

Abstract
Resistance to platinum-based chemotherapy becomes a major obstacle in non-small-cell lung cancer (NSCLC) treatment. Overexpression of the excision repair cross-complementing 1 (ERCC1) gene is reported to negatively influence the effectiveness of cisplatin-based therapy for NSCLC cells. In this study, we confirm that high ERCC1 expression correlates with cisplatin resistance in NSCLC cells. Importantly, histone deacetylase inhibitors (HDACis) re-sensitize ERCC1-high NSCLC cells to cisplatin both in vitro and in vivo. Mechanistically, the HDACi induces the expression of miR-149 by acetylation and activation of E2F1, which directly targets ERCC1 and inhibits ERCC1 expression. Inhibition of miR-149 reverses the promotion effect of HDACis on cisplatin-induced DNA damage and cell apoptosis in ERCC1-high NSCLC cells. In conclusion, this study reveals a novel mechanism by which HDACis re-sensitizes ERCC1-high NSCLC cells to cisplatin via regulation of the E2F1/miR-149/ERCC1 axis, and we propose that combination of HDACis and cisplatin might hold promise to be a more effective therapeutic paradigm for ERCC1-high NSCLCs.
Keywords: NSCLC, HDACi, cisplatin resistance, ERCC1, miR-149
Graphical Abstract
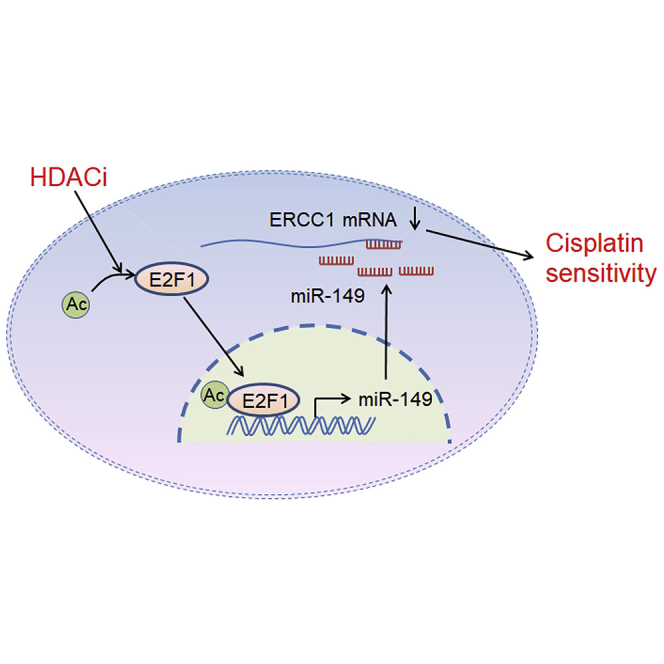
Overexpression of ERCC1 is reported to negatively influence the effectiveness of cisplatin-based therapy for NSCLC cells. Liu and colleagues demonstrate that HDACis could re-sensitize ERCC1-high NSCLC cells to cisplatin via regulation of the E2F1/miR-149/ERCC1 axis, and they propose that the combination of HDACis and cisplatin might hold promise to be a more effective therapeutic strategy for ERCC1-high NSCLCs.
Introduction
Lung cancer is a leading cause of cancer-related deaths worldwide, and non-small-cell lung cancer (NSCLC) accounts for approximately 80% of all cases of lung cancer.1 Cisplatin-based therapy is one of the most effective chemotherapeutic treatments for NSCLC.2 However, the unavoidable development of drug resistance significantly hinders its efficacy in therapy.3 The mechanism of action of cisplatin involves induction of DNA damage that leads to cell cycle arrest and apoptosis. Accordingly, increased DNA repair has been proposed to represent a major mechanism underlying cisplatin resistance.4
Excision repair cross-complementation group 1 (ERCC1), an important mediator of nucleotide excision repair (NER), plays a key role in excision of the damaged DNA.5 Increased ERCC1 expression is positively correlated with DNA repair capacity, which may serve as a target for therapy to increase sensitivity to cisplatin.6,7 Indeed, low levels of ERCC1 expression predict enhanced overall survival in NSCLC patients treated with cisplatin, and inhibition of ERCC1 expression could potentially increase the sensitivity of cisplatin-based therapy for NSCLC.
Histone deacetylases (HDACs), the key component of the epigenetic machinery regulating gene expression, are frequently overexpressed in tumors and have been shown to play vital roles in tumor proliferation, invasion, metastasis, and angiogenesis.8 HDAC inhibitors (HDACis) decrease HDAC activity and exert suppressive effects against various tumor cells via inhibiting cell proliferation and inducing cell cycle arrest and apoptosis.9,10 Therefore, HDACis are considered as a promising novel therapeutic approach in the light of their potent tumor-selective effects, some of which are currently the focus of clinical trials, such as vorinostat (suberoylanilide hydroxamic acid [SAHA]) and trichostatin A (TSA).11,12 Recently, HDACis have been continuously explored for use in combination with other antitumor agents for optimized efficacy and minimized toxicity.13 Combining HDACis with primary chemotherapeutic agents that induce DNA damage or apoptosis has shown promising results in preclinical research studies.14, 15, 16
In this study, we investigated the effect of combining HDACis with cisplatin on ERCC1-high NSCLC cells. Our results demonstrated that HDACis can sensitize ERCC1-high NSCLC cells to cisplatin both in vitro and in vivo. Mechanistically, the HDACi induces the expression of miR-149, which directly targets ERCC1 and inhibits ERCC1 expression. Furthermore, our data showed that inhibition of miR-149 reverses the promotion effect of HDACis on cisplatin-induced cell apoptosis in ERCC1-high NSCLC cells. This study reveals a novel mechanism by which the HDACi re-sensitizes ERCC1-high NSCLC cells to cisplatin via upregulation of miR-149 expression, and we propose that combination of HDACis and cisplatin might hold promise to be a more effective therapeutic paradigm for the treatment of ERCC1-high NSCLC cells.
Results
High ERCC1 Expression Correlates with Cisplatin Resistance in NSCLC Cells
ERCC1 has been shown to be crucial in predicting cisplatin resistance and can be used for tailoring cisplatin-based chemotherapy.17 Using a dataset of syngeneic NSCLC cells, we found that NSCLC cells with higher ERCC1 expression exhibited more resistance to cisplatin as determined by their half-maximal inhibitory concentration (IC50) (Figures 1A and 1B). We further determined ERCC1 expression in 21 cisplatin-sensitive NSCLC tissues and 18 cisplatin-resistant NSCLC tissues by immunohistochemistry analysis and found that ERCC1 was significantly more overexpressed in cisplatin-resistant NSCLC tissues than in cisplatin-sensitive NSCLC tissues (Figures 1C and 1D). Furthermore, we focused on whether the ERCC1 expression level is correlated with chemotherapy efficacy; accordingly, the association between ERCC1 expression and poor prognosis of NSCLC patients receiving cisplatin-based chemotherapy was analyzed using the online Kaplan-Meier survival analysis of expression data (probe 203720_s_at, probe 228131_at) (http://www.kmplot.com/lung). The results showed that overall survival was shorter in patients with high expression of ERCC1 than in those with low expression in the chemotherapy group (probe 203720_s_at, p = 0.018; probe 228131_at, p = 0.045; log rank test) (Figure 1E). These results confirmed that upregulation of ERCC1 is involved in cisplatin resistance of NSCLC cells.
Figure 1.

High ERCC1 Expression Correlates with Cisplatin Resistance in NSCLC Cells
(A) The expression of ERCC1 in NSCLC cells was analyzed by western blotting. β-Actin was used as an inner control. (B) IC50 values of cisplatin for NSCLC cells. (C) Representative immunohistochemical staining examples of ERCC1 protein expression in cisplatin-resistant NSCLC tissues and cisplatin-sensitive NSCLC tissues (scale bar, 50 μm). The lung cancer tissue sections were quantitatively scored according to the percentage of positive cells and staining intensity as described in Materials and Methods. The percentage and intensity scores were multiplied to obtain a total score (range, 0–12), and the tumors were finally determined as follows: negative (−), score 0; lower expression (+), score ≤4; moderate expression (++), score 5–8; and high expression (+++), score ≥9. (D) ERCC1 expression scores in cisplatin-resistant NSCLC tissues and cisplatin-sensitive NSCLC tissues. (E) Kaplan-Meier overall survival (OS) curves (http://kmplot.com/analysis/) of lung cancer patients administered chemotherapy relative to different expression levels of ERCC1. ∗p < 0.05.
We then evaluated the role of ERCC1 in the maintenance of cisplatin resistance in cisplatin-resistant A549/DDP and H1299 cells. An MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay showed that knockdown of ERCC1 significantly increased cisplatin sensitivity of A549/DDP cells and H1299 cells in comparison with control short hairpin RNA (shRNA) (Figures 2A and 2B). As a complement to RNAi experiments, we assessed the impact of ERCC1 overexpression on cisplatin sensitivity of A549 and H460 cells. We found that overexpression of ERCC1 significantly increased cisplatin resistance of A549 and H460 cells (Figures 2C and 2D). Moreover, we found that overexpression of ERCC1 decreased γ-H2AX foci in cisplatin-treated A549 cells (Figure 2E), whereas knockdown of ERCC1 increased γ-H2AX foci in cisplatin-treated A549/DDP cells (Figure 2F). We further investigated the effect of ERCC1 knockdown on cisplatin resistance in a xenograft tumor model. We found that knockdown of ERCC1 moderately inhibited tumor growth. Notably, knockdown of ERCC1 significantly increased cisplatin sensitivity (Figure 2G), in which the tumor volumes in A549/DDP/ERCC1 shRNA-bearing mice were significantly lower than those in A549/DDP/control shRNA-bearing mice (Figure 2H).
Figure 2.

ERCC1 Enhances Cisplatin Resistance in NSCLC Cells
(A) H1299 and A549/DDP cells were transfected with control shRNA (shControl) or ERCC1 shRNA (shERCC1), and the expression of ERCC1 was analyzed by western blotting. (B) H1299 and A549/DDP cells were transfected with ERCC1 shRNA and then treated with cisplatin at the indicated concentration for 48 h; cell viability was measured by an MTS assay. (C) A549 and H460 cells were transfected with ERCC1-expressing vector (LV-ERCC1), and the expression of ERCC1 was analyzed by western blotting. (D) A549 and H460 cells were transfected with LV-ERCC1and then treated with cisplatin at the indicated concentration for 48 h; cell viability was measured by an MTS assay. (E) A549 cells were transfected with LV-ERCC1 and then treated with 5 μM cisplatin for 48 h; cells were fixed and labeled with anti-γ-H2AX antibodies. The γ-H2AX foci were analyzed by immunofluorescence microscopy. (F) A549/DDP cells were transfected with ERCC1 shRNA and then treated with 20 μM cisplatin for 48 h; cells were fixed and labeled with anti-γ-H2AX antibodies. The γ-H2AX foci were analyzed by immunofluorescence microscopy. (G and H) A549/DDP cells transfected with control shRNA or ERCC1 shRNA were injected to the right shoulder of nude mice. Tumor-bearing mice were treated with either PBS or cisplatin (3 mg/kg body weight per day) for 4 weeks. (G) At the end of treatment, tumors were excised. (H) Tumor sizes were measured at every 3 days. ∗p < 0.05, ∗∗p < 0.01.
HDACis Sensitize ERCC1-High NSCLC Cells to Cisplatin In Vitro and In Vivo
HDACis are approved for clinical use in many cancers, including SAHA and TSA.18 Previous studies have demonstrated that HDACis modulate the expression and function of DNA repair proteins, thereby enhancing the sensitivity of cancer cells to DNA-damaging agents.19,20 To determine whether the HDACi SAHA would synergize with cisplatin in ERCC1-high NSCLC cells, we exposed A549/DDP and H1299 cells to either 10 μM cisplatin or 0.5, 1, and 2 μM SAHA and observed minimal cytotoxicity (Figure 3A). However, the combination of 10 μM cisplatin with increasing concentrations of SAHA up to 2 μM synergistically inhibited cell viability of A549/DDP and H1299 cells (Figure 3A). Similar results were observed in combination of cisplatin and another HDACi, TSA (Figure S1). Moreover, the combination of cisplatin and SAHA also decreased cell viability of A549 cells as well as ERCC1-overexpressed A549 cells compared to cisplatin alone (Figure 3B). We further chose the highest concentration of SAHA (2 μM) that was synergistic with cisplatin in a cell apoptosis assay. Treatment with SAHA significantly increased cisplatin-induced cell apoptosis in A549/DDP and H1299 cells (Figure 3C). Furthermore, we observed that HDACis significantly increased cisplatin-induced DNA damage and γ-H2AX foci formation in ERCC1-high NSCLC cells (Figure 3D).
Figure 3.

The HDACi SAHA Sensitizes ERCC1-High NSCLC Cells to Cisplatin In Vitro
(A) H1299 and A549/DDP cells were co-treated with cisplatin and SAHA at the indicated concentration for 48 h, and cell viability was measured by an MTS assay. (B) A549 cells transfected with LV-ERCC1 were co-treated with cisplatin and SAHA at the indicated concentration for 48 h, and cell viability was measured by an MTS assay. (C) H1299 and A549/DDP cells were co-treated with 10 μM cisplatin and 2 μM SAHA for 48 h, cells were stained with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide, and cell apoptosis was analyzed by flow cytometry. (D) H1299 and A549/DDP cells were co-treated with 10 μM cisplatin and 2 μM SAHA for 48 h, and cells were fixed and labeled with anti-γ-H2AX antibodies. The γ-H2AX foci were analyzed by immunofluorescence microscopy. ∗p < 0.05, ∗∗p < 0.01.
We next evaluated the effect of the combination of cisplatin and HDACis on tumor growth in mouse xenograft models. H1299 cells were subcutaneously injected into nude mice to establish tumor xenografts. When the tumor sizes reached ∼60 mm3, the mice were randomly grouped and received i.p. (intraperitoneal) injection of PBS (0.2 mL), cisplatin (5 mg/kg), SAHA (60 mg/kg), or a combination of cisplatin and SAHA. Tumor growth curves showed that treatment with cisplatin or SAHA alone led to a slight decrease in tumor volume compared to vehicle control (Figure 4A). Importantly, SAHA combined with cisplatin led to a sustained and significant inhibition on tumor growth (Figure 4A), as well as a significant reduction in tumor sizes (Figure 4B) and tumor weights (Figure 4C). Moreover, the combination of cisplatin and SAHA also significantly inhibited the growth of A549/DDP tumor xenografts (Figures 4D–4F). These differences were unlikely to be a result of generalized toxicity of drug treatment, since body weight loss in mice with combination therapy was not significantly greater than that in mice with monotherapy. Furthermore, the combination treatment reduced the expression of proliferation marker Ki-67 and increased the expression of apoptosis marker cleaved caspase-3 in H1299-derived tumors (Figure 4G). In addition, SAHA treatment significantly increased cisplatin sensitivity in nude mice bearing A549/ERCC1 cells (Figure S2). Taken together, these results suggested that the combination of SAHA and cisplatin has therapeutic potential, especially in ERCC1-high NSCLC cells.
Figure 4.

The HDACi SAHA Sensitizes ERCC1-High NSCLC Cells to Cisplatin In Vivo
H1299 (2 × 106) and A549/DDP (2 × 106) cells were inoculated subcutaneously into severe combined immunodeficiency (SCID) mice, and palpable tumors were allowed to develop for 7 days. Mice were randomly allocated into four groups: vehicle control (0.01% DMSO in PBS, n = 4), cisplatin (10 mg/kg/d, n = 4), SAHA (50 mg/kg/d, n = 4), and the cisplatin/SAHA combination (n = 4) via i.p. injection for 3 weeks. (A and D) The tumor size was measured at indicated time intervals and calculated. The tumor volume (V) was calculated using the formula: V = (½ × larger diameter) × (smaller diameter)2, and growth curves were plotted using average tumor volume within each experimental group at the set time points. (B and E) At the end of treatment, tumors were excised and subjected to further analyses. (C and F) Tumor weights were measured. (A–C) H1299 cells. (D–F) A549/DDP cells. (G) H1299-derived tumor tissues were resected, fixed, sectioned, and placed on slides. Tumor specimens were subjected to immunohistochemical staining with antibodies specific to Ki-67 and caspase-3. A two-tailed Student’s t test was used for statistical analysis. ∗p < 0.05, ∗∗p < 0.01.
HDACis Modulate miR-149 Expression in ERCC1-High NSCLC Cells
Antagonizing the action of the HDACs leads to a reversal of silencing of a large number of genes, including those of microRNAs (miRNAs).21, 22, 23 To determine whether miRNAs were induced by HDACis in ERCC1-high NSCLC cells, the human 384 SeraMir miRNA profiler was used to assess H1299 cells treated with SAHA (Figure 5A). 380 miRNAs were measured and the top miRNAs, most significantly (>8-fold) increased/decreased miRNAs in the SAHA-treated cells relative to control cells, are shown in Figure 5B. miR-149, miR-141, miR-379, miR-301a, miR-29c, miR-520e, miR-504, miR-432, miR-376a, miR-495, miR-516b, miR-514, miR-518b, and miR-205 were induced by SAHA, but miR-302c, miR-214, miR-298, miR-126, miR-28-3p, miR-337-5p, miR-192, miR-487a, and miR-423-5p were significantly decreased by SAHA. miR-149 was the most highly induced by SAHA treatment in H1299 cells. Quantitative reverse transcription PCR (qRT-PCR) further confirmed that treatment of SAHA significantly increased miR-149 expression in H1299 and A549/DDP cells (Figure 5C). Moreover, treatment of the HDACi TSA induced miR-149 in a concentration-dependent manner in H1299 and A549/DDP cells (Figure S3).
Figure 5.

The HDACi SAHA Modulates miR-149 Expression in ERCC1-High NSCLC Cells
(A) miRNA profiles were examined in SAHA-treated H1299 cells. (B) Differentially expressed miRNAs (>8-fold) in the SAHA-treated cells relative to the control. (C) H1299 and A549/DDP cells were treated with different concentrations of SAHA, and the expression of miR-149 was detected with qRT-PCR. (D) Sequence analysis of the promoter region revealed two conserved E2F1-binding sites at the core promoter region of miR-149. (E) Total protein extracts of H1299 cells in SAHA-treated cells or control cells were subjected to IP using E2F1 antibody or control IgG, followed by immunoblotting (IB) with acetyl-lysine antibody (left panels). Reciprocal IP was done using acetyl-lysine antibody or control IgG, followed by IB with the E2F1 antibody (right panels). (F) The binding of E2F1 on the miR-149 promoter region was measure by ChIP analysis. (G) H1299 cells were transfected with human cytomegalovirus promoter (pCMV)-E2F1 expression plasmid, together with the miR-149 promoter luciferase reporter construct or the E2F1-binding site mutant (mt) promoter luciferase reporter construct. Luciferase activity was measured after 48 h. (H) H1299 cells were treated with SAHA for 48 h, and the binding of E2F1 on the miR-149 promoter region was measure by ChIP analysis. (I) H1299 cells were transfected with E2F1 siRNA and then treated with SAHA; miR-149 promoter luciferase activity was measured after 48 h. (J) H1299 and A549/DDP cells were transfected with E2F1 siRNA and then treated with SAHA; the mRNA levels of miR-149 were measured by qRT-PCR. ∗∗p < 0.01.
To further analyze the underlying mechanism by which HDACis affect miR-149 expression, the sequence of the miR-149 promoter region was examined with the TFSEARCH program for potential transcription factor binding sites. We identified two potential E2F1 binding motifs in the proximal region of the miR-149 promoter (Figure 5D). Previous studies showed that HDACis induced acetylation of E2F1 and increased E2F1 transcriptional activity.24,25 Indeed, an immunoprecipitation (IP) assay demonstrated that SAHA significantly increased acetylation of lysine of E2F1 (Figure 5E). We next investigated whether E2F1 mediated HDACi-induced miR-149 expression. We found that E2F1 directly bound to the promoter region of miR-149 (Figure 5F) and promoted miR-149 promoter activity (Figure 5G). More importantly, we found that treatment of SAHA resulted in increased E2F1 binding to the miR-149 promoter (Figure 5H). Treatment with SAHA markedly increased miR-149 promoter activity, which was reversed by E2F1 shRNA transfection (Figure 5I). Accordingly, treatment with SAHA increased miR-149 expression, whereas knockdown of E2F1 led to a significant decrease in the expression of miR-149 in SAHA-treated cells (Figure 5J). Taken together, these results suggested that HDACis modulate miR-149 expression via activating transcription factor E2F1 in ERCC1-high NSCLC cells.
ERCC1 Is the Direct Target of miR-149
We further investigated whether miR-149 mediates the effect of HDACis on cisplatin sensitivity in ERCC1-high NSCLC cells. TargetScan 7.2 (http://www.targetscan.org/) predicted that the 3′ UTR of ERCC1 mRNA contains a putative miR-149 binding site (Figure 6A). To determine whether miR-149 regulates ERCC1 by binding to the corresponding 3′ UTR, we cloned the 3′ UTR from ERCC1 into the pmirGLO luciferase reporter vector and co-transfected this vector with miR-149 mimics or scramble control into HEK293T cells. Co-transfection of the pmirGLO-ERCC1 3′ UTR-wild-type-luc vector with miR-149 mimics resulted in lower luciferase activity in cells than that with miR scramble control (miR-SCR) (Figure 6B). Similar results were also observed in ERCC1-high NSCLC cells (Figure 6C). To confirm that miR-149 specifically regulates ERCC1 expression, we generated pmirGLO-ERCC1-3′ UTR mutant constructs, in which the sequence for miR-149 binding on the 3′ UTR was mutated (Figure 6A). Mutation of the miR-149 binding site abolished the effect of miR-149 on luciferase activity (Figures 6B and 6C). Furthermore, we found that overexpression of miR-149 decreased the levels of ERCC1 mRNA in H1299 and A549/DDP cells (Figure 6D). In contrast, the expression of ERCC1 in A549 and H460 cells transfected with miR-149 inhibitor (anti-miR-149) was higher than that transfected with anti-miR negative control (anti-NC). The results were confirmed by western blotting analysis (Figure 6E). Moreover, the expression levels of ERCC1 and miR-149 were quantified in total RNA derived from eight NSCLC cell lines, and the results generally showed a negative correlation between ERCC1 and miR-149 levels (Figure 6F). Taken together, these results provided evidence that miR-149 specifically targets the 3′ UTR region of ERCC1 and thus inhibits its expression.
Figure 6.

ERCC1 Is the Direct Target of miR-149
(A) TargetScan 7.2 predicted miR-149-binding sites in the 3′ UTR of ERCC1. Wild-type (WT) and mt 3′ UTR of ERCC1 were cloned into a luciferase reporter plasmid, respectively. (B) HEK293T cells were co-transfected with WT or mt ERCC1-3′ UTR-luciferase reporter constructs and miR-scramble control (miR-SCR) or miR-149 mimics and anti-miR negative control (anti-NC) or anti-miR-149, respectively; the relative luciferase activities were measured 48 h after transfection. Firefly luciferase activity of the reporters was normalized to the internal Renilla luciferase activity. (C) H1299 and A549/DDP cells were co-transfected with WT or mt ERCC1-3′ UTR-luciferase reporter constructs and miR-SCR or miR-149 mimics for 48 h; the relative luciferase activities were measured 48 h after transfection. (D) H1299 and A549/DDP cells were transfected with miR-SCR or miR-149 mimics, A549 and H460 cells were transfected with anti-NC or anti-miR-149, and the mRNA levels of ERCC1 were measured by real-time RT-PCR. (E) H1299, A549/DDP, A549, and H460 cells were transfected with miR-SCR or miR-149 mimics (left) and anti-NC or anti-miR-149 (right), and the expression levels of ERCC1 were measured by western blotting. (F) Correlation of ERCC1 and miR-149 expression levels in eight NSCLC cell lines. ∗p < 0.05, ∗∗p < 0.01.
The HDACi SAHA Sensitizes ERCC1-High NSCLC Cells to Cisplatin via Regulating miR-149
We next examined whether the HDACi SAHA re-sensitizes ERCC1-high NDCLC cells to cisplatin via miR-149 targeting of ERCC1. Western blotting analysis showed that treatment with SAHA significantly decreased the ERCC1 protein levels, whereas inhibition of miR-149 restored ERCC1 expression (Figure 7A). Using MTS assays, we found that transfection with anti-miR-149 attenuated the cytotoxic effects of SAHA in H1299 and A549/DDP cells (Figure 7B). Moreover, inhibition of miR-149 significantly reversed the promotion effect of SAHA on cisplatin-induced cell apoptosis (Figure 7C, D). Additionally, H1299 and A549/DDP cells transfected with anti-miR-149 showed a significant decrease in DNA damage and γ-H2AX expression induced by SAHA (Figure 7E; Figure S4). Collectively, these data suggested that miR-149 plays an important role in HDACi-increased cisplatin sensitivity of ERCC1-high NSCLC cells.
Figure 7.

The HDACi SAHA Sensitizes ERCC1-High NSCLC Cells to Cisplatin via Regulating miR-149
(A) H1299 and A549/DDP cells were transfected with anti-NC or anti-miR-149 and then treated with 2 μM SAHA for 48 h; the expression of ERCC1 was measured by western blotting. (B) H1299 and A549/DDP cells were transfected with anti-NC or anti-miR-149 and then treated with 2 μM SAHA for 48 h; cell viability was measured by an MTS assay. (C and D) H1299 and A549/DDP cells were transfected with anti-NC or anti-miR-149 and then treated with 2 μM SAHA for 48 h; cells were stained with annexin V-FITC and propidium iodide, and the cell apoptosis was analyzed by flow cytometry (C); Portions of the apoptotic cells are presented (D). (E) H1299 cells were transfected with anti-NC or anti-miR-149 and then treated with 2 μM SAHA for 48 h; cells were fixed and labeled with anti-γ-H2AX antibodies. The γ-H2AX foci were analyzed by immunofluorescence microscopy. ∗∗p < 0.01.
Discussion
With different treatment for advanced NSCLC, the cisplatin-based therapy is still taken for the foundation of treatment for most patients with advanced NSCLC.2 However, development of drug resistance becomes an increasingly significant clinical problem.3 Several molecular mechanisms could induce resistance to cisplatin, such as altered DNA repair, acquired genetic mutation, and cytosolic inactivation of the drug.26,27 Considering the prominent role in NER-mediated repair of cisplatin-caused DNA lesions, ERCC1 has been evaluated as a promising biomarker to predict the response of patients to platinum-based chemotherapy.7,28 In accordance with these reports, our studies showed that high ERCC1 levels are associated with increased resistance to cisplatin, and drug discovery targeting ERCC1 could thus result in the development of cisplatin-enhancing combination therapy.
HDACis have been considered as a promising novel therapeutic approach in the light of their potent tumor-selective effects.11 However, the use of these inhibitors for treatment of NSCLC has thus far demonstrated limited success as a monotherapy. Recently, several studies focused on the anti-tumor effect of the combination with chemotherapeutic drug.14,16 Since induction of DNA damage is the most critical mechanism of cisplatin action, and HDACis also alter gene expression of cancer cells with respect to the DNA damage response,29 the combination of cisplatin and HDACis exhibits synergistic antiproliferative effects in several cancer cells, including NSCLC cells.15 Importantly, HDACis have also been shown to overcome resistance to cisplatin.30,31 Although we have incomplete understanding of the molecular mechanisms, the impact of oncogenes, and thus the key pathways through which HDACis overcome cisplatin resistance, ERCC1 might have a predictive role for the efficacy of combined HDACis and cisplatin treatment.32,33 Accordingly, our studies proposed that HDACis significantly inhibited ERCC1 expression, which in turn increased cisplatin sensitivity in ERCC1-high NSCLC cells both in vitro and in vivo. These important results may potentially expand more clinical applications of HDACis and benefit patients with ERCC1-high NSCLC by increasing cisplatin sensitivity and reducing the required cisplatin dosage to minimize its side effects.
HDACis function as modifiers of histone and non-histone proteins via inhibiting deacetylation and then trigger the re-expression of certain genes, which play important roles in antitumor activity. Recent studies suggested that HDACis can also alter miRNA levels to inhibit cancer cell proliferation and induce cell apoptosis. miRNAs (non-coding RNAs of about 19–25 nt) target specific mRNAs to induce mRNA degradation or inhibit translation, thereby regulating a variety of cellular processes, such as proliferation, differentiation, apoptosis, invasion, and metastasis, as well as drug resistance. Nevertheless, the exact mechanisms by which HDACis overcome cisplatin resistance through miRNAs remain unknown. Using a miRNA profiler, we identified that miR-149 was the most upregulated miRNA induced by HDACis. In order to elucidate the mechanism behind it, we highlight the role of E2F1 in HDACi-induced miR-149 expression. E2F1, a positive regulator of cell cycle progression and also a potent inducer of apoptosis,34 was found to transcriptionally regulate miRNA expression.35,36 It is known that E2F1-dependent transcription is regulated by associated histone modifications, which influence gene expression through changes of the chromatin context.37 E2F1 is a non-histone target of HDACs.38 Several studies have shown that HDACs modulated E2F1-mediated transcription by directly deacetylating E2F1 and suppressing its transcription activity.39,40 Inhibition of HDACs causes accumulation of acetylated forms of E2F1, altering its function.24 In accordance with these findings, we found that treatment with HDACis induced acetylation of E2F1, which resulted in increased E2F1 binding to the miR-149 promoter and increased miR-149 promoter activity. Thus, the E2F1-miR-149 axis represents a novel mechanism by which HDACis overcome cisplatin resistance.
Recent studies have implicated the essential role of miR-149 in cancer progression.41 However, depending on the cancer type, miR-149 can behave either as a tumor suppressor or as an “onco-miR” that promotes tumor progression, suggesting that this miRNA has diverse functions.42,43 miR-149 has been shown to target GSK3α, in turn resulting in increased expression of Mcl-1 and resistance to apoptosis in melanoma cells.44 In contrast, Chan et al.45 found that a low level of miR-149 was significantly associated with advanced stages of breast cancer, and miR-149 targeted GIT1 and small GTPases Rap1a and Rap1b to suppress breast cancer cell invasion and metastasis.45,46 In NSCLC, miR-149 was reported to inhibit cell invasion and reverse the epithelial-to-mesenchymal transition (EMT) phenotype by inhibiting FOXM1.47 Moreover, studies have shown that miR-149 participated in regulating drug sensitivity and resistance.48 For example, miR-149 negatively regulated polymerase β (polβ) expression by binding to its 3′ UTR, thereby increasing sensitivity of esophageal cancer cells to cisplatin.49 He et al.50 reported that miR-149 was downregulated in doxorubicin (Adriamycin)-resistant human breast cancer cells and involved in chemoresistance by targeting GlcNAc (N-acetylglucosamine) N-deacetylase/N-sulfotransferase-1 (NDST1). These previous findings are similar to our observation that miR-149 increased cisplatin sensitivity in NSCLC cells. Furthermore, we found that miR-149 negatively regulated ERCC1 expression by directly binding to its 3′ UTR. Inhibition of miR-149 reversed the pro-apoptotic effect of HDACis and cisplatin sensitivity in ERCC1-high NSCLC cells. Therefore, the finding that miR-149 directly represses ERCC1 provides a rationale for the treatment of ERCC1-high NSCLC.
In summary, our results reveal a novel mechanism by which HDACis re-sensitize ERCC1-high NSCLC cells to cisplatin via regulation of E2F1-miR-149-ERCC1 axis, and we propose that the combination of HDACis and cisplatin might hold promise to be a more effective therapeutic paradigm for the treatment of ERCC1-high NSCLC.
Materials and Methods
Cell Lines and Cell Culture
A549 and cisplatin-resistant A549/DDP cells were obtained from the Cancer Institute & Hospital, Chinese Academy of Medical Sciences (Beijing, China). H460, H1299, H1975, H272, H1650, and HCC827 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The cell lines were subjected to short tandem repeat (STR) analysis. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were maintained in a 5% CO2-humidified atmosphere at 37°C. To maintain drug resistance, cisplatin (at a final concentration of 1 μM) was added for the culture of A549/DDP cells.
Cell Viability Assay
Cell viability was assessed by an MTS assay using the CellTiter 96 AQueous One solution cell proliferation assay (Promega, Madison, WI, USA), according to the manufacturer’s protocol.
Cell Apoptosis Assay
Cell apoptosis was determined by an annexin V and PI (propidium iodide) kit (KeyGEN, Nanjing, China) and analyzed using a FACSCanto II flow cytometer (BD Biosciences, USA) according to the manufacturers’ instructions.
Western Blotting
Total protein was isolated from the cells using radioimmunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology, China) that was supplemented with a protease inhibitor cocktail. Protein extracts were separated via 8%–12% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were then blocked in 5% fat-free milk and incubated with primary antibodies overnight at 4°C. Following washing with Tris-buffered saline in Tween 20 (TBST), the membranes were incubated with secondary antibodies at room temperature. Signal was visualized via enhanced chemiluminescence (ECL) using an ECL kit (Thermo Fisher Scientific, USA). Antibodies against β-actin (catalog #4970), ERCC1 (catalog #12345), and E2F1 (catalog #3742) were from Cell Signaling Technology (Beverly, MA, USA).
miRNA Profiling Using Multiplex qRT-PCR
Expression profiles of miRNA in H1299 cells treated with SAHA were analyzed by the human SeraMir 384 miRNA profiler (System Biosciences, Mountain View, CA, USA). miRNA extraction and cDNA synthesis were performed using a complete SeraMir exosome RNA amplification kit (System Biosciences). The kit contains 380 mature human miRNA qRT-PCR primers. qRT-PCR reactions were determined by the ABI 7900 HT Fast real-time PCR system (Applied Biosystems, Foster City, CA, USA).
Real-Time RT-PCR
Total cellular RNA was extracted using TRIzol reagent (Invitrogen, CA, USA). For miR-149 detection, qRT-PCR was carried out by using an all-in-one miRNA first-strand cDNA synthesis kit and miRNA qRT-PCR detection kit (GeneCopoeia, Rockville, MD, USA). For mRNA detection, expression of ERCC1 mRNA was determined by a first-strand cDNA synthesis kit and SYBR Green master mix according to the manufacturer’s instructions (Applied Biosystems). A quantitative real-time PCR reaction was conducted using an ABI 7500 Fast real-time PCR system (Applied Biosystems, Foster City, CA, USA). Gene expression levels were calculated by the 2−ΔΔCt method. Gene-specific primers used are as follows: miR-149, forward, 5′-CATCCTTTCTGGCTCCGTGT-3′, reverse, 5′-GCGTGATTCGTGCTCGTATATC-3′; U6, forward, 5′-CTCGCTTCGGCAGCACA-3′, reverse, 5′-ACGCTTCACGAATTTGCGT-3′; ERCC1, forward, 5′-CTGGAGCCCCGAGGAAGC-3′, reverse, 5′-CACTGGGGGTTTCCTTGG-3′; β-actin, forward, 5′-ACACTGTGCCCATCTACGAGG-3′, reverse, 5′-AGGGGCCGGACTCGTCATACT-3′.
ChIP Analysis
The chromatin IP (ChIP) analysis was carried out using a Millipore EZ-Magna ChIP kit (Millipore) following the manufacturer’s protocol. Briefly, 5 × 106 cells were collected and fixed with 1% formaldehyde. After quenching in 0.125 M glycine, cells were washed in PBS and resuspended in ChIP lysis buffer. Cells were then sonicated by Bioruptor sonication system UCD-300. The chromatin was immunoprecipitated by either anti-immunoglobulin G (IgG) or anti-E2F1 antibody. Precipitated DNA samples and inputs were subjected to PCR. The primers used to target the amplification of E2F1 binding site in miR-149 promoter are 5′-GAGCCCAGCGCGAGAC-3′ (sense) and 5′-CGAGGCAGGGCTTCCC-3′ (antisense).
Luciferase Assays
The miR-149 promoter (−1000 to +1 regions) and E2F1 binding site-mutated miR-149 promoter were inserted into the pGL3-basic vector (Promega, Madison, WI, USA). All constructs were validated by sequencing. Cells were co-transfected with pGL3-miR-149 or mut-pGL3-miR-149 using Lipofectamine 3000 reagent. Cells were harvested at 48 h after transfection and then assayed by the Dual-Luciferase reporter assay system (Promega, Madison, WI, USA). The relative firefly luciferase activity was normalized to Renilla luciferase activity.
Animals
All animal work was approved by the Animal Experimentation Ethics Committee of Guangzhou Medical University and conducted in accordance with protocols of the committee.
Immunodeficient mice (5–6 weeks old) were used in assays for tumor growth in a subcutaneous xenograft model. Suspensions of H1299 cells (2 × 106) or A549/DDP cells (2 × 106) were injected subcutaneously into the right flank of mice. When tumors reached a diameter of 4 mm in size, mice were randomly allocated into four groups: vehicle control (0.01% DMSO in PBS, n = 4), cisplatin (10 mg/kg/d, n = 4), SAHA (50 mg/kg/d, n = 4), and the cisplatin/SAHA combination (n = 4) via i.p. injection for 3 weeks. Tumor growth and body weights were measured every 3 days. Tumor volume (V) was calculated by the following formula: V = (½ × larger diameter) × (smaller diameter)2, and growth curves were plotted using mean tumor volume within each group at the set time points. Mice were sacrificed at the end of the study, and tumors were removed for immunohistochemical staining or western blotting analysis.
Immunofluorescence
Cells were grown on 20-mm coverglass-bottom dishes, washed with PBS, fixed with 4% paraformaldehyde, and permeabilized in 0.1% Triton X-100. After blocking with 10% normal goat serum, cells were incubated with phosphorylated (phospho-)histone H2AX (Ser139) primary antibody (#9718, Cell Signaling Technology, USA) at a dilution of 1:200 overnight at 4°C. The cells were incubated with Alexa Fluor 594-conjugated anti-rabbit IgG (1:1,000 dilution) for 1 h in the dark at room temperature, and then nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. The images were captured using a Zeiss LSM710 confocal microscope (Zeiss, Germany).
Immunohistochemistry and Scoring System
This study was approved by the Medical Ethics Committee of the Affiliated Tumor Hospital of Guangzhou Medical University and performed in accordance with the Declaration of Helsinki. Primary tumor specimens from 39 patients diagnosed with NSCLC who had undergone complete resection were obtained in the Affiliated Tumor Hospital of Guangzhou Medical University between 2008 and 2013. Twenty-one patients with NSCLC for whom cisplatin-based chemotherapy was effective following surgery (patients who were cisplatin-sensitive), and 18 patients with NSCLC for whom cisplatin-based chemotherapy was ineffective following surgery (patients who were cisplatin-resistant) were included in the study.
The immunohistochemistry staining of ERCC1 was accomplished using the streptavidin-peroxidase complex method (UltraSensitive, Maixin, Fuzhou, China). The sections were deparaffinized in xylene and rehydrated with graded alcohol. 3% H2O2/methanol was applied to block endogenous peroxide activity for 10 min. Next, sections were boiled in 0.01 M citrate buffer (pH 6.0) for 2 min with an antigen retrieval. After incubating with 10% normal goat serum to reduce non-specific antibody binding, tissue sections were incubated with primary antibody (1:100 dilution) in a moist chamber overnight at 4°C. The sections were then sequentially incubated with biotinylated goat anti-mouse serum IgG and streptavidin-biotin conjugated with horseradish peroxidase. Finally, the color reaction was developed by 3,3′-diaminobenzidine tetrahydrochloride (DAB).
The intensity of ERCC1 staining was graded as follows: 0, no signal; 1, weak; 2, moderate; and 3, marked. Tumor cell percentage scores were assigned as follows: 1, 1%–25%; 2, 26%–50%; 3, 51%–75%; and 4, 76%–100%. A final score of 0–12 was calculated by the staining intensity multiplied by tumor cell percentage score. The tumors were finally determined as follows: negative (−), score 0; lower expression (+), score ≤4; moderate expression (++), score 5–8; and high expression (+++), score ≥9. An optimal cutoff value was identified: a staining index score of ≥5 was used to indicate tumors of high ERCC1 expression, and ≤4 for low expression.
Bioinformatics Analysis
The prognostic value of the ERCC1 was analyzed by a web-based Kaplan-Meier plotter (http://www.kmplot.com/lung), which is a meta-analysis tool of gene expression and survival data of 2,437 lung cancer patients (2015 version) using multiple microarray data.51,52
Statistical Analysis
Data were analyzed with SPSS v21.0 software (IBM, Armonk, NY, USA) and GraphPad Prism 6 software (GraphPad, San Diego, CA, USA). Relative gene expression levels were analyzed by Student’s t test. Results represent mean ± SD of three independent experiments, with p < 0.05 considered statistically significant. Agilent human miRNA array images generated by the Agilent scanner and Feature Extraction software (Agilent Technologies, v10.7.1.1) were used to acquire raw data, which were analyzed by GeneSpring GX software (Agilent Technologies, v13.1). For these experiments, ANOVA or t tests were used to evaluate differences between groups. The p values were two-sided, with p < 0.05 considered statistically significant.
Author Contributions
H.L. and G.J. designed the experiments, analyzed the data, and drafted the manuscript and figures. H.L., Y.Y., and S.L. reviewed and edited the paper. Y.H., D.C, and Y.Y. detected the cells’ biological function, conducted the real-time qPCR assays, carried out the western blot and luciferase reporter assays, and performed immunohistochemistry analysis. S.Z. carried out the animal experiments. P.L., H.X., and P.Y. helped to acquire the experimental data. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81503103, 81402497, 81772825, 81800153); the China Postdoctoral Science Foundation (2018M633032); grants from Guangdong Natural Science Funds (2017A030313500); the Science and Technology Program of Guangzhou (201707010381); the Innovation Project of Guangdong Education Department (Grant 2016KTSCX117); and by the Scientific Research Project of Guangzhou Municipal Colleges and Universities (grant 1201630143).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.05.001.
Contributor Information
Guanmin Jiang, Email: jianggm3@mail.sysu.edu.cn.
Hao Liu, Email: haoliu2020@163.com.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Fennell D.A., Summers Y., Cadranel J., Benepal T., Christoph D.C., Lal R., Das M., Maxwell F., Visseren-Grul C., Ferry D. Cisplatin in the modern era: the backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016;44:42–50. doi: 10.1016/j.ctrv.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71:3–10. doi: 10.1016/j.lungcan.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Pearl L.H., Schierz A.C., Ward S.E., Al-Lazikani B., Pearl F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer. 2015;15:166–180. doi: 10.1038/nrc3891. [DOI] [PubMed] [Google Scholar]
- 5.Stingele J., Bellelli R., Boulton S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017;18:563–573. doi: 10.1038/nrm.2017.56. [DOI] [PubMed] [Google Scholar]
- 6.Deans A.J., West S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNeil E.M., Melton D.W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012;40:9990–10004. doi: 10.1093/nar/gks818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glozak M.A., Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 9.Eckschlager T., Plch J., Stiborova M., Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016;6:a026831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane A.A., Chabner B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 12.Marks P.A. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs. 2010;19:1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suraweera A., O’Byrne K.J., Richard D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 2018;8:92. doi: 10.3389/fonc.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carraway H.E., Gore S.D. Addition of histone deacetylase inhibitors in combination therapy. J. Clin. Oncol. 2007;25:1955–1956. doi: 10.1200/JCO.2006.09.8293. [DOI] [PubMed] [Google Scholar]
- 15.Diyabalanage H.V., Granda M.L., Hooker J.M. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013;329:1–8. doi: 10.1016/j.canlet.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carew J.S., Giles F.J., Nawrocki S.T. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 17.Reed E. ERCC1 and clinical resistance to platinum-based therapy. Clin. Cancer Res. 2005;11:6100–6102. doi: 10.1158/1078-0432.CCR-05-1083. [DOI] [PubMed] [Google Scholar]
- 18.Halsall J.A., Turner B.M. Histone deacetylase inhibitors for cancer therapy: an evolutionarily ancient resistance response may explain their limited success. BioEssays. 2016;38:1102–1110. doi: 10.1002/bies.201600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.To K.K., Tong W.S., Fu L.W. Reversal of platinum drug resistance by the histone deacetylase inhibitor belinostat. Lung Cancer. 2017;103:58–65. doi: 10.1016/j.lungcan.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Roos W.P., Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016;44:10017–10030. doi: 10.1093/nar/gkw922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott G.K., Mattie M.D., Berger C.E., Benz S.C., Benz C.C. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66:1277–1281. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh T.H., Hsu C.Y., Tsai C.F., Long C.Y., Wu C.H., Wu D.C., Lee J.N., Chang W.C., Tsai E.M. HDAC inhibitors target HDAC5, upregulate microRNA-125a-5p, and induce apoptosis in breast cancer cells. Mol. Ther. 2015;23:656–666. doi: 10.1038/mt.2014.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun W., Yi Y., Xia G., Zhao Y., Yu Y., Li L., Hua C., He B., Yang B., Yu C. Nrf2-miR-129-3p-mTOR axis controls an miRNA regulatory network involved in HDACi-induced autophagy. Mol. Ther. 2019;27:1039–1050. doi: 10.1016/j.ymthe.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y., Tan J., Zhuang L., Jiang X., Liu E.T., Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA. 2005;102:16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kachhap S.K., Rosmus N., Collis S.J., Kortenhorst M.S., Wissing M.D., Hedayati M., Shabbeer S., Mendonca J., Deangelis J., Marchionni L. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE. 2010;5:e11208. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makovec T. Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019;53:148–158. doi: 10.2478/raon-2019-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 28.Olaussen K.A., Dunant A., Fouret P., Brambilla E., André F., Haddad V., Taranchon E., Filipits M., Pirker R., Popper H.H., IALT Bio Investigators DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 29.Robert C., Rassool F.V. HDAC inhibitors: roles of DNA damage and repair. Adv. Cancer Res. 2012;116:87–129. doi: 10.1016/B978-0-12-394387-3.00003-3. [DOI] [PubMed] [Google Scholar]
- 30.Lin C.T., Lai H.C., Lee H.Y., Lin W.H., Chang C.C., Chu T.Y., Lin Y.W., Lee K.D., Yu M.H. Valproic acid resensitizes cisplatin-resistant ovarian cancer cells. Cancer Sci. 2008;99:1218–1226. doi: 10.1111/j.1349-7006.2008.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ong P.S., Wang X.Q., Lin H.S., Chan S.Y., Ho P.C. Synergistic effects of suberoylanilide hydroxamic acid combined with cisplatin causing cell cycle arrest independent apoptosis in platinum-resistant ovarian cancer cells. Int. J. Oncol. 2012;40:1705–1713. doi: 10.3892/ijo.2012.1354. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y.C., Chang P.Y., Chao C.C. CITED2 silencing sensitizes cancer cells to cisplatin by inhibiting p53 trans-activation and chromatin relaxation on the ERCC1 DNA repair gene. Nucleic Acids Res. 2015;43:10760–10781. doi: 10.1093/nar/gkv934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai Y., Yan X., Zhang G., Zhao W., Jiao S. The predictive value of ERCC1 and p53 for the effect of panobinostat and cisplatin combination treatment in NSCLC. Oncotarget. 2015;6:18997–19005. doi: 10.18632/oncotarget.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Z., Yu Q. E2F1-mediated apoptosis as a target of cancer therapy. Curr. Mol. Pharmacol. 2009;2:149–160. doi: 10.2174/1874467210902020149. [DOI] [PubMed] [Google Scholar]
- 35.Lizé M., Pilarski S., Dobbelstein M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Differ. 2010;17:452–458. doi: 10.1038/cdd.2009.188. [DOI] [PubMed] [Google Scholar]
- 36.Deng M., Zeng C., Lu X., He X., Zhang R., Qiu Q., Zheng G., Jia X., Liu H., He Z. miR-218 suppresses gastric cancer cell cycle progression through the CDK6/Cyclin D1/E2F1 axis in a feedback loop. Cancer Lett. 2017;403:175–185. doi: 10.1016/j.canlet.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Blais A., Dynlacht B.D. E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glozak M.A., Sengupta N., Zhang X., Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Martínez-Balbás M.A., Bauer U.M., Nielsen S.J., Brehm A., Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marzio G., Wagener C., Gutierrez M.I., Cartwright P., Helin K., Giacca M. E2F family members are differentially regulated by reversible acetylation. J. Biol. Chem. 2000;275:10887–10892. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- 41.Zhi Y., Zhou H., Mubalake A., Chen Y., Zhang B., Zhang K., Chu X., Wang R. Regulation and functions of microRNA-149 in human cancers. Cell Prolif. 2018;51:e12465. doi: 10.1111/cpr.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Guo X., Xiong L., Yu L., Li Z., Guo Q., Li Z., Li B., Lin N. Comprehensive analysis of microRNA-regulated protein interaction network reveals the tumor suppressive role of microRNA-149 in human hepatocellular carcinoma via targeting AKT-mTOR pathway. Mol. Cancer. 2014;13:253. doi: 10.1186/1476-4598-13-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He Y., Yu D., Zhu L., Zhong S., Zhao J., Tang J. miR-149 in human cancer: a systemic review. J. Cancer. 2018;9:375–388. doi: 10.7150/jca.21044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin L., Hu W.L., Jiang C.C., Wang J.X., Han C.C., Chu P., Zhang L.J., Thorne R.F., Wilmott J., Scolyer R.A. MicroRNA-149∗, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc. Natl. Acad. Sci. USA. 2011;108:15840–15845. doi: 10.1073/pnas.1019312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan S.H., Huang W.C., Chang J.W., Chang K.J., Kuo W.H., Wang M.Y., Lin K.Y., Uen Y.H., Hou M.F., Lin C.M. MicroRNA-149 targets GIT1 to suppress integrin signaling and breast cancer metastasis. Oncogene. 2014;33:4496–4507. doi: 10.1038/onc.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischoff A., Huck B., Keller B., Strotbek M., Schmid S., Boerries M., Busch H., Müller D., Olayioye M.A. miR149 functions as a tumor suppressor by controlling breast epithelial cell migration and invasion. Cancer Res. 2014;74:5256–5265. doi: 10.1158/0008-5472.CAN-13-3319. [DOI] [PubMed] [Google Scholar]
- 47.Ke Y., Zhao W., Xiong J., Cao R. miR-149 inhibits non-small-cell lung cancer cells EMT by targeting FOXM1. Biochem. Res. Int. 2013;2013:506731. doi: 10.1155/2013/506731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ow S.H., Chua P.J., Bay B.H. miR-149 as a potential molecular target for cancer. Curr. Med. Chem. 2018;25:1046–1054. doi: 10.2174/0929867324666170718102738. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y., Chen J., Zhang M., Zhang W., Li M., Zang W., Dong Z., Zhao G. miR-149 sensitizes esophageal cancer cell lines to cisplatin by targeting DNA polymerase β. J. Cell. Mol. Med. 2018;22:3857–3865. doi: 10.1111/jcmm.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He D.X., Gu X.T., Li Y.R., Jiang L., Jin J., Ma X. Methylation-regulated miR-149 modulates chemoresistance by targeting GlcNAc N-deacetylase/N-sulfotransferase-1 in human breast cancer. FEBS J. 2014;281:4718–4730. doi: 10.1111/febs.13012. [DOI] [PubMed] [Google Scholar]
- 51.Győrffy B., Surowiak P., Budczies J., Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE. 2013;8:e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie K., Liang C., Li Q., Yan C., Wang C., Gu Y., Zhu M., Du F., Wang H., Dai J. Role of ATG10 expression quantitative trait loci in non-small cell lung cancer survival. Int. J. Cancer. 2016;139:1564–1573. doi: 10.1002/ijc.30205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.