

Partial loss of SWI/SNF BAF removes a tumor suppressor but not its transcriptional function required for proliferation.
Abstract
SWI/SNF (switch/sucrose nonfermenting) complexes regulate transcription through chromatin remodeling and opposing gene silencing by Polycomb group (PcG) proteins. Genes encoding SWI/SNF components are critical for normal development and frequently mutated in human cancer. We characterized the in vivo contributions of SWI/SNF and PcG complexes to proliferation-differentiation decisions, making use of the reproducible development of the nematode Caenorhabditis elegans. RNA interference, lineage-specific gene knockout, and targeted degradation of SWI/SNF BAF components induced either overproliferation or acute proliferation arrest of precursor cells, depending on residual protein levels. Our data show that a high SWI/SNF BAF dosage is needed to arrest cell division during differentiation and to oppose PcG-mediated repression. In contrast, a low SWI/SNF protein level is necessary to sustain cell proliferation and hyperplasia, even when PcG repression is blocked. These observations show that incomplete inactivation of SWI/SNF components can eliminate a tumor-suppressor activity while maintaining an essential transcription regulatory function.
INTRODUCTION
During development and tissue homeostasis, proliferating stem and progenitor cells ultimately give rise to daughter cells that acquire specialized functions. The terminal differentiation of such cells coincides with a permanent withdrawal from the cell division cycle. This cell cycle arrest is achieved by a combination of cell cycle regulators that include the retinoblastoma tumor suppressor (Rb) protein family of transcriptional corepressors, cyclin-dependent kinase (CDK)–inhibitory proteins (CKIs) that bind and block CDKs, and E3 ubiquitin ligases such as the anaphase-promoting complex in association with the coactivator Cdh1/FZR1 (APC/C-FZR1) that promote protein degradation (1). In addition to these general regulators of the cell cycle, lineage-specific transcription factors and chromatin regulators coordinate the arrest of cell division with terminal differentiation. In particular, SWI/SNF (switch/sucrose nonfermenting) chromatin remodeling complexes have been found to play an important role in this process (2–4).
The multisubunit SWI/SNF chromatin remodeling complexes were initially identified as positive regulators of gene expression in yeast [for review, see (5)]. Independent studies in Drosophila identified SWI/SNF components as antagonists of Polycomb-mediated transcriptional repression, with homology searches revealing evolutionary conservation in mammals. Extensive biochemical characterizations support that multiple SWI/SNF subcomplexes are modularly assembled from a variety of different subunits. These SWI/SNF complexes contain an adenosine triphosphatase (ATPase) core subunit and use the energy generated by ATP hydrolysis to alter nucleosome occupancy at gene regulatory regions, to evict Polycomb-repressor complexes, and to participate in additional cellular processes such as DNA repair (6–8). SWI/SNF complexes can be divided into BRG1/BRM-associated factor (BAF) and Polybromo-associated BAF (PBAF) variants. These complexes consist of one of two mutually exclusive ATPase subunits (BRM/SMARCA2 or BRG1/SMARCA4), additional highly conserved “core” subunits (SNF5/SMARCB1, BAF155/SMARCC1, and BAF170/SMARCC2), an array of variable “accessory” subunits, and BAF- or PBAF-specific variant subunits (ARID1A/B, or ARID2, PBRM1, and BRD7, respectively) (9) (Fig. 1, A and B). While sophisticated in vitro studies have described the nature and function of SWI/SNF complexes at a biochemical level, in vivo characterizations of SWI/SNF complex functions and interactions with Polycomb group (PcG) proteins have been limited.
Fig. 1. The SWI/SNF BAF complex promotes cell cycle exit.

(A) Schematic representation of the two conserved SWI/SNF subcomplexes (using C. elegans nomenclature). (B) Table of C. elegans and mammalian homolog names for SWI/SNF subunits. The SWI/SNF complex consists of core subunits (green), accessory (blue), and BAF- (purple) and PBAF-specific (orange) signature subunits. (C) Lineage of the C. elegans mesoblast (M). The M cell is born during early embryogenesis and initiates proliferation halfway through the first larval stage (L1), forming 14 striated muscle cells (BWM), two scavenger cells [coelomocytes (CC)], and two ventral muscle precursor cells [sex myoblasts (SM)]. The SMs remain quiescent and migrate anteriorly to the vulva, resuming proliferation late in the third larval stage (L3), and differentiate to form 16 muscle cells required for egg laying. (D) Design of the lineage-tracing reporter, single-copy integrated into the C. elegans genome. A universal promoter (Peft-3) drives expression of tagBFP2 flanked by two LoxP sites and followed by the let-858 untranslated region (UTR). Excision of tagBFP2 leads to mCherry expression, providing a visible switch from blue-to-red fluorescence in cells where CRE is expressed and all daughter cells. (E) Representative image of mesoblast lineage descendants marked by the lineage tracing construct in an L4 larva (lateral view, ventral down; arrowheads point to BWM, brackets indicate egg-laying muscle precursors). (F) Representative images of the vulva region of RNAi-treated larvae. Anterior to the left, ventral down; scale bars, 10 μm in all images. (G) Quantification of mesoblast lineage descendants per animal at the L4 stage following RNAi by feeding of synchronized L1 larvae for the indicated genes, in wild-type or fzr-1 mutant backgrounds. Twenty to 30 animals were scored for each condition.
Understanding in vivo function is particularly important because mammalian SWI/SNF complexes act as tumor suppressors and are altered in a wide variety of cancers. Mutations in the collective set of SWI/SNF subunit–encoding genes have been found in 20% of examined human cancers (7, 10, 11). The broad spectrum of the identified genetic alterations makes it difficult to understand their exact oncogenic effects. While the BAF-specific subunit ARID1A is most frequently mutated, alteration of specific SWI/SNF subunits is associated with specific cancer types (7, 10, 11). Moreover, cancer-associated SWI/SNF missense mutations or deletions are often heterozygous or affect subunits for which paralogs exist [(6, 12), www.cbioportal.org]. Heterozygous mutations in genes encoding SWI/SNF subunits are also associated with intellectual disability disorders such as the Coffin-Siris syndrome (12). While haploinsuffiency likely explains the prevalence of genetically dominant loss-of-function SWI/SNF mutations in neurologic diseases, the unusual mutation spectrum of SWI/SNF genes in human cancer remains puzzling.
In this study, we characterize how partial versus complete loss of function of various SWI/SNF subunits affects the in vivo proliferation and differentiation of muscle precursor cells. We take advantage of the invariant cell lineage and advanced possibilities for controlled manipulation of the nematode Caenorhabditis elegans (13). Using lineage-specific gene knockout and protein degradation technologies, we demonstrate that various subunits of the SWI/SNF BAF complex contribute strong dosage-dependent functions in cell proliferation. As such, partial loss of function of BAF subunits leads to hyperplasia, which is enhanced by loss of negative cell cycle regulators. This indicates a tumor-suppressive function of SWI/SNF BAF, which resides, in part, on PcG protein opposition. Notably, we found that in the same cells, low levels of the SWI/SNF complex are required for cell proliferation, independently of the presence of PcG proteins or negative cell cycle regulators. Our single-molecule fluorescence in situ hybridization (smFISH) and RNA-sequencing (RNA-seq) studies show that acute inactivation of SWI/SNF BAF in muscle precursor cells rapidly alters the transcript levels of several hundred genes, including cyd-1 cyclin D, demonstrating that the complex is continuously required for the regulation of gene expression. Thus, in the same cell type and developmental decisions, a high dosage of SWI/SNF BAF subunits is needed for temporal arrest of cell division and PcG opposition, while a low level is required to sustain proliferation. We propose that similar dosage-dependent effects contribute to the selection of SWI/SNF partial loss-of-function mutations during carcinogenesis.
RESULTS
The SWI/SNF BAF subcomplex is crucial for cell division arrest during development
To investigate how the SWI/SNF complex regulates cell proliferation, we exploited the fact that cell divisions in the nematode C. elegans follow a well-characterized invariant pattern throughout development. Abnormalities resulting from aberrant regulation of proliferation-differentiation processes can therefore be readily recognized, monitored, and quantified on the basis of in vivo observations. Previously, we observed that a lineage-specific temperature-sensitive mutation in the SWI/SNF core subunit gene swsn-1 (SMARCC1/2) gives rise to hyperplasia during C. elegans postembryonic mesoderm development. When combined with loss of negative cell cycle regulators, this mutation induces a unique tumorous overproliferation phenotype (2).
To examine the role of specific SWI/SNF subunits in the regulation of proliferation, we performed RNA interference (RNAi) experiments for C. elegans genes predicted to encode components of the BAF and PBAF subcomplexes. These complexes share core subunits and several additional proteins, while differing in a few specific factors (Fig. 1, A and B). We focused on the mesoblast (M) lineage, which includes two sequential periods of cell cycle quiescence, proliferation, and muscle differentiation (Fig. 1C) (13). An integrated “tagBFP2-to-mCherry” lineage-tracing reporter and hlh-8 Twist promoter-CRE recombinase transgene (Phlh-8::CRE) facilitated the quantification of mCherry-positive mesoblast daughter cells (Fig. 1, D and E). Using this background, we observed that knockdown of the core ATPase subunit swsn-4 BRM/BRG1, the core subunit snfc-5 SNF5, and the BAF-specific subunit swsn-8 ARID1 increased the number of M descendants. Knockdown of either one of three PBAF-specific SWI/SNF subunits had limited effects (Fig. 1, F and G, and fig. S1, C and D). Simultaneous inhibition of negative regulators of the cell cycle further emphasized the different contributions of BAF versus PBAF subunits. Single knockout of the APC/C activator fzr-1 Cdh1, an inhibitor of cell cycle entry, did not alter the M lineage division pattern. However, fzr-1 loss enhanced the hyperplasia of M descendants when combined with knockdown of SWI/SNF core subunits and swsn-8 ARID1, but not when combined with PBAF-specific subunits (Fig. 1, F and G, and fig. S1, C and D). These data indicate that the SWI/SNF BAF complex contributes critically to the cell division arrest of muscle precursor cells.
SWI/SNF gene knockout induces two opposite cell proliferation defects
RNAi of one of the core subunits, swsn-1 (SMARCC1/2), led to an unexpectedly variable number of mesoblast descendants, with animals showing a range from fewer to more than the normal number of cells (Fig. 1, F and G, and fig. S1, C and D). To test whether this reflects variability in RNAi-induced loss of function, we created conditional knockout alleles, as SWI/SNF null mutations are lethal. Using CRISPR-Cas9–mediated genome editing, we introduced Lox sites in endogenous genes encoding the SWSN-1 and SWSN-4 core components, the BAF-specific SWSN-8 subunit, and the accessory subunit SWSN-2.1 BAF60 (Fig. 2A). We combined these loxed SWI/SNF alleles with the Phlh-8::CRE and tagBFP2-to-mCherry integrated transgenes to induce M lineage–specific gene deletion and reporter expression.
Fig. 2. Knockouts of endogenous SWI/SNF genes induce cell division phenotypes that differ greatly from the corresponding RNAi phenotypes.
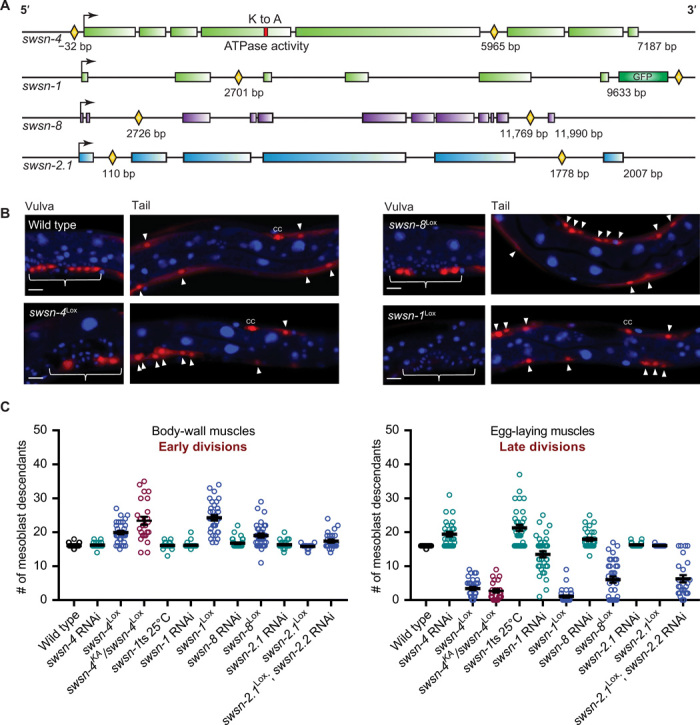
(A) Schematic of Lox sites (yellow diamonds) integrated into the endogenous SWI/SNF genes indicated. The swsn-4 ATPase-dead lysine-to-alanine mutation (K to A) is shown as a red block. Transcriptional start sites are indicated with arrows, introns are shown as black lines, and exons as colored blocks. (B) Representative images of mesoblast lineage descendants in wild-type and indicated SWI/SNF gene knockout animals. Arrowheads point at body-wall muscle (BWM), and brackets indicate egg-laying muscle precursors. Scale bar, 10 μm. (C) Quantifications of mesoblast lineage descendants for the indicated genotypes, in the tail area (early dividing body wall muscles), and around the vulva (late-dividing egg-laying muscles). Note that, in contrast to RNAi, SWI/SNF gene knockouts lead to overproliferation of the early dividing BWM precursors and cell division arrest of the late-dividing egg-laying muscle precursor cells. Twenty-six to 35 animals were scored per condition.
The swsn-1, swsn-4, and swsn-8 knockout phenotypes differed greatly from those resulting from RNAi knockdown of the same genes and from the temperature-sensitive swsn-1 phenotype (Fig. 2, B and C, and fig. S2) (2). Specifically, instead of the RNAi-induced extra M descendants in the vulva region, the gene knockouts resulted in fewer late muscle precursors. Moreover, early-formed M cell descendants, which showed normal cell cycle arrest after RNAi treatment, overproliferated in the knockout strains (Fig. 2, B and C). Simultaneous inactivation of the fzr-1 cell cycle inhibitor synergistically increased the number of extra M lineage divisions in early development but did not suppress the reduced number of late M lineage divisions (fig. S2). On the basis of these knockout data, the SWI/SNF BAF complex appears to exert a critical function in cell number expansion, in addition to promoting cell cycle arrest and differentiation.
In contrast to the other conditional SWI/SNF mutants, swsn-2.1 knockout larvae remained normal. Two paralogous C. elegans genes, swsn-2.1 and swsn-2.2, encode BAF60-related SWI/SNF subunits, compared to three paralogs in mammals (14). When combined with swsn-2.2 RNAi, the swsn-2.1 knockout closely resembled the other conditional SWI/SNF gene knockouts (Fig. 2C). This indicates that swsn-2.1 and swsn-2.2 BAF60 act redundantly, and likely in combination with core subunits as well as SWSN-8 ARID1, in M-lineage control (Fig. 2C).
ATPase activity of the SWI/SNF complex is required both for cell division arrest and cell proliferation
To assess whether the knockout phenotypes result from loss of the ATPase-dependent functions of the complex, we created an ATPase-dead swsn-4 allele by introducing a lysine-to-alanine (KA) mutation of a conserved residue that is essential for ATP hydrolysis (15) (Fig. 2A and fig. S2A). Because this mutant dies soon after hatching, we maintained the swsn4KA mutation in a trans-heterozygous combination with a wild-type or swsn-4Lox allele. Following M lineage–specific CRE expression, the swsn-4KA/swsn-4Lox mutant showed similar or somewhat stronger cell division abnormalities, compared with homozygous swsn-4Lox knockout animals (Fig. 2C and fig. S2). These data support that ATPase activity of the SWI/SNF BAF complex is required to promote both the cell cycle arrest of early body wall muscle (BWM) precursors and the expansion of egg-laying muscle precursor cells in late development.
SWI/SNF BAF subunit loss–induced hyperplasia coincides with delayed differentiation
We used promoter-fusion reporters and single-molecule FISH (smFISH) experiments to examine the proliferation-differentiation status of the SWI/SNF knockout cells. This showed residual hlh-8 Twist expression, which is normally restricted to undifferentiated muscle precursors (fig. S3, A and B) (16). Moreover, expression of the differentiation-specific myo-3 myosin heavy chain reporter was reduced at the time of normal BWM differentiation (17) (fig. S3, C and D). Further, expression of the S-phase cyclin cye-1 cyclin E was increased, and expression of the CDK inhibitor cki-1 Kip1 decreased compared with wild type, based on quantification of the number of mRNA copies per cell in smFISH experiments (fig. S3E). These data support that the extra cells in the conditional SWI/SNF knockout strains result from a prolonged proliferation-competent, not fully differentiated state compared with wild-type mesoblast descendants.
The SWI/SNF cell cycle arrest function is dosage sensitive
We considered whether different levels of SWI/SNF BAF may explain the opposite overproliferation and proliferation-arrest phenotypes. Dosage sensitivity of SWI/SNF complex functions has been implied by the spectrum of mutations detected in human cancer and intellectual disability disorders (11, 12, 18). When examined as the only alteration, a heterozygous swsn-4KA/wt BRG1/BRM mutation did not alter the mesoblast proliferation-differentiation pattern in our C. elegans model, with both the early-formed BWM and late-formed egg-laying muscle cells being normal in number and appearance (Fig. 3A and fig. S2D). By contrast, loss of the G1 inhibitors lin-23 β-TrCP or cki-1 Kip1 leads to hyperplasia (2), which was enhanced by inactivation of a single swsn-4 allele. Specifically, we combined lin-23 RNAi and lineage-specific cki-1Lox knockout with a single inactive swsn-4 allele (swsn-4KA/wt) and observed significant increases in the number of M cell descendants (Fig. 3A). This haploinsufficiency indicates that the cell cycle arrest function of SWI/SNF BAF is highly sensitive to the expression level of the complex.
Fig. 3. Opposite and acute cell division phenotypes depending on the dosage of residual SWI/SNF activity.

(A) Quantification of total number of mesoblast descendants for the indicated genotypes. *P ≤ 0.05, ****P ≤ 0.0001. (B) Quantification of mesoblast descendants for the indicated genotypes in the tail (early divisions) and around the vulva (late divisions). (C) Expression of SWSN-1::GFP in the M cell at 4 and 7 hours of larval development following gene knockout or gene knockout combined with protein degradation (+Prot. Deg.). Scale bars, 10 μm. Arrows indicate mesoblast cells, outlined in zoom images (scale bars, 1 μm). (D) Quantification of SWSN-1::GFP by fluorescence intensity in M for indicated times and genotypes (normalized to wild-type levels). (E) Quantifications of total numbers of mesoblast descendants for the indicated genotypes. (F) Representative images of strong overproliferation following SWSN-1 protein degradation and lin-23 RNAi (top), and of the one-cell arrest (arrow) after swsn-1 gene knockout together with protein degradation in lin-23 RNAi (bottom). Scale bars, 10 μm. (G) Quantifications of mesoblast descendants for indicated genotypes, in the tail (early) and around the vulva (late). (H) Quantification of SWSN-1::GFP by fluorescence intensity in M descendants for indicated times and genotypes (normalized to wild-type levels). (I) Quantifications of total numbers of mesoblast descendants for the indicated genotypes and times. n numbers of worms scored for all panels in fig. S4D. A.U., arbitrary units.
A low level of SWI/SNF BAF is required for cell proliferation
More effectively than missing one allele of swsn-4, extra cell divisions result from RNAi of SWI/SNF BAF subunits, the swsn-1ts mutation (2), and, initially, SWI/SNF gene knockout. We hypothesized that in each of these situations, hyperplasia is associated with incomplete loss of SWI/SNF function. Following CRE-Lox–mediated gene excision, residual mRNA and protein are initially present and will be depleted over time and with additional cell divisions. This means that a true null phenotype will follow gene excision after a certain delay and could be manifested as cell division arrest. To test this hypothesis, we sought to advance and increase SWI/SNF inactivation. First, we combined the swsn-1(os22ts) mutation with lineage-specific swsn-1 RNAi. Here, we expressed a swsn-1 double-stranded RNA (dsRNA) hairpin in the embryonic mesoblast, controlled by the hlh-8 Twist promoter. Compared with swsn-1(os22ts) and RNAi by feeding L1 larvae, the hairpin RNAi animals, with or without the swsn-1ts mutation, showed a phenotype more similar to the swsn-1 gene knockout, both in overproliferation of the early BWM precursors and in cell division arrest of the late-dividing egg-laying muscle precursors (Fig. 3B). Further, we also combined the lineage-specific hairpin RNAi with swsn-1 knockout to deplete swsn-1 mRNA earlier in development. This led to decreased numbers of both the early-dividing and late-dividing cells compared with the knockout alone. This illustrates that loss of SWI/SNF function may lead to either increased or reduced cell numbers, with stronger interference apparently resulting in fewer cells.
To further examine and control the degree of SWI/SNF gene inactivation, we made use of the combined insertion of a green fluorescent protein (GFP) tag and Lox sites into the endogenous swsn-1 gene (Fig. 2A). Following lineage-specific gene knockout, SWSN-1::GFP expression was still detectable in the mesoblast before (4 hours) and at the time (7 hours) of the first postembryonic cell division, although at progressively lower levels than observed in the wild type (Fig. 3, C and D). These observations support that despite SWI/SNF gene knockout, residual protein remains present during the early mesoblast divisions.
To create an acute null phenotype, we combined the conditional gene knockout with lineage-specific protein degradation. This was achieved by expressing an anti-GFP nanobody fusion protein (nanobody::ZIF-1) that targets GFP to a CUL-2–based E3 ubiquitin ligase (19) and thereby triggers efficient SWSN-1::GFP proteolysis. To create a system for lineage-specific protein degradation, we integrated a transgene with a STOP-Lox cassette in between the ubiquitous eft-3 promoter and nanobody::ZIF-1 coding sequences (fig. S4, A and B). This way, CRE expression in the mesoblast can be used to induce protein degradation in parallel to swsn-1::gfp gene excision. This double inactivation approach reduced SWSN-1::GFP expression to undetectable levels before the first larval division (Fig. 3C). Nanobody-mediated degradation alone, without gene knockout, also led to strong SWSN-1::GFP depletion before the time of the first mesoblast cell division (Fig. 3D).
Notably, the effects of SWSN-1::GFP degradation alone versus degradation plus gene knockout were completely opposite. The combined SWSN-1::GFP protein degradation and gene knockout resulted in a complete block of cell division of the mesoblast in most animals (Fig. 3E). Even when combined with lin-23 knockdown, the single embryonic mesoblast did not enter cell division during larval development (Fig. 3, E and F, bottom). This result demonstrates that a certain level of the SWI/SNF complex is required for mesoblast proliferation. By contrast, lineage-specific SWSN-1::GFP protein degradation by itself—without gene knockout—resulted in strong overproliferation of early and late muscle precursor cells (Fig. 3G). The overproliferation of both early and late precursors was further enhanced by simultaneously interfering with negative cell cycle regulation (lin-23 RNAi; Fig. 3, E and F, top).
To further characterize the relationship between SWI/SNF levels and cell proliferation, we quantified SWSN-1::GFP levels over time during the course of the mesoblast divisions in the L1/L2 larval stages. We performed these experiments for three genetic backgrounds: the control reporter strain (wild type), the strains with the swsn-1::gfpLox conditional knockout alone, or the SWSN-1::GFP protein degradation alone (Fig. 3H and fig. S4C; note: combined knockout with protein degradation was excluded because of the complete arrest of M division). In parallel, we counted the numbers of mesoblast descendants over the same time period (Fig. 3I). We found that SWSN-1::GFP was initially invisible in the protein degradation alone strain (7 to 10 hours; Fig. 3, D and H), which corresponded to a temporary delay in cell division (Fig. 3I). At 14 hours of larval development, SWSN-1::GFP was reexpressed at low levels, and cell proliferation had resumed. Low levels of SWSN-1::GFP were maintained over the subsequent time points. Thus, SWSN-1 was present at a low level when protein degradation interfered with division arrest and induced overproliferation of M descendants (Fig. 3, H and I).
Upon swsn-1 knockout alone, SWSN-1::GFP levels declined only gradually and remained detectable throughout the initial rounds of cell division, which occurred at times similar to wild-type cell divisions (Fig. 3, H and I). At the time that wild-type cells normally exit the cell cycle, swsn-1 knockout cells briefly continued proliferation to reach cell quantities that resemble those scored in later stages (L4), and proliferation arrested during the time window in which SWSN-1::GFP reached undetectable levels (Fig. 3, B, H, and I). Together, these data demonstrate that the SWI/SNF complex exerts dosage-sensitive functions: A low amount is essential to allow proliferation, while a higher level is needed for temporal cell division arrest (fig. S4E).
The SWI/SNF complex is continuously required in epidermal precursor cells
To expand our analysis to a different cell type, we tested how different levels of SWI/SNF function affect the proliferation of epithelial cells. We chose to examine vulva formation, which is formed by descendants of the ventral cord precursor cells (Fig. 4A). The first postembryonic division of these cells generates a neuroblast and epithelial precursor. The central epithelial precursor cells acquire the potential to contribute cells to the vulva [vulval precursor cells (VPCs)]. Dependent on multiple integrated signal transduction pathways, these VPCs invariably generate 22 vulval cells during late larval development, which form a functional connection between the uterus and the outside by the adult stage (Fig. 4A) (20).
Fig. 4. The SWI/SNF complex is continuously required in epidermal precursor cells.
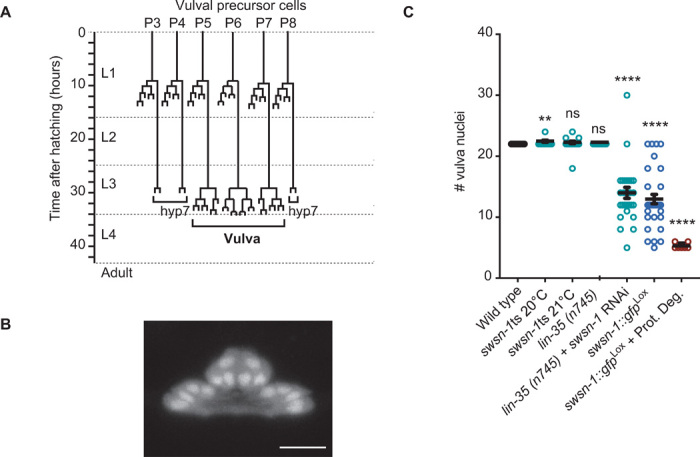
(A) The lineage of vulval development. The y axis indicates the time (hours) of larval development after hatching; vertical lines represent vulval precursor cells (VPCs), horizontal lines are cell divisions, and hyp7 denotes hypodermal fusion fate. (B) Maximum projection of the vulva after 40 hours of development, at the time when quantifications are carried out. Vulval nuclei express mCherry from the lineage-tracing reporter after Plin-31::CRE activity. Individual nuclei are easily identifiable. Scale bar, 10 μm. See movie S1 for Z-stack. (C) Quantification of the number of vulval nuclei for the indicated genotypes. The lin-35(n745) mutation did not affect the VPC division pattern but was included to increase the efficiency of RNAi, as the neuroblast derived precursor (P) cells are relatively resistant to RNAi. Eleven to 90 animals were scored per condition. **P ≤ 0.01, ****P ≤ 0.0001. ns, not significant.
We combined our lineage-tracing reporter (Fig. 1D) with CRE expression from the lin-31 FOXB1 promoter, which is active in the VPCs. As expected, this resulted in mCherry-positive VPCs from the L2 stage onward (Fig. 4B). The most anterior of the six VPCs fuses with the surrounding epidermis (hypodermis) in 50% of the animals. We observed an equal distribution of animals with 5 or 6 VPCs, which always expanded to 22 cells by the end of the vulval cell divisions (Fig. 4, B and C, wild type). These cells expressed SWSN-1::GFP, as did all other cells throughout development (fig. S5A, top). Notably, weak inactivation of swsn-1, using the os22ts allele at a semipermissive temperature (20° or 21°C) or RNAi, occasionally resulted in extra vulva cells, which was never observed in the wild type (Fig. 4C). In contrast, following swsn-1 RNAi or lineage-specific knockout, VPC daughter cell numbers were variably reduced. Complete inactivation through combined SWSN-1::GFP gene knockout and protein degradation resulted in a complete cell proliferation arrest of the VPCs (Fig. 4C; note: only five or six cells, corresponding to the undivided VPC number). We conclude that the SWI/SNF complex probably contributes to proliferation inhibition during vulva cell differentiation and that the essential SWI/SNF function in sustaining proliferation is conserved across multiple tissues in C. elegans.
SWI/SNF BAF exerts Polycomb-dependent and independent functions in proliferation
SWI/SNF complexes oppose gene silencing by Polycomb repressor complexes PRC1 and PRC2 (11, 21, 22). Therefore, unrestrained PcG-mediated gene silencing might underlie the overproliferation and cell division arrest phenotypes of SWI/SNF knockout cells. To address this, we examined the contribution of MES-2, a H3K27 methyltransferase similar to EZH2, which is the critical catalytic component of the PRC2 complex (11, 21, 22). We generated GFP-tagged and mCherry-tagged endogenous alleles to visualize in vivo expression of MES-2 EZH2 and SWI/SNF BAF subunits. This revealed coexpression of PcG and SWI/SNF components throughout C. elegans development, and in all cell types, including the M lineage (fig. S5, A and B). Thus, removal of the antagonistic SWI/SNF complexes could, in principle, lead to prolonged or abnormal PcG-mediated gene silencing.
To be able to test whether unopposed PcG activity contributes to the SWI/SNF knockout phenotypes, we created conditional alleles of mes-2, again using CRE-Lox–based recombination of the endogenous gene, with or without a GFP tag (Fig. 5A). Nanobody::ZIF-induced protein degradation alone reduced MES-2::GFP below the detectable level (fig. S6A). As expected for PcG-mediated epigenetic repression, gene knockout of mes-2Lox, or even mes-2::gfpLox knockout combined with nanobody::ZIF-1 expression, did not immediately alter the M lineage. However, after four to seven rounds of cell division, mes-2 PRC2 inactivation resulted in a variable and partially penetrant premature arrest of cell division—and possibly premature initiation of differentiation (fig. S6B).
Fig. 5. Simultaneous knockout of Polycomb PRC2 and SWI/SNF subunit genes rescues mesoblast descendant overproliferation but not cell division arrest.

(A) Schematic of Lox site integrations into the endogenous EZH2-related Polycomb gene mes-2, with Lox sites indicated by yellow diamonds. The transcriptional start site is indicated with an arrow, introns are shown as black lines, and exons as colored blocks. (B) Quantifications of mesoblast lineage descendants in the indicated genotypes, in the tail area (early-dividing BWM), and around the vulva (late-dividing egg-laying muscles). Thirteen to 27 animals were scored per condition. (C) Quantifications of total numbers of mesoblast lineage descendants in the indicated genotypes. Sixteen to 35 animals were scored per condition. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001.
When combined with knockout of mes-2, the overproliferation of early muscle precursor cells in SWI/SNF gene knockout animals was substantially reduced. The double knockout animals often showed close to wild-type BWM numbers (Fig. 5B). In agreement with previous RNAi experiments (2), these data support that SWI/SNF BAF promotes cell cycle arrest and differentiation of early muscle precursors, in part, by antagonizing Polycomb-mediated transcriptional repression.
Contrary to this early effect, the mes-2Lox knockout did not suppress the arrest of late mesoblast descendants in SWI/SNF mutants (Fig. 5B). The removal of mes-2 exacerbated the cell division arrest of SWI/SNF mutant late egg-laying muscle precursor cells (Fig. 5B). Because mes-2 and swsn-1 knockout both lead to reduced proliferation of late muscle precursors, we also examined the one-cell mesoblast arrest in L1. Acute double knockout of mes-2::gfp and swsn-1::gfp in the mesoblast gave rise to the single-cell arrest phenotype (Fig. 5C). Thus, PcG PRC2 loss, supported by complete absence of MES-2::GFP, did not alleviate the SWI/SNF complex requirement in cell proliferation. These data imply that the essential SWI/SNF complex function is separate from its antagonism of PcG-mediated gene repression.
Acute SWI/SNF depletion leads to cell division arrest before DNA replication
The immediate arrest of swsn-1 null mesoblasts indicates that a SWI/SNF complex activity is continuously required. The arrested cells did not undergo cell death and remained present even in old adults. These observations appear consistent with SWI/SNF BAF requirement in inducing or sustaining cell proliferation. A previous study reported that SWI/SNF BAF promotes chromosome decatenation by promoting chromatin binding of topoisomerase IIA (23). While this function would be critical in mitosis, our quantitative measurements of DNA content showed that the arrested SWI/SNF null mesoblasts stopped the cell cycle before, or very early in, the S phase (Fig. 6A). We considered the possibility of a DNA damage or intra–S phase checkpoint–induced arrest, as SWI/SNF complexes have also been implicated in DNA damage repair and replication (6, 24). To test whether such checkpoints are responsible for the one-cell arrest, we added high concentrations of exogenous deoxynucleotide triphosphates (dNTPs), which has been reported to bypass DNA damage–mediated arrests (25). Further, we also performed RNAi of chk-1 Chk1 and double RNAi of lin-35 Rb and cep-1 p53, conditions that also should prevent DNA damage–mediated checkpoint arrest (26). As none of these conditions affected the single-cell arrest phenotype, evidence for checkpoint arrest was not obtained (Fig. 6B).
Fig. 6. Transcriptome RNA-seq demonstrates a continuous requirement for SWI/SNF BAF complexes in normal transcription and proliferation control.

(A) Quantification of M cell DNA content in synchronized L1 larvae after 7 hours of development for the indicated genotypes. Wild-type M cells have undergone the S phase but not yet divided, leading to a 4C DNA content, whereas M cells in swsn-1::gfpLox + Prot. Deg. animals show an approximately 2C DNA content, indicative of cells arresting in the G1/early S phase. DNA was stained with propidium iodide, and DNA content normalized to that of differentiated embryonic BWM cells (2C). (B) Quantifications of mesoblast descendants for the indicated genotypes and treatments in L1 larvae. Fourteen to 28 animals were quantified per condition. (C) Principal components analysis (PCA) indicating clustering of replicate RNA-seq libraries, prepared from fluorescence-activated cell sorting (FACS)–sorted 2000-cell samples from wild-type and swsn-1::gfpLox + Prot. Deg. L1 larvae at 5.5 hours of development. Samples “A” and “B” are true biological replicates, with RNA-seq libraries prepared from different starting populations of synchronized worms. Within A and B, duplicate/triplicate RNA-seq libraries were prepared from different 2000-cell populations, isolated from the same starting worm population, and can thus be considered “semibiological” replicates. (D) Volcano plot indicating differentially expressed genes between swsn-1::gfpLox + Prot. Deg. and wild-type isolated mesoblast cells at 5.5 hours of development. (E) Quantifications of the number of mRNA molecules per cell in smFISH experiments for the indicated genes, in synchronized L1 larvae at 6.5 hours of larval development (just before the usual first M division), and in wild-type compared with swsn-1::gfpLox + Prot. Deg. Twenty to 27 animals were scored per condition. **P ≤ 0.05.
The SWI/SNF complex is continuously required and promotes expression of cyd-1 cyclin D1
We performed whole transcriptome RNA-seq to further characterize the arrested cells. We used pools of 2000 wild-type or acute swsn-1 knockout mesoblasts, isolated from synchronous cultures of L1 larvae at 5.5 hours of development (1 to 1.5 hours before the normal time of the first mitosis). Principal components analysis (PCA) showed a clear separation of the wild-type and mutant sequence data sets (Fig. 6C). Nevertheless, only a limited number of genes showed significantly different expression, of which the large majority were reduced in the SWSN-1–depleted mesoblasts (213 genes; table S2). Among those, cell cycle genes were well represented, in particular presumed E2F targets (e.g., cdc-25.2, cdk-1, cyb-1 cyclin B1, and cyb-3 cyclin B3) (Fig. 6D, red boxes). These genes encode regulators of the G2/M transition and are expected to be expressed in wild-type cells, which at this stage are preparing for mitosis, but not in G1-arrested acute swsn-1 knockout cells. Therefore, the reduced transcript levels of these cell cycle genes may result indirectly from the early cell cycle arrest of swsn-1 mutant mesoblasts.
Regulators of the G1/S transition, such as cdk-4 CDK4/6, cye-1 cyclin E, cki-1 p21, and lin-35 Rb, showed similar expression in wild-type and arrested mesoblasts (Fig. 6D, gray boxes). As a possible exception, cyd-1 cyclin D transcripts were significantly reduced in one of the two biological replicates of swsn-1 mutant mesoblasts. As cyclin D transcription is an important regulator of cell cycle entry, we followed up on this finding by examining transcript numbers with single-cell resolution. Using smFISH, we observed that the number of cyd-1 mRNA molecules was much lower in swsn-1 mutant mesoblasts, compared with normal mesoblasts before the first cell division (Fig. 6E). These observations point to cyd-1 as a candidate for being a direct SWI/SNF target. RNAi of critical cyd-1 downstream targets, lin-35 Rb, and fzr-1 FZR1/Cdh1 (27), did not suppress the one-cell arrest, which indicates that cyd-1 is not the only critical gene to depend on the SWI/SNF complex. Together, our data demonstrate a continuous requirement for SWI/SNF BAF complexes in normal transcription and proliferation control.
DISCUSSION
In this study, we examined SWI/SNF and PcG complex functions in an in vivo system that provides a well-defined cellular context and reproducible developmental decisions. Our gene knockdown and knockout experiments demonstrate that in the same cell type and developmental stage, reducing the level of SWI/SNF core subunits or C. elegans ARID1 interferes with cell cycle exit, while complete inactivation of the identical subunits is incompatible with cell proliferation. Our quantifications of residual SWSN-1 protein amounts and cell numbers over time, following different treatments, indicate that hyperplasia is consistently associated with a reduced but detectable level of SWSN-1. The timing of overproliferation and gene expression studies support that the reduced SWI/SNF levels result in suspended cell cycle withdrawal associated with precursor cell differentiation. By contrast, a complete SWI/SNF inactivation appeared capable of arresting cell division at any point in the mesoblast and VPC lineages. Therefore, we conclude that a low level of SWI/SNF activity is continuously needed, independently of cellular context, to support cellular proliferation (fig. S4E). Together with EZH2 knockout studies, our data imply that the SWI/SNF BAF ATPase exerts a tumor suppressor function in the mesoblast lineage that requires a relatively high functional level and involves PcG opposition, while a low level is essential and sufficient to sustain cell proliferation. Our data are consistent with the model that partial loss-of-function SWI/SNF gene mutations are selected during carcinogenesis because they reduce a differentiation-promoting tumor suppressor activity without inactivating the critical requirement for the SWI/SNF complex.
That the function of SWI/SNF complexes is dosage sensitive is also indicated by the heterozygous and sometimes subtle mutations in SWI/SNF subunits identified in human cancer and neurologic disease (6, 12). Several mechanisms have been proposed to explain the remarkable mutation spectrum. Mutation or deletion of specific SWI/SNF subunits could lead to the formation of complexes with alternative or aberrant composition and activities (28, 29). In certain cases, mutations appear to lead to neomorphic gain of function or dominant-negative inhibition and thereby promote cancer formation or neurologic disease (12, 30). Moreover, mutations could affect the fidelity of DNA repair or chromosome segregation and indirectly contribute to tumorigenesis. Alternatively and more in line with our observations, various subunits of the SWI/SNF complex may be haploinsufficient if a single wild-type allele is not sufficient to maintain normal tumor suppression or development (6–8).
In our system, incomplete inactivation of multiple different BAF components, including the single ATPase and BAF-specific ARID1 subunit, results in very similar hyperplasia phenotypes. This indicates that the reduced SWI/SNF function, rather than the activity of complexes with an abnormal subunit composition, leads to overproliferation. Hyperplasia was observed early and reproducibly after SWI/SNF BAF gene knockout or protein degradation. This implies that the hyperplastic response in our system does not depend on a dominant-negative mechanism or on the generation of secondary mutations. In addition to dominant loss-of-function mutations, cancer type–specific gain of function and dominant-negative mutations affect specific SWI/SNF subunits. As such, common heterozygous mutations in the BRG1 ATPase selected in human cancer are likely to act in a dominant-negative way (30). Nevertheless, such mutations do not fully inactivate the wild-type allele, and residual BRG1 function is thus retained in the heterozygotes. The importance of residual SWI/SNF function seen in our system was previously revealed in synthetic lethal screens. Such screens and follow-up experiments demonstrated that ARID1B is essential in ARID1A mutant cancers, while tumors with mutations in the BRG1 (SMARCA4) ATPase depend on BRM (SMARCA2) (28, 31, 32).
Some cancer cells, however, appear to survive without SWI/SNF function. Malignant rhabdoid tumors are well known for their homozygous loss of SNF5 (SMARCB1), one of the core subunits of SWI/SNF complexes. Although initially expected to fully disable SWI/SNF complexes, SNF5-deficient cancer formation was found to depend on the presence of BRG1 (29). Recent studies revealed the existence of ncBAF complexes that do not contain SNF5, while removal of non-canonical BAF (ncBAF)–specific subunits induces synthetic lethality in cancer cells lacking SNF5 (9, 33). Thus, in the best-described examples of homozygous SWI/SNF core subunit loss, cell proliferation still depends on the presence of residual SWI/SNF complex activity, in this case provided by the atypical SWI/SNF ATPase ncBAF (9, 33). In a specific small cell carcinoma of the ovary and a subset of cancer-derived cell lines, biallelic mutation of BRG1 coincides with transcriptional silencing of the BRM locus (6, 10, 34). Although it is difficult to exclude that a trace amount of residual BRM permits the proliferation of these cells, activation of compensatory mechanisms may allow complete loss of SWI/SNF ATPase activity in some cell types.
In our study, interfering with any one of the BAF complex subunits induced hyperplasia or cell proliferation arrest, depending on partial or complete removal. We characterized this thoroughly for SWNF-1, the sole homolog of the scaffold subunits BAF155/170. This subunit is shared between all SWI/SNF complexes, and without it, no version of the complex can assemble (9). Therefore, rather than changing the SWI/SNF subunit composition and allowing the formation of aberrant residual complexes, we affected the global levels of SWI/SNF activity. As such, our data support that the complete loss of all SWI/SNF ATPase activity is incompatible with cell proliferation and that there are perhaps overlapping core functions for all of the different SWI/SNF variants in promoting a proliferation-competent state.
Several mechanisms could underlie the dosage sensitivity of SWI/SNF functions. In our system, a high dosage is needed when cells transition to a differentiated state, at which time many loci are transcriptionally activated or silenced. Compared with altering the chromatin state during gene activation or silencing, the maintenance of gene expression may require lower levels of the SWI/SNF complex. If true, then partial SWI/SNF inactivation will interfere with differentiation but not continued proliferation, which would provide a mechanism promoting tumorigenesis. Possibly in support of such a mechanism, SMARCB1 or SMARCA4 gene knockout in mouse embryonic fibroblasts (MEFs) most significantly reduced the transcript numbers from genes with gene ontology (GO) terms associated with development and differentiation (35).
In our experiments, the essential SWI/SNF function appeared independent from PRC2-mediated gene silencing. Other activities of the complex, such as chromatin remodeling, could be critical for mesoblast and VPC proliferation. Both knockout MEF cells and embryonic stem cells with heterozygous dominant-negative alleles of BRG1 showed a broad reduction in chromatin accessibility at active enhancers, which was remarkably associated with loss of H3K27Ac rather than increased PcG protein binding (30, 35). These studies did not reveal whether widespread transcriptional deregulation, reduced expression of some critical genes, or other defects are incompatible with cellular proliferation when the SWI/SNF activity falls below a critical level. Our RNA-seq analysis showed that the acute arrest of mesoblast proliferation occurred when the expression of a limited number of genes was significantly altered. We identified C. elegans cyclin D as one of the genes whose transcription is acutely sensitive to SWI/SNF inactivation. In contrast to cyd-1 mutants, the proliferation arrest associated with strong SWI/SNF loss was insensitive to knockdown of cell cycle inhibitors (27, 36). Therefore, we expect that the down-regulation of cyd-1 and other critical genes is responsible for the tight cell cycle arrest. Two recent human cancer studies also concluded that SWI/SNF ATPases promote cyclin D1 expression (37, 38).
That loss of function of SWI/SNF subunits can lead to opposite phenotypes (hyperplasia versus division arrest), depending on residual complex levels, provides support for the clinical exploration of cancer cell vulnerabilities that result from SWI/SNF gene mutations (39). At the same time, the delicate balance between dosage-dependent SWI/SNF and PcG regulators observed in our system illustrates that the outcome of targeted therapies will be difficult to predict and highly context dependent. The many parallels between observations in our system and human cancer cells support that efficient genetic screens in C. elegans may help identify synthetic lethal interactions that are broadly associated with SWI/SNF loss and cause little toxicity in normal cells.
MATERIALS AND METHODS
Strains and culture
Genotypes of all strains used in this study are listed in table S1. C. elegans was cultured on nematode growth media (NGM) plates seeded with OP50 bacteria and generally maintained at 20°C. Strains containing the pha-1(e2123) mutation were maintained at 15°C and shifted to 25°C for mutant phenotype analysis.
RNA-mediated interference
Bacterial cultures of Escherichia coli HT115 containing L4440 empty vector or vector with genomic or open reading frame (ORF) gene inserts were grown overnight, induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 1 hour, concentrated 2.5 times, and seeded onto NGM plates containing tetracycline (12.5 μg/ml), ampicillin (100 μg/ml), and 2 mM IPTG. Early L1 larvae were exposed to feeding RNAi for knockdown of SWI/SNF components, and the number of mesoblast descendants was analyzed in late L3/early L4 animals of the same generation. swsn-1, swsn-4, swsn-8, swsn-7, swsn-9, swsn-2.2, swsn-3, swsn-6, and phf-10 RNAi vectors were cloned by ligating a ~1000-bp complementary DNA (cDNA) fragment into the L4440 vector. When examining contributions of lin-23, chk-1, lin-35, or cep-1, L4 animals were placed on RNAi plates and the F1 was analyzed.
Molecular cloning and genome editing
Single-copy integration of the recombination reporter (readoutlox; Peft-3::LoxP::egl-13NLS::tagBFP2::tbb-2UTR::LoxP::egl-13NLS::mCherry::tbb-2UTR) into chromosomes III and V standard MosSCI sites was performed as described in (2). The Phlh-8::swsn-1::unc-54 untranslated region (UTR) hairpin construct was generated by inserting a ~1100-bp swsn-1 cDNA fragment, followed by the antisense sequence of the same ~1100-bp swsn-1 cDNA fragment including a stop codon, after the Phlh-8 promoter. See fig. S5 for details on the anti-GFP nanobody–ZIF-1 construct.
All Lox insertions, as well as the ATPase dead swsn-4 mutant, were generated by temperature-sensitive pha-1 coconversion (40) using single-stranded DNA oligonucleotides with 40-bp homology arms as repair templates. For pha-1 coconversion, seven times outcrossed pha-1(e2123) young adults, grown at the permissive temperature of 15°C, were injected into the gonads with the following injection mix: U6::gRNA target construct (50 ng/μl), pJW1285 (60 ng/μl; U6::gRNA pha-1 and Cas9 construct), and single-stranded DNA repair templates (50 ng/μl) for pha-1 as well as the appropriate target. Injected worms were immediately placed at 25°C. F1 rescued pha-1 animals were analyzed by polymerase chain reaction (PCR) amplification, using primers flanking the Lox insertion site, for the presence of a reduced-mobility DNA band indicative of Lox site insertion. The swsn-4 K-to-A mutation was generated in a strain with a GFP-expression cassette, eft-3::GFP::2×NLS::tbb-2 3′UTR, integrated on chromosome IV close to the swsn-4 locus. By selecting GFP-positive animals, the homozygous lethal swsn-4 ATPase-dead mutation can be easily maintained. All mutations were sequence verified. The swsn-1::egfpLoxP strain was generated by inserting enhanced GFP (eGFP) using the self-excising cassette (SEC) method (41) into wild-type N2 worms, which leaves a LoxP scar. The injection mix contained the following: U6::gRNA target construct (100 ng/μl), pDD268 eGFP SEC vector (20 ng/μl) with 150-bp swsn-1 left homology arm and swsn-1 600-bp right homology arm, Peft-3::Cas9 (50 ng/μl; Addgene 46168), and Pmyo-2::tdTomato (2.5 ng/μl). The second LoxP site was introduced by crossing the strain with pha-1(e2123) and by temperature-sensitive pha-1 coconversion as described above. The mes-2::egfpLoxN strain was made by inserting eGFP with a LoxN site in one of the GFP introns into a strain that already contained a LoxN site in the first intron of mes-2.
Microscopy and quantification of cell numbers
For analysis and quantification, animals were mounted on 3% agarose slides, using tetramisole (10 mM) in M9 plus 0.05% Tween (36). Combining Phlh-8::CRE with the lineage-tracing reporter allowed rapid identification of cells in the mesoblast lineage (mCherry positive), whereas Plin-31::CRE was used to visualize VPC daughter cells. The numbers of M descendants and VPC descendants were quantified by counting the number of mCherry-positive cells at specific developmental times. Images of the proliferation phenotypes were obtained using a Zeiss LSM700 confocal microscope. SWSN-1::GFP fluorescence intensities were quantified by using the ImageJ measurement tool, selecting the region of interest (ROI; M cell nucleus), and subtracting the background signal (same ROI, not including GFP-positive cells in the same larva). At least 16 larvae per condition were measured.
To quantify vulval nuclei, animals were synchronized using hypochlorite treatment and grown for 40 (tracing-reporter controls, swsn-1ts, or knockout) or 50 hours [strains containing lin-35(n745) mutation and protein degradation strains) at 20°C unless indicated otherwise. Still images and Z-stacks were taken using a 63×/1.4 numerical aperture lens on a Zeiss Axioplan microscope or a Zeiss confocal microscope, with a slice interval of 0.32 μm for Z-stacks. Vulval cell quantifications were performed on the basis of Z-stacks.
Single-molecule FISH
smFISH was performed essentially as described in (2). Cy5-coupled probes against mRNAs of interest were ordered from Stellaris (http://singlemoleculefish.com/), with 23 to 48 probes per gene of interest ranging from 18 to 22 bp in length. L1 or L4 animals were fixed for 30 min at room temperature (RT) in 400 μl of Bouin’s fixative + 400 μl of methanol and 10 μl of β-mercaptoethanol, three times freeze thawed and again tumbled for 30 min in fixative at RT. For permeabilization, the fixative was removed and exchanged for borate–Triton–β-mercaptoethanol (BTB; 1× borate buffer, 0.5% Triton, and 2% β-mercaptoethanol) solution. Animals were tumbled three times for 1 hour in BTB solution at RT. BTB solution was then replaced with wash buffer A (Stellaris) containing 20% formamide, and then with 100 μl of hybridization solution containing smFISH probes to a final concentration of 0.25 to 0.5 μM, and incubated overnight at 32°C. Samples were washed in wash buffer A without formamide and incubated with 0.05 ng of 4′,6-diamidino-2-phenylindole (DAPI) in wash buffer A for 30 min at 32°C. After a final wash in wash buffer B (Stellaris), animals were mounted on slides with Vectashield mounting medium and imaged within 4 hours. Images were acquired using a Nikon Eclipse Ti Spinning Disk confocal microscope, using a 100× objective. The tetramethylrhodamine (TMR) (mCherry) spots were used to draw an ROI around (mCherry-positive) M lineage descendants in ImageJ, in which the number of Cy5 fluorescent mRNA spots was quantified using the ComDet plugin in ImageJ [https://imagej.net/Spots_colocalization_(ComDet)].
Statistical analysis
Sample sizes were not predetermined; instead, all available animals of the right stage and genotype were counted. smFISH data are included from at least 8 independent animals, and reporter expression and cell numbers from at least 10 independent animals. Graphs and data analysis were produced using GraphPad Prism 6.05. Plots indicate all data points and the mean (average) ± SEM. As the data essentially fit normal distributions, unpaired two-tailed Student’s t tests were used to examine statistical significance of the difference between means.
Propidium iodide staining
Propidium iodide staining was carried out after Carnoys fixation as previously described (42). For DNA quantification, Z-stacks were acquired using a Zeiss LSM700 confocal microscope. Maximum projections (SUM) were made in ImageJ from all the stacks in which the M cell DNA was visible, and pixels were quantified using ImageJ. Postmitotic, differentiated BWM cells (2C) were quantified in the same manner and used as a reference.
Larval cell isolation
The C. elegans L1 larval cell isolation protocol was adapted from (36). To generate large amounts of synchronized L1 larvae, worms were grown in S medium in liquid culture for two generations (to enrich for gravid adults) and bleached. Eggs were hatched overnight (for 18 to 22 hours) in S medium without food, and starved L1 larvae were split into three aliquots and put back into S medium with OP50 for 5.5 hours. Cultures were put on ice for 15 min, spun down at 1300g, and washed two times in M9 and once in H20. L1 larvae were then transferred to 1.5-ml Eppendorf tubes (20 to 40 μl of L1 pellet per Eppendorf) and spun down at 16,000g. Larvae were treated with SDS–dithiothreitol (DTT) solution (20 mM Hepes, pH 8.0, 0.25% SDS, 200 mM DTT, and 3% sucrose) for 2 min, washed six times in egg buffer (25 mM Hepes, pH 7.3, 118 mM NaCl, 48 mM KCl, 2 mM CaCl2, and 2 mM MgCl2, adjusted to 340 ± 5 mOsm with H2O), and then treated with pronase E (20 mg/ml) in L15/fetal bovine serum (FBS) buffer [10% FBS and 1% penicillin-streptomycin (Sigma, P4458) in L15 insect medium, adjusted to 340 ± 5 mOsm with 60% sucrose] for 30 to 40 min. After 10- and 20-min incubation in pronase E, a pellet pestle motor with a pellet pestle adapted to 1.5-ml microtubes (Sigma, Z359971 and Z359947) was used for 1 min on each sample. Last, cell preparations were washed three times in L15/FBS, spun down at 9600g for 5 min between each wash, and resuspended in 1 ml of L15/FBS.
Fluorescence-activated cell sorting
Cell preparations were allowed to settle on ice for 30 min, and the top 850 μl of the supernatant was removed and transferred to a new Eppendorf tube for fluorescence-activated cell sorting (FACS). Cells were sorted according to mCherry-positive signal using a BD FACSAria III (BD Biosciences). For each sample, 2000 cells were sorted into L15/FBS buffer. In one session, three wild-type and three mutant samples were sorted. Immediately after sorting, cells were spun down at 12,000g for 5 min, resuspended in TRIzol, and frozen at −80°C.
cDNA library preparation
cDNA libraries were prepared according to a combination of the CEL-Seq and CEL-Seq2 protocols with some modifications (43, 44). RNA was precipitated using chloroform/isopropanol precipitation at −20°C for 48 to 72 hours and washed once in 75% ethanol. CEL-Seq2 primers were used (one unique primer per sample), with each primer containing an anchored polyT, a 6-bp unique barcode, 6-bp Unique molecular identifier (UMI), a 5′ Illumina adaptor, and a T7 promoter. The CEL-Seq1 protocol was followed for a first round of reverse transcription and cDNA cleanup followed by in vitro transcription, as well as for fragmentation of amplified RNA (aRNA), as described (43). aRNA was run on an Agilent bioanalyzer [RNA pico–chromatin immunoprecipitation (ChIP)] for quality control and quantification. The CEL-Seq2 protocol was followed for a second round of reverse transcription and PCR amplification, as described (44). cDNA was amplified for 11 to 15 cycles depending on aRNA amounts, run on an Agilent bioanalyzer (DNA pico-ChIP), quantified using a Qubit, and 1 to 2 ng was sequenced with 5% coverage on an Illumina NextSeq500.
RNA-seq data analysis
Data analysis was carried out in R version 3.4.4. PCA was performed with the plotPCA function after carrying out variance-stabilized transformation on the data. Differential gene expression was analyzed using DESeq2 (default settings) using a padj cutoff of 0.1 (45).
Supplementary Material
Acknowledgments
We are grateful to members of the Korswagen and Van Oudenaarden groups at the Hubrecht Institute for help with RNA-seq experiments, and to P. Verrijzer and members of the Van den Heuvel and Boxem groups for input, discussion, and comments on the manuscript. Several strains were provided by the CGC, which is funded by the NIH National Center for Research Resources (NCRR). Funding: This work was supported by Worldwide Cancer Research (WCR) grant 14-1294, and M.G. was supported by the EMBO Long-Term Fellowship ALTF 991-2016. Author contributions: A.v.d.V. performed most of the SWI/SNF knockdown and knockout experiments, analyzed the results, and cowrote the paper. M.G. generated cki-1Lox and mes-2::gfpLox strains; performed the smFISH, DNA and protein quantifications, and RNA-seq experiments; analyzed the results; and cowrote the paper. V.P. generated the anti-GFP nanobody::ZIF-1 construct and strains, performed the VPC experiments, and contributed to the experimental design. S.v.d.H. conceived the study, acquired funding, provided guidance with experimental design, and cowrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay3823/DC1
REFERENCES AND NOTES
- 1.Ruijtenberg S., van den Heuvel S., Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 15, 196–212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruijtenberg S., van den Heuvel S., G1/S inhibitors and the SWI/SNF complex control cell-cycle exit during muscle differentiation. Cell 162, 300–313 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Yu Y., Chen Y., Kim B., Wang H., Zhao C., He X., Liu L., Liu W., Wu L. M. N., Mao M., Chan J. R., Wu J., Lu Q. R., Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albini S., Coutinho Toto P., Dall’Agnese A., Malecova B., Cenciarelli C., Felsani A., Caruso M., Bultman S. J., Puri P. L., Brahma is required for cell cycle arrest and late muscle gene expression during skeletal myogenesis. EMBO Rep. 16, 1037–1050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathur R., Roberts C. W. M., SWI/SNF (BAF) complexes: Guardians of the epigenome. Annu. Rev. Cancer Biol. 2, 413–427 (2018). [Google Scholar]
- 6.Kadoch C., Crabtree G. R., Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson B. G., Roberts C. W. M., SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kadoch C., Copeland R. A., Keilhack H., PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry 55, 1600–1614 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Mashtalir N., D’Avino A. R., Michel B. C., Luo J., Pan J., Otto J. E., Zullow H. J., McKenzie Z. M., Kubiak R. L., St. Pierre R., Valencia A. M., Poynter S. J., Cassel S. H., Ranish J. A., Kadoch C., Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shain A. H., Pollack J. R., The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLOS ONE 8, e55119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., Crabtree G. R., Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bögershausen N., Wollnik B., Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front. Mol. Neurosci. 11, 252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sulston J. E., Horvitz H. R., Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156 (1977). [DOI] [PubMed] [Google Scholar]
- 14.Ertl I., Porta-de-la-Riva M., Gómez-Orte E., Rubio-Peña K., Aristizábal-Corrales D., Cornes E., Fontrodona L., Osteikoetxea X., Ayuso C., Askjaer P., Cabello J., Cerón J., Functional interplay of two paralogs encoding SWI/SNF chromatin-remodeling accessory subunits during Caenorhabditis elegans development. Genetics 202, 961–975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richmond E., Peterson C. L., Functional analysis of the DNA-stimulated ATPase domain of yeast SWI2/SNF2. Nucleic Acids Res. 24, 3685–3692 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harfe B. D., Branda C. S., Krause M., Stern M. J., Fire A., MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development 125, 2479–2488 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Kosho T., Miyake N., Carey J. C., Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: Historical review and recent advances using next generation sequencing. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 241–251 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Wang S., Tang N. H., Lara-Gonzalez P., Zhao Z., Cheerambathur D. K., Prevo B., Chisholm A. D., Desai A., Oegema K., A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development 144, 2694–2701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternberg P. W., Vulval development. WormBook 25, 1–28 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuettengruber B., Bourbon H.-M., Di Croce L., Cavalli G., Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Kia S. K., Gorski M. M., Giannakopoulos S., Verrijzer C. P., SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 28, 3457–3464 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dykhuizen E. C., Hargreaves D. C., Miller E. L., Cui K., Korshunov A., Kool M., Pfister S., Cho Y.-J., Zhao K., Crabtree G. R., BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brownlee P. M., Meisenberg C., Downs J. A., The SWI/SNF chromatin remodelling complex: Its role in maintaining genome stability and preventing tumourigenesis. DNA Repair 32, 127–133 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Bester A. C., Roniger M., Oren Y. S., Im M. M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D. S., Kerem B., Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalogeropoulos N., Christoforou C., Green A. J., Gill S., Ashcroft N. R., chk-1 is an essential gene and is required for an S-M checkpoint during early embryogenesis. Cell Cycle 3, 1196–1200 (2004). [PubMed] [Google Scholar]
- 27.The I., Ruijtenberg S., Bouchet B. P., Cristobal A., Prinsen M. B. W., van Mourik T., Koreth J., Xu H., Heck A. J. R., Akhmanova A., Cuppen E., Boxem M., Muñoz J., van den Heuvel S., Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat Commun. 6, 5906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helming K. C., Wang X., Wilson B. G., Vazquez F., Haswell J. R., Manchester H. E., Kim Y., Kryukov G. V., Ghandi M., Aguirre A. J., Jagani Z., Wang Z., Garraway L. A., Hahn W. C., Roberts C. W. M., ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20, 251–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X., Sansam C. G., Thom C. S., Metzger D., Evans J. A., Nguyen P. T. L., Roberts C. W. M., Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 69, 8094–8101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges H. C., Stanton B. Z., Cermakova K., Chang C.-Y., Miller E. L., Kirkland J. G., Ku W. L., Veverka V., Zhao K., Crabtree G. R., Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 25, 61–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman G. R., Rahal R., Buxton F., Xiang K., McAllister G., Frias E., Bagdasarian L., Huber J., Lindeman A., Chen D., Romero R., Ramadan N., Phadke T., Haas K., Jaskelioff M., Wilson B. G., Meyer M. J., Saenz-Vash V., Zhai H., Myer V. E., Porter J. A., Keen N., McLaughlin M. E., Mickanin C., Roberts C. W. M., Stegmeier F., Jagani Z., Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. U.S.A. 111, 3128–3133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson B. G., Helming K. C., Wang X., Kim Y., Vazquez F., Jagani Z., Hahn W. C., Roberts C. W. M., Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell. Biol. 34, 1136–1144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michel B. C., D’Avino A. R., Cassel S. H., Mashtalir N., McKenzie Z. M., McBride M. J., Valencia A. M., Zhou Q., Bocker M., Soares L. M. M., Pan J., Remillard D. I., Lareau C. A., Zullow H. J., Fortoul N., Gray N. S., Bradner J. E., Chan H. M., Kadoch C., A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 20, 1410–1420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karnezis A. N., Wang Y., Ramos P., Hendricks W. P., Oliva E., D’Angelo E., Prat J., Nucci M. R., Nielsen T. O., Chow C., Leung S., Kommoss F., Kommoss S., Silva A., Ronnett B. M., Rabban J. T., Bowtell D. D., Weissman B. E., Trent J. M., Gilks C. B., Huntsman D. G., Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 238, 389–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alver B. H., Kim K. H., Lu P., Wang X., Manchester H. E., Wang W., Haswell J. R., Park P. J., Roberts C. W. M., The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Heuvel S., Kipreos E. T., C. elegans cell cycle analysis. Methods Cell Biol. 107, 265–294 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Xue Y., Meehan B., Macdonald E., Venneti S., Wang X. Q. D., Witkowski L., Jelinic P., Kong T., Martinez D., Morin G., Firlit M., Abedini A., Johnson R. M., Cencic R., Patibandla J., Chen H., Papadakis A. I., Auguste A., de Rink I., Kerkhoven R. M., Bertos N., Gotlieb W. H., Clarke B. A., Leary A., Witcher M., Guiot M.-C., Pelletier J., Dostie J., Park M., Judkins A. R., Hass R., Levine D. A., Rak J., Vanderhyden B., Foulkes W. D., Huang S., CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat Commun. 10, 558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue Y., Meehan B., Fu Z., Wang X. Q. D., Fiset P. O., Rieker R., Levins C., Kong T., Zhu X., Morin G., Skerritt L., Herpel E., Venneti S., Martinez D., Judkins A. R., Jung S., Camilleri-Broet S., Gonzalez A. V., Guiot M.-C., Lockwood W. W., Spicer J. D., Agaimy A., Pastor W. A., Dostie J., Rak J., Foulkes W. D., Huang S., SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 10, 557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierre R. St., Kadoch C., Mammalian SWI/SNF complexes in cancer: Emerging therapeutic opportunities. Curr. Opin. Genet. Dev. 42, 56–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward J. D., Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickinson D. J., Pani A. M., Heppert J. K., Higgins C. D., Goldstein B., Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035–1049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boxem M., Srinivasan D. G., van den Heuvel S., The Caenorhabditis elegans gene ncc-1 encodes a cdc2-related kinase required for M phase in meiotic and mitotic cell divisions, but not for S phase. Development 126, 2227–2239 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Hashimshony T., Wagner F., Sher N., Yanai I., CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666–673 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K. J., Rozenblatt-Rosen O., Dor Y., Regev A., Yanai I., CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay3823/DC1