

Graphical Abstract
A light-activated gRNA allows for precise spatiotemporal control of gene editing in both mammalian cells and zebrafish embryos, with excellent off to on switching. This highly programmable approach can be easily adapted to any desired target sequence and can be delivered to a variety of organisms in the form of ribonucleoprotein complexes.
Keywords: caged compounds, gene editing, optical control, CRISPR/Cas9, zebrafish
Introduction
Adapted from the prokaryotic acquired immune system, CRISPR/Cas9 has been extensively studied and meticulously developed for efficient and precise genome editing in a customizable fashion.[1] As an RNA-guided DNA endonuclease, Cas9 protein first binds to a guide RNA (gRNA), which then enables target site recognition through Watson-Crick base pairing between the 5’ 20-nucleotide protospacer region of the gRNA and the DNA sequence. Subsequent cleavage of the target locus is then carried out by the nuclease domains HNH and RuvC of Cas9.[2] Recent developments of the CRISPR/Cas9 system include broad genomic targetability through Cas9 variants with PAM promiscuity,[3] gene activation and repression,[4] nucleobase editing,[5] genomic loci imaging,[6] and epigenetic modifications.[7] However, concerns of off-target genomic manipulation[8] and the desire for synchronization of CRISPR/Cas9 activity with precisely orchestrated genetic networks need to be addressed.
Aiming for higher genomic editing precision by limiting the window of CRISPR/Cas9 activity as well as probing spatiotemporally controlled gene function, researchers have endeavored to broaden the CRISPR/Cas9 toolkit for conditional control of its activity.[9] Such efforts include small molecule-induced Cas9 protein activation[10] or reassembly,[11] light-activation of Cas9,[12] reconstitution of single-chain Cas9[13] and split-Cas9,[14] NIR-controllable release of CRISPR/Cas9,[15] dimerization-based switchable Cas9 activation,[16] as well as optically controlled recruitment of transcription factors to catalytically dead Cas9 (dCas9).[14;17] Amongst these developments, most efforts were dedicated to the regulation of the Cas9 protein to restore its function upon external stimulation. These methods inevitably require protein engineering, including the screening of mutations and split sites,[11; 14] directed evolution,[10a] or unnatural amino acid mutagenesis.[12] We anticipate that conditional control of chemically modified gRNA will not only circumvent the need for protein engineering, but will also provide a direct path to regulating the interaction between Cas9:gRNA ribonucleoprotein (RNP) complex and the target dsDNA. Several previous reports have shed light on this path, including the use of cleavable antisense-DNA as a protector for gRNA activity,[18] ligand-dependent RNA cleavage and deprotection,[19] ligand-dependent recruitment of transcriptional activators to dCas9,[20] and small molecule-induced reassembly of the Cas9:gRNA complex.[21] These designs, however, are still limited by the requirement for a third cellular component[18–19] or have reduced gRNA stability due to the inability to form an RNP complex before activation.[21–22] This is of particular importance, as RNP delivery has been established as a universal approach for gene editing in different cells, tissues, and organisms with high efficiency and specificity, compared to alternative editing modalities.[23]
Results and Discussion
We henceforth introduce a photocaged gRNA design for the direct regulation of the interaction between RNP and dsDNA using light and demonstrate its application in an animal model. 6-Nitropiperonyloxymethylene (NPOM)-caged nucleobases have been successfully applied in the light-triggering of nucleic acid base-pairing in many biological systems.[24] Here, we employed NPOM-caged uridine and guanosine (Figure 1a) in the generation of light-activated gRNA for the targeting of genomic loci in both mammalian cells and zebrafish embryos (Figure 1b, Supporting Information, Figure S1–Figure S4). By replacing regular nucleobases with NPOM-caged nucleobases within the protospacer region of the gRNA, we anticipated that Cas9:gRNA:dsDNA ternary complex formation is inhibited until photolysis restores the native base-pairing capability of the gRNA, while Cas9:caged gRNA interactions remain undisturbed (Figure 1c). Our past experience has shown that very little background activity and excellent off → on switching upon light exposure is achieved by installing one caging group every 5–6 nucleobases, evenly distributed throughout the oligonucleotide.[25] As a distinct advantage, we further envisioned that the Cas9:caged gRNA RNP can be pre-assembled and delivered as a complex for improved gRNA stability,[22b] facilitating convenient cross-species application in both cultured mammalian cells as well as aquatic embryos by lipid-mediated transfection[26] and microinjection,[13] respectively. We pursued a single caged gRNA because several studies have demonstrated enhanced stability compared to the combination of crRNA (CRISPR RNA) and tracrRNA (transactivating crRNA), when complexed with Cas9 protein.[27]
Figure 1.
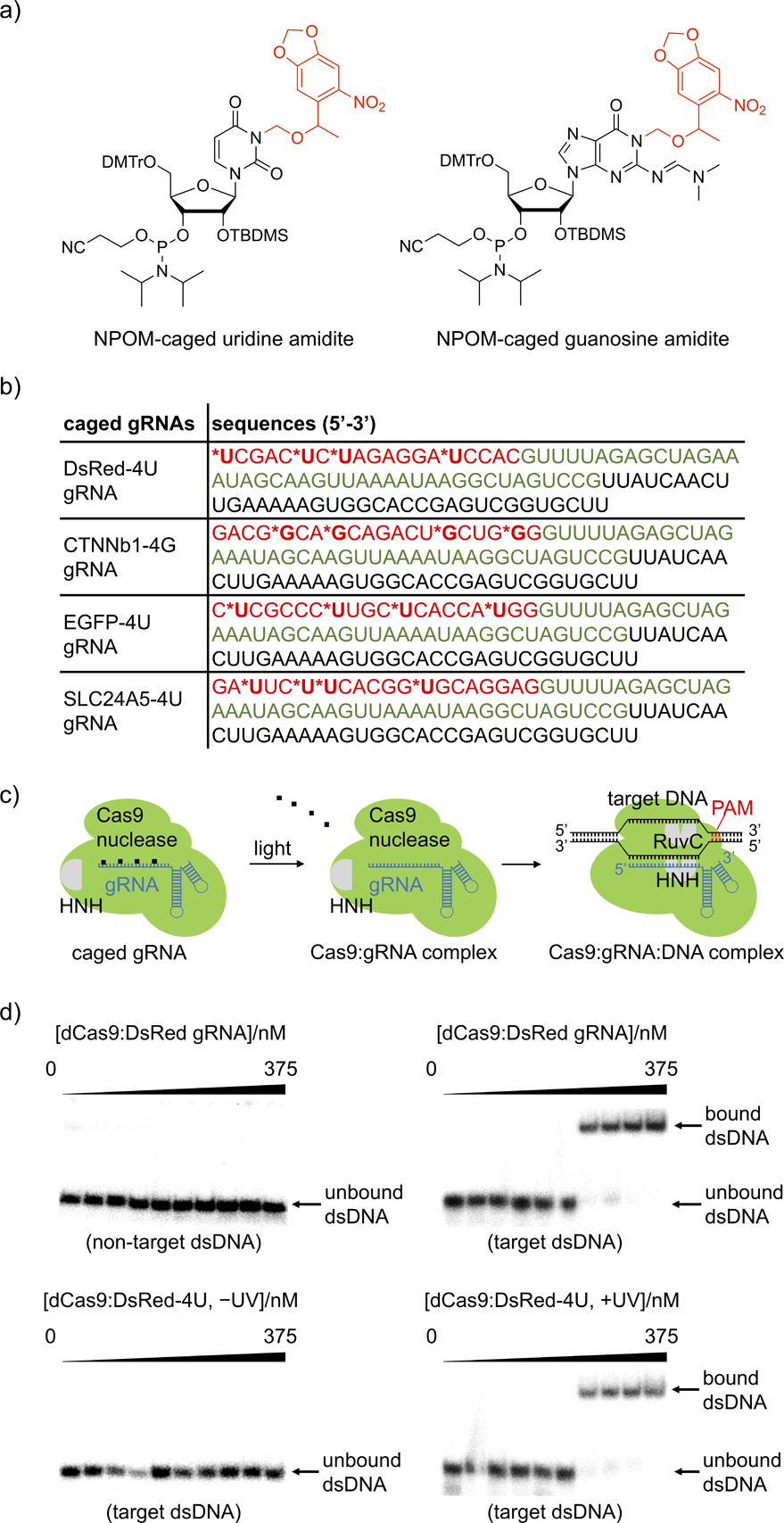
a) Structure of NPOM-protected uridine and guanosine amidites with photocleavable caging groups shown in red. b) Sequences of the photocaged gRNAs. The photocaged nucleotides are labelled by asterisks and the 20 nt base-pairing region of the gRNA is shown in red. The Cas9 binding region is shown in green and the S. pyogenes terminator region is shown in black. The corresponding non-caged gRNAs (DsRed, CTNNb1, EGFP, and SLC24A5) have the exact same sequences without the nucleobase caging groups. c) The NPOM-photocaging groups are designed to abolish RNP binding to the target dsDNA until they are photochemically cleaved, thereby generating an active Cas9:gRNA complex. d) Autoradiography of gel shift assays demonstrate that the photocaged gRNA effectively blocked the binding affinity of Cas9 to target 32P-labelled dsDNA and that binding is fully restored upon light activation, matching negative (non-target dsDNA) and positive (target dsDNA) controls. The exact concentrations of dCas9:gRNA used are 0, 0.0375, 0.1125, 0.375, 1.125, 3.75, 11.25, 37.5, 112.5, and 375 nM.
As a proof of concept, we first substituted four uridines evenly distributed within the 20 nucleotide base-pairing region of the DsRed gRNA with photocaged uridines for effective blockade of gRNA:dsDNA hybridization, generating DsRed-4U gRNA (nomenclature is used similarly for all other genes).[28] Catalytically dead Cas9 (dCas9) was recombinantly expressed in E. coli (Supporting Information, Figure S5) and non-caged control gRNAs were generated via in vitro transcription (Supporting Information, Figure S6). The decaging of the DsRed-4U gRNA was verified by HPLC which showed >90% caging group photolysis within 3 minutes of irradiation (Supporting Information, Figure S7). To test if caged gRNA can suppress base pairing and restore gRNA:dsDNA hybridization upon irradiation with UV light, gel shift assays of the RNP complex binding to 32P-labelled dsDNA (55 bp, Supporting Information, Table 1) were conducted. The binding ability of dCas9:caged gRNA RNP to target dsDNA was fully suppressed, matching a non-target dsDNA and the complete absence of any gRNA (Supporting Information, Figure S8a) as negative controls. Importantly, light-induced decaging was shown to completely restore this interaction to the same level as the RNP containing the non-caged gRNA (Figure 1d). The disrupted interaction between dsDNA and the RNP complex is the result of blocked Watson-Crick base-pairing by the caging groups,[25] thus preventing further DNA binding after recognition of the PAM sequence and subsequent cleavage of the DNA target.[29] Notably, both non-caged and caged gRNAs bind to the Cas9 protein with similar affinity, demonstrating that the caging of the protospacer region of the gRNA does not interfere with formation of the Cas9:gRNA RNP complex (Supporting Information, Figure S8b).
After successful optical control of the interaction between the RNP and the dsDNA, we designed photocaged gRNAs targeting different loci in both mammalian cells and zebrafish embryos following the developed strategy (Figure 1b). We first tested the optical triggering of CRISPR/Cas9 activity in mammalian cells harboring a dual-fluorescence reporter plasmid (Supporting Information, Figure S9b).[30] Targeted cleavage by Cas9 endonuclease both at the beginning and at the end of the DsRed-polyA gene cassette results in cells switching from expressing DsRed to expressing EGFP (Figure 2a). HEK293T cells were transfected with Cas9:EGFP gRNA together with Cas9:DsRed gRNA or Cas9:DsRed-4U gRNA RNPs, and were incubated for 6 hours before irradiation with 365 nm light. It should be noted that only one caged gRNA is needed in combination with a non-caged EGFP gRNA to achieve optical control over DsRed gene excision and activation of EGFP expression. The cells were then incubated for 72 hours, followed by imaging. EGFP expression was only observed after light exposure, indicating activation of DsRed excision, while caged RNP-transfected cells that were kept in the dark showed only minimal background activation (Figures 2b and 2c). DsRed fluorescence is visible in all cells, since the fluorescent protein expressed before the temporally controlled activation of CRISPR/Cas9 is stable and still present at the imaging timepoint. Insufficient editing of the transiently transfected pRG reporter could also contribute to the observed DsRed fluorescence. Fluorescent protein expression levels were quantified (ImageJ). First, background was subtracted based on a fixed value determined by the fluorescence intensity of non-transfected cells.[31] Then the fluorescence intensity of each channel for all the cells in one well was integrated to represent the expression level of the fluorescent protein (Figure 2c).[32] Furthermore, cell viability was analyzed after irradiation with 365 nm light, showing no phototoxicity with up to 30 minutes of irradiation (Supporting Information, Figure S10).
Figure 2.
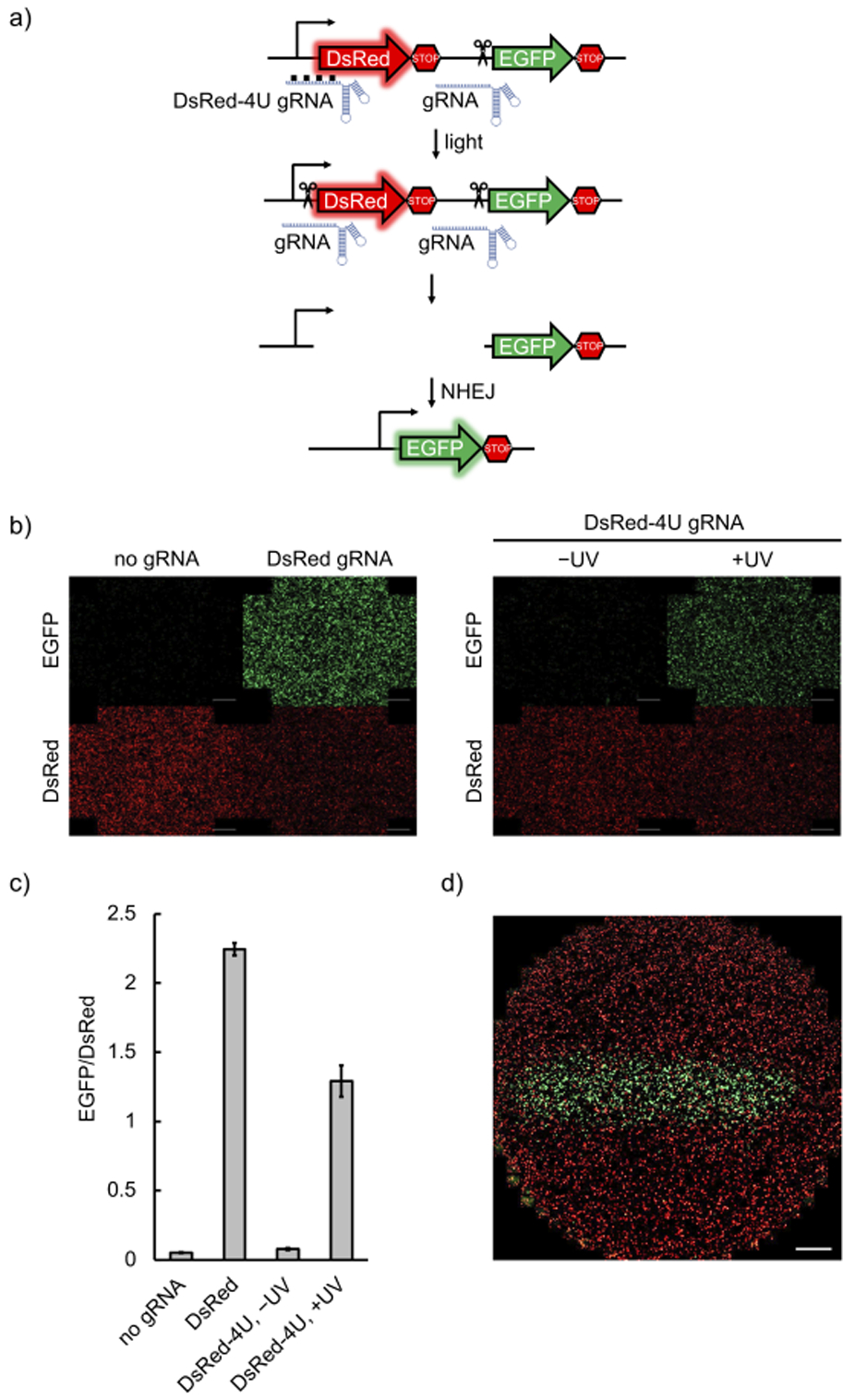
a) Schematic of the pRG reporter plasmid. Upon light activation, the presence of both gRNAs leads to excision of the DsRed-terminator cassette and NHEJ activates expression of EGFP. b) HEK293T cells transfected with the pRG reporter plasmid, followed by delivery of Cas9:gRNA RNP complexes, were irradiated or kept in the dark. EGFP expression was only observed with non-caged gRNAs or when the caged gRNA was photochemically activated (scale bars = 100 μm). c) EGFP and DsRed fluorescence was quantified by integration of fluorescence intensity in three independently transfected and treated wells for each condition using ImageJ. d) Spatial control of light-activated Cas9:gRNA function through patterned irradiation. HEK293T cells transfected with the pRG reporter and the Cas9:caged gRNA complex were exposed to 365 nm irradiation through a 2 mm-wide slit in a mask (scale bar = 100 μm).
Optical control of caged gRNA presents an opportunity for precise spatial activation of gene editing. Indeed, only cells exposed to 365 nm light through a slit-containing mask produced EGFP fluorescence, while a minimal degree of background activity was observed in non-exposed cells, potentially due to accidental exposure to ambient light (Figure 2d).
To demonstrate applicability of the developed optical tool to editing of the mammalian genome, we used a reported gRNA sequence (Figure 1b) to target a human genomic locus within the CTNNb1 gene.[33] Here, NPOM-caged guanosine (Figure 1a) was used instead of NPOM-caged uridine in order to achieve an even distribution of caged nucleobases throughout the gRNA sequence for efficient blocking of RNP:dsDNA interaction. Either CTNNb1 gRNA or caged CTNNb1–4G gRNA were delivered to HEK293T cells as Cas9 RNP complexes. Exposure to 365 nm light was performed at 6 hours after delivery and cells were incubated for 72 h, before lysis and amplification of the genomic target site by nested PCR. Sanger sequencing of the amplicon and TIDE (Tracking of Indels by DEcomposition) analysis[34] (Supporting Information, Figure S11) showed indel formation with 16.0 ± 4.0% frequency for CTNNb1 gRNA and 26.2 ± 8.9% frequency for light-activated CTNNb1–4G gRNA, while virtually no background editing was detected in the absence of irradiation of CTNNb1–4G gRNA (0.9 ± 0.6% frequency). TIDE analyses were conducted in triplicate, values were averaged, and errors represent standard deviations. These results demonstrate that we are efficiently editing the mammalian cell genome with light-activated CTNNb1–4G gRNA to a similar extent as the non-caged gRNA, while only limited background editing was observed in the absence of irradiation – showcasing the excellent off → on switching of our caged gRNA methodology.
The Cas9:gRNA RNP complex is an excellent tool for gene editing in aquatic embryos, due to ease of assembly and injection into the fertilized oocyte.[13] Furthermore, optical control is a powerful approach for conditional gene editing in zebrafish, because the embryos are transparent during the early stages of development, allowing for irradiation of all tissues. To demonstrate the utility of photocaged gRNAs to control Cas9 gene editing in zebrafish, we first injected RNPs assembled with a caged gRNA targeting the start codon of EGFP (EGFP-4U) in the genome of a transgenic fish line (Tg(ubi:loxP-EGFP-loxP-mCherry)) (Figure 3a).[35] Disruption of the start codon prevents EGFP expression, as demonstrated in representative micrographs after injection of a non-caged gRNA:Cas9 complex (Figure 3b). Optical activation of EGFP-4U RNP complexes in embryos at 1 hour post-fertilization (hpf) had similar editing efficiency as the non-caged gRNA containing RNP, significantly reducing EGFP expression in all animals, as determined by fluorescent imaging at 48 hpf (Figure 3c) and fluorescence intensity quantification (Supporting Information, Figure S12). While the images show complete disappearance of green fluorescence, TIDE analysis shows indel rates of 82.5 ± 5.2% for EGFP gRNA, 69.4 ± 7.3% for light-activated EGFP-4U gRNA, and 3.6 ± 1.3% for caged EGFP-4U gRNA in the absence of irradiation (Supporting Information, Figure S13a). TIDE analyses were conducted on three independently injected embryos, values were averaged and errors represent standard deviations. Mosaic EGFP expression is still present as indicated by TIDE analysis, but is not detected by the imaging conditions. This mosaicism is reported in other zebrafish RNP injection experiments as well.[36] No toxicity was observed from exposure of the embryos to 365 nm light for up to 30 minutes, well exceeding our 5 min irradiation for gRNA activation (Supporting Information, Figure S14).
Figure 3.
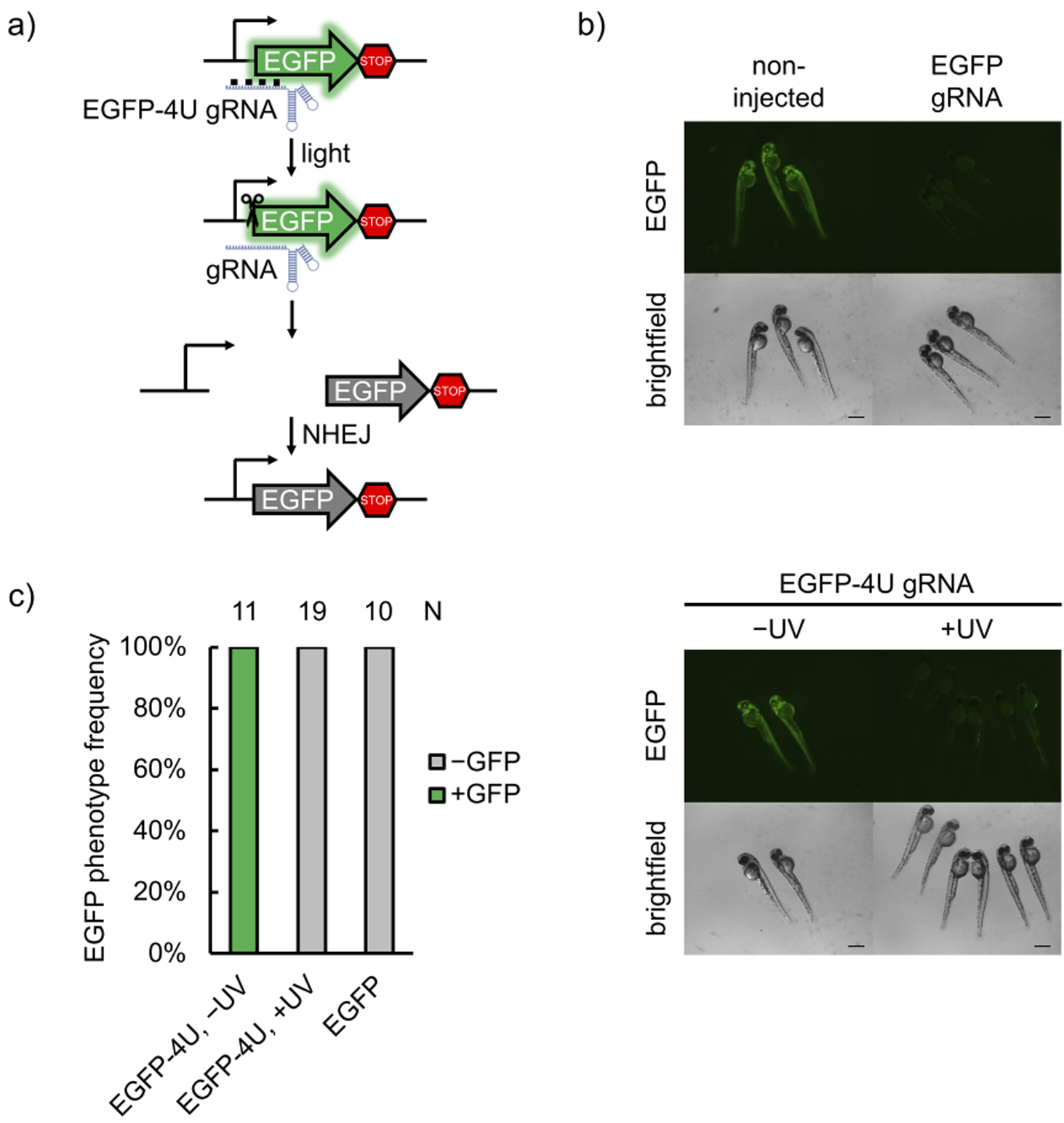
a) Schematic of the transgenic fish line fluorescent reporter. The gRNA recognizes the start codon region of EGFP, mutates it upon editing, and thus abolishes expression of the fluorescent protein. b) Representative micrographs of zebrafish at 48 hpf. Non-injected embryos demonstrate strong EGFP expression, while Cas9 RNP-injected embryos show drastically less EGFP expression (scale bars = 300 μm). c) Phenotype frequencies of the injected embryos at 48 hpf are shown. Editing ability is blocked in caged EGFP-4U gRNA until irradiation with 365 nm light at 1 hpf, indicating optical control of gene editing in embryos.
In order to further validate the universal applicability of this methodology, we next targeted the slc24a5 gene,[37] an endogenous gene in zebrafish which is important for development of pigmentation by 48 hpf. slc24a5 has been edited through Cas9 RNP injection before,[36] and the lack of pigmentation induced by indel introduction at this locus is commonly referred to as the golden phenotype, most robustly observed as pigment loss in the retina of the developing animal. We used the same gRNA sequence that has been previously used for disrupting slc24a5 function.[36a] Four uridine bases in the protospacer region were replaced with NPOM-caged uridines that led to inhibition of Cas9 editing ability until it was restored upon irradiation with 365 nm light. The resulting gene editing led to loss of retinal pigmentation (Figure 4a). Phenotype frequency was tunable through increasing light exposure (30 sec, 1 min, and 5 min), which also led to increases in indel rates (14.6 ± 6.3% for 30 sec, 27.9 ± 7.4% for 1 min, and 75.4 ± 4.7% for 5 min irradiation), reaching non-caged RNP phenotype frequencies and indel rates (71.8 ± 2.1%, Supporting Information, Figure S13b). This is similar to the reported editing efficiencies with this gRNA (87.3 ± 8.1%).[36a] This demonstrates the ability for tuning of editing efficiency and that full optical activation of Cas9 RNP function can be achieved with a reasonable irradiation time. Meanwhile, background editing in the absence of light-activation remained low (indel rate 3.1 ± 1.6%).
Figure 4.
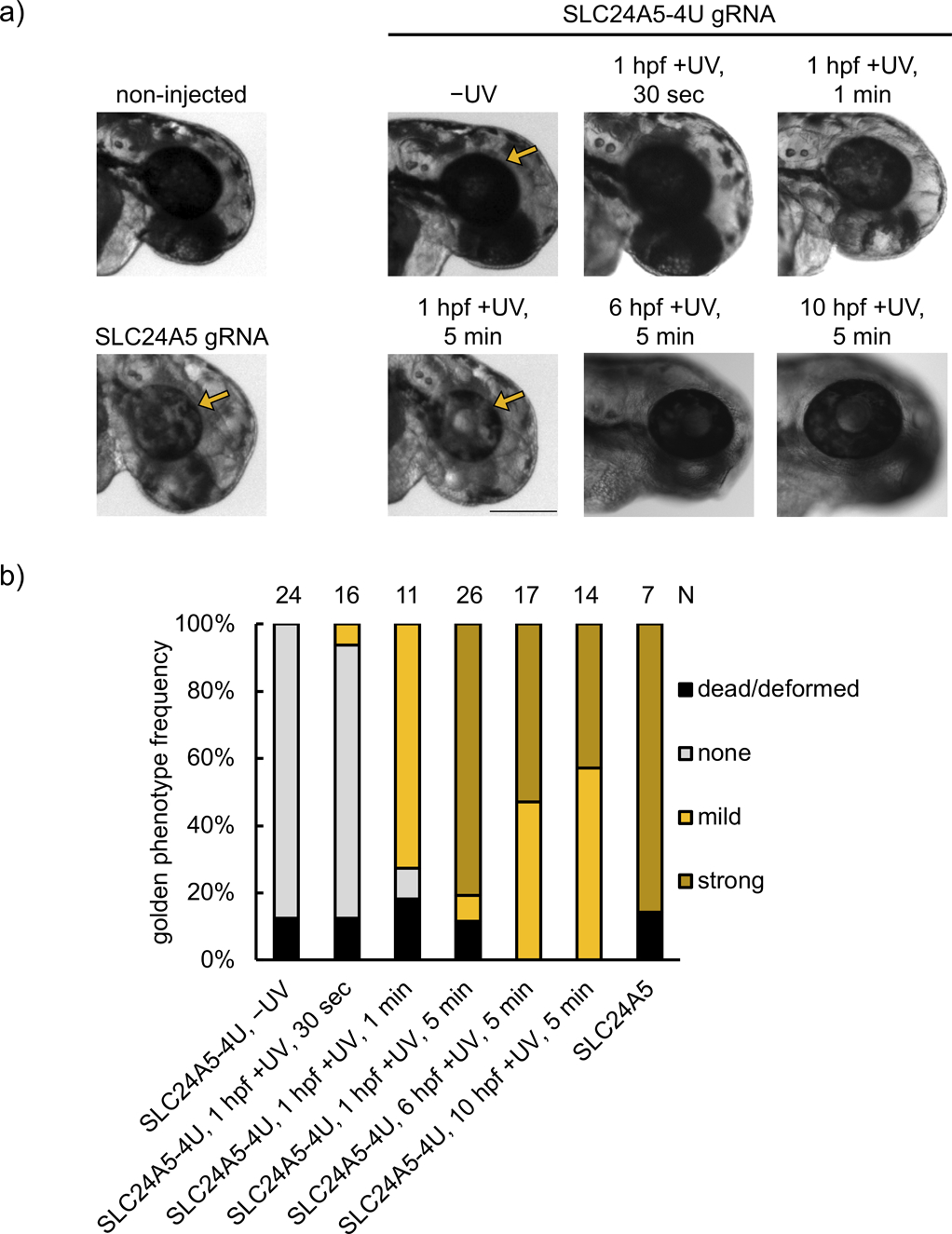
a) Representative images of embryos injected with RNPs assembled from non-caged SLC24A5 gRNA or caged SLC24A5–4U gRNA, and either kept in the dark or irradiated (365 nm) for 30 sec, 1 min, or 5 min at 1 hpf. Embryos were also irradiated at later timepoints (6 hpf and 10 hpf) for 5 min. Images were recorded at 48 hpf. Arrows point to the retina, demonstrating loss of pigmentation through successful editing of the slc24a5 locus (scale bar = 300 μm). b) Phenotype frequencies of the injected embryos at 48 hpf. The golden phenotype was determined based on the level of retinal pigmentation, with mild representing small patches of pigment loss, and strong representing a majority or complete retinal pigment loss.
As an external trigger, light provides a unique opportunity to convey temporal control, which was demonstrated by SLC24A5–4U gRNA activation at later timepoints in zebrafish development. We activated editing at the beginning of gastrulation (6 hpf, indel rate 63.1 ± 8.0%) and at the end of gastrulation (10 hpf, indel rate 21.3 ± 2.6%), representing critical timepoints when early cell populations become established and migrate (Figure 4b, Supporting Information, Figure S15). The lower indel rates seen with later activation could be caused by a lower concentration of the RNP complex due to dilution as the organism grows, an increasing number of cells that need to be edited, and/or RNP complex degradation over time.[38] Notably, indel rates between the strong and mild phenotype embryos irradiated at 1 hpf were similar (71.8 ± 2.1% in the strong phenotype versus 70.4 ± 8.5% in the mild phenotype), suggesting consistent editing efficiency between embryos in the same condition. The discrepancy in phenotype is likely due to the difference in editing efficiency specifically of melanocytes between embryos scored as mild or strong, which only represent a small proportion of total embryo cells. Overall, mosaicism increases with shorter irradiation time or later timepoint of activation, trending with the average strength of the observed phenotype.
Conclusion
In summary, we developed a new method to optically control CRISPR/Cas9 activity through nucleobase-caged gRNAs, thereby further expanding the tool set available for conditional control of gene editing with spatial and temporal resolution.[39] Flexibility in the synthesis of caged gRNAs is ensured through the use of both caged uridine and caged guanosine nucleotides, while allowing for standard solid-phase protocols. We successfully applied this optically controlled gene editing approach in both mammalian cells and zebrafish embryos with high efficiency for both transiently transfected plasmid DNA and genomic loci targets. NPOM-caged gRNAs expand the gene editing toolbox with unique features, including 1) rapid and non-invasive activation of CRISPR/Cas9 activity, 2) precise temporal and spatial control, 3) modularity and programmability of the light-activated gRNA sequence, 4) formation of a stable Cas9:gRNA complex from commercially available protein, 5) broad applicability for delivery into cells and organisms in the form of RNP complexes, and 6) capability for tuning of gene editing efficiency through simple modification of light exposure duration. We expect that NPOM-caged gRNAs will find utility in the dissection of regulatory networks in the developing zebrafish embryo in a temporally and spatially sensitive manner. Furthermore, the light-activated Cas9:gRNA RNP system can be tuned to red-shifted activation wavelengths by using alternative caging groups, is expected to be functional in other cell lines and (aquatic) embryos, and should be easily adaptable to other Cas systems without the need for protein engineering.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R21HD085206 and R01GM112728 to AD; WB was supported by T32GM088119).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Adli M, Nat Commun 2018, 9, 1911; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang H, Russa M. La, Qi LS, Annu Rev Biochem 2016, 85, 227–264; [DOI] [PubMed] [Google Scholar]; c) Wright AV, Nunez JK, Doudna JA, Cell 2016, 164, 29–44; [DOI] [PubMed] [Google Scholar]; d) Barrangou R, Doudna JA, Nat Biotechnol 2016, 34, 933–941; [DOI] [PubMed] [Google Scholar]; e) Komor AC, Badran AH, Liu DR, Cell 2017, 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jiang F, Doudna JA, Annu Rev Biophys 2017, 46, 505–529. [DOI] [PubMed] [Google Scholar]
- [3].Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR, Nature 2018, 556, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F, Nature 2015, 517, 583–588; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) L. A Gilbert MA. Horlbeck B. Adamson J. Villalta E, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, Cell 2014, 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Eid A, Alshareef S, Mahfouz MM, Biochem J 2018, 475, 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B, Cell 2013, 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA, Nat Biotechnol 2015, 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Norris AL, Lee SS, Greenlees KJ, Tadesse DA, Miller MF, Lombardi H, BioRxiv 2019, 715482. [Google Scholar]
- [9].a) Zhou W, Deiters A, Angew Chem Int Ed Engl 2016, 55, 5394–5399; [DOI] [PubMed] [Google Scholar]; b) Nunez JK, Harrington LB, Doudna JA, ACS Chem Biol 2016, 11, 681–688; [DOI] [PubMed] [Google Scholar]; c) Gangopadhyay SA, Cox KJ, Manna D, Lim D, Maji B, Zhou Q, Choudhary A, Biochemistry 2019, 58, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) M. K, Pattanayak V, Thompson DB, Zuris JA, Liu DR, Nat Chem Biol 2015, 11, 316–318; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu KI, Ramli MN, Woo CW, Wang Y, Zhao T, Zhang X, Yim GR, Chong BY, Gowher A, Chua MZ, Jung J, Lee JH, Tan MH, Nat Chem Biol 2016, 12, 980–987; [DOI] [PubMed] [Google Scholar]; c) L. B, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, Savage DF, Nat Biotechnol 2016, 34, 646–651; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rose JC, Stephany JJ, Valente WJ, Trevillian BM, Dang HV, Bielas JH, Maly DJ, Fowler DM, Nat Methods 2017, 14, 891–896; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gao Y, Xiong X, Wong S, Charles EJ, Lim WA, Qi LS, Nat Methods 2016, 13, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zetsche B, Volz SE, Zhang F, Nat Biotechnol 2015, 33, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A, J Am Chem Soc 2015, 137, 5642–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xin Y, Duan C, Methods Mol Biol 2018, 1742, 205–211. [DOI] [PubMed] [Google Scholar]
- [14].Nihongaki Y, Kawano F, Nakajima T, Sato M, Nat Biotechnol 2015, 33, 755–760. [DOI] [PubMed] [Google Scholar]
- [15].Pan Y, Yang J, Luan X, Liu X, Li X, Yang J, Huang T, Sun L, Wang Y, Lin Y, Song Y, Sci Adv 2019, 5, eaav7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou XX, Zou X, Chung HK, Gao Y, Liu Y, Qi LS, Lin MZ, ACS Chem Biol 2018, 13, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Polstein LR, Gersbach CA, Nat Chem Biol 2015, 11, 198–200; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shao J, Wang M, Yu G, Zhu S, Yu Y, Heng BC, Wu J, Ye H, Proc Natl Acad Sci U S A 2018, 115, E6722–E6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jain PK, Ramanan V, Schepers AG, Dalvie NS, Panda A, Fleming HE, Bhatia SN, Angew Chem Int Ed Engl 2016, 55, 12440–12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ferry QR, Lyutova R, Fulga TA, Nat Commun 2017, 8, 14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maji B, Moore CL, Zetsche B, Volz SE, Zhang F, Shoulders MD, Choudhary A, Nat Chem Biol 2017, 13, 9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kundert K, Lucas JE, Watters KE, Fellmann C, Ng AH, Heineike BM, Fitzsimmons CM, Oakes BL, Savage DF, El-Samad H, Doudna JA, Kortemme T, 2018.
- [22].a) Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J, Elife 2013, 2, e00471; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, S. J, Mazagova M, Choi SH, Yun HY, Xiao H, Yang P, Luo X, Lim RK, Pugh HM, Wang F, Kazane SA, Wright TM, Kim CH, Schultz PG, Young TS, Angew Chem Int Ed Engl 2016, 55, 7520–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) DeWitt MA, Corn JE, Carroll D, Methods 2017, 121–122, 9–15; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, Urnes C, Munares GA, Ghosh A, Doudna JA, Nat Biotechnol 2017, 35, 431–434; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Glass Z, Lee M, Li Y, Xu Q, Trends Biotechnol 2018, 36, 173–185; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lin S, Staahl BT, Alla RK, Doudna JA, Elife 2014, 3, e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Liu Q, Deiters A, Acc Chem Res 2014, 47, 45–55; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hemphill J, Govan J, Uprety R, Tsang M, Deiters A, J Am Chem Soc 2014, 136, 7152–7158; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Govan JM, Young DD, Lusic H, Liu Q, O. M, Deiters A, Nucleic Acids Res 2013, 41, 10518–10528; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Deiters A, Lusic H, Synthesis 2006, 2006, 2147–2150. [Google Scholar]
- [25].a) Lusic H, Young DD, Lively MO, Deiters A, Organic letters 2007, 9, 1903–1906; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Young DD, Edwards WF, Lusic H, Lively MO, Deiters A, Chem Commun (Camb) 2008, 462–464; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Young DD, Lusic H, Lively MO, Yoder JA, Deiters A, Chembiochem 2008, 9, 2937–2940. [DOI] [PubMed] [Google Scholar]
- [26].Yu X, Liang X, Xie H, Kumar S, Ravinder N, Potter J, X. d. M. du Jeu, J. D. Chesnut, Biotechnology letters 2016, 38, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, Bacchetta R, Tsalenko A, Dellinger D, Bruhn L, Porteus MH, Nat Biotechnol 2015, 33, 985–989; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ryan DE, Taussig D, Steinfeld I, Phadnis SM, Lunstad BD, Singh M, Vuong X, Okochi KD, McCaffrey R, Olesiak M, Roy S, Yung CW, Curry B, Sampson JR, Bruhn L, Dellinger DJ, Nucleic Acids Res 2018, 46, 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Connelly CM, Uprety R, Hemphill J, Deiters A, Mol Biosyst 2012, 8, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang Q, Wen F, Zhang S, Jin J, Bi L, Lu Y, Li M, Xi XG, Huang X, Shen B, Sun B, Sci Adv 2019, 5, eaaw9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].De Gasperi R, Rocher AB, Sosa MA, Wearne SL, Perez GM, Friedrich VL Jr., Hof PR, Elder GA, Genesis 2008, 46, 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Furia L, Pelicci PG, Faretta M, Cytometry A 2013, 83, 333–343. [DOI] [PubMed] [Google Scholar]
- [32].Higuchi-Sanabria R, Garcia EJ, Tomoiaga D, Munteanu EL, Feinstein P, Pon LA, PLoS One 2016, 11, e0146120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, Doench JG, Nat Commun 2018, 9, 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brinkman EK, Chen T, Amendola M, van Steensel B, Nucleic Acids Res 2014, 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI, Development 2011, 138, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Jao L-E, Wente SR, Chen W, Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 13904–13909; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Burger A, Lindsay H, Felker A, Hess C, Anders C, Chiavacci E, Zaugg J, Weber LM, Catena R, Jinek M, Robinson MD, Mosimann C, Development 2016, 143, 2025. [DOI] [PubMed] [Google Scholar]
- [37].Lamason RL, Mohideen MA, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, Sinha S, Moore JL, Jagadeeswaran P, Zhao W, Ning G, Makalowska I, McKeigue PM, O’Donnell D, Kittles R, Parra EJ, Mangini NJ, Grunwald DJ, Shriver MD, Canfield VA, Cheng KC, Science 2005, 310, 1782–1786. [DOI] [PubMed] [Google Scholar]
- [38].Kim S, Kim D, Cho SW, Kim J, Kim J-S, Genome Res 2014, 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].a) Ankenbruck N, Courtney T, Naro Y, Deiters A, Angewandte Chemie 2018, 57, 2768–2798; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kowalik L, Chen JK, Nat Chem Biol 2017, 13, 587–598; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) O’Banion CP, Lawrence DS, Chembiochem 2018, 19, 1201–1216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.