

Abstract
Patients with rheumatoid arthritis (RA) have very different outcomes, particularly with regard to bone erosions. Since osteoclasts are responsible for bone destruction adjacent to rheumatoid synovium, profiling osteoclasts from circulating precursors in RA could help identify patients at risk for bone destruction. In this study, we sought to determine whether the functional characteristics of osteoclasts generated from their blood precursors were modified by RA activity or were intrinsic to osteoclasts and associated with the RA phenotype (erosive or not).
Osteoclasts were generated in vitro from peripheral blood mononuclear cells (PBMCs) of subjects with RA (n = 140), as well as sex- and age-matched healthy controls (n = 101). Osteoclastic parameters were analyzed at baseline and during the follow-up for up to 4 years, with regular assessment of RA activity, bone erosions, and bone mineral density (BMD). As a validation cohort, we examined RA patients from the Early Undifferentiated PolyArthritis (EUPA) study (n = 163).
The proportion of CD14+ PBMC was higher in RA than in control subjects, but inversely correlated with the 28-joint disease activity score (DAS28). Also surprisingly, in osteoclast cultures from PBMCs, active RA was associated with lower osteoclastogenic capacity, while in vitro bone resorption per osteoclast and resistance to apoptosis were similar in both active and quiescent RA. In a small subgroup analysis, osteoclasts from subjects with recent RA that had progressed at four years to an erosive RA exhibited at baseline greater resistance to apoptosis than those from patients remaining non-erosive.
Our findings establish that when RA is active, circulating monocytes have a reduced potential to generate osteoclasts from PBMCs in vitro. In addition, osteoclasts associated with erosive disease had resistance to apoptosis from the start of RA.
Keywords: Rheumatoid arthritis, Erosion, Osteoclast, CD14+ monocytes
Highlights
-
•
Osteoclasts are derived in vitro from circulating monocytes in rheumatoid arthritis.
-
•
Blood CD14+ monocytes (%) are higher but inversely correlated with disease activity.
-
•
Active rheumatoid arthritis is associated with reduced osteoclast formation in vitro.
-
•
Inflammation alters the ability to generate osteoclasts from circulating monocytes.
-
•
Osteoclast resistance to apoptosis is stable and associated with long-term erosions.
1. Introduction
Rheumatoid arthritis (RA) is associated with a high risk of progressive bone and joint destruction caused by chronic synovitis, and still remains a leading cause of disability (McInnes and Schett, 2011). In experimental models of arthritis and in RA, multinucleated giant cells are observed at the synovium-bone interface (Roux and Brown, 2009). These cells express phenotypic markers of osteoclasts, and it is now widely accepted that osteoclasts are responsible for the bone destruction in RA (Schett and Gravallese, 2012). Mouse models harboring defective osteoclastogenesis have provided new insights into the mechanisms of arthritis-related bone destruction. Indeed, osteopetrotic mice lacking osteoclasts, such as RANKL- or RANK-deficient mice (Tnfsf11−/− and Tnfrsf11a−/−, respectively) or Fos−/− transgenic mice, can develop arthritis (collagen-induced or TNFα-mediated arthritis) but do not have the expected bone erosions (Li et al., 2004; Pettit et al., 2001; Redlich et al., 2002). The presence of activated osteoclasts at the sites of bone resorption is a result of the recruitment and differentiation of osteoclast precursors arising from newly recruited blood monocytes within the inflamed synovium (Hasegawa et al., 2019), which provides a suitable microenvironment for the formation of fully differentiated osteoclasts (Gravallese et al., 2000; Lubberts et al., 2002).
Early control of RA activity leads to better outcomes (Goekoop-Ruiterman et al., 2008). Moreover, an overall correlation exists between joint inflammation and resulting joint destruction at the population level, although this is far less true at the individual level (Miossec et al., 2011). Joint inflammation by itself is insufficient to cause bone erosions, as evidenced by patients with systemic lupus erythematosus and some RA patients with decades of chronic joint inflammation who never develop bone erosions. So far, no prognostic biomarker of erosiveness has been validated in RA.
Osteoclasts are derived from CD14+ precursors belonging to the monocyte-macrophage lineage. In a mouse model of arthritis, a particular subpopulation of osteoclastic precursors has been identified as the source of the osteoclasts that will develop in the inflamed synovium and cause resorption of periarticular bone (Hasegawa et al., 2019). There might be clinical value in identifying these CD14+ osteoclastic precursors in the blood of subjects with RA to identify cases that will have a propensity to destroy joints and to differentiate these forms of arthritis from those that will not be destructive.
Osteoclasts formed from their circulating precursors in RA patients show increased lacunar bone resorption in vitro compared to osteoclasts generated from healthy controls (Gengenbacher et al., 2008; Hirayama et al., 2002). Similarly, in a previous publication, we compared the formation of osteoclasts from peripheral blood mononuclear cells (PBMCs) in 3 groups: RA (active or not), osteoarthritis, and age- and sex-matched controls. CD14+ peripheral precursors were more numerous in RA patients, with a greater capacity to form mature osteoclasts, which displayed higher numbers of nuclei and greater resistance to apoptosis (Durand et al., 2011). However, the generation of osteoclasts from their PBMC-derived precursors could be altered by the systemic inflammation that would exist at time of blood sampling in a patient with active arthritis.
In this new study, we assessed the impact of inflammation (RA activity) on the number of PBMC-CD14+ cells, and on their ability to differentiate into mature osteoclasts in vitro, as well as on the survival and bone resorption capacity of the PBMC-derived osteoclasts. Parameters associated with the development of erosion without being influenced by inflammation or time (“intrinsic” characteristics) might be good biomarker candidates for a destructive outcome in RA. Likewise, the osteoclast precursor profile could also influence the development of systemic bone loss (BMD). For this study, we used the same cohort of RA patients and controls used in our previous study (Durand et al., 2011). The RA cohort was monitored up to 4 years, with regular collection of data on RA activity, severity (erosive status), bone mineral density (BMD), and PBMC-derived osteoclast parameters (CD14+ precursors, in vitro osteoclastogenesis, bone resorption and apoptosis). We also validated our findings in an independent cohort of RA patients from the Early Undifferentiated PolyArthritis (EUPA) study (Carrier et al., 2009).
2. Patients and methods
2.1. Clinical investigation and phenotype classification
The In Vitro Osteoclast Differentiation in Arthritis (IODA) study involved subjects with RA recruited from outpatient clinics in one of our institutions, the Centre Hospitalier Universitaire de Sherbrooke (CHUS) (n = 140). Sex- and age-matched healthy controls were also recruited locally through newspaper advertisements (baseline cohort described in (Durand et al., 2011)). All participants signed an informed-consent form before entering the study. This study was approved by the Ethics Review Board of the CHUS.
RA diagnosis was based on the 2010 American College of Rheumatology criteria (Aletaha et al., 2010). The clinical assessment consisted of a physical examination and a questionnaire, the Health Assessment Questionnaire Disability Index (HAQ). Blood samples were obtained at the initial visit (V1) for all patients and controls, and also for patients only at subsequent annual visits (V2, V3 and V4). Rheumatoid factor (RF) and anti–cyclic citrullinated peptide (anti-CCP2) were determined by a clinical laboratory (CHUS) using immuno-turbidimetric measurement (RF) or enzyme-linked immunosorbent assay (ELISA) (anti-CCP2); serum 25 (OH) D levels were determined as well. Erosion scores from the Sharp/van der Heijde modified method (SHS) (van der Heijde, 2000) were available for RA patients, as were BMD values at baseline and annual visits.
Osteoclasts were derived from PBMCs in in vitro cultures at each annual visit. RA activity was defined using the Disease Activity Score in 28 joints (DAS28) expressed as a continuous variable. For baseline analyses only, two groups of patients were constituted according to their level of RA activity (remission or active RA) using DAS28 as a categorical variable, with a standard cut-off of 2.6 above which RA was considered as active (Fransen et al., 2004). Erosive disease was defined as a score of at least 5 on the erosion component of the SHS. Concomitant malignancy, immune dysfunction or any other autoimmune disease were considered as exclusion criteria.
2.2. Validation cohort: EUPA
The prospective recruitment of recent-onset polyarthritis in an ongoing longitudinal cohort of patients with early arthritis (the EUPA study) has been described in detail previously (Carrier et al., 2009). Briefly, clinical and biological variables were assessed regularly (at inclusion, 12, 18, 30, 42 and 60 months), including demographic features, DAS28, modified HAQ, levels of serum C-reactive protein, complete blood count, serum levels of RF and anti-CCP2 antibodies, and medications. Hand and foot X-rays were obtained at each visit and scored according to SHS (van der Heijde, 2000). For the validation study, we included 163 consecutive patients at time of their inclusion in the observational EUPA cohort. Osteoclast cultures from PBMCs were generated at inclusion and at 30 months.
2.3. Reagents
Fetal bovine serum (FBS) was purchased from Gibco (Thermo Fisher Scientific, Waltham, MA), macrophage-colony stimulating factor (M-CSF) from MJS BioLynx Canada (Brockville, ON, Canada), and fluorescent-dye-coupled anti-human CD14, CD16 and osteoclast-associated receptor (OSCAR) antibodies from BioLegend (San Diego, CA). Soluble human RANKL (sRANKL)- glutathione S-transferase (GST) fusion protein was generated in one of our labs as described elsewhere (Manolson et al., 2003). Tunnel assay was performed using the TACS Blue Label kit from R&D Systems (Minneapolis, MN). All other reagents were purchased from Sigma-Aldrich Canada, Ltd.
2.4. Staining for cell surface markers and flow cytometry
PBMCs were counted using standard flow cytometry to detect the expression of CD14 and CD16. PBMCs were incubated for 30 min in the dark in a 1:10 solution of fluorescently tagged monoclonal antibodies against human CD14, CD16 and OSCAR in phosphate-buffered saline plus 1% bovine albumin. Cells were washed once, then analyzed using a FACScan from Becton Dickinson (Mississauga, ON, Canada).
2.5. In vitro osteoclastogenesis
Mature human osteoclasts were generated in vitro from overall PBMCs isolated by density-gradient centrifugation, washed, and suspended in α-MEM with antibiotics, glutamine, and 10% FBS. After incubating overnight, the cells were washed to remove non-adherent cells. The selected PBMCs were then seeded at a density of 1.6 × 106 cells/cm2 in plates or on bone slices and cultured for 21 days in the same medium supplemented with M-CSF (10 ng/mL) and GST-sRANKL (50 ng/mL). The medium was changed every 2–3 days. We had previously shown that under these conditions fully-differentiated osteoclasts are formed, which are able to resorb bone, the only specific marker of osteoclast terminal differentiation (Durand et al., 2011; Durand et al., 2013).
2.6. Bone resorption
PBMCs were settled on devitalized bone slices (two slices per individual), and then cultured for 3 weeks under the same conditions as above, and then in presence of 10% CO2 for 10 additional days. The bone slices were then removed, washed, and stained with 0.2% toluidine blue. Optical light microscopy with epi-illumination was used to determine bone resorption (resorbed area quantified by ImageJ).
2.7. TRAP staining
At the end of the cultures, mature osteoclasts were fixed in formaldehyde (3.7%) for 15 min and stained for tartrate-resistant acid phosphatase (TRAP) activity using a leukocyte acid phosphatase kit according to the manufacturer's protocol (Sigma-Aldrich Canada), which produces a purple cytoplasmic stain. TRAP-positive multinucleated cells (≥3 nuclei) were counted as osteoclasts. The results are the mean of five random fields of view.
2.8. Apoptosis assay
Osteoclast apoptosis was assessed using the TACS Blue Label kit (R&D Systems). This TUNEL technique allows in situ visualization of DNA fragmentation at the single cell level. Briefly, cells were deprived of M-CSF and RANKL survival factors and FBS was reduced to 1% 24 h prior to apoptosis assessment. Cells were then fixed and permeabilized. Biotinylated nucleotides were incorporated using a terminal deoxynucleotidyl transferase enzyme (TdT). Streptavidin-HRP conjugate was then added, followed by the substrate, TACS Blue Label. The resulting enzymatic reaction generated an insoluble blue-colored precipitate where DNA fragmentation had occurred (Chamoux et al., 2008). The stained samples were examined using a light microscope. Osteoclasts with two or more positive nuclei were counted as apoptotic.
2.9. Statistical analyses
All statistical analyses were undertaken using SPSS 16.0 software and presented as median ± error type. Baseline demographics, and clinical and biological variables were compared using Chi-squared, Fisher's exact, or nonparametric Kruskall-Wallis tests, where appropriate. Pearson or Spearman tests (in non-normal distributions) were used for correlation analyses. Significance was set at p < 0.05, using the Bonferroni correction for multiple comparisons (padj < 0.05), when appropriate.
Analysis using the general linear model (GLM) with continuous outcomes, established correlations between initial osteoclastic parameters and the evolution of RA. Osteoclastic parameters and RA evolution parameters were evaluated in 4 consecutive yearly visits from baseline to 4 years, which allowed the use of the model of generalized estimating equations (GEE) analysis of repeated measures with categorical variables for outcomes (Hanley et al., 2003). For the correlation analysis between osteoclast profile and RA activity over time, the “Last observation carried forward” (LOCF) method was used to compensate for missing data by imputing the last observed value to subsequent time points where data are missing.
3. Results
3.1. Demographics of the cohort and RA characteristics
We used a well described cohort of RA patients and their age- and sex- matched controls (Durand et al., 2011) to probe for the influence of disease activity on the in vitro formation and survival of osteoclasts generated from subjects' PBMCs. In addition, we explored potential correlations between the number of PBMC-CD14+ cells or in vitro osteoclastic parameters and bone erosions. All 140 individuals with RA and 101 healthy controls were included in the present study. All individuals were evaluated at V1, 80 patients with RA were evaluated at V2 13.2 ± 1.7 months later, 76 patients at V3 after 25.6 ± 2.5 months, and 102 patients at V4 after 41.6 ± 3.3 months (mean ± SD). The median of follow-up was 40 months (IQR: 29.3–43.2, range: 0–48.2).
Demographics and the characteristics of both RA groups (active disease and remission), as well as those of the control group are shown in Table 1. According to DAS28 (cut-off 2.6), RA was active in 75 of 139 patients at baseline; DAS28 status could not be calculated in 1 patient at baseline. More than two thirds of patients were females, half of whom were postmenopausal. Anti-CCP2 antibodies were positive in 75% of RA patients. Patients with active RA were less physically active and most often female. Median duration of the disease at baseline was 9.5 years, higher in the group with active RA. As well, most patients were taking disease-modifying antirheumatic drugs (DMARDs) in both groups, but prednisone use was greater in patients with active RA (25%) as was the use of bisphosphonates (40%). There were no other significant differences between the two groups of RA, except for parameters of disease activity (PGA, PhGA, morning stiffness, HAQ), and joint erosive damage.
Table 1.
Patient demographics and baseline disease characteristics.
| Non-active RA (n = 64) |
Active RA (n = 75) |
Total RA (n = 140)a |
Controls (n = 101) |
|
|---|---|---|---|---|
| Age (years), mean (SD) | 59 (11.6) | 62.2 (11) | 60.6 (11.4) | 61.1 (6.1) |
| Female n (%) | 37(57.8) | 57 (76)⁎ | 94 (67.6) | 58 (57.4) |
| BMI kg/m2, mean (SD) | 27 (5.4) | 26.1 (4.9) | 26.5 (5.1) | 26.5 (4.5) |
| Activity (min/week) Mdn (IQR) | 240 (0–360) | 30 (0–240)⁎ | 180 (0−300) | 360 (225–480) |
| Ethnicity - Caucasian | 64 (100) | 72 (96) | 136 (97.8) | 97 (98.0) |
| Menopause n (% of all patients) | 19 (29.7) | 32 (42.7) | 51 (36.7) | 27 (35.5) |
| Tobacco use (ever smoked), n (%) | 31 (48.4) | 30 (40.0) | 62 (44.3) | 21 (20.8) |
| Alcohol use (yes/no) n (%) | 33 (51.6) | 33 (44) | 66 (47.1) | 83 (82.2) |
| Serum 25OH D, nmol/L Mdn (IQR) | 70.6 (22.1) | 75.6 (34.4) | 73.4 (29.3) | 69.8 (18.7) |
| RA duration years, Mdn (IQR) | 4 (7.5–14) | 12 (5–23)⁎ | 9.5 (4.3–17) | N/A |
| PGA (mm), Mdn (IQR) | 10 (4.25–19.75) | 39 (21–61)⁎⁎⁎ | 22 (10–48) | N/A |
| PhGA (mm), Mdn (IQR) | 12.5 (4.25–29.75) | 42 (25–63)⁎⁎⁎ | 28.5 (8.25–54.25) | N/A |
| Morning stiffness (min), Mdn (IQR) | 0 (0−10) | 25 (0–60)⁎⁎⁎ | 5 (0−30) | N/A |
| HAQ, Mdn (IQR) | 0.125 (0–0.594) | 1.125 (0.5–1.5)⁎⁎⁎ | 0.625 (0–1.25) | N/A |
| DAS28, mean (SD) | 2.0 (0.5) | 3.9 (1.1)⁎⁎⁎ | 3.0 (1.1) | N/A |
| Total SHS, Mdn (IQR) | 13 (2.5–36.6) | 31.3 (5.3–74.1)⁎ | 18.5 (3.8–60.8) | N/A |
| Erosion SHS, Mdn (IQR) | 5.8 (1–24.3) | 16.3 (2.5–37.3)⁎ | 10 (1.5–31.0) | N/A |
| Rheumatoid factor (titer), Mdn (IQR) | 80 (0−320) | 40 (0–320) | 80 (0–320) | N/A |
| Anti-CCP (titer), Mdn (IQR) | (n = 49) 163 (38.5–275) |
(n = 56) 181.5 (16.75–267) |
(n = 106) 164.5 (20.25–269.5) |
N/A |
| DMARD use, n (%) | 54 (84.4) | 68 (90.7) | 122 (87.8) | N/A |
| Biologic use, n (%) | 16 (25) | 22 (29.3) | 38 (27.3) | N/A |
| Prednisone use, n (%) | 7 (10.3) | 16 (25.3) ⁎ | 26 (18.7) | N/A |
| Bisphosphonate use, n (%) | 15 (23.4) | 30 (40.0) ⁎ | 45 (32.4) | N/A |
Mdn: median, SD: Standard deviation, IQR: Interquartile Range (25th–75th percentiles).
Anti-CCP: Anti-cyclic citrullinated peptide; DAS28: disease activity score (28 joints); DMARDs: Disease-modifying antirheumatic drugs; HAQ: health assessment questionnaire; PhGA: Physician global assessment; PGA: Patient global assessment; RA: Rheumatoid arthritis; Total/Erosion SHS: Total/Erosion score from the Sharp/van der Heijde modified method.
Non-Active RA and Active RA groups were compared
p < 0.05.
p < 0.001.
DAS28 status could not be evaluated at baseline in 1 patient.
More than 5 erosions (destructive RA) were present at the end of the study (V4) in 82 patients of the 98 (83%) who had X-rays in this long-term visit. Baseline characteristics of patients with erosive damage at 4 years included an older age, a longer disease duration with erosions already present at V1 in 70.7%, and a greater likelihood of being positive for anti-CCP2 (Suppl. Table S1).
3.2. Differences in CD14+ monocyte fractions of PBMCs (osteoclast precursors) associated with RA activity
Osteoclast precursors are included in the CD14+ monocyte fraction of PBMCs (Nicholson et al., 2000). Therefore, we sought to determine the relative abundance of CD14+ PBMCs in patients with RA and control subjects. At baseline, higher numbers of CD14+ monocytes/104 PBMCs were found in patients with active RA (median [IQR]: 1605 [1159–2266]) and with non-active RA (1812 [1257–2626]) compared to healthy controls (1353 [850–1752] CD14+/104 PBMCs, p = 0.0001, all RA vs. controls). When RA activity was evaluated as a categorical value, the proportion of CD14+ monocytes did not differ whether RA was active or not (p = 0.24) (Fig. 1a). However, a significant inverse correlation was found with DAS28 used as a continuous variable (r = −0.191, p = 0.025) (Fig. 1b and Suppl. Table S2). Similar results were obtained after adjustment for baseline treatments (prednisone, DMARDs, biologics or bisphosphonates) (data not shown).
Fig. 1.
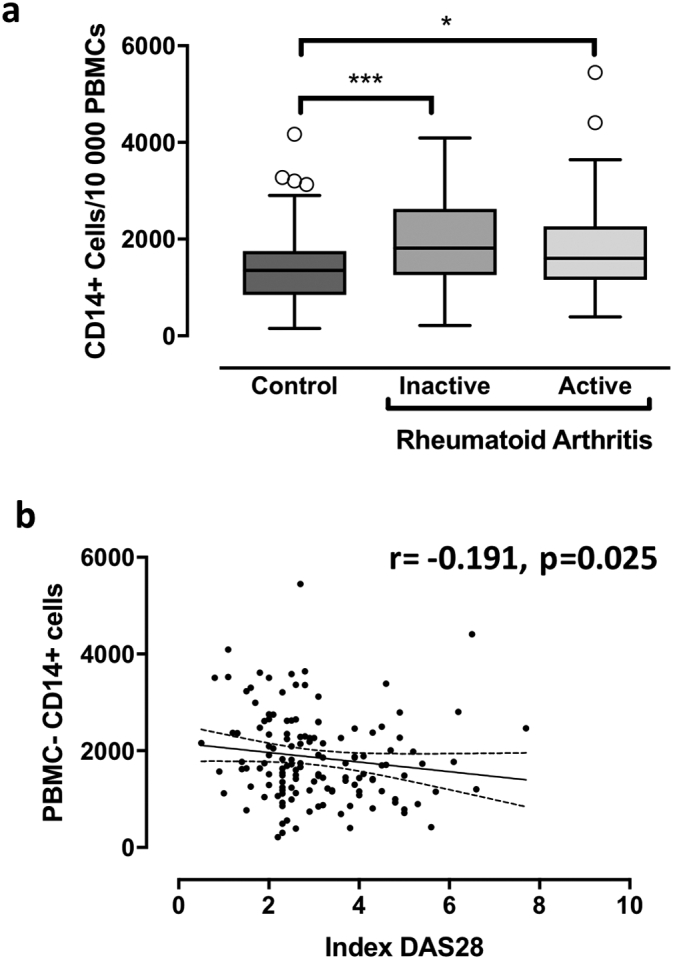
CD14+ cells in peripheral blood. PBMCs from patients with active or non-active RA or healthy controls were collected at baseline. 1a: CD14+cells: From PBMCs, CD14+ mononuclear cells were counted using standard flow cytology. Results are expressed as the number of CD14+ cells/10000 cells (mean ± SD). All graphs are presented as Box-and-Whisker Plots [median, IQR (25–75)]. *p < 0.05, ***p < 0.001 (comparisons as indicated). 1b: Correlation analysis: Correlation between RA activity (DAS 28) and the number of CD14+ cells/10000 cells was presented graphically (95% confident bands of the best fit diagonal line are shown).
The observation that, at baseline, fewer CD14+ precursors were present in PBMCs as RA activity increased, prompted us to evaluate the proportion of CD16+ cells in peripheral monocytes in addition to CD14+ cells at the following visits. Indeed, the proportion of CD14+ monocytes expressing CD16 may expand during inflammation and CD14 expression may decrease, giving rise to cell states less capable of osteoclast differentiation (Seeling et al., 2013). Although an inverse correlation was found at baseline between the proportion of CD14+ monocytes and DAS28 (Fig. 1a), the proportion of CD14+ or CD16+ positive cells were not associated with concomitant DAS28 over time (Suppl. Table S3). Nevertheless, both CD14+ and CD16+ single positive cells were highly correlated with each other and with CD14+CD16+ double-positive cells at all follow-up visits (Suppl. Table S3).
3.3. Differences in osteoclastogenic potential associated with RA activity at baseline
The potential for osteoclast differentiation from in vitro cultured precursors was quantified as the number of multinucleated TRAP-positive cells per culture well (Fig. 2a). We previously reported that osteoclast formation was significantly greater in cultures from patients with RA than controls (Durand et al., 2011). In the present study, our results indicated a greater number of osteoclasts generated by PBMCs from patients with non-active RA compared to those with active RA (median [IQR]: 458 [232–778] vs. 267 [114–477], respectively, padj = 0.006) and compared to control cultures (248 [57–463], padj < 0.001) (Fig. 2b). Furthermore, an inverse correlation was observed at baseline between the number of osteoclasts formed in culture and the DAS28 score used as a continuous variable (r = −0.202, p = 0.017) (Fig. 2c and Suppl. Table S2). Similar results were obtained after adjustment for baseline treatments (prednisone, DMARDs, biologics or bisphosphonates) (data not shown).
Fig. 2.

Osteoclastic phenotype at baseline: TRAP staining. PBMCs from patients with active or non-active RA or healthy controls were differentiated for 21 days with RANKL and M-CSF. 2a: TRAP-staining: Representative micrograph showing TRAP-stained osteoclast cultures, with arrows pointing at positive multinucleated cells (scale bar 50 μm). 2b: Number of TRAP-positive MNCs: the number of multinucleated cells (MNCs) (three or more nuclei) TRAP positive cells per well was evaluated in all groups. All graphs are presented as Box-and-Whisker Plots [median, IQR (25–75)]. **p < 0.01, ***p < 0.001 (comparisons as indicated). 2c: Correlation analysis: Correlation between RA activity (DAS 28) and the number of TRAP-positive MNCs was presented graphically (95% confident bands of the best fit diagonal line are shown).
Taken together, these results suggested a decreased potential to generate osteoclasts from PBMC-derived CD14+ cells from patients with active RA compared to those with inactive disease. In fact, the number of CD14+ cells required to generate osteoclasts in vitro was 6.6 [3–14.7] CD14+cells/osteoclast in active RA compared to only 4 [1.8–9] in non-active RA (active vs. non-active padj = 0.04), and 4.7 [2.6–14.6] in controls (active RA vs. controls padj = 1.0). Thus, at baseline, active RA was associated with the formation of fewer osteoclasts from peripheral precursors compared to non-active RA, and this low potential was not different to that observed in cells from controls.
3.4. Relationship of osteoclast apoptosis and bone resorption with RA activity at baseline
Cells from both active and non-active RA patients exhibited intrinsic resistance to serum starvation-induced apoptosis when compared to controls, with no difference between active and non-active states of RA (Fig. 3). After 24 h, apoptosis reached 22% [18-30] in healthy controls compared to 10% [4.25–18] in RA patients (p < 0.001), with apoptosis rates of 9% [4-18] in active RA and 12% [5–19.5] in non-active RA (active vs. non-active padj = 1.0). Finally, bone resorption by PBMC-derived osteoclasts was evaluated (Fig. 4a) and expressed as the area resorbed per osteoclast. There were no differences in the resorptive capacity of osteoclasts from subjects with active or non-active RA, compared to that of controls (Fig. 4b).
Fig. 3.
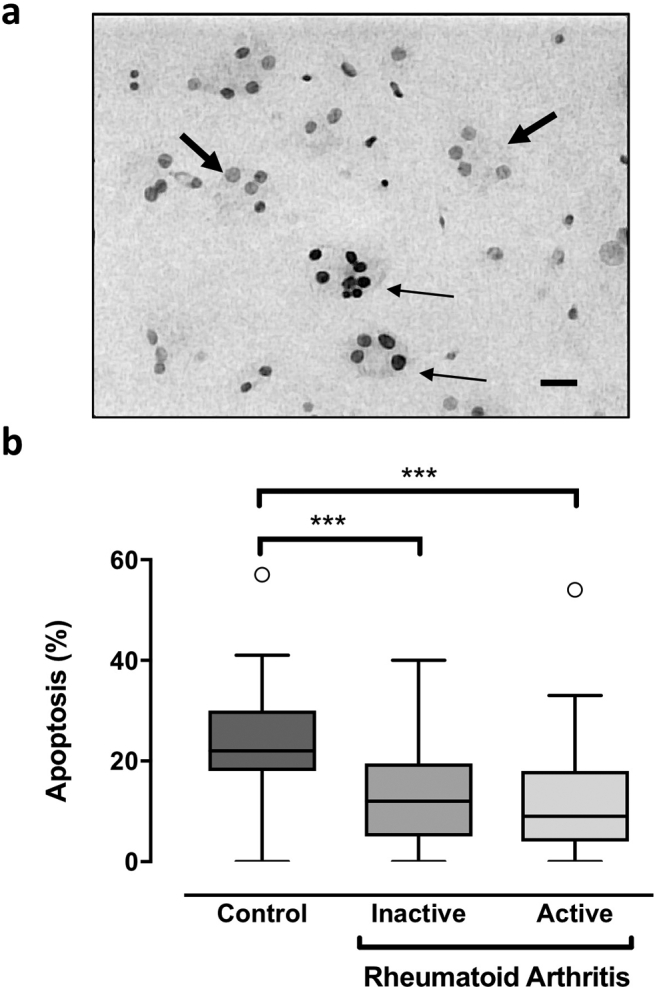
Osteoclastic phenotype at baseline: Apoptosis. PBMCs from patients with active or non-active RA or healthy controls were differentiated for 21 days with RANKL and M-CSF. 3a: Apoptosis: At the end of the PBMC cultures, 24 h after the M-CSF and RANKL had been removed, osteoclast apoptosis was evaluated using a TUNEL-derived method (TACS Blue). Nuclei appeared dark in apoptotic cells (fine arrows), clear in non-apoptotic cells (thick arrows) (scale bar 50 μm). 3b: Number of apoptotic MNCs: Results are expressed as the number of apoptotic MNCs over total MNCs. All graphs are presented as Box-and-Whisker Plots [median, IQR (25–75)]. ***p < 0.001 (comparisons as indicated).
Fig. 4.

Osteoclastic phenotype at baseline: Bone resorption. PBMCs from patients with active or non-active RA or healthy controls were differentiated for 21 days with RANKL and M-CSF. 4a: Bone resorption was assessed at the end of the differentiation period by Toluidine Blue staining of the bone slices (arrows) (scale bar 200 μm). 4b: Quantification: The resorbed surface area was quantified (ImageJ), and reported graphically to the number of osteoclasts (μm2/osteoclast). All graphs are presented as Box-and-Whisker Plots [median, IQR (25–75)]. 4c: Correlation analysis: Correlation between BMD and bone resorption was presented graphically (95% confident bands of the best fit diagonal line are shown).
Thus, at baseline, RA activity was associated with the formation of fewer osteoclasts from peripheral blood precursors compared to non-active RA, while in vitro bone resorption per osteoclast was similar, and osteoclasts were found to be resistant to apoptosis in RA regardless its activity.
Since peripheral inflammation may contribute to systemic bone loss (Kleyer et al., 2014; Tanaka and Ohira, 2018), we also assessed the association of inflammation and osteoclastic markers with BMD. We observed a negative correlation between in vitro bone resorption and 25-OH vitamin D serum levels (r = −0.191, p = 0.028) (Suppl. Table S2). Although no correlation was found between inflammation (DAS28) and BMD (T-score or gr/cm2) at the spine or femoral neck, we observed at baseline that BMD at the lumbar spine correlated positively with the resorption area obtained in vitro (r = 0.288, p = 0.001) (Fig. 4c and Suppl. Table S2).
3.5. No predictive value of osteoclastic parameters (PBMC CD14+ cells, PBMC-derived osteoclasts) on RA activity
Since RA activity had an impact on some osteoclastic parameters (decrease in the proportion of PBMC-CD14+ and in osteoclast formation in vitro), we investigated whether these parameters, like other clinical and biological factors, could predict the activity of the disease at the next visit. In a multivariate analysis, age, BMI and being treated with prednisone were independent predictors of active RA at the next visit. RA activity at time of assessment was the best predictor of the risk for active RA at the next visit (p < 0.001) (Table 2).
Table 2.
Univariate and multivariate model to evaluate the predictive value of having active RA at the subsequent visit.
| Univariate analysis |
Multivariate analysis |
|||
|---|---|---|---|---|
| RR (95% CI) | p-Value | RR (95% CI) | p-Value | |
| RA activity | 3.35 (2.48–4.52) | <0.001 | – | – |
| Osteoclasts (OCs) > 248/well | 1.07 (0.89–1.28) | 0.479 | – | – |
| PBMC CD14+ | 1.12 (0.90–1.38) | 0.304 | – | – |
| RA activity and in vitro OC formation (median 248a) | ||||
| Remission and OCs ≤ 248/well | 1 | 1 | ||
| Remission and OCs > 248/well | 2.06 (1.03–4.13) | 0.042 | 1.84 (0.93–3.63) | 0.079 |
| Active and OCs ≤ 248/well | 5.41 (2.87–10.17) | <0.001 | 4.88 (2.65–8.96) | <0.001 |
| Active and OCs > 248/well | 5.21 (2.79–9.73) | <0.001 | 4.56 (2.48–8.40) | <0.001 |
| RA activity and PBMC CD14+ (median 13.5a) |
||||
| Remission and CD14+ ≤ 13.5% | 1 | 1 | ||
| Remission and CD14+ > 13.5% | 0.96 (0.54–1.71) | 0.896 | 0.94 (0.51–1.70) | 0.826 |
| Active and CD14+ ≤ 13.5% | 2.82 (1.75–4.55) | <0.001 | 2.72 (1.65–4.51) | <0.0001 |
| Active and CD14+ > 13.5% | 3.37 (2.06–5.51) | <0.001 | 3.28 (1.96–5.51) | <0.0001 |
RA: Rheumatoid arthritis; OC: Osteoclasts; PBMCs: peripheral blood mononuclear cells.
Median set as cut-off to differentiate between high and low values of the parameter of interest.
When combining RA current activity with in vitro osteoclast formation (osteoclast number/well) or proportion of CD14+ cells in PBMCs (%), RA activity remained strongly predictive of RA activity at the subsequent visit regardless of the number of osteoclasts in uni- and multivariate analyses (p < 0.001). In the case of non-active disease/remission (DAS28 < 2.6), a high level of osteoclast formation from PBMCs in vitro was also associated with the risk of having active RA at the next visit (RR [95% CI] = 2.06 [1.03–4.13], p = 0.042) (Table 2).
3.6. Association between osteoclast parameters and RA bone phenotype (erosions)
While RA activity fluctuated with time at the individual level, the average cohort disease activity scores (DAS28) remained stable. However, RA progression was illustrated by the accumulation of bone erosions over time (erosion SHS scores) (Suppl. Table S4). Analyses of osteoclastic parameters at baseline and each visit up to a median of 40-months confirmed earlier data (Durand et al., 2011) that, among those parameters, in vitro bone resorption and apoptosis rates of mature osteoclasts remain constant over time. On the other hand, the number of PBMC-derived CD14+ osteoclast precursors and the number of osteoclasts formed in PBMC cultures, which were shown to be inversely related to RA activity, varied independently over time (Suppl. Table S4). Therefore, some osteoclastic parameters may reflect intrinsic characteristics of osteoclasts that remain stable over time, whereas other characteristics may change in parallel with the activity of RA, such as the number of osteoclasts formed in vitro and the proportion of CD14+ cells among PBMCs.
Analyses using the GLM or GEE models comparing the data with respect to baseline values and those at the previous visit, found inverse correlations of the number of PBMC-derived CD14+ cells and the number of osteoclasts differentiated in vitro with erosion SHS scores. Similarly, in vitro bone resorption was inversely correlated with changes in erosion SHS scores over time, as was apoptosis. In the analysis using the GLM model, changes in femoral neck BMD and T-score correlated negatively with the number of osteoclasts formed in vitro (increasing bone density was associated with the formation of fewer osteoclasts) (Suppl. Table S5).
To verify whether some osteoclastic parameters were associated with long-term bone erosions and a destructive RA phenotype, we then compared the profile of the osteoclast parameters obtained initially (V1) to the erosive (erosion score more of at least 5 at V4) or non-erosive (erosion score <5 at V4) status of RA patients at 4 years. Of the 102 patients evaluated at V4, 98 had X-rays, 82 had erosive RA and 16 had non-erosive RA. There was no association between the osteoclastic profile at baseline and the erosive status at V4 (Table 3).
Table 3.
Osteoclast parameters and the erosive progression in RA.
| Erosion SHS < 5 at V4 |
Erosion SHS ≥ 5 at V4 |
p | p-adj⁎ | |||
|---|---|---|---|---|---|---|
| n | median (IQR) | n | median (IQR) | |||
| All | n = 16 | n = 82 | ||||
| OCs/well V1 | 16 | 286.5 (210.0–744.5) | 82 | 372.5 (153.0–668.0) | 0.5446 | 0.5307 |
| CD14% PBMCs V1 | 16 | 15.0 (9.9–19.6) | 82 | 17.4 (12.6–23.7) | 0.1440 | 0.2792 |
| Resorption (μm2) V1 | 15 | 82,484 (0–1,264,508) | 82 | 0 (0–836,372) | 0.3865 | 0.7158 |
| Apoptosis (%) V1 | 13 | 17.0 (12.0–18.0) | 58 | 7.5 (4.0–15.0) | 0.1081 | 0.2531 |
| RA > 5 years | ||||||
| OCs/well V1 | 5 | 592.0 (306.0–783.0) | 64 | 334.5 (124.0–682.5) | 0.2467 | 0.4193 |
| CD14% PBMCs V1 | 5 | 19.1 (12.3–21.6) | 64 | 17.3 (12.5–23.7) | 0.9631 | 0.8562 |
| Resorption (μm2) V1 | 4 | 85,782 (33411–684,625) | 64 | 0 (0–404,559) | 0.5374 | 0.6845 |
| Apoptosis (%) V1 | 4 | 0.0 (0.0–8.5) | 44 | 8.0 (4.5–16.5) | 0.1144 | 0.1957 |
| RA ≤ 5 years | ||||||
| OCs/well V1 | 11 | 258.0 (191.0–706.0) | 18 | 487.0 (229.0–535.0) | 0.6691 | 0.9243 |
| CD14% PBMCs V1 | 11 | 14.9 (7.7–18.6) | 18 | 17.8 (12.9–25.9) | 0.1265 | 0.1817 |
| Resorption (μm2) V1 | 11 | 82,484 (0–2,733,971) | 18 | 482,357 (0–1,410,990) | 0.7816 | 0.9137 |
| Apoptosis (%) V1 | 9 | 18.0 (17.0–21.0) | 14 | 5.0 (3.0–12.0) | 0.0036 | 0.0287 |
IQR: Interquartile Range (25th–75th percentiles).
Erosion SHS: Erosion score from the Sharp/van der Heijde modified method; OC: osteoclast.
p adjusted for DAS28.
However, since bone erosions typically occur more rapidly during the course of RA (Barra et al., 2014), we analyzed the correlation between baseline osteoclast parameters and the final RA phenotype (erosive or not) separately in subjects with RA of <5 years duration at baseline (“early” RA) and in subjects with RA of longer duration. Interestingly, in subjects who did evolve to long-term erosive status at V4, osteoclasts were already more resistant to apoptosis at baseline compared to those of subjects who remained non-erosive, even after DAS28 adjustment. Osteoclast resistance to apoptosis was thus already present at baseline, defining de facto a more aggressive phenotype in patients with recent RA (<5 years). None of the other baseline osteoclast parameters (proportion of CD14+ cells in PBMCs, in vitro osteoclast number and resorptive activity) were associated with long-term erosive status (Table 3).
3.7. Validation cohort: EUPA study
Considering our result showing a decrease in osteoclast formation from PBMCs of subjects with active RA versus those with non-active RA, we wondered whether this result would be reproduced in a cohort of patients with early RA. To this end, we studied the formation of osteoclasts from PBMCs of subjects included in the EUPA study, an independent cohort with recent RA (Suppl. Table S6). At inclusion in the EUPA study, with a median of 4 months after onset of inflammatory symptoms, all patients had active RA (Carrier et al., 2009).
Using a GEE or a GLM model, there was an inverse correlation between the number of osteoclasts derived from PBMCs at baseline (V1) and the occurrence of erosions or Sharp score variations over follow-up (Table 4). We also observed an increase in the number of osteoclasts derived from PBMCs between baseline (V1) and 30 months, concomitant with a general decrease in disease activity (DAS28 remission present in 70% at 30 months), further supporting the observations made in the IODA cohort.
Table 4.
Osteoclast numbers derived from PBMCs in the EUPA cohort.
| GLM model | Osteoclast number V1 |
Osteoclast number > 248 V1a |
|---|---|---|
| Estimate (SE) | Estimate (SE) | |
| Erosion SHS (log) | −0.0001 (0.0001)⁎ | −0.2287 (0.0575)⁎⁎⁎ |
| ∆Erosion SHS from V1 | −0.0006 (0.0002)⁎⁎ | −0.7558 (0.2067)⁎⁎⁎ |
| DAS28-CRP (log) | −0.000004 (0.00001) | 0.0009 (0.0161) |
| GEE model | Osteoclast number V1 |
Osteoclast number > 248 V1a |
|---|---|---|
| RR (95% CI) | RR (95% CI) | |
| Erosion SHS ≥ 5 | 1 (1–1)⁎ | 0.18 (0.07–0.47)⁎⁎⁎ |
| ∆Erosion SHS ≥ 5 from V1 | 1 (0.99–1) | 0.21 (0.03–1.66) |
| DAS28-CRP remission | 1 (1–1) | 1.18 (0.95–1.47) |
Erosion SHS: Erosion score from the Sharp/van der Heijde modified method.
Analysis using the general linear model (GLM) with continuous outcomes, established correlations between initial osteoclastic parameters (V1) and the progression of RA (activity, erosion). Osteoclastic parameters and RA progression (DAS28, erosions) were evaluated during yearly visits from baseline to 4 years, a design allowing the use of generalized estimating equations (GEE) analysis with repeated measures using categorical variables for outcomes.
p adjusted for DAS28.
p < 0.05.
p < 0.01.
p < 0.001.
Median set as cut-off to differentiate between high and low values of the parameter of interest.
4. Discussion
In the rheumatoid synovium, proinflammatory mediators encompass potent osteoclastogenic factors, acting through an increase in RANKL expression by stromal/osteoblastic cells or lymphoid cells (Roux and Brown, 2009), or synergistically with or independently of RANKL, as has been reported for IL-1, TNFα (Kobayashi et al., 2000; Lam et al., 2000), IL-6 and IL-11 (Kudo et al., 2003). Noteworthy, some anti-inflammatory mediators can also promote osteoclastogenesis, such as TGF-β, which can act via RANKL expression or independently (Itonaga et al., 2004). Nevertheless, effective in vivo osteoclastogenesis requires far more than just appropriate cytokines; it requires a spatially and timely orchestrated recruitment of CD14+ circulating osteoclast precursors to the bone, their activation and their fusion to produce mature osteoclasts. The egress of the CD14+ osteoclast precursors from blood is directed by a myriad of factors diffusing from within the joints, including TNF-α (Ming et al., 1987), MIP-1α (Oba et al., 2005) and RANKL (Breuil et al., 2003), which are among the cardinal mediators of inflammation and osteoclast-mediated periarticular bone destruction.
We previously found that the phenotype of osteoclast precursors isolated from PBMCs of RA patients carries a “rheumatoid imprint”, with a higher proportion of CD14+ cells and a greater potential for their differentiation into mature osteoclasts, which are more numerous and resistant to apoptosis, compared to controls (Durand et al., 2011). In the current study, we investigated correlations between disease activity and PBMC-derived osteoclast parameters. While in vitro bone resorption and osteoclast apoptosis were not influenced by variation in the RA disease activity score, we observed that at baseline the number of osteoclasts formed in vitro was inversely correlated to the DAS28 (fewer osteoclasts in active RA compared to non-active RA), and a similar trend emerged when looking at the proportion of CD14+ cells in PBMCs.
Thus, the formation of osteoclasts in vitro was decreased when blood was sampled in an inflammatory context (active RA), with a decrease in the potential to generate osteoclasts from PBMC-derived CD14+ cells. One explanation for our findings could be related to the recruitment of osteoclast precursors from the blood to the periarticular tissues when disease is active, leading to depletion of osteoclast precursors in the blood, although only a small proportion of blood monocytes are ultimately recruited to the inflamed synovium. The lower potential to generate in vitro osteoclasts could also be related to a different commitment of peripheral CD14+ monocytes during rheumatoid inflammation with an expansion of CD16+ monocytes and an increase in a subpopulation of CD14lowCD16+ cells whose ability to produce osteoclasts is diminished (Seeling et al., 2013; Kawanaka et al., 2002; Wong et al., 2012). However, we did not refine our analysis with CD14low/high and CD16 expression at baseline, while an inverse correlation of CD14+ proportion and osteoclast formation with RA activity was observed at that time.
It is also possible that, in active RA, a systemic proinflammatory factor such as IL-12 could inhibit the osteoclastogenic capacity of peripheral monocytes (Furst and Emery, 2014; Horwood et al., 2001). Alternatively, when RA is more quiescent, a systemic factor with anti-inflammatory potential may bias monocyte development toward the osteoclastic lineage. TGF-ß could be a candidate cytokine in this context. Indeed, TGF-ß is increased in the blood of patients with RA (Mieliauskaite et al., 2009) as well as in the synovium (Gonzalo-Gil et al., 2013) and has the intrinsic capacity to promote osteoclastogenesis independently of RANKL (Itonaga et al., 2004). Interestingly, TGF-ß has also been shown to modulate the expression of CD16, with an induction of CD16 on CD14+ monocytes in RA synovial fluid (Yoon et al., 2014).
In the prospective analysis of the cohort, the vast majority of associations between osteoclastic parameters and a later progression of RA activity or erosion scores went in an unexpected direction, with fewer precursors, less in vitro osteoclast formation or bone resorption predicting increases in activity or erosive scores later. This “anti-osteoclast” profile was associated with RA activity, and most likely induced by rheumatoid inflammation, as discussed above. Indeed, and as expected, RA activity at time of evaluation was the best predictor of the risk of having active RA at a subsequent visit. Importantly, our results were validated in an independent longitudinal cohort from the EUPA study, in which all subjects had active polyarthritis at inclusion, and marked decrease in disease activity at each subsequent follow-up (Carrier et al., 2009). In these subjects, the number of osteoclasts formed from PBMCs was inversely correlated with erosion scores and disease activity at subsequent visits.
In a Canadian multicenter study, bone erosions were present in a quarter of patients at first presentation and new erosions developed in >20% of seropositive patients over the next 12 months (Barra et al., 2014). Early identification of patients at risk of evolution toward an erosive phenotype is crucial as structural damage is strongly associated with loss of function. Among the different osteoclast parameters evaluated, we found that resistance to apoptosis observed in mature osteoclasts of patients with recent RA onset correlated with a long-term erosive phenotype. However, the number of patients in the non-erosive IODA subgroup was low, limiting the strength of the results, that should be to confirmed in a larger study. An increase in osteoclast survival could explain at least in part a more aggressive (erosive) profile of RA and could be related to the underlying rheumatoid process affecting already genetically or epigenetically primed osteoclasts. Indeed, the inflammatory microenvironment in arthritis can alter the sensitivity of osteoclasts to apoptosis; for example, TNFα can stimulate anti-apoptotic Bcl-xL protein expression in osteoclasts via the transcription factor Est-2 and thus modulate apoptotic threshold (Zhang et al., 2005).
Our results also support an impact of the osteoclast phenotype on BMD at the femoral neck or lumbar spine, although they show some inconsistencies. At baseline, lumbar spine BMD correlated positively with in vitro osteoclast bone resorption, a surprising relation, nonetheless fully consistent with the inverse correlation observed at baseline between inflammation and in vitro osteoclast formation. However, in the GLM model, establishing correlations between initial osteoclastic parameters and systemic or local bone loss during the evolution of RA, a negative correlation was observed between femoral neck BMD and the number of osteoclasts formed in vitro. While systemic inflammation contributes to systemic bone loss (Kleyer et al., 2014), it is likely that additional factors play a major role (i.e., the classic factors of osteoporosis such as hypogonadism or corticosteroid therapy). Conversely, postmenopausal status could contribute to periarticular bone loss (Guler-Yuksel et al., 2009).
Nonetheless, our in vitro study also has some limitations. We did not evaluate cells at multiple times during osteoclast differentiation, which could have provided information on the early stages of osteoclast formation and the impact of disease activity on apoptosis and osteoclast number. In addition, although the ability to resorb bone represents a defining marker for terminal osteoclastic differentiation, its quantification is challenging, particularly in primary culture of human cells. Indeed, variability among cells from individual donors could have accounted for the absence of a major impact of disease activity on bone resorption in vitro.
5. Conclusions
We studied several parameters of osteoclasts derived in vitro from peripheral blood of patients with RA and controls. We observed fewer osteoclasts generated in vitro from PBMCs, and less PBMC CD14+ precursors when blood was sampled in active RA, and this anti-osteoclastic profile was associated with subsequent RA flare-ups. This reduced potential to generate osteoclasts could be linked to rheumatoid inflammation itself. As expected, the strongest predictive factor of RA activity at the next annual visit was RA activity itself, limiting the utility of these osteoclast parameters to predict resistance to treatment and subsequent erosions. However, the erosive phenotype of RA was associated with resistance to apoptosis of PBMC-derived osteoclasts, a parameter not influenced by RA activity and stable over time in the patients. Thus, resistance to apoptosis is a potential biomarker for predicting erosiveness in RA. Special care should be paid during the development of biomarkers derived from peripheral blood cells. Indeed, the relationship between in vitro osteoclast parameters and in vivo erosiveness can be altered by factors such as inflammation that influence the distribution and phenotypes of peripheral cells. Therefore, the utility of osteoclast characteristics in the definition of the long-term RA phenotypes, alone or as part of a composite index, remains to be confirmed.
The following are the supplementary data related to this article.
Comparison of baseline characteristics in RA patients remaining nonerosive versus those who became erosive over 4 years.
Correlation at baseline between osteoclast parameters, demographics, RA characteristics and bone phenotype.
Proportion of CD14- and CD16-positive cells over time and RA activity.
Changes in osteoclast parameters, RA activity and bone loss (erosion or BMD) over time.
Analyses over time using GLM (general linear model) and GEE (generalized estimating equations) models.
Patient demographics and baseline disease characteristics (EUPA cohort).
Author contributions
AJF, MFM, SVK, GB, RER, SJD devised the study; HAC, MD, PhD conducted the experiments; NC, AF, HAC, MD, PhD, GB, SR acquired and analyzed the data, and HAC and SR drafted the paper. All authors reviewed critically the data and revised the manuscript. All authors read and approved the final version.
Funding
This work was supported by CIHR (Canadian Institutes of Health Research) IMHA (Institute of Musculoskeletal Health and Arthritis) New Emerging Team Grant (FRN #QNT-83330), the Canadian Arthritis Network (2006- SRID-IJD-01) and Pfizer Canada. The sponsors were not involved in the study and had no influence on the publishing of data.
Transparency document
Transparency Document
CRediT authorship contribution statement
H. Allard-Chamard:Investigation, Formal analysis, Writing - original draft, Writing - review & editing.N. Carrier:Methodology, Formal analysis, Writing - review & editing.P. Dufort:Investigation, Formal analysis, Writing - review & editing.M. Durand:Investigation, Formal analysis, Writing - review & editing.A.J. de Brum-Fernandes:Conceptualization, Methodology, Formal analysis, Writing - review & editing, Funding acquisition.G. Boire:Conceptualization, Methodology, Formal analysis, Writing - review & editing.S.V. Komarova:Conceptualization, Methodology, Writing - review & editing.S.J. Dixon:Conceptualization, Methodology, Writing - review & editing.R.E. Harrison:Conceptualization, Methodology, Writing - review & editing.M.F. Manolson:Conceptualization, Methodology, Writing - review & editing.S. Roux:Formal analysis, Writing - original draft, Writing - review & editing.
Declaration of competing interest
SR reports personal fees from Amgen Canada, outside the submitted work; Dr. Boire reports personal fees and grants from Amgen, BMS, Celgene, Eli Lilly, Pfizer, Merck, and grants from Abbvie, Novartis, Roche, and UCB, outside the submitted work; all other authors confirm that they have no conflicts of interest.
Acknowledgements
We are especially indebted to our study coordinator Noémie Poirier for her continuing involvement and dedication.
Footnotes
The Transparency document associated with this article can be found, in online version.
References
- Aletaha D., Neogi T., Silman A.J., Funovits J., Felson D.T., Bingham C.O., 3rd, Birnbaum N.S., Burmester G.R., Bykerk V.P., Cohen M.D., Combe B., Costenbader K.H., Dougados M., Emery P., Ferraccioli G., Hazes J.M., Hobbs K., Huizinga T.W., Kavanaugh A., Kay J., Kvien T.K., Laing T., Mease P., Menard H.A., Moreland L.W., Naden R.L., Pincus T., Smolen J.S., Stanislawska-Biernat E., Symmons D., Tak P.P., Upchurch K.S., Vencovsky J., Wolfe F., Hawker G. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–2581. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- Barra L., Pope J.E., Orav J.E., Boire G., Haraoui B., Hitchon C., Keystone E.C., Thorne J.C., Tin D., Bykerk V.P., Investigators C. Prognosis of seronegative patients in a large prospective cohort of patients with early inflammatory arthritis. J. Rheumatol. 2014;41(12):2361–2369. doi: 10.3899/jrheum.140082. [DOI] [PubMed] [Google Scholar]
- Breuil V., Schmid-Antomarchi H., Schmid-Alliana A., Rezzonico R., Euller-Ziegler L., Rossi B. The receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) is a new chemotactic factor for human monocytes. FASEB J. 2003;17(12):1751–1753. doi: 10.1096/fj.02-1188fje. [DOI] [PubMed] [Google Scholar]
- Carrier N., Cossette P., Daniel C., de Brum-Fernandes A., Liang P., Menard H.A., Boire G. The DERAA HLA-DR alleles in patients with early polyarthritis: protection against severe disease and lack of association with rheumatoid arthritis autoantibodies. Arthritis Rheum. 2009;60(3):698–707. doi: 10.1002/art.24353. [DOI] [PubMed] [Google Scholar]
- Chamoux E., Houde N., L’Eriger K., Roux S. Osteoprotegerin decreases human osteoclast apoptosis by inhibiting the TRAIL pathway. J. Cell. Physiol. 2008;216(2):536–542. doi: 10.1002/jcp.21430. [DOI] [PubMed] [Google Scholar]
- Durand M., Boire G., Komarova S.V., Dixon S.J., Sims S.M., Harrison R.E., Nabavi N., Maria O., Manolson M.F., Mizianty M., Kurgan L., de Brum-Fernandes A.J. The increased in vitro osteoclastogenesis in patients with rheumatoid arthritis is due to increased percentage of precursors and decreased apoptosis - the In Vitro Osteoclast Differentiation in Arthritis (IODA) study. Bone. 2011;48(3):588–596. doi: 10.1016/j.bone.2010.10.167. [DOI] [PubMed] [Google Scholar]
- Durand M., Komarova S.V., Bhargava A., Trebec-Reynolds D.P., Li K., Fiorino C., Maria O., Nabavi N., Manolson M.F., Harrison R.E., Dixon S.J., Sims S.M., Mizianty M.J., Kurgan L., Haroun S., Boire G., de Fatima Lucena-Fernandes M., de Brum-Fernandes A.J. Monocytes from patients with osteoarthritis display increased osteoclastogenesis and bone resorption: the In Vitro Osteoclast Differentiation in Arthritis study. Arthritis Rheum. 2013;65(1):148–158. doi: 10.1002/art.37722. [DOI] [PubMed] [Google Scholar]
- Fransen J., Creemers M.C., Van Riel P.L. Remission in rheumatoid arthritis: agreement of the disease activity score (DAS28) with the ARA preliminary remission criteria. Rheumatology (Oxford) 2004;43(10):1252–1255. doi: 10.1093/rheumatology/keh297. [DOI] [PubMed] [Google Scholar]
- Furst D.E., Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine-associated cell targets. Rheumatology (Oxford) 2014;53(9):1560–1569. doi: 10.1093/rheumatology/ket414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M., Sebald H.J., Villiger P.M., Hofstetter W., Seitz M. Infliximab inhibits bone resorption by circulating osteoclast precursor cells in patients with rheumatoid arthritis and ankylosing spondylitis. Ann. Rheum. Dis. 2008;67(5):620–624. doi: 10.1136/ard.2007.076711. [DOI] [PubMed] [Google Scholar]
- Goekoop-Ruiterman Y.P., de Vries-Bouwstra J.K., Allaart C.F., van Zeben D., Kerstens P.J., Hazes J.M., Zwinderman A.H., Ronday H.K., Han K.H., Westedt M.L., Gerards A.H., van Groenendael J.H., Lems W.F., van Krugten M.V., Breedveld F.C., Dijkmans B.A. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum. 2008;58(2 Suppl):S126–S135. doi: 10.1002/art.23364. [DOI] [PubMed] [Google Scholar]
- Gonzalo-Gil E., Criado G., Santiago B., Dotor J., Pablos J.L., Galindo M. Transforming growth factor (TGF)-beta signalling is increased in rheumatoid synovium but TGF-beta blockade does not modify experimental arthritis. Clin. Exp. Immunol. 2013;174(2):245–255. doi: 10.1111/cei.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravallese E.M., Manning C., Tsay A., Naito A., Pan C., Amento E., Goldring S.R. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43(2):250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Guler-Yuksel M., Allaart C.F., Goekoop-Ruiterman Y.P., de Vries-Bouwstra J.K., van Groenendael J.H., Mallee C., de Bois M.H., Breedveld F.C., Dijkmans B.A., Lems W.F. Changes in hand and generalised bone mineral density in patients with recent-onset rheumatoid arthritis. Ann. Rheum. Dis. 2009;68(3):330–336. doi: 10.1136/ard.2007.086348. [DOI] [PubMed] [Google Scholar]
- Hanley J.A., Negassa A., Edwardes M.D., Forrester J.E. Statistical analysis of correlated data using generalized estimating equations: an orientation. Am. J. Epidemiol. 2003;157(4):364–375. doi: 10.1093/aje/kwf215. [DOI] [PubMed] [Google Scholar]
- Hasegawa T., Kikuta J., Sudo T., Matsuura Y., Matsui T., Simmons S., Ebina K., Hirao M., Okuzaki D., Yoshida Y., Hirao A., Kalinichenko V.V., Yamaoka K., Takeuchi T., Ishii M. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat. Immunol. 2019;20(12):1631–1643. doi: 10.1038/s41590-019-0526-7. [DOI] [PubMed] [Google Scholar]
- Hirayama T., Danks L., Sabokbar A., Athanasou N.A. Osteoclast formation and activity in the pathogenesis of osteoporosis in rheumatoid arthritis. Rheumatology (Oxford) 2002;41(11):1232–1239. doi: 10.1093/rheumatology/41.11.1232. [DOI] [PubMed] [Google Scholar]
- Horwood N.J., Elliott J., Martin T.J., Gillespie M.T. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J. Immunol. 2001;166(8):4915–4921. doi: 10.4049/jimmunol.166.8.4915. [DOI] [PubMed] [Google Scholar]
- Itonaga I., Sabokbar A., Sun S.G., Kudo O., Danks L., Ferguson D., Fujikawa Y., Athanasou N.A. Transforming growth factor-beta induces osteoclast formation in the absence of RANKL. Bone. 2004;34(1):57–64. doi: 10.1016/j.bone.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Kawanaka N., Yamamura M., Aita T., Morita Y., Okamoto A., Kawashima M., Iwahashi M., Ueno A., Ohmoto Y., Makino H. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46(10):2578–2586. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- Kleyer A., Finzel S., Rech J., Manger B., Krieter M., Faustini F., Araujo E., Hueber A.J., Harre U., Engelke K., Schett G. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann. Rheum. Dis. 2014;73(5):854–860. doi: 10.1136/annrheumdis-2012-202958. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Takahashi N., Jimi E., Udagawa N., Takami M., Kotake S., Nakagawa N., Kinosaki M., Yamaguchi K., Shima N., Yasuda H., Morinaga T., Higashio K., Martin T.J., Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000;191(2):275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo O., Sabokbar A., Pocock A., Itonaga I., Fujikawa Y., Athanasou N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32(1):1–7. doi: 10.1016/s8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- Lam J., Takeshita S., Barker J.E., Kanagawa O., Ross F.P., Teitelbaum S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 2000;106(12):1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Schwarz E.M., O’Keefe R.J., Ma L., Boyce B.F., Xing L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J. Bone Miner. Res. 2004;19(2):207–213. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- Lubberts E., Oppers-Walgreen B., Pettit A.R., Van Den Bersselaar L., Joosten L.A., Goldring S.R., Gravallese E.M., Van Den Berg W.B. Increase in expression of receptor activator of nuclear factor kappaB at sites of bone erosion correlates with progression of inflammation in evolving collagen-induced arthritis. Arthritis Rheum. 2002;46(11):3055–3064. doi: 10.1002/art.10607. [DOI] [PubMed] [Google Scholar]
- Manolson M.F., Yu H., Chen W., Yao Y., Li K., Lees R.L., Heersche J.N. The a3 isoform of the 100-kDa V-ATPase subunit is highly but differentially expressed in large (>or=10 nuclei) and small (<or= nuclei) osteoclasts. J. Biol. Chem. 2003;278(49):49271–49278. doi: 10.1074/jbc.M309914200. [DOI] [PubMed] [Google Scholar]
- McInnes I.B., Schett G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011;365(23):2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- Mieliauskaite D., Venalis P., Dumalakiene I., Venalis A., Distler J. Relationship between serum levels of TGF-beta1 and clinical parameters in patients with rheumatoid arthritis and Sjogren’s syndrome secondary to rheumatoid arthritis. Autoimmunity. 2009;42(4):356–358. doi: 10.1080/08916930902831977. [DOI] [PubMed] [Google Scholar]
- Ming W.J., Bersani L., Mantovani A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J. Immunol. 1987;138(5):1469–1474. [PubMed] [Google Scholar]
- Miossec P., Verweij C.L., Klareskog L., Pitzalis C., Barton A., Lekkerkerker F., Reiter S., Laslop A., Breedveld F., Abadie E., Flamion B., Dere W., Mpofu S., Goel N., Ethgen D., Mitlak B., Ormarsdottir S., Rao R., Tsouderos Y., Reginster J.Y., E. Group for Respect of, S. Excellence in Biomarkers and personalised medicine in rheumatoid arthritis: a proposal for interactions between academia, industry and regulatory bodies. Ann. Rheum. Dis. 2011;70(10):1713–1718. doi: 10.1136/ard.2011.154252. [DOI] [PubMed] [Google Scholar]
- Nicholson G.C., Malakellis M., Collier F.M., Cameron P.U., Holloway W.R., Gough T.J., Gregorio-King C., Kirkland M.A., Myers D.E. Induction of osteoclasts from CD14-positive human peripheral blood mononuclear cells by receptor activator of nuclear factor kappaB ligand (RANKL) Clin Sci (Lond) 2000;99(2):133–140. [PubMed] [Google Scholar]
- Oba Y., Lee J.W., Ehrlich L.A., Chung H.Y., Jelinek D.F., Callander N.S., Horuk R., Choi S.J., Roodman G.D. MIP-1alpha utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp. Hematol. 2005;33(3):272–278. doi: 10.1016/j.exphem.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Pettit A.R., Ji H., von Stechow D., Muller R., Goldring S.R., Choi Y., Benoist C., Gravallese E.M. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 2001;159(5):1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redlich K., Hayer S., Ricci R., David J.P., Tohidast-Akrad M., Kollias G., Steiner G., Smolen J.S., Wagner E.F., Schett G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Invest. 2002;110(10):1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Brown J. Osteoclast apoptosis in rheumatic diseases characterized by a high level of bone resorption (osteoporosis, rheumatoid arthritis, myeloma and Paget’s disease of bone) Curr Rev Rheumatol. 2009;5(2):98–110. [Google Scholar]
- Schett G., Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012;8(11):656–664. doi: 10.1038/nrrheum.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeling M., Hillenhoff U., David J.P., Schett G., Tuckermann J., Lux A., Nimmerjahn F. Inflammatory monocytes and Fcgamma receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc. Natl. Acad. Sci. U. S. A. 2013;110(26):10729–10734. doi: 10.1073/pnas.1301001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Ohira T. Mechanisms and therapeutic targets for bone damage in rheumatoid arthritis, in particular the RANK-RANKL system. Curr. Opin. Pharmacol. 2018;40:110–119. doi: 10.1016/j.coph.2018.03.006. [DOI] [PubMed] [Google Scholar]
- van der Heijde D. How to read radiographs according to the sharp/van der Heijde method. J. Rheumatol. 2000;27(1):261–263. [PubMed] [Google Scholar]
- Wong K.L., Yeap W.H., Tai J.J., Ong S.M., Dang T.M., Wong S.C. The three human monocyte subsets: implications for health and disease. Immunol. Res. 2012;53(1–3):41–57. doi: 10.1007/s12026-012-8297-3. [DOI] [PubMed] [Google Scholar]
- Yoon B.R., Yoo S.J., Choi Y., Chung Y.H., Kim J., Yoo I.S., Kang S.W., Lee W.W. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA) PLoS One. 2014;9(10) doi: 10.1371/journal.pone.0109775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Badell I.R., Schwarz E.M., Boulukos K.E., Yao Z., Boyce B.F., Xing L. Tumor necrosis factor prevents alendronate-induced osteoclast apoptosis in vivo by stimulating Bcl-xL expression through Ets-2. Arthritis Rheum. 2005;52(9):2708–2718. doi: 10.1002/art.21236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of baseline characteristics in RA patients remaining nonerosive versus those who became erosive over 4 years.
Correlation at baseline between osteoclast parameters, demographics, RA characteristics and bone phenotype.
Proportion of CD14- and CD16-positive cells over time and RA activity.
Changes in osteoclast parameters, RA activity and bone loss (erosion or BMD) over time.
Analyses over time using GLM (general linear model) and GEE (generalized estimating equations) models.
Patient demographics and baseline disease characteristics (EUPA cohort).
Transparency Document