vis, several efforts were made to identify, and, if possible, isolate and characterize the effective catalyst responsible for this reaction. Two possible scenarios under which the reaction could be catalyzed involve either a neutral [Co(I)] species or a corresponding cationic [Co(I)]+ intermediate. To support the need for cationic cobalt, parallel reactions were executed with one using ‘(dppp)Co(I)Br’ (isolated from reduction of (dppp)Co(II)Br2 using Zn) and the other containing dpppCo(I)Br and ZnBr2. The (dppp)Co(I)Br reaction in the absence of ZnBr2 showed no product formation (See SI p. S12). The (dppp)Co(I)Br and ZnBr2 reaction showed diene conversion to product albeit in low yield (p. S12). This indicated that only under conditions that favor the formation of cationic cobalt did the reaction proceed. We had previously observed this behavior in a number of other ethylene/1,3-dodecadiene and methyl acrylate/1,3-dodecadiene dimerization reactions where several isolated neutral Co(I) complexes (e. g., (COD)Co(η3-cycloheptenyl), (dppp)Co(η3-cycloheptenyl), Zn-reduced (dppp)CoBr2 free of ZnBr2) failed to affect the reaction in the absence of a Lewis acid.4 Addition of one equivalent of NaBArF (a bromide sequestering agent better than ZnBr2) dramatically accelerates these reactions. We have seen similar behavior also in the reaction under study (2,3-dimethylbuta-1,3-diene + methyl acrylate, See Supporting Information for details, p. S14–15, See Figure S3). This strongly supports the hypothesis that the cationic cobalt is the active catalyst.
In an attempt to isolate a cationic cobalt complex that was a competent catalyst, recrystallized Co(I) complex (dppp)3Co2Cl2 (2) was further reacted with NaBArF in ether in the presence of 1,4-cyclooctadiene with the expectation to produce a substitutionally labile Co(I) complex (6, Eq 6). The reaction did not yield the expected product, but gave instead, another 16e-complex [(dppp)2Co]+ [BArF]− (7, Eq 7), identified by X-ray crystallography (Figure 10).23 This complex, had a UV-Vis spectrum (675 nm, 960 nm) distinctly different from the species that was observed when (dppp)3Co2Cl2 was treated with NaBArF in that it lacked the prominent peak around 500 nm. This complex did not catalyze the heterodimerization reaction under the reaction conditions. Incidentally, we had also isolated a related [Co(I)]+ complex, 8, from a medium containing (dppp)CoBr2, Zn and ZnBr2. Collectively the above listed experiments strongly suggested a hitherto unknown substitutionally labile cationic cobalt (I) species that was present under the reaction conditions, (dppp)CoBr2 + Zn + ZnBr2 or (dppp)CoBr2 + Zn + NaBArF, was responsible for initiation of the catalytic process.24
Figure 10.

Solid -state structures of [(dppp)2Co]+ [BArF]− (7) and [(dppp)2Co]+ [ZnBr3.THF]− (8). Hydrogens are omitted for clarity. The THF molecule in the second structure contains a disordered carbon atom and the two positions for this disordered atom are shown in the attached ORTEP plot. See Supporting Information for cif.
As stated before, the three species of cobalt, (dppp)Co(II)Br2, ‘(dppp)Co(I)Br’ and the presumed ‘[(dppp)Co(I)]+’ intermediate were present under the reaction conditions (Figures 4 and 7). Notice also that the generation of this species (‘[(dppp)Co(I)]+), presumably responsible for the 510 nm peak in the UV spectrum, appeared to be cleaner under the Zn/NaBArF reduction conditions (Figure 8) as compared to Zn/ZnBr2 conditions. Although the major absorbance originated from the [Co(I)]+ peak, small peaks attributed to the Co(II) and Co(I) species were still observed (especially under the latter reaction conditions), suggesting that all three species were present at the start of the reaction. This suggested that the catalyst went through two transformations prior to being able to affect the reaction described in Eq 5: (dppp)Co(II)Br2 to (dppp)Co(I)Br, and, (dppp)Co(I)Br to [(dppp)Co(I)]+, with only the [(dppp)Co(I)] + being the active species. If insufficient time was given to allow for these transformations, the reaction appeared initially sluggish, and the kinetic measurements were not reproducible. This behavior was exactly what was observed for short stir times (<2 hours) with the induction period, but not at the longer stir times (two hours and beyond). The reproducible reaction at stir times of two hours (or greater) in conjunction with slow evolution of a catalytic species as determined by the initial UV-Vis analysis strongly supported the presence of an induction period during which the active catalytic species, [(dppp)Co(I)]+ was produced. Thus, to obtain the highest TON and rate of reaction, the sufficient stir time of the mixture containing the pre-catalyst, reducing agent, and activator must be allowed.17
Same excess experiments
After establishing a robust and reliable protocol for the catalytic reaction, the mechanistic investigations were continued by performing “same excess” experiments. The “same excess” experiments were used to probe catalyst activation and product inhibition. For the “same excess” experiments, the difference between the molarities of diene and acrylate were maintained constant across multiple experiments, but the starting concentrations were changed, as listed in Table 2. More details of experimental protocols including conversions and starting concentrations can be found in the supporting information (p. S7-S8). Figure 11 shows the concentration profiles of acrylate for each of the same excess experiments. As has commonly been done to mitigate experimental artifacts associated with sampling, the data for each experiment were time shifted in Figure 11, such that the molar concentration of acrylate of 0.25 M (i.e., 15% conversion of the [acrylate]) lined up with one another (unshifted data located in the SI, p. S9, Figure S2 (b)). From the four experiments, it was observed that the concentration of acrylate over time decreased similarly regardless of the initial substrate concentrations. Yet, the experiment revealed the possibility of catalyst deactivation or product inhibition. The overlap seen from the same excess experiments demonstrate that it was not the catalyst turning over to form product that was causing it to deactivate, but instead, the low TON was an issue related to the relative concentration of the starting material to the product. This suggested that at low concentrations of diene the catalyst deactivated or remain coordinated to the product, limiting the TON of the reaction. This observation was critical in further work to increase the TON (vide infra). Because of these observations, product inhibition studies were performed. The product inhibition experiments showed at large excess of product (1.2 eq) the reaction is hampered in terms of rate. This suggest that the product displacement is involved in the turnover limiting step (TOLS) or affected the catalyst resting state (See SI page S9-S10, Figure S2 (a) for experimental and visual analysis). When the addition of a large excess of product (58 equivalence) is made to the active catalyst, an increase in the peak at ca. 960 nm is observed. This demonstrates that the product is able to bind to the active catalyst (The UV-Vis spectra can be found in the supporting information p. S28 – S30). Further supporting that product is able to inhibit the catalytic pathway.
Table 2.
‘Same Excess’ Experimental Conditions and Resultsa
| Excess Experiment | Acrylate (M) | (dppp)CoBr2 (mmol) | Zn (mmol) | ZnBr2 (mmol) | Diene Conver. (%)b |
|---|---|---|---|---|---|
| 1 | 0.26 | 0.152 | 1.52 | 1.58 | 90 |
| 2 | 0.28 | 0.152 | 1.53 | 1.58 | 91 |
| 3 | 0.31 | 0.152 | 1.52 | 1.57 | 98 |
| 4 | 0.36 | 0.152 | 1.52 | 1.58 | 89 |
See Supporting Information for full details on the experimental procedure and characterization of the products.
Determined by GC-FID analysis.
Figure 11.

Acrylate concentration vs time for same excess reactions, with time shift to 15% conversion of 0.36 M acrylate and 0.10 M diene. The catalyst concentration for all of these reactions were 40 mmole. –The diene concentration was set to 0.10 M, 0.12 M, 0.15 M, and 0.20 M while the acrylate concentration was set to 0.26 M, 028 M, 0.31 M, and 0.36 M respectively.
Different excess experiments
The different excess experiments were used to investigate the order of diene and acrylate in the reaction. They were performed by varying the difference in molarity of diene and acrylate (p. S9). The different conditions and results were recorded in Table 3. The different excess experiments were best analyzed by plotting reaction rate as a function of concentration, as shown in Figure 12. The standard conditions were 0.31 M acrylate and 0.15 M diene, which was performed in duplicates to determine the standard deviation of the experiments and correspondingly the limitations of the analysis.
Table 3.
‘Different Excess’ Experimental Conditions and Resultsa
| Excess Experiment | Diene (M) | Acrylate (M) | (dppp)CoBr2 (mmol) | Zn (mmol) | ZnBr2 (mmol) | Conversion of Diene (%)a |
|---|---|---|---|---|---|---|
| 1 | 0.151 | 0.17 | 0.152 | 1.52 | 1.58 | 97 |
| 2 | 0.145 | 0.20 | 0.153 | 1.53 | 1.58 | 92 |
| 3 | 0.152 | 0.31 | 0.151 | 1.55 | 1.57 | 94 |
| 4 | 0.151 | 0.31 | 0.152 | 1.52 | 1.57 | 98 |
| 5 | 0.302 | 0.32 | 0.152 | 1.54 | 1.56 | 98 |
See supporting information for full details on the experimental procedure and characterization of the products.
Determined by GC-FID analysis.
Figure 12.

Different excess experiments in which the diene and catalyst concentration was maintained constant at 0.152 M and 40 mmole respectively where the acrylate concentration was varied 0.17 M (black), 0.20 M (red) and 0.31 M (blue). (a) Acrylate rate vs acrylate concentration for different excess test. (b) Diene rate vs diene concentration for different excess experiments. (c) Diene rate vs diene concentration for change in diene concentration experiments.
In the different excess experiments, the concentration of only one of the starting materials was changed to obtain the order of each reactant. For these experiments, the acrylate concentration was varied from 0.17 to 0.31 M while keeping the diene concentration constant at 0.15 M across all three experiments. The 0.20 M and the 0.17 M acrylate different excess experiments, red and black lines respectively in Figure 12b, were within the standard deviations of the standard conditions (0.31 M acrylate and 0.15 M diene). These results indicated that as the acrylate changes in concentration that the consumption of diene was not changed, which indicated that the acrylate was zeroth order in the rate law. This statement was further supported by data in Figure 12a, where each of the three different excess experiments were parallel and shifted to the right as the initial amount of acrylate increased. If the reaction rate was dependent on the concentration of the acrylate, one would expect the data from the different experiments to have different slopes. Both figures support the statement that acrylate was zeroth order in the reaction. The diene order was directly tested by varying the concentration of diene while keeping the acrylate constant (Figure 12c). These results show that doubling the diene concentration doubled the rate of the reaction. This supports the results that the diene is first order in the reaction rate law. Coupled with the results from the same excess experiments, it can be concluded that the reaction was first order in the diene. Variable Time Normalization Analysis (VTNA)15 was also performed to aid in determining the order of both the acrylate and diene. For experiments changing the diene concentration, the reaction was first order in terms of the diene. Upon changing the concentration of the acrylate the results support that the reaction was zeroth order in the acrylate. The graphical representation and experimental procedure for VTNA can be seen in the SI p. S55.
Order in the catalyst
An important element of the overall catalytic cycle is the order of the catalyst. The effect of different catalyst concentrations were investigated using a range from 0.2 mM, 0.30 mM, and 0.40 mM. The values for the diene and acrylate concentration were kept at 0.15 M and 0.31 M respectively in the model and the values that were used for the catalyst were 0.20 mM, 0.30 mM, and 0.40 mM. From the experimental data, the reaction rate was determined as a function of diene concentration. As would be expected the rate decreases when less catalyst was used. The experimental data for 0.20 mM catalyst was used to determine an experimental reaction rate constant (k). The rate constant was found to be 79.6 min−1. Using this rate constant, a model fit assuming that the catalyst was 1st order was calculated, as shown in Figure 13. Excellent agreement is observed between the experimental values and the model for the 0.20 mM, 0.30 mM, and 40 mM catalyst trials. The results were consistent with the reaction being first order in the catalyst. VTNA was also used to verify the order of the catalyst. VTNA showed overlap between the three reactions upon normalization of the time axis (X-axis). This strongly supports the conclusion that the catalyst is first order. The experimental data can be found in the SI p. S55.
Figure 13.

Rate of diene conversion vs diene concentration with first order catalyst model overlaid. The model was prepared by graphing first order rates using the rate law: rate = k[diene][cat], where the diene concentration and catalyst loading were used as appropriate. Acrylate and diene concentration were held constant (0.31 M and 0.15 M respectively) while the catalyst concentration was varied 20 mM catalyst (blue), 30 mM catalyst (black), and 40 mM (red).
Catalyst deactivation
The stability of the reduced catalyst precursor (in the absence of substrates, coordinating solvent or excess reducing agent) was examined by monitoring the peak at 517 nm, Figure 6. This peak was formed under reaction conditions, as shown in Figure 7 and appears only after treatment of the Co(I) bromide with a Lewis acid, causing us to attribute the peak at 517 nm peak to the [(dppp)Co(I)]+ species. Even under oxygen and moisture free conditions, the peak at 517 nm decreased with time, which can be seen in Figure 6. Although this seemed counterintuitive with the previously mentioned induction period and same excess experiments, this can be explained partly. Under the reaction conditions, there was a large excess of both Zn and ZnBr2. Both of these species were responsible for the formation of the active [(dppp)Co(I)]+. Since Zn and ZnBr2 were present in large excess, at least part of the catalyst that goes to produce Co(II)-species (possibly by redox processes/ disproportionation)19b, 22 could be immediately activated again. This was not the case for the results seen in Figure 6, where only the supernatant was analyzed via UV-Vis, after filtering off the insoluble Zn and ZnBr2. In these tests, only the catalyst itself existed in solution, meaning it could not be re-activated. Therefore, the decay of the 517 nm peak was considered characteristic of an intermediate species.
While the cationic cobalt was unstable in pure solvent,21 it was possible that the catalyst could be stabilized by the presence of the diene and/or the acrylate. The stability of the catalyst was evaluated in the presence of the different substrates, as shown in Figures 14a and 14b. Addition of the acrylate showed a sharp decrease in the peak corresponding to the [Co(I)]+ species (517 nm) and an increase in the peaks corresponding to the Co(II) species (660 and 735 nm). Note the peak at 660 nm seemed unchanged, but recall the solution was being diluted during addition of a solution of the acrylate. This suggested that the acrylate was not able to stabilize the active catalytic species. Addition of the diene (Figure 14b) showed a decrease in the peak (517 nm) corresponding to the [Co(I)]+ species, but at a slower rate than when the acrylate was added. More notably, the addition of diene caused the peaks corresponding to the Co(II) species (660 and 735 nm) to decrease in intensity. This suggested that the diene was able to stabilize the active low-valent catalytic species and prevent degradative pathways that led to the formation of potentially nonactive Cospecies. Approximate time constants were found for the effect on the absorbance by acrylate and diene. The time constant for diene was calculated as 5.9 min/Abs and the time constant for acrylate was found to be 3.7 min/Abs (see SI p. S25 for the calculations of the time constants).
Figure 14.

a. The supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 was stirred for two hours after which, the following was added: Methyl acrylate in DCE (0.11 mmoles, 0.22 M, 58 eq with respect to cobalt species) added to [(dppp)Co(I)]+ (0.0038 mmole, 1.9 mM). Spectra were taken for 3 min with 1 min interval.
Figure 14b. The supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 was stirred for two hours after which, the following was added:. 2,3-dimethyl-1,3-butadiene in DCE added (0.044 mmoles, 0.044 M, 11.63 eq with respect to cobalt species) added to [(dppp)Co(I)]+ (0.0038 mmole, 19 mM). Spectra taken for 5 min with 1 min interval.
Figure 14c. The supernatant of a reaction mixture of (dppp)Co(II)Br2, Zn, and ZnBr2 was stirred for two hours after which, the following was carried out: methyl acrylate in DCE (0.11 mmoles, 0.22 M, 58 eq with respect to cobalt species) was added to [(dppp)Co(I)] + (0.0038 mmole, 1.9 mM). Readings were taken for 4 minutes. Then 2,3-dimethyl-1,3-butadiene in DCE added (0.044 mmoles, 0.044 M, 11.63 eq with respect to cobalt species) was added to [(dppp)Co(I)]+ (0.0038 mmole, 19 mM), readings were taken for another 5 minutes.
 |
(9) |
Reversibility and summary of catalytic cycle and rate law
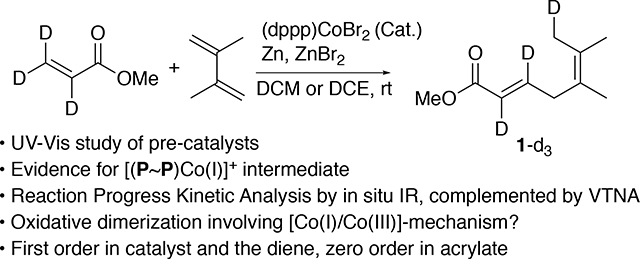
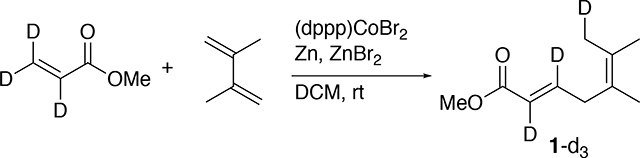
To investigate the reversibility of the reductive elimination step in Figure 2 (the catalytic cycles A/B), a deuterium labeling experiment was performed (Eq 9). The product of methyl acrylate-d3 with the diene was characterized by 13C and 1H NMR. Using an inverse gated decoupling experiment (13C{1H}), the 13C NMR was used to obtain quantitative information on the incorporation of deuterium. The deuterium attachment to the methyl group in product-d3 shifted the corresponding 13C NMR peak by approximately 0.3 δ and caused a triplet to form (20.24 ppm to 19.96 ppm).25 Using this information, it was found that the CH2D peak, when compared to the intensity of its neighboring CH3 peak, integrated as 0.96 to 1.00, suggesting at least 96% deuterium incorporation. This strongly suggested that the β-hydride elimination step in the proposed mechanistic cycles was not reversible.
A mechanism that is consistent with all the available experimental observations is the oxidative dimerization route, shown in Figure 2 B. More details of this mechanism are shown in Figure 15. It is hypothesized that the cationic Co(I) (A) reacts with the diene to form B, a Co(I) complex coordinated to the diene.26 The diene undergoes oxidative cyclization giving a metallacyclic species (C) in which cobalt is in the +3 oxidation state. The formation of the cobaltacycle is followed by acrylate coordination to quickly form the intermediate D. Subsequently a migratory insertion takes place to give yet another metallacycle E. A β-hydride elimination occurred to give F. Reductive elimination returns the Co to oxidation state (I), giving the complex G. The experimental observations would be consistent with the rate determining step being the displacement of the product 1 by 2,3-methyl-1,3-butadiene. While we have not been able to isolate any of the ‘in-cycle’ species, the order of the catalyst (1st order), the diene (1st order) and the acrylate (0sth order), and the observed inhibition of the reaction near completion of the reaction (Figure 11 and Figure S2(a)) are all consistent with this mechanistic scheme. The exclusive formation of the 5,6-(Z)- adduct in 4-substituted 1,3-dienes (Eq 3 and 4, 9) also support this scheme. A more detailed kinetic model is included in the Supporting Information (p. S 56). Various scenarios for turnover-limiting steps are considered in this analysis to reach the conclusion we have made. At this point we cannot exclude that the catalytic cycle could involve free cationic Co(I) [Figure S21, p. S59], but our chemical intuition would indicate that the data were consistent with the reaction mechanism proposed in Figure 15.
Figure 15.

A proposed mechanism consistent with experimental observations (S is solvent).
Reaction engineering
From the above kinetic and mechanistic investigation two major findings were made. One, the catalyst deactivation was not attributed to how many turnovers it performed, but instead, stability of the putative [Co]+ intermediate. The reaction was first order in diene and the catalyst, with only diene being represented in the rate law of the reaction. This signified that to increase the rate of the reaction the concentration of diene needed to be increased and, this could be done without any deleterious effect on the reaction (see Figure 14c). Three, compared to acrylate, diene was a stronger chelator to the active catalyst, ‘[(dppp)Co(I)]+’. Near the end of the reaction there appears to be an inhibition by the product as judged by the kinetic profile with 1.2 equivalents of purified product added to the reaction at the beginning along with the rest of the reagents (Figure S2).
With this information in mind, the reaction parameters were changed in an effort to increase the TON. A high concentration of diene and acrylate were used in a batch reaction since a high concentration was likely to both stabilize the catalyst while at the same time increase the rate of the reaction. Thus, instead of a 0.15 M diene and a 0.31 M acrylate, a 3.0 M diene and 3.2 M acrylate reaction was performed under otherwise similar concentration of the catalyst. Under these conditions, the product was formed with a yield of 48% in 18 h, improving the TON by nearly a factor of 10. These studies suggested that the cationic Co(I) catalyst was inherently prone to deactivation by hither to unknown mechanisms, including possible formation of a tetraligated Co(I) species I (Figure 10) that does not catalyze the reaction. Improvements in the design of ligands such as the use of a hemilabile ligand27 might alleviate this situation.
CONCLUSIONS
1,3-Dienes and alkyl acrylates, two of the most abundant feedstock starting materials, undergo highly selective co-dimerization in the presence of Zn-reduced (bis-phosphine)CoX2 complexes and a halide sequestering agent such as ZnBr2. Recent advances in synthetic methodology have allowed highly enantioselective coupling (enantiomeric ratios >95:5) of these relevant partners; yet, the efficiency as measured by the TON in the catalyst for these reactions remained modest. For any large-scale applications, more efficient processes have to be developed. Understanding the mechanism of the reaction is the first step in this process towards increasing the catalytic efficiency. Through isolation and characterization of possible low-valent cobalt precursors involved and a study of the reaction profile using reaction progress kinetic analysis (RPKA), several important details of the mechanism have emerged. For example, it was found that the reaction had an induction period that required at least two hours of stir time to generate the competent catalyst. Further investigation of the induction period using UV-Vis analysis revealed that transformation of Co(II) to Co(I), and subsequently to a cationic [Co(I)]+ species was responsible for this delay for the onset of the reaction. and the low TON observed was a result of the inherent instability of the putative cationic Co(I)-species that catalyzes the reaction. Different excess experiments provided insights that the rate law for the reaction that was first order in the diene and zeroth order in the acrylate. Catalyst loading experiments showed that the catalyst was first order. The orders in the various regents were further confirmed by Variable Time Normalization Analysis (VTNA). From this detailed understanding of the reaction, a mechanism was proposed and based on the conclusions of this study it was possible to increase the TON by a factor of 10 by conducting the reaction at an increased concentration of the starting materials. Isolation and identification of a catalytically inactive tetra-ligated cationic Co(I) complex suggested that the formation of such a species may be one mode of deactivation of the catalyst.
Supplementary Material
SYNOPSIS.
Coupling reactions of feedstock alkenes are promising, but few of these reactions area practiced industrially. Even though recent advances in the synthetic methodology have led to excellent regio- and enantioselectivies in the dimerization reactions between 1,3-dienes and acrylates, the efficiency as measured by the TON in the catalyst has remained modest. Through a combination of reaction progress kinetic analysis (RPKA) of a prototypical dimerization reaction characterization of isolated low-valent cobalt catalyst precursors involved, in the reaction several important details of the mechanism of this reaction have emerged. (i) The prototypical reaction has an induction period that requires at least two hours of stir time to generate the competent catalyst. (ii) Reduction a Co(II) complex to a Co(I) complex, and subsequent generation of a cationic [Co(I)]+ species are responsible for this delay. (iii) Through RPKA using in situ IR spectroscopy, same excess experiments reveal inhibition by the product towards the end of the reaction and no catalyst deactivation is observed as long as diene is present in the medium. The low TON observed is most likely the result of the inherent instability of the putative cationic Co(I)-species that catalyzes the reaction. (iv) Different excess experiments suggest that the reaction is first order in the diene and zero order in the acrylate. (v) Catalyst loading experiments show that the catalyst is first order. The orders in the various regents were further confirmed by Variable Time Normalization Analysis (VTNA). (vi) A mechanism based on oxidative dimerization [via Co(I)/Co(III)-cycle] is proposed. Based on the results of this study it is possible to increase the TON by a factor of 10 by conducting the reaction at an increased concentration of the starting materials, especially, the diene, which seems to stabilize the catalytic species.
ACKNOWLEDGMENTS
We like to acknowledge Dr. Stanley Jing for the preparation of the complex 8 and Dr. J. C. Gallucci and Dr. Curtis E. Moore for determination of the solid-state structures of 7 and 8 by X-ray crystallography. Financial assistance for this research provided by the U.S. National Institutes of Health (R01 GM108762, to TVR) and the U.S. National Science Foundation (CHE-1362095 and CHE-1900141 to TVR) is gratefully acknowledged. NAB acknowledges assistance from National Science Foundation CBET-1645126, CBET-1605037 and OSU IMR-FG0211. MG was a recipient of the Mayers Undergraduate Scholarship (OSU) and ACS Summer Undergraduate Research Fellowship (2019). We also acknowledge Professor Shiyu Zhang and his students for their help in the use of the UV-Vis spectrometer.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Crystallographic Information for Co-complex 7 (CIF). Crystallographic Information for Co-complex 8 (CIF). Data have been deposited at the Cambridge Crystallographic Data Centre under accession numbers CCDC 1945796 and 1953991 respectively.
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).(a) Cornils B; Herrmann WA Applied Homogeneous Catalysis with Organometallic Compounds; VCH: New York, 1996; Vol. 1 and 2; [Google Scholar]; (b) Schaub T; Hashmi ASK; Paciello RA Tackling Challenges in Industrially Relevant Homogeneous Catalysis: The Catalysis Research Laboratory (CaRLa), an Industrial–Academic Partnership. J. Org. Chem. 2018, 84, 4604–4614.; For some recent notable examples, see: [DOI] [PubMed] [Google Scholar]; (c) Shi SL; Wong ZL; Buchwald SL Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 2016, 532, 353–356; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liao K; Negretti S; Musaev DG; Bacsa J; Davies HML Site-selective and stereoselective functionalization of unactivated C-H bonds. Nature 2016, 533, 230–234; [DOI] [PubMed] [Google Scholar]; (e) Zhang W; Wang F; McCann SD; Wang DH; Chen PH; Stahl SS; Liu GS Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mlynarski SN; Schuster CH; Morken JP Asymmetric synthesis from terminal alkenes by cascades of diboration and cross-coupling. Nature 2014, 505, 386–390; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C-H crotylation of primary alcohols via hydrohydroxyalkylation of butadiene. Science 2012, 336, 324–327; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zuend SJ; Coughlin MP; Lalonde MP; Jacobsen EN Scaleable catalytic asymmetric Strecker syntheses of unnatural-amino acids. Nature 2009, 461, 968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Schaus SE; Brandes BD; Larrow JF; Tokunaga M; Hansen KB; Gould AE; Furrow ME; Jacobsen EN Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)Co(III) complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [DOI] [PubMed] [Google Scholar]
- (2).(a) Hirano M Recent advances in the catalytic linear cross-dimerizations. ACS Catalysis 2019, 9, 1408–1430; [Google Scholar]; (b) Chauvin Y; Olivier H Dimerization and Codimerization In Applied Homogeneous Catalysis with Organometallic Compounds; Cornils B, Herrmann WA, Eds.; VCH: New York, 1996; Vol. 1, pp. 258–268. [Google Scholar]; (c) RajanBabu TV; Cox GA; Lim HJ; Nomura N; Sharma RK; Smith CR; Zhang A Hydrovinylation Reactions in Organic Synthesis in Comprehensive Organic Synthesis, 2nd Edition; Molander GA,, Knochel P, Eds.; Elsevier: Oxford, 2014; Vol. 5, pp 1582–1620. [Google Scholar]; (d) Hilt G; Luers S Cobalt(I)-catalyzed 1,4-hydrovinylation reactions of 1,3-dienes with functionalized terminal alkenes under mild conditions. Synthesis 2002, 609–618. [Google Scholar]; (e) Lo JC; Kim D; Pan C-M; Edwards JT; Yabe Y; Gui J; Qin T; Gutiérrez S; Giacoboni J; Smith MW; Holland PL; Baran PS Fe-Catalyzed C-C bond construction from olefins via radicals. J. Am. Chem. Soc. 2017, 139, 2484–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Koltzenburg S; Maskos M; Nuyken O Polymer Chemistry; Springer-Verlag: Berlin, 2017. [Google Scholar]; (b) For two outstanding examples of commercial processes that use olefin oligomerization including homo-dimerization of alkenes, see, [Google Scholar]; (c) Dimersol technology (propylene dimerization): Bogdanovi’c B; Henc B; Löser A; Meister B; Pauling H; Wilke G Use of cationic nickel for olefin dimerization. Angew. Chem. Int. Ed. Engl. 1973, 12, 954–964. [Google Scholar]; (d) Shell Higher Olefin Process via ethylene oligomerization: Keim W Oligomerization of ethylene to α-olefins: Discovery and development of the Shell higher olefin process (SHOP). Angew. Chem., Int. Ed. 2013, 52, 12492–12496. [DOI] [PubMed] [Google Scholar]; (e) Speiser F; Braunstein P; Saussine L Catalytic ethylene dimerization: Recent developments with nickel complexes containing P,N-chelating ligands. Acc. Chem. Res. 2005, 38, 784–793. [DOI] [PubMed] [Google Scholar]
- (4).Jing SM; Balasanthiran V; Pagar V; Gallucci JC; RajanBabu TV Catalytic enantioselective hetero-dimerization of acrylates and 1,3-dienes. J. Am. Chem. Soc. 2017, 139, 18034–18043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Su ACL Codimerization of ethylene and butadiene. Adv. Organomet. Chem. 1979, 17, 269–318. [Google Scholar]
- (6).Wittenberg D Novel dienylation reactions. Angew. Chem. Int. Ed. 1964, 3, 153. [Google Scholar]
- (7).(a) Feldman KS; Grega KC Cobalt mediated alkene diene coupling - documentation of scope, limitations, and regioselectivity. J. Organomet. Chem. 1990, 381, 251–260; [Google Scholar]; (b) Mitsudo T-a..; Zhang SW; Kondo T; Watanabe Y. Ruthenium complex-catalyzed selective syntheses of 3,5-dienoic acid derivatives by coupling of 1,3-dienes or allylic carbonates with acrylic compound. Tetrahedron Lett. 1992, 33, 341–344. [Google Scholar]
- (8).(a) Hilt G; du Mesnil FX; Luers S An efficient cobalt(I) catalyst system for the selective 1,4-hydrovinylation of 1,3-dienes. Angew. Chem. Int. Ed. 2001, 40, 387–389; [DOI] [PubMed] [Google Scholar]; (b) Erver F; Hilt G Multi-component regio- and diastereoselective cobalt-catalyzed hydrovinylation/allylboration reaction sequence. Org. Lett. 2011, 13, 5700–5703. [DOI] [PubMed] [Google Scholar]
- (9).(a) Hiroi Y; Komine N; Komiya S; Hirano M Regio- and enantioselective linear cross-dimerizations between conjugated dienes and acrylates catalyzed by new Ru(0) complexes. Organometallics 2014, 33, 6604–6613; [Google Scholar]; (b) Hirano M; Tanaka Y; Komine N Synthesis of and catalytic linear cross-dimerizations by an electron-deficient cyclic diene complex of ruthenium(0). Organometallics 2018, 37, 4173–4176. [Google Scholar]
- (10).See Supporting Information page S7 for details.
- (11).Joseph J; RajanBabu TV; Jemmis ED A Theoretical investigation of the Ni(II)-catalyzed hydrovinylation of styrene. Organometallics 2009, 28, 3552–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Hilt G; Treutwein J Cobalt-catalysed hydrovinylation as the key step in a short synthesis of moenocinol. Chem. Commun. 2009, 1395–1397. [DOI] [PubMed] [Google Scholar]; (b) Timsina YN; Sharma RK; RajanBabu TV Cobalt-catalyzed asymmetric hydrovinylation of 1,3-dienes. Chem. Sci. 2015, 6, 3994–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Hirano M; Ueda T; Komine N; Komiya S; Nakamura S; Deguchi H; Kawauchi S Mechanistic insights into catalytic linear cross-dimerization between conjugated dienes and styrenes by a ruthenium(0) comple. J. Organomet. Chem. 2015, 797, 174–184; [Google Scholar]; (b) Hirano M; Komiya S Oxidative coupling reactions at ruthenium(0) and their applications to catalytic homo- and cross-dimerizations. Coord. Chem. Rev. 2016, 314, 182–200. [Google Scholar]; (c) For an oustanding investigation of the mechanism of Fe-catalyzed heterodimerization of 1,3-dienes and n-alkenes, see: Schmidt VA; Kennedy CR; Bezdek MJ; Chirik PJ Selective [1,4]-hydrovinylation of 1,3-dienes with unactivated olefins enabled by iron diimine catalysts. J. Am. Chem. Soc. 2018, 140, 3443–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) See also ref 2(e) for mechanistic studies of an Fe-catalyzed olefin cross-coupling reactions. [Google Scholar]
- (14).(a) Blackmond DG Reaction progress kinetic analysis: A powerful methodology for mechanistic studies of complex catalytic reactions. Angew. Chem. Int. Ed. 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG Kinetic profiling of catalytic organic reactions as a mechanistic tool. J. Am. Chem. Soc. 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- (15).Nielson C; Bures J Visual kinetic analysis. Chem. Sci. 2010, 10 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Analysis of the IR spectrum of methyl acrylate: Carmona P.; Moreno J. The infrared spectra and structure of methyl acrylate. J. Mol. Struct. 1982, 82, 177–185. [Google Scholar]; (b) Analysis of the IR spectrum of 2,3-dimethylbutadiene: Durig JR; Compton DAC. Analysis of torsional spectra of molecules with two internal C3v rotors. 16. Infrared and Raman spectra, vibrational assignment, methyl torsional potential function, and gas phase thermodynamic functions of 2,3-dimethylbuta-1,3-diene. J. Phys. Chem. 1979, 83, 2879–2886. [Google Scholar]
- (17).For a discussion of how ligands affect the reduction of Co(II) complexes and thus the induction periods in another Co(I)-catalyzed reaction (Diels-Alder reaction between an alkyne and isoprene), see: Hilt G.; Janikowski J; Schwarzer M; Burghaus O; Sakow D; Bröring M; Drüschler M; Huber B; Roling B; Harms K; Frenking G, Studies of electronic effects of modified pyridine-imine ligands utilized in cobalt-catalyzed meta-selective Diels–Alder reaction. J. Organomet. Chem. 2014, 749, 219–223. [Google Scholar]
- (18).We place the quotation marks around the formula for these species since the exact identity of this coordinately unsaturated complex is not known, and it may involve coordination to a reagent or solvent, or intermolecular bridging through dppp ligands (See text).
- (19).(a) Aresta M; Rossi M; Sacco A Tetrahedral complexes of cobalt(I). Inorg. Chim. Acta 1969, 3, 227–231 [Google Scholar]; (b) Krzystek J; Ozarowski A; Zvyagin SA; Telser J High spin Co(I): High-frequency and -field EPR spectroscopy of CoX(PPh3)3 (X = Cl, Br). Inorg. Chem. 2012, 51, 4954–4964. [DOI] [PubMed] [Google Scholar]; (c) Heinze K; Huttner G; Zsolnai L; Schober P Complexes of cobalt(II) chloride with the tripodal trisphosphane triphos: Solution dynamics, spin-crossover, reactivity, and redox activity. Inorg. Chem. 1997, 36, 5457–5469. [Google Scholar]
- (20).Preparation and characterization including solid-state structures of [(dppp)3Co2Cl2] (2), CCDC accession number 1873380 and [(dppp)3Co2Br2] (3), CCDC accession number 1814337 have been reported. See ref 24(a) and ref 4 respectively.
- (21).Rupp R; Frick A; Huttner G; Rutsch P; Winterhalter U; Barth A; Kircher P; Zsolnai L eta-(4)-coordination of dienes and heterodienes to the tripod-cobalt(I) template [CH3C(CH2PPh2)3(Co)]+: Synthesis, structure, and dynamics. Eur. J. Inorg. Chem. 2000, 523–536. [Google Scholar]
- (22).Rose MJ; Bellone DE; Di Bilio AJ; Gray HB Spectroscopic and magnetic properties of an iodo Co-I tripodal phosphine complex. Dalton Transactions 2012, 41, 11788–11797. [DOI] [PubMed] [Google Scholar]
- (23).Bis-phosphine cobalt-complexes are known to undergo ligand exchange reactions in gas phase as well as in solution, see:; (a) Fiebig L; Kuttner J; Hilt G; Schwarzer MC; Frenking G; Schmalz HG.; Schäfer M, Cobalt Catalysis in the Gas Phase: Experimental Characterization of Cobalt(I) Complexes as Intermediates in Regioselective Diels-Alder Reactions. J. Org. Chem. 2013, 78, 10485–10493. [DOI] [PubMed] [Google Scholar]; (b) Kiefer G; Vrubel H; Scopelliti R; Severin K, The Intricate Chemistry of Cobalt(II) Halides and Bis(diphenylphosphanyl)ethane. Eur. J. Inorg. Chem. 2013, 2013, 4916–4921. [Google Scholar]
- (24).Use of NaBArF gives the best reactions in cross-dimerizations, hydroboration and [2+2]-cycloadditions.; (a) Duvvuri K; Dewese KR; Parsutkar MM; Jing SM; Mehta MM; Gallucci JC; RajanBabu TV Cationic Co(I)-intermediates for hydrofunctionalization reactions: regio- and enantioselective cobalt-catalyzed 1,2-hydroboration of 1,3-dienes. J. Am. Chem. Soc. 2019, 141, 7365–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parsutkar MM; Pagar VV; RajanBabu TV Catalytic enantioselective synthesis of cyclobutenes from alkynes and alkenyl derivatives. J. Am. Chem. Soc. 2019, 141, 15367–15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).MacDougal J, Simpson M, Cole-Hamilton D: Comparison of carbon-13 nuclear magnetic resonance methods for the analysis of multiple partially deuterated products from catalytic reactions: heptan- 1-ol and 2-methylpropanol. J. Chem. Soc. Dalton Trans. 1994, 20,3061–3065. [Google Scholar]
- (26).For a related cationic complex of Co(I), see ref. 21.
- (27).(a) Nandi M, Jin J, and, and RajanBabu TV Synergistic effects of hemilabile coordination and counterions in homogeneous catalysis: New tunable monophosphine ligands for hydrovinylation reactions. J. Am. Chem. Soc. 1999, 121, 9899–9900. [Google Scholar]; (b) Smith CR; RajanBabu TV Efficient, selective, and green: catalyst tuning for highly enantioselective reactions of ethylene. Org. Lett. 2008, 10, 1657–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.