

Abstract
Despite the relevance of complex root microbial communities for plant health, growth and productivity, the molecular basis of these plant-microbe interactions is not well understood. Verrucomicrobia are cosmopolitans in the rhizosphere, nevertheless their adaptations and functions are enigmatic since the proportion of cultured members is low. Here we report four cultivated Verrucomicrobia isolated from rice, putatively representing four novel species, and a novel subdivision. The aerobic strains were isolated from roots or rhizomes of Oryza sativa and O. longistaminata. Two of them are the first cultivated endophytes of Verrucomicrobia, as validated by confocal laser scanning microscopy inside rice roots after re-infection under sterile conditions. This extended known verrucomicrobial niche spaces. Two strains were promoting root growth of rice. Discovery of root compartment-specific Verrucomicrobia permitted an across-phylum comparison of the genomic conformance to life in soil, rhizoplane or inside roots. Genome-wide protein domain comparison with niche-specific reference bacteria from distant phyla revealed signature protein domains which differentiated lifestyles in these microhabitats. Our study enabled us to shed light into the dark microbial matter of root Verrucomicrobia, to define genetic drivers for niche adaptation of bacteria to plant roots, and provides cultured strains for revealing causal relationships in plant-microbe interactions by reductionist approaches.
Subject terms: Soil microbiology, Bacteria, Symbiosis
Introduction
In land plants, roots are the primary site for interactions with diverse microbes, which are recruited from soil and filtered by the plant1. Plants have probably evolved along with their associated microbes for millions of years. The fossil record suggests that bacteria were already endophytic2 in plants with simple rhizoid-based rooting systems, during the early evolution of roots 400 million years ago3. Current results of reverse genetics and community structure illustrate that endophytic colonization is an active process requiring specific traits of bacteria to establish inside1,4. It is known for eukaryotes5 and prokaryotes6 that exposure of different populations to similar environments may result in similar features because of parallel selection in their genomes. Thus, it is conceivable that bacteria of different taxonomic background may share characteristics for a plant-associated or endophytic lifestyle.
However, how bacteria adapt to and interact with plants is a fundamental question for understanding the molecular basis of root microbiome functions. To discover causal relationships of underlying principles, reductionist studies use single isolates or reconstitution experiments from synthetic communities (large pure culture collections isolated from plants) as promising tools7. Yet, in the domain Bacteria many phyla have no or few cultivated representatives8, with more than 88% of all isolates belonging to only four phyla9. Also in the highly diverse phylum Verrucomicrobia10, the view on bacterial capacities is skewed because cultured strains are strongly underrepresented. The generally poor cultivation success of Verrucomicrobia has impaired progress in understanding their ecology and capacities11.Culture-independent methods suggest verrucomicrobial communities in many diverse biomes including soils11, where they drive biogeographical pattern changes12. Even in these studies, primer bias in commonly used PCR primers may lead to an underestimation of 16S rRNA genes during amplification13. With improved primer sets, a high relative abundance of Verrucomicrobia was found in grassland soils (35%) and even 52% in subsoils of a coniferous forest13. Verrucomicrobia are also cosmopolitans in the rhizosphere: in addition to Arabidopsis thaliana14, poplar15, and maize16,17 rhizosphere soils, rice roots18,19 harbour a considerable verrucomicrobial population (4–6%)20, and Verrucomicrobia were recently reported to constitute even 25% of the endorhizosphere population21. Due to the lack of isolates, only few studies focused on the possible ecological importance or niche adaptation of Verrucomicrobia in microbe-plant interactions22,23, and no endophytic strains are available. Concomitantly, in published synthetic communities of plant-associated microbes Verrucomicrobia are lacking, for example for Arabidopsis24 or poplar and maize25. Therefore, we attempted to isolate root-associated Verrucomicrobia in order to evaluate their root colonization capacity, plant promotion characteristics, and putative genomic adaptations to root-associated lifestyle in this phylum, as it is distant to common root endophytes.
Here we report the isolation of four novel, divergent strains of Verrucomicrobia from rice roots or rhizomes and thus extend the cultivated root microbiota to this phylum. Two strains were demonstrated for the first time to be root endophytic in gnotobiotic cultures, a novel feature of Verrucomicrobia. Characteristics that are common to other plant growth-promoting bacteria such as Proteobacteria, were surprisingly also detected in the isolates of the distant phylum Verrucomicrobia, which suggests widespread similar genetic drivers of niche adaption and functional diversification. Across-phylum comparison of protein domains encoded in genomes of well-studied reference endophytes, root surface and soil bacteria, allowed to identify novel signature protein domains for compartments, and thus provided a broader insight in adaptations for plant-associated lifestyle that are common in distant phyla.
Results and Discussion
Isolation of novel Verrucomicrobia from rice, Oryza spp
In order to obtain cultivable Verrucomicrobia, we subjected roots of O. sativa and roots and rhizomes of an undomesticated species of rice, O. longistaminata, to a large scale cultivation approach. Different compartments were targeted - surface and endosphere after effective surface sterilization. Among a larger culture collection of roughly 250 strains of mainly Proteobacteria, Actinobacteria and Firmicutes (not shown), four divergent Verrucomicrobia were detected that had been isolated under aerobic culture conditions: Small colonies of slow-growing bacteria were found among fast-growing strains upon extended incubation of agar plates. Strains LW23, ER46 and LR76 were obtained from the rhizoplane, rhizome endosphere, and rhizome surface of O. longistaminata, respectively, whereas strain EW11 was isolated from the root endosphere of O. sativa. Phylogenetic analysis of almost complete 16S rRNA gene sequences placed strain LR76 in subdivision 2, and EW11 and ER46 in subdivision 4 (Fig. 1a), out of seven known subdivisions proposed for Verrucomicrobia10,26. Strain LW23 clustered as sister lineage to methylotrophs of subdivision 6, without cultured bacteria being closely related (Fig. 1a). Phylogenetic, phylogenomic (Fig. 1a,b) and physiological features suggest that strain LW23 belongs to a novel subdivision 8 (Supplementary Note 1, Supplementary Tables S1,S2). The putative new subdivision consists entirely of uncultured bacteria from diverse ecological habitats, with exception of strain LW23 and Verrucomicrobiaceae bacterium GAS474 from forest soil: for example intestine of the earthworm Eisenia fetida27 (97% identity), a low-level radioactive waste site28 (91% identity), marine (coral) and aquatic moss, freshwater lake, as well as terrestrial habitats. Therefore, it is difficult to predict which physiological requirements are needed for isolation of more cultured strains belonging to this ecologically diverse new subdivision. Previous subdivision numbers 5 and 7 were meanwhile assigned to the rank of distinct phyla closely related to Planctomycetes, Verrucomicrobia and Chlamydiae, namely ‘Lentisphaerae’29,30 and ‘Kiritimatiellaeota’31; therefore these subdivision numbers were not re-used by us for the novel subdivision.
Figure 1.

Molecular phylogenetic (a), phylogenomic (b) and cell morphology (c) analysis of the novel Verrucomicrobia isolates. (a) Molecular phylogeny based on based 16S rRNA gene sequences. Novel isolates of Verrucomicrobia in bold, putative novel subdivision 8 in purple. Almost complete 16S rRNA gene sequences were aligned with reference sequences from representatives of every subdivision of the phylum Verrucomicrobia. The evolutionary history was inferred by using the Maximum Likelihood method implemented in IQ-TREE. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. There were a total of 1228 positions in the final dataset, and Chlamydia trachomatis (NR 025888) was used as outgroup. (b) Phylogenomic relationships of Verrucomicrobia inferred by the whole-genome-based CVTree approach. Genomes or metagenomes covering all known subdivisions were selected, and novel isolates (purple) included. Analysis by CVTree3 with K = 6. (c) Cell morphology of strains EW11(1), ER46(2), LR76(3), and LW23(4). (A, B) Transmission electron micrographs of thin sections. (C, D) Images of cells stained with SYBR green to reveal condensed nucleic acids inside cells by Confocal Laser Scanning Microscopy. White arrowhead, electron-dense material. Scale bars indicate 500 nm (1–2 A + B, 3–4 B), 1000 nm (3–4 A) or 2 µm (C,D). Endospores were not observed. Specimen preparation through conventional fixation and dehydration lead to an irregular, scattered shape of some cells (2B) as described for other Verrucomicrobia88.
Cultivated root-colonizing rhizosphere soil isolates were described only within Verrucomicrobia subdivision 1 up to now23,32. In subdivision 4, “Opitutae” were detected in the rhizosphere soil of pine by culture-independent methods33, but we report the first cultivated root-associated members: strain ER46, related to a paddy soil isolate Opitutus sp. VeSm1334, and strain EW11, more distantly related to Verrucomicrobia sp. TSB47 from termite gut. Spartobacteria (subdivision 2) were most commonly found in terrestrial environments13, including in rhizosphere soil33. The strictly anaerobic Terrimicrobium sacchariphilum from rice paddy soil35 was the closest cultured relative to strain LW76. Thus our study extended the range of cultivated root inhabitants to three other subdivisions, suggesting a more widespread close association with roots among Verrucomicrobia.
A 16S rRNA gene sequence identity below 98.7–99% has been defined as threshold value for species delineation36. Above this value, the analysis of the average nucleotide identity (ANI)37,38 between genomes is used to compare the genetic relatedness among putative species. ANI values of ~94% correspond to the traditional 70% DNA-DNA hybridization boundary differentiating species from each other39. Comparison of genomes of the novel strains to those of known related species yielded less than 89% (Supplementary Table S3), confirming the rank of at least novel species. As 16S rRNA gene identities less than 94.5% suggest distinct genera40, all strains except ER46 probably constitute novel genera (Supplementary Table S3). Formal description of the new taxa will be submitted elsewhere, as names Astrumicrobium roseum LW23, Opitutus oryzae ER46, Spartobacter rhizophilus LR76, and Albicoccus flocculans EW11 are proposed.
Cell compartmentalization in Verrucomicrobia isolates extends to the novel subdivision
Morphological analysis suggested that cell compartmentalization in Verrucomicrobia extends to the novel subdivision. Some members of the Verrucomicrobia possess a compartmentalized cell plan analogous to that found in phylum Planctomycetes31,41, detected in cells of subdivision 1, 2 and 341. SYBR green staining of nucleic acids showed clearly distinct small zones of fluorescence in confocal laser scanning microscopy (CLSM) analysis of Astrumicrobium roseum LW23 (Fig. 1c), indicating that nucleoid-containing cell compartments also occur in the novel proposed subdivision 8. In Spartobacter rhizophilus LR76 from subdivision 2, compartmentalized staining was observed as well, but it was not evident in cocci EW11 and ER46 from subdivision 4 (Fig. 1c).
Endophytic colonization pattern of novel Verrucomicrobia
Root-associated bacteria may occupy different niches - the rhizosphere soil, root surface (rhizoplane), or endorhizosphere - and thus show increasing intimacy of the interaction with plants as well as genomic adaptations to their niche1. Microscopic validation is most important for the assessment of a truly endophytic lifestyle1,42. Therefore, we tested the isolate’s colonization capacities for rice in a hydroponic cultivation system under gnotobiotic conditions. Differential colonization patterns were detected for the novel Verrucomicrobia by fluorescence microscopy (Supplementary Fig. 1) and CLSM (Fig. 2); microbial contaminations were not detected in any of the uninoculated controls (Supplementary Fig. 1). S. rhizophilus LR76 colonized the root surface, while A. roseum LW23 attached to roots and root hairs in large aggregates. For none of both, endophytic colonization was observed. Only isolates originating from the endosphere were proven to be endophytes: O. oryzae ER46 attached only weakly to the root surface. Nonetheless, bacteria colonized roots endophytically, clearly visible inside rhizodermis cells and root hairs (Fig. 2e,f; Supplementary Videos 1, 2). In contrast, A. flocculans EW11 colonized the root surface very evenly, densely, and in some cases covered roots in large biofilms (Fig. 2g,h). Endophytic colonization was detected in intercellular spaces and inside root cells (Fig. 2i,j and Supplementary Videos 3,4). This is the first microscopic proof of endophytic colonization by verrucomicrobia and clearly extends the niche of Verrucomicrobia to the root endophytic compartment.
Figure 2.

Confocal Laser Scanning Microscopy of rice roots at 8 days post inoculation in hydroponic gnotobiotic culture. Rice variety O. sativa ssp. japonica cv. Nipponbare, inoculated under gnotobiotic conditions and incubated for 7d. Roots were stained with SYBR Green to visualize bacteria. (a) Colonization of strain LR76, of strain LW23 (b,c), of strain EW11 (e,f), and of strain ER46 (d,g–j). Bars indicate 100μm (a), 50 μm (b–e,g,h) or 20 μm (f), respectively. Streaking of plant medium on RSA agar did not reveal any contamination. The non-inoculated control pants showed microscopically no bacterial colonization (Supplementary Fig. 1), and generally in this seed batch, intrinsic endophytes were not detected in any experiment.
Protein domains significantly enriched in root-associated bacteria
Validation of verrucomicrobia strains as endophytes or root surface colonizers permits to elucidate features which are characteristic for a plant-associated or endophytic lifestyle, if they are shared among root-associated but not soil bacteria across diverse phyla. Thus, draft genome sequences were generated for our strains (Supplementary Table S1). For comparison, we chose genomes and metagenomes of distantly related bacteria with well-documented typical lifestyles in endorhizosphere or rhizoplane of various plants, or in soil (Supplementary Table S4). Particularly endophytic lifestyle must be unambiguously demonstrated by microscopic means42,43, as commonly used techniques to remove root surface bacteria in culture-independent studies44 often fail1, and thus the true endomicrobiome picture will be blurred. Rhizoplane bacteria were included, since previous studies for example on Spartina45 or Kallar grass46 had indicated that rhizoplane and endorhizosphere populations may be different from each other; thus they might be differentiated by specific bacterial traits of endophytes that are required for ingress and establishment inside the host1. Tested bacteria comprised of diverse phyla, Verrucomicrobia, Proteobacteria, Acidobacteria, and Gemmatimonadetes.
Our across-phylum quantification of Pfam-domain diversity per genome revealed significant differences for all three compartments (Fig. 3a). The number of different domains per genome was highest in true endophytes, followed by rhizoplane colonizers, and lowest in soil bacteria. This suggests an increased degree of complexity of protein functions with increased intimacy of plant-microbe interaction, as it has been speculated1. Niche specialization was also indicated in Principle Component Analysis, which clearly separated Pfam domains of endophytes, rhizoplane bacteria and soil bacteria (Fig. 3b, Supplementary Table S5), with P < 0.018 in pairwise ANOSIM comparisons of the compartments. Despite the higher diversity of phyla among soil bacteria selected in our reference collection, soil-related protein domains clustered well and appeared less diverse (Fig. 3b), suggesting high functional similarities. Functional relatedness of endosphere and rhizoplane microbiomes was indicated by a larger overlap of protein domains with each other (188, Fig. 3c) in comparison to soil microbiomes (73 or 93, respectively).
Figure 3.

Protein domains differentiating bacterial lifestyles in endophere, rhizoplane and soil across bacterial phyla. Protein domains in (meta)genomes of well-established model organisms typically colonizing endosphere, rhizoplane or soil were compared. (a) Abundance of unique protein domains in (meta)genomes of bacteria from different compartments. Box-and-whisker plots for median (center lines), 25th and 75th percentiles (box edges), extreme data points (whiskers), and mean value (plus). Horizontal lines: statistical significance according to analysis of variance (ANOVA) followed by Holm-Sidak’s multiple comparison test, P ≤ 0.05 (*) or P ≤ 0.001 (***). (b) Between-group principal component analysis (bgPCA) of all Pfam protein domains in (meta)genomes of bacteria from the three compartments (P < 0.005 for all 3 pairwise comparisons according to PERMANOVA). (c) Venn diagram indicating differences and commonalities in Pfam domains of structural proteins encoded in the (meta)genomes from different compartments. The inner circle indicates the number of core protein families shared between all strains. The number of domains shared by all strains of one compartment are given in the outer circles (based on data from Supplementary Table S5). (d) Compartment signatures of protein domains. Examples of Pfam domains are given that are significantly enriched in (meta)genomes from endosphere or rhizoplane bacteria; for a given Pfam domain, bars are labelled by different letters if they are significantly different from each other at P < 0.05 using the Holm-Sidak method without assuming equal variance. Functions related to the respective domains are given below. Compartments depicted in the same colour in A-D. Model organisms were for soil: Verrucomicrobia bacterium isolates AV21, AV32, AV80, Acidobacteria bacterium isolate gp1 AA112, Gemmatimonadetes bacterium isolate AG11 [soil metagenomes obtained by whole genome shotgun sequencing (WGS)], Opitutus terrae PB90-1, Opitutus sp. GAS368, Azoarcus aromaticum EbN1 (complete genomes); for rhizoplane: Verrucomicrobia Astrumicrobium roseum LW23 and Spartobacter rhizophilus LR76, Azospirillum halopraeferans Au4, Azoarcus communis SWuB3, Azospirillum brasilense Sp7 (all WGS), and endophytes: Opitutaceae Albicoccus flocculans EW11 and Opitutus terrae ER46 (WGS), Azoarcus olearius BH72, Herbaspirillum seropedicae Z67, Gluconacetobacter diazotrophicus PAl 5, Serratia proteamaculans 568, Azospirillum sp. B510, Pseudomonas putida W619, Pseudomonas stutzeri A1501, Klebsiella variicola 342 (previously: K. pneumoniae 342), Enterobacter sp. 638, Methylobacterium populi BJ001, and Stenotrophomonas maltophilia R551-3 (complete genome). Based on data from Supplementary Table 7 where details are given. Graphs done with GraphPad Prism.
Therefore, a robust quantitative comparison of protein domain occurrence was carried out for genomes representative for the three compartments. It revealed putative functions differentiating all three the lifestyles from each other (Supplementary Table S4). Several domains are discussed which matched also domain occurrence in our novel Verrucomicrobia (Fig. 3d). Presence in phylogenetically very distant root-associated phyla, Proteobacteria and Verrucomicrobia, are a strong indication of lifestyle-adapted traits. Enriched in endosphere compared to soil microbiomes, the most striking group with 19 domains was related to motility by flagella, across phyla. They were almost absent from the soil microbiome but also abundant in many rhizoplane bacteria, exemplified by cytoplasmic chaperone FliJ, basal rod protein domain Flg_bbr_C, and flagellar hook-basal body complex protein FliE (Fig. 3d). Swarming assays corroborated motility of Verrucomicrobia endophytes EW11 and ER46. Motility allows the bacteria to move towards the host guided by root exudates and other gradients, and was proven to convey an advantage for endophytic colonization47. Significantly enriched in both, endosphere and rhizoplane over soil, were domains related to transport (SBP_bac_11, HupE_UreJ), transcriptional regulation (ArsC), but also synthesis of cobalamin (CobW_C) and putative siderophores (Isochorismatase). In the legume symbiont Ensifer meliloti, cobalamin synthesis was proven to play a critical role for survival in the plant host48. Biocontrol against plant pathogens may be mediated though competition for iron49, by synthesis, secretion and uptake of iron-chelating siderophores. Endophyte EW11 and epiphyte LW23 showed siderophore production in bioassays (Supplementary Note 2, Supplementary Table S6), and also harboured TonB-dependent siderophore receptor genes (fiu). Putative TonB-receptor genes were present in all four verrucomicrobial isolates. These domains may thus be regarded as typical for root-associated bacteria.
Several domains which were significantly enriched in rhizoplane versus soil microbiomes but were rare in endophytes indicate that the root surface is a special microniche: it may be a hotspot for DNA exchange, require pH-regulation to combat acidification by roots, and provide carbohydrates: (a) TfoX_N is found in of transcriptional regulators TfoX that positively regulate genes required for DNA transformation (late competence-specific genes); DUF87, now Her_A, is present in proteins related to bacterial conjugation like TraD (transferosome) or TraC (assembly of F-pilins). (b) Rhizoplane microbiomes harboured also significantly more domains related to Na+/H+ and K+/H+ antiporters (PhaG_MnhG_YufB, MNHE, MNHB), and proteins involved in pH regulation (MrpF_PhaF) in comparison to soil microbiomes. (c) Close vicinity to roots and their exuded polysaccharides is likely to promote significant enrichment of domains related to sugar and cellobiose transporters or glycolase hydrolases (PTS_EIIC, PTS_IIB/Glyco_hydro_4C, Glyco_hydro_4).
Quantitative domain analysis was even able to functionally differentiate typical endophytes from rhizoplane colonizers. Endophytes appear to specialize towards certain nitrogenous metabolites, because domains of asparaginase, oligopeptide transporters (OPT) and adenosine deaminase (A_deaminase) were significantly enriched and virtually absent in rhizoplane bacteria. Specific for rhizoplane bacteria were several domains related to regulatory proteins which were virtually absent in the endophyte and soil bacteria set: Ankyrin repeats (Ank_3) mediating protein–protein interactions in signal transduction, are far more common in eukaryotes than in bacteria, supporting the recent finding that plant-associated bacteria share proteins that resemble plant proteins25; VapB_antitoxin domains of VapB proteins that control plasmid stabilization, often to avoid loss of virulence genes, are supportive of the assumption of active genetic exchange on the root surface. Enriched over endophytes and almost absent in soil bacteria were ADP-ribosylglycohydrolases (ADP_ribosyl_GH) to remove covalent protein modifications of ADP-ribose moieties to alter protein activities; they were particularly abundant in rhizoplane verrucomicrobia; the FIST C domain (FIST_C) is a novel sensory domain present in signal transduction proteins putatively related to amino acid metabolism.
Our approach using well-defined model bacteria provided new insights in niche-specific signature proteins. In contrast, most domains identified as plant-specific in a previous study were only taxa-specific and not significantly enriched across phyla in stringent analyses25, which might be entailed by inclusion of a plethora of strains that are not sufficiently niche-specific.
Other traits for plant growth promotion and colonization in Verrucomicrobia
As these are the first Verrucomicrobia isolated from roots or rhizomes, additional features related to plant colonization and interaction were of particular interest. Evidence accumulates that Verrucomicrobia play critical roles in environmental carbon cycling and (poly)saccharide degradation, as they can have high coding densities for glycoside hydrolase genes50,51. According to our analysis, they add a large set of carbohydrate hydrolysing enzymes to the endomicrobiome. Carbohydrate- active enzymes annotated by the dbCAN2 annotation tool52 were abundant in the plant isolates, with endophytes EW11 and ER46 harbouring the highest gene density (7.5% or 7.0%, respectively) in comparison to root surface bacteria LR76 and LW23 (6% or 4.4%, respectively). For 163 or 77 of these enzymes of the endophytes, respectively, signal peptides were predicted, suggesting the bacteria are equipped with a large set of exoenzymes. In contrast, genomes of diazotrophic, proteobacterial model endophytes such as Azoarcus olearius BH7249, Klebsilla pneumoniae 34253, or Methylobacterium populi BJ00154, harboured a much smaller gene density (2,3%, 2.1%, or 2.3%, with 11–23 predicted signal peptides, respectively). The rice Verrucomicrobia appear to participate also in several steps of nitrogen cycling (Supplementary Note 2, Supplementary Table S7).
Combining culture dependent (Supplementary Tables S6,S8) and –independent data (Supplementary Tables S6,S7), we evaluated further possible plant-host related characteristics of our strains. Two-component chemosensory systems including putative methyl-accepting proteins (MCP) and chemotaxis proteins (Che) were encoded in all four genomes. These proteins have an important role for root colonization by movement toward root exudate components55. An alternative bacterial movement on surfaces through twitching motility56, and also adherence to the plant host57, can be mediated by type IV pili. Several components for the pilus apparatus were encoded in all four genomes, particularly PilT which is responsible for retraction of the pilus in twitching motility and essential for endophytic rice root colonization by Azoarcus olearius BH7258. In addition, our isolates carried genes for agglutination proteins like hemagglutinin or putative adhesins. Also Type III secretion systems which commonly secrete effector proteins into host cells were traced in the genomes of the good colonizers EW11 and LW23 (Supplementary Note 2).
Particularly significant is the detection of traits for plant growth promotion in our isolates (Supplementary Tables S6,S7). Bacterial synthesis of plant hormones such as the auxin indole-3-acetic acid (IAA) may increase root development. Hints for either weak or strong synthesis of indole-related compounds, such as IAA (indole acetic acid) and its precursors IAM (indole-acetamide) or IPA (indole-3-pyruvic acid)59, were detected for all strains in a colorimetric bioassay. Phosphate is often a limiting factor in soil60. Mineralization or solubilisation by bacteria can occur through the production and secretion of organic acids or phosphatases61,62. All strains showed activity of acid phosphatases for the solubilisation of organic phosphates63, and possessed also genes for citrate synthase; two strains were capable of solubilizing phosphate in a plate assay based on tricalcium phosphate (Supplementary Tables S6,S7), however this test is only indicative of phosphate solubilisation in soils and does not prove a direct contribution of phosphorous to the plant64. In order to obtain hints whether root-associated verrucomicrobia can indeed affect plant growth, short-term inoculation assays (7 d) were carried out for rice seedlings grown under gnotobiotic conditions. In comparison to the sterile, uninoculated control, an endophytic (ER46) and a surface strain (LR76) originating from O. longistaminata increased fresh weight of roots (Fig. 4), which was not significantly affected by strains LW23 or EW11 under these conditions.
Figure 4.
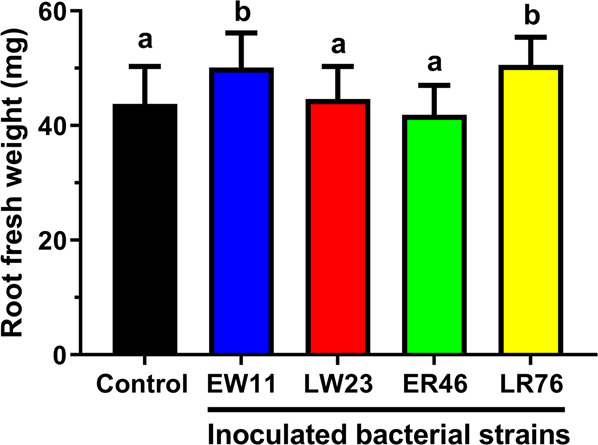
Root growth promotion by verrucomicrobial isolates. Sterile seedlings of O. sativa cv. Nipponbare were grown in hydroponic culture without (control) or with inoculation of single verrucomicrobial strains, and root fresh weight was evaluated 7 d post inoculation. Error bars above columns indicate standard deviations from three independent experiments (6–12 plants each). Columns headed by different letters are significantly different from each other at P < 0.05 according to analysis of variance (ANOVA) followed by uncorrected Fisher’s LSD.
Concluding Remarks
There is a lack of fundamental knowledge on principles underlying assembly of a complex root microbial community and its interplay with hosts for plant fitness. Defined collections of plant microbes will help to address these questions in reconstitution experiments under sterile conditions, however current collections like for Arabidopsis thaliana that cover roughly 60% of the root community24 or other collections25, miss Verrucomicrobia entirely. Endophytes, that deeply ingress into the tissue and are probably most tightly associated with plants, have previously never been cultivated among Verrucomicrobia. As Verrucomicrobia were recently estimated to account for up to 25% of rice root endophytes21, they are likely to play an important but hidden role in the root interactome. Thus, our novel root-associated isolates provide a valuable new resource to disclose functions of these slow-growing, but often highly abundant bacteria in complex root microbiota.
Despite their phylogenetic divergence from commonly known root bacteria, Verrucomicrobia share a combination of characteristics typical for other beneficial root microbes that were previously unknown to Verrucomicrobia15 (Fig. 5). Comparative analysis of signature proteins did not indicate a recent gene transfer to our plant colonizers, consistent with an ancient origin of bacterial plant colonization. This enabled us to define deep-rooting core features across phyla for association with plants, and thus gives fundamental insights into functional adaptations to the specific underground niches provided by the earth’s most important primary producers, plants.
Figure 5.

Overview of selected features of plant-colonizing Verrucomicrobia strains based on genomic data or experimental tests. Features common to all four isolates are shown in blue, colour codes for occurrence in Spartobacter rhizophilus LR76, Astrumicrobium roseum LW23, or endophytes Albicoccus flocculans EW11 or Opitutus terrae ER46 are given in the figure. In addition to carbohydrate metabolism, selected transporters and nitrogen cycling pathways, features likely related to plant-microbe interactions are shown.
Materials and Methods
Bacterial isolation and cultivation conditions
Rice grains (Oryza sativa ssp. japonica cv. Nipponbare and O. longistaminata) were dehusked, surface sterilized, washed and germinated as described previously65,66. After germination, the plants were grown in moistened rice paddy soil obtained from Vercelli (Italy) in a phytotron (30 °C, 80% humidity, 14 h/10 h day/night cycle at 16 000 lx). The plants were irrigated with distilled water regularly. Root systems from 2 – 10 cm soil depth were taken from 8 week-old plants, washed with tap water, and shaken in wash buffer (SM-medium46) without carbon and nitrogen source) together with sterile glass beads (Ø 1 mm, Roth, Karlsruhe, Germany) in Erlenmeyer flasks on a reciprocal shaker46, to obtain the rhizoplane bacteria in the supernatant.To obtain putative endophytic bacteria, washed roots were surface-sterilized by shaking 2 min in 5% sodium hypochlorite solution and five washing steps with sterile water67. To check for surface contamination, the samples were dipped on R2A medium68 agar plates and incubated as described below, and no colonies were visible after incubation. Surface-sterilized roots were homogenized with sterile quartz sand in a sterile mortar. Serial dilutions were plated on R2A agar supplemented with vitamin solution69, and incubated at 28 °C.
Amplification of 16S rDNA sequence and phylogenetic analysis
As template for the amplification of 16S rRNA gene (rDNA) sequences, 2 µl of bacterial cell lysate in a 50 µl reaction was used. To obtain the lysate, cells were incubated in 1 X Tris-EDTA buffer pH 8.0 containing 0.01% Tween20 [v/v] at 95 °C for 15 min. PCR amplification of almost complete 16S rDNA genes was performed using 2.5 U Taq Polymerase (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 50 µM of each dNTP and the primer pair Bac8uf (5′-AGAGTTTGATNHTGGYTCAG-3′) and Univ1492r (5′-GGNTCCTTGTTACGACTT-3′)70,71 at 95 °C for 5 min, 40 cycles of 95 °C for 1 min, 52 °C for 30 sec, 72 °C for 2 min and a final elongation of 10 min at 72 °C in a Biometra thermocycler. Sequencing of PCR amplicons was carried out at the LGC Genomics Sequencing Service (Berlin, Germany). Quality of the sequences obtained was checked manually using BioEdit 7.2.572. Sequences were aligned using a secondary- structure based Infernal aligner obtained through the Ribosomal Database project73. Alignments were reviewed in MEGA 774 and if necessary manually refined. Phylogenetic trees were based on 1228 positions and were reconstructed by the maximum likelihood method implemented in IQ-TREE75 under the best-fit model (GTR + F + R6) according to the Bayesian information criterion (BIC) model finder.
Whole-genome-based phylogenetic comparisons were done with a composition vector approach using CVTree376,77 with a K’mer size of 6 (CVTree3, http://cvtree.big.ac.cn/cvtree/cvtree/).
Tests of growth characteristics
All tests of growth characteristics were carried out at 28 °C under aerobic conditions, except for temperature tolerance or anaerobic growth, respectively. Tolerance to extreme pH conditions was tested in R2A medium adjusted to pH 4 or 5 with homopiperazine-1,4-bis(2-ethanesulfonic acid) (Homo-Pipes) as buffer system. For alkaline conditions at pH 8.5 or 9.5, 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid (CAPSO) buffer was used. Cultures were incubated at 28 °C until growth for positive controls on neutral R2A agar became visible.
Anaerobic growth was tested on agar plates incubated for 14 days at 28 °C in an anaerobic reaction chamber (Merck Millipore, Darmstadt, Germany) supplemented with GasPak indicator stripes (Becton, Dickinson and Company, New Jersey, USA) and Oxoid AneroGen Sachets (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Testing plant growth promotion characteristics and colonization competence
For the evaluation of inorganic phosphate solubilisation, Pikovskaya agar plates were used78. Bacteria were grown in liquid culture before transfer to the plates in droplets. Incubation was done at 28 °C for 7 days. The formation of clear halo around the bacterial droplets was evaluated, with Bacillus subtilis as positive control. Production of siderophores was tested according to79 on Chrome azurol S agar plates with modifications. Chrome azurol S agar plates without piperazine and deferrization were overlaid with a solid layer of R2A medium, before transferring droplets of bacterial culture. A formation of orange halos around the bacterial droplets was rated as positive for siderophore production, with Azospirillum brasilense Sp7 as positive control. The survey for indole-3-acetic acid (IAA) production and indole-related compounds was performed according to80 after growth in presence of L-tryptophan. Briefly, the cultures as well as the positive control (A. brasilense Sp7) were grown in liquid R2A Medium supplemented with 6 mM L-tryptophan. After centrifugation, the supernatant was incubated with salkowski reagent (0.01 M FeCl3 in 35% perchloric acid) in the dark at RT, and a colour change from yellow to red with an absorbance peak at 530 nm indicated a positive result.
For evaluation of colonization competence and plant growth promotion, O. sativa cv. Nipponbare was grown in a hydroponic test system. Dehusked rice grains were surface-sterilized, washed and germinated in magenta vessels (Sigma-Aldrich, Darmstadt, Germany) as described above. Instead of transfer to pots, the seedlings were grown in sterile glass tubes (100 ml) containing stainless-steel mesh as carrier, in 50 ml plant medium without nitrogen source (as described in65). Iron-citrate in the medium was replaced by iron-EDTA. Bacteria were grown in liquid R2A medium or R2A agar at 28 °C, washed with plant medium and inoculated to OD 0.067. After incubation in the phytotron for 7 days, roots were inspected for colonization, and plant medium was streaked on R2A agar to detect possible microbial contaminants. In three separate experiments on plant growth promotion, root fresh weight was determined after 7 days.
Microscopy
For evaluation of root colonization, roots were stained at room temperature with SYBR Green I nucleic acid stain (Sigma-Aldrich, Darmstadt, Germany) in a 1000x working solution. For detection of cellular compartments, 500x working solution was applied. Fluorescence of the root material was evaluated using an epifluorescence microscope (Zeiss Axioplan 2, Carl Zeiss AG, Oberkochen, Germany) with a Zeiss Axiocam 503 colour camera. For detection of green fluorescence at 522 nm, the Zeiss filter set 09 was used. To visualize the plant auto-fluorescence, the Zeiss filter set 43 He was used.
For imaging by confocal laser scanning microscopy (CLSM), a Zeiss 880 Microscope (Carl Zeiss, Jena, Germany) with the high resolution Airyscan detector was used, equipped with an argon/neon-laser with a wavelength λ = 488, 543 nm. A 20× dry objective lens (Zeiss NEONFLUAR 20×, 0.5 HD) and 40x oil objective lens (Zeiss NEONFLUAR 40×, 0.5 HD) were used. For the excitation and emission the following settings were used: 488 nm laser (100 mW) and beam splitters BP495-575 for the green dye; 543 nm laser (100 mW) and beam splitters BP570-650 for auto-fluorescence. The images were reconstructed using ZEN software (Black Edition, 2011, Carl Zeiss, Germany) based on the structured illumination algorithm. Further analysis was performed on reconstructed super-resolution images in ZEN software blue edition (2012, Carl Zeiss, Jena, Germany). 3D images of the infected roots were constructed from the Z-stack images. The z-stack ZEN file was first exported as multi TIFF files, and then the multiple TIFF files were converted to a single 3D image by ImageJ (ImageJ bundled with 64-bit Java 1.8.0_112).
Before transmission electron microscopy cells were grown under micro-aerobic conditions (for strains LR76, EW11 and ER46) or under aerobic conditions (strain LW23) in liquid R2A medium (supplemented with vitamins, and fructose and maltose at 0.5 g/l). Cells were fixed in 2% glutaraldehyde in cacodylate buffer (50 mM, pH 7.2) for 45 minutes on ice followed by washing in cacodylate buffer and HEPES buffer (50 mM, pH 7.2). The cells were then embedded in agarose blocks and dehydrated in increasing concentrations of ethanol at −20 °C before stepwise infiltrations in LR White resin (Sigma Aldrich, Missouri, USA)65. Agar blocks were transferred to gelatine-capsules before polymerization at 65 °C for 48 hours. Ultra-thin sections were prepared with an Ultramicrotome Ultracut E (Reichert-Jung, Leica, Wetzlar, Germany) and stained on grids with filtered uranyl acetate in distilled water (3%) followed by lead citrate in CO2-free distilled water (2.5%) for 1–2 minutes, respectively. Sections were viewed in a Zeiss 900 A Transmission Electron Microscope (Carl Zeiss AG, Oberkochen, Germany) operated at 80 kV.
Draft Genome sequencing and bioinformatic analyses
For genomic DNA extraction bacterial cell pellets were washed in Tris-EDTA Saline (TES, pH 8) buffer and DNA was isolated by using a phenol/chloroform extraction protocol81. DNA was dissolved in TE buffer and stored at −80 °C. DNA concentration was determined with a NanoDrop 2000 spectrophotometer. Library preparation was performed as described by the manufacturer using the TruSeq PCR-Free Library Preparation Kit (Illumina, San Diego, California, USA) after mechanical fragmentation of the genomic DNA into 550 bp fragments by application of the Covaris M220 focused-ultrasonicator (ThermoFisher Scientific, Waltham, Massachusetts, USA). The bacterial genomes were sequenced (2 × 300 bp) using an Illumina MiSeq platform (Illumina, San Diego, California, USA). We assembled the sequencing reads for each of the four genomes using the CLC Genomics Workbench 9.0 De Novo Assembly 1.3 with default settings (Qiagen, Venlo, Netherlands). After assembly, contig sequences with a minimum size of 1 kb were included for further analyses.
Draft genomes of the described strains were annotated by the Rapid Annotation tool using the Subsystems Approach (RAST) Server82,83 and compared to results of the annotation derived by the NCBI prokaryotic genome annotation pipeline (PGAP)84. Functional annotation and automatic pathway reconstruction were generated by the Kyoto Encyclopaedia of Genes and Genomes (KEGG) Automatic Annotation Server (KAAS) by GHOSTX comparisons85,86. PFAM domain searches were carried out with CLC Genomics Workbench version 11.0 using the Pfam-A family database (version 31.0) and profile’s gathering cutoff. The abundance of Pfam domains in bacteria from the three compartments was compared by multiple T-tests not assuming a consistent standard deviation using GraphPad Prism v7.05 (Graphpad software). This software was also used for one-way ANOVA (analysis of variance) followed by Holm-Sidak’s multiple comparison test. P > 0.05 was considered not significant. Venn diagrams were drawn with unique Pfam domain accessions using a Venn diagram drawing tool (http://bioinformatics.psb.ugent.be/webtools/Venn/). To visualize the relationship between protein domain group means of the three compartments, a between-group principal component analysis (bgPCA) was carried out. Protein domain differences between compartments was assessed by non-parametric multivariate analysis of variance (PERMANOVA) based on 9999 permutations using a Bray-Curtis similarity index. Multivariate analysis was performed with Past 387.
Carbohydate-active enzymes were identified in the dbCAN2 annotation tool52 using HMMER search at E-Values < 1e-15, coverage >0.35.
The annotated complete genome sequences were deposited in GenBank: strain LW23 (Accession No. QAZA00000000), strain LR76 (Accession No. QAYZ00000000), strain EW11 (Accession No. QAYY00000000) and strain ER46 (Accession No. QAYX00000000).
Supplementary Information
Acknowledgements
We gratefully acknowledge Thea Fründt and Ute Helmboldt-Caesar for their help and advice during TEM sample preparation. Andrea Schaffrath and Rolf Nimzyk are acknowledged for technical and bioinformatic assistance during genome sequencing. Financial support was provided by a grant of The Deutsche Forschungsgemeinschaft DFG (Re756/16-2 and Re756/18-1) and of the Federal Ministry of Education and Research (BMBF, 0315967) to B.R.-H. and T.H.
Author contributions
W.B., T.H. and B.R.-H. designed the experiments. W.B. isolated and characterized isolates, carried out genome analysis and most colonization experiments, some done by B.R.-H. X.J. carried out CLSM studies. J.M. carried out tests on PGPR characteristics. T.H. designed and performed protein domain analysis. B.R.-H., W.B. and T.H. wrote the manuscript with contributions of X.J.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-65277-6.
References
- 1.Reinhold-Hurek B, Bünger W, Burbano CS, Sabale M, Hurek T. Roots shaping their microbiome: Global hot spots for microbial activity. Annu. Rev. Phytopathol. 2015;53:403–424. doi: 10.1146/annurev-phyto-082712-102342. [DOI] [PubMed] [Google Scholar]
- 2.Krings M, et al. Endophytic cyanobacteria in a 400-million-yr-old land plant: A scenario for the origin of a symbiosis? Rev. Paleaobot. Palynol. 2009;153:62–69. [Google Scholar]
- 3.Kenrick P, Strullu-Derrien C. The origin and early evolution of roots. Plant Physiol. Bioch. 2014;166:570–580. doi: 10.1104/pp.114.244517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroll S, Agler MT, Kemen E. Genomic dissection of host-microbe and microbe-microbe interactions for advanced plant breeding. Curr. Opin. Plant Biol. 2017;36:71–78. doi: 10.1016/j.pbi.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Foll M, Gaggiotti OE, Daub JT, Vatsiou A, Excoffier L. Widespread signals of convergent adaptation to high altitude in Asia and America. Am. J. Hum. Gen. 2014;95:394–407. doi: 10.1016/j.ajhg.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li SJ, et al. Microbial communities evolve faster in extreme environments. Sci. Rep. 2014;4:6205. doi: 10.1038/srep06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vorholt JA, Vogel C, Carlström CI, Müller DB. Establishing causality: Opportunities of synthetic communities for plant microbiome research. Cell Host Microbe. 2017;22:142–155. doi: 10.1016/j.chom.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Hugenholtz P, Kyrpides NC. A changing of the guard. Environ. Microbiol. Rep. 2009;11:551–553. doi: 10.1111/j.1462-2920.2009.01888.x. [DOI] [PubMed] [Google Scholar]
- 9.Rinke C, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 10.Schlesner H, Jenkins C, Staley JT. The phylum Verrucomicrobia: A phylogenetically heterogeneous bacterial group. Prokaryotes. 2006;7:881–896. [Google Scholar]
- 11.Kielak A, et al. Phylogenetic and metagenomic analysis of Verrucomicrobia in former agricultural grassland soil. FEMS Microbiol Ecol. 2010;71:23–33. doi: 10.1111/j.1574-6941.2009.00785.x. [DOI] [PubMed] [Google Scholar]
- 12.Fierer N, et al. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science. 2013;342:621–624. doi: 10.1126/science.1243768. [DOI] [PubMed] [Google Scholar]
- 13.Bergmann GT, et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 2011;43:1450–1455. doi: 10.1016/j.soilbio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebeis SL, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 15.Utturkar SM, et al. Enrichment of root endophytic bacteria from Populus deltoides and single-cell-genomics analysis. Appl Environ Microbiol. 2016;82:5698–5708. doi: 10.1128/AEM.01285-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peiffer JA, et al. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA. 2013;110:6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguirre-von-Wobeser E, Rocha-Estrada J, Shapiro LR, de la Torre M. Enrichment of Verrucomicrobia, Actinobacteria and Burkholderiales drives selection of bacterial community from soil by maize roots in a traditional milpa agroecosystem. PLoS One. 2018;13:e0208852. doi: 10.1371/journal.pone.0208852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sessitsch A, et al. Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol. Plant-Microbe Interact. 2012;25:28–36. doi: 10.1094/MPMI-08-11-0204. [DOI] [PubMed] [Google Scholar]
- 19.Hardoim PR, et al. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015;79:293–320. doi: 10.1128/MMBR.00050-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards J, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci USA. 2015;112:E911–920. doi: 10.1073/pnas.1414592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng J, et al. Assembly of root-associated microbiomes of typical rice cultivars in response to lindane pollution. Environ. Int. 2019;131:104975. doi: 10.1016/j.envint.2019.104975. [DOI] [PubMed] [Google Scholar]
- 22.da Rocha UN, Plugge CM, George I, van Elsas JD, van Overbeek LS. The rhizosphere selects for particular groups of Acidobacteria and Verrucomicrobia. PLoS One. 2013;8:16–20. doi: 10.1371/journal.pone.0082443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Rocha UN, van Elsas JD, van Overbeek LS. Verrucomicrobia subdivision 1 strains display a difference in the colonization of the leek (Allium porrum) rhizosphere. FEMS Microbiol Ecol. 2011;78:297–305. doi: 10.1111/j.1574-6941.2011.01156.x. [DOI] [PubMed] [Google Scholar]
- 24.Bai Y, et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature. 2015;528:364–369. doi: 10.1038/nature16192. [DOI] [PubMed] [Google Scholar]
- 25.Levy A, et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2018;50:138–150. doi: 10.1038/s41588-017-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rattray RM, Perumbakkam S, Smith F, Craig AM. Microbiomic comparison of the intestine of the earthworm Eisenia fetida Fed Ergovaline. Curr. Microbiol. 2010;60:229–235. doi: 10.1007/s00284-009-9530-8. [DOI] [PubMed] [Google Scholar]
- 28.Field EK, et al. Application of molecular Ttechniques to elucidate the influence of cellulosic waste on the bacterial community structure at a simulated low-level-radioactive-waste site. Appl Environ Microbiol. 2010;76:3106–3115. doi: 10.1128/AEM.01688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta RS, Bhandari V, Naushad HS. Molecular signatures for the PVC clade (Planctomycetes, Verrucomicrobia, Chlamydiae, and Lentisphaerae) of bacteria provide insights into their evolutionary relationships. Front Microbiol. 2012;3:1–19. doi: 10.3389/fmicb.2012.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho JC, Vergin KL, Morris RM, Giovannoni SJ. Lentisphaera araneosa gen. nov., sp nov, a transparent exopolymer producing marine bacterium, and the description of a novel bacterial phylum, Lentisphaerae. Environ Microbiol. 2004;6:611–621. doi: 10.1111/j.1462-2920.2004.00614.x. [DOI] [PubMed] [Google Scholar]
- 31.Spring S, et al. Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J. 2016;10:2801–2816. doi: 10.1038/ismej.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.da Rocha UN, Andreote FD, de Azevedo JL, van Elsas JD, van Overbeek LS. Cultivation of hitherto-uncultured bacteria belonging to the Verrucomicrobia subdivision 1 from the potato (Solanum tuberosum L.) rhizosphere. J. Soil. Sediment. 2010;10:326–339. [Google Scholar]
- 33.Chow ML, Radomski CC, McDermott JM, Davies J, Axelrood PE. Molecular characterization of bacterial diversity in Lodgepole pine (Pinus contorta) rhizosphere soils from British Columbia forest soils differing in disturbance and geographic source. FEMS Microbiol Ecol. 2002;42:347–357. doi: 10.1111/j.1574-6941.2002.tb01024.x. [DOI] [PubMed] [Google Scholar]
- 34.Janssen PH, Schuhmann A, Mörschel E, Rainey FA. Novel anaerobic ultramicrobacteria belonging to the Verrucomicrobiales lineage of bacterial descent isolated by dilution culture from anoxic rice paddy soil. Appl Environ Microbiol. 1997;63:1382–1388. doi: 10.1128/aem.63.4.1382-1388.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu YL, Kuang XZ, Shi XS, Yuan XZ, Guo RB. Terrimicrobium sacchariphilum gen. nov., sp nov., an anaerobic bacterium of the class ‘Spartobacteria’ in the phylum Verrucomicrobia, isolated from a rice paddy field. Int. J. Syst. Evol. Microbiol. 2014;64:1718–1723. doi: 10.1099/ijs.0.060244-0. [DOI] [PubMed] [Google Scholar]
- 36.Stackebrandt E, Ebers J. Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today. 2006;33:152–155. [Google Scholar]
- 37.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richter M, Rosselló-Móra R, Glöckner FO, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–931. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 40.Yarza P, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014;12:635–645. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 41.Lee KC, et al. Phylum Verrucomicrobia representatives share a compartmentalized cell plan with members of bacterial phylum Planctomycetes. BMC Microbiol. 2009;9:5. doi: 10.1186/1471-2180-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reinhold-Hurek B, Hurek T. Life in grasses: diazotrophic endophytes. Trends Microbiol. 1998;6:139–144. doi: 10.1016/s0966-842x(98)01229-3. [DOI] [PubMed] [Google Scholar]
- 43.Reinhold-Hurek B, Hurek T. Living inside plants: bacterial endophytes. Curr. Opin. Plant Biol. 2011;14:435–443. doi: 10.1016/j.pbi.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Bulgarelli D, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- 45.McClung CR, Van Berkum P, Davis RE, Sloger C. Enumeration and localization of N2-fixing bacteria associated with roots of Spartina alterniflora Loisel. Appl. Environ. Microbiol. 1983;45:1914–1920. doi: 10.1128/aem.45.6.1914-1920.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reinhold B, Hurek T, Niemann E-G, Fendrik I. Close association of Azospirillum and diazotrophic rods with different root zones of Kallar grass. Appl. Environ. Microbiol. 1986;52:520–526. doi: 10.1128/aem.52.3.520-526.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buschart A, et al. Flagella mediate endophytic competence rather than act as MAMPS in rice - Azoarcus sp. strain BH72 interactions. Mol. Plant-Microbe Interact. 2012;25:191–199. doi: 10.1094/MPMI-05-11-0138. [DOI] [PubMed] [Google Scholar]
- 48.Taga ME, Walker GC. Sinorhizobium meliloti requires a cobalamin-dependent ribonucleotide reductase for symbiosis with its plant host. Mol. Plant-Microbe Interact. 2010;23:1643–1654. doi: 10.1094/MPMI-07-10-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krause A, et al. Complete genome of the mutualistic, N2-fixing grass endophyte Azoarcus sp. strain BH72. Nat. Biotechnol. 2006;24:1385–1391. doi: 10.1038/nbt1243. [DOI] [PubMed] [Google Scholar]
- 50.He, S. et al. Ecophysiology of freshwater Verrucomicrobia inferred from metagenome-assembled genomes. mSphere2 (2017). [DOI] [PMC free article] [PubMed]
- 51.Pold, G. et al. Genome sequence of Verrucomicrobium sp. strain GAS474, a novel bacterium isolated from soil. Genome Announc. 6 (2018). [DOI] [PMC free article] [PubMed]
- 52.Zhang H, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fouts DE, et al. Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Biol. 2008;4:e1000141. doi: 10.1371/journal.pgen.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Aken B, Peres CM, Doty SL, Yoon JM, Schnoor JL. Methylobacterium populi sp. nov., a novel aerobic, pink-pigmented, facultatively methylotrophic, methane-utilizing bacterium isolated from poplar trees (Populus deltoides x nigra DN34) Int. J. Syst. Evol. Microbiol. 2004;54:1191–1196. doi: 10.1099/ijs.0.02796-0. [DOI] [PubMed] [Google Scholar]
- 55.De Weert S, et al. Flagella-driven chemotaxis towards exudate components is an important trait for tomato root colonization by Pseudomonas fluorescens. Mol. Plant-Microbe Interact. 2002;15:1173–1180. doi: 10.1094/MPMI.2002.15.11.1173. [DOI] [PubMed] [Google Scholar]
- 56.Strom MS, Lory S. Structure-function and biogenesis of the type IV pili. Annu. Rev. Microbiol. 1993;47:565–596. doi: 10.1146/annurev.mi.47.100193.003025. [DOI] [PubMed] [Google Scholar]
- 57.Dörr J, Hurek T, Reinhold-Hurek B. Type IV pili are involved in plant-microbe and fungus-microbe interactions. Mol. Microbiol. 1998;30:7–17. doi: 10.1046/j.1365-2958.1998.01010.x. [DOI] [PubMed] [Google Scholar]
- 58.Böhm M, Hurek T, Reinhold-Hurek B. Twitching motility is essential for endophytic rice colonization by the N2- fixing endophyte Azoarcus sp. strain BH72. Mol. Plant-Microbe Interact. 2007;20:526–533. doi: 10.1094/MPMI-20-5-0526. [DOI] [PubMed] [Google Scholar]
- 59.Gilbert S, et al. Bacterial production of indole related compounds reveals their role in association between duckweeds and endophytes. Front. Chem. 2018;6:265. doi: 10.3389/fchem.2018.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vassilev N, Vassileva M. Biotechnological solubilization of rock phosphate on media containing agro-industrial wastes. Appl. Microbiol. Biotechnol. 2003;61:435–440. doi: 10.1007/s00253-003-1318-3. [DOI] [PubMed] [Google Scholar]
- 61.Reyes I, Bernier L, Simard RR, Antoun H. Effect of nitrogen source on the solubilization of different inorganic phosphates by an isolate of Penicillium rugulosum and two UV-induced mutants. FEMS Microbiol Ecol. 1999;28:281–290. [Google Scholar]
- 62.Miller SH, et al. Biochemical and genomic comparison of inorganic phosphate solubilization in Pseudomonas species. Environ. Microbiol. Rep. 2010;2:403–411. doi: 10.1111/j.1758-2229.2009.00105.x. [DOI] [PubMed] [Google Scholar]
- 63.Walpola BC, Yoon MH. Phosphate solubilizing bacteria: Assessment of their effect on growth promotion and phosphorous uptake of mung bean (Vigna radiata [L.] R. Wilczek). Chil. J. Agr. Res. 2013;73:275–281. [Google Scholar]
- 64.Bashan Y, Kamnev AA, de-Bashan LE. A proposal for isolating and testing phosphate-solubilizing bacteria that enhance plant growth. Biol. Fertil. Soils. 2013;49:1–2. [Google Scholar]
- 65.Hurek T, Reinhold-Hurek B, Van Montagu M, Kellenberger E. Root colonization and systemic spreading of Azoarcus sp. strain BH72 in grasses. J. Bacteriol. 1994;176:1913–1923. doi: 10.1128/jb.176.7.1913-1923.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X, et al. Rice responds to endophytic colonization which is independent of the common symbiotic signaling pathway. New Phytol. 2015;208:531–543. doi: 10.1111/nph.13458. [DOI] [PubMed] [Google Scholar]
- 67.Hardoim PR, et al. Rice root-associated bacteria: insights into community structures across 10 cultivars. FEMS Microbiol Ecol. 2011;77:154–164. doi: 10.1111/j.1574-6941.2011.01092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reasoner DJ, Geldreich EE. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49:1–7. doi: 10.1128/aem.49.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hurek T, Van Montagu M, Kellenberger E, Reinhold-Hurek B. Induction of complex intracytoplasmic membranes related to nitrogen fixation in Azoarcus sp. BH72. Mol. Microbiol. 1995;18:225–236. doi: 10.1111/j.1365-2958.1995.mmi_18020225.x. [DOI] [PubMed] [Google Scholar]
- 70.Lane, D. J. In Nucleic Acids Tech. Bact. Syst. Vol. 115–175 (eds. Stackebrandt, E. & Goodfellow, M.) (Wiley, 1991).
- 71.Edwards U, Rogall T, Blocker H, Emde M, Böttger EC. Isolation and direct complete nucleotide determination of entire genes - characterization of a gene coding for 16S-ribosomal RNA. Nucleic Acids Res. 1989;17:7843–7853. doi: 10.1093/nar/17.19.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hall T. BioEdit: An important software for molecular biology. GERF: Bul. Biosc. 2011;2:60–61. [Google Scholar]
- 73.Cole JR, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zuo GH, Hao BL. CVTree3 Web Server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy. Genom. Proteom. Bioinf. 2015;13:321–331. doi: 10.1016/j.gpb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuo G, Li Q, Hao B. On K-peptide length in composition vector phylogeny of prokaryotes. Comput. Biol. Chem. 2014;53:166–173. doi: 10.1016/j.compbiolchem.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 78.Gupta RC, Singal R, Shankar A, Kuhad RC, Saxena RK. A modified plate assay for screening phosphate solubilizing microorganisms. J. Gen. Appl. Microbiol. 1994;40:255–260. [Google Scholar]
- 79.Schwyn B, Neilands JB. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987;160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
- 80.Gordon SA, Weber RP. Colorimetric estimation of indolacetic acid. Plant Physiol. 1951;26:192–195. doi: 10.1104/pp.26.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hurek T, Burggraf S, Woese CR, Reinhold-Hurek B. 16S rRNA-targeted polymerase chain reaction and oligonucleotide hybridization to screen for Azoarcus spp., grass-associated diazotrophs. Appl. Environ. Microbiol. 1993;59:3816–3824. doi: 10.1128/aem.59.11.3816-3824.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Overbeek R, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aziz RK, et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics. 2008;9:1–15. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tatusova T, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suzuki, S., Kakuta, M., Ishida, T. & Akiyama, Y. GHOSTX: An improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS One9 (2014). [DOI] [PMC free article] [PubMed]
- 87.Hammer O, Harper DAT, Ryan PD. Past: Paleontological statistics software package for education and data analysis. Palaeont. Electr. 2001;4:1–9. [Google Scholar]
- 88.Tegtmeier D, Belitz A, Radek R, Heimerl T, Brune A. Ereboglobus luteus gen. nov. sp. nov. from cockroach guts, and new insight into the oxygen relationship of the genera Optitutus and Didymococcus (Verrucomicrobia: Optitutaceae) Syst. Appl. Microbiol. 2018;41:101–112. doi: 10.1016/j.syapm.2017.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.