

Abstract
The identification of activating mutations in NOTCH1 in 50% of T cell acute lymphoblastic leukemia has generated interest in elucidating how these mutations contribute to oncogenic transformation and in targeting the pathway. A phenotypic screen identified compounds that interfere with trafficking of Notch and induce apoptosis via an endoplasmic reticulum (ER) stress mechanism. Target identification approaches revealed a role for SLC39A7 (ZIP7), a zinc transport family member, in governing Notch trafficking and signaling. Generation and sequencing of a compound-resistant cell line identified a V430E mutation in ZIP7 that confers transferable resistance to the compound NVS-ZP7–4. NVS-ZP7–4 altered zinc in the ER, and an analog of the compound photoaffinity labeled ZIP7 in cells, suggesting a direct interaction between the compound and ZIP7. NVS-ZP7–4 is the first reported chemical tool to probe the impact of modulating ER zinc levels and investigate ZIP7 as a novel druggable node in the Notch pathway.
Advances in genomics have led to many new medicines through target-based approaches1,2. But not all proteins have validated as good drug targets and not all target-based screens identify ligands. For example, many oncogenes in cancer, for example, MYC and RAS, remain undrugged3, and identifying ligands does not guarantee a path to molecules with the desirable disease modulation properties. The Notch signaling family of proteins is a good example of both challenges. Notch signaling is a conserved cell–cell communication pathway important in development and differentiation4. Identification of oncogenic activating Notch1 mutations in over 50% of T cell acute lymphoblastic leukemia (T-ALL)5 and constitutive activation of the pathway in mantle cell lymphoma and chronic lymphocytic leukemia as well as some solid tumors has generated interest in this pathway6,7. When a Notch receptor interacts with a ligand on an adjacent cell, the receptor is cleaved at the S2 site in the negative regulatory region by an ADAM metalloprotease. This cleavage generates the substrate for the subsequent cleavage by the gamma-secretase complex and release of the intracellular domain (ICD) of Notch for translocation to the nucleus8–10. Targeting of Notch signaling with gamma-secretase inhibitors has met with challenges in the clinic11,12, mostly due to dose-limiting side effects13,14. In addition, the pathophysiology of Notch alterations in cancer is complex and not fully understood7. Therefore, we sought to identify additional modulators of Notch signaling through phenotypic screening and use these tools to provide insights into Notch regulation. The screening of small-molecule libraries against cellular pathways has re-emerged as a credible drug discovery strategy15,16, particularly when coupled with rigorous hit triage and follow-up on molecular targets and mechanism of action17,18. Phenotypic screening can yield chemical probes, new druggable nodes, new connections between biological targets and disease as well as accelerate testing of these connections in translational models19–21.
We describe the discovery of a low molecular weight probe of SLC39A7, a Zrt-, Irt-like protein zinc transport family member ZIP722,23, through a phenotypic screen for inhibitors of the Notch signaling pathway. Treatment of cells with the NVS-ZP7–4 class of compounds, led to ER stress, interfered with trafficking of Notch to the cell surface, and induced apoptosis in T-ALL cells. Multiple approaches validated ZIP7 as the target of these compounds, including generation of a compound-resistant cell line and photoaffinity labeling of ZIP7. NVS-ZP7–4 increased ER zinc levels, suggesting functional modulation of ZIP7. Two families of zinc transporters control zinc homeostasis in cells24,25. The SLC30 (ZnT) proteins transport zinc from the cytoplasm to organelles or extracellular space. The SLC39 (ZIP) transporters are responsible for zinc transport into the cytoplasm from the extracellular space or out of organelles. The ZIP and ZnT families of zinc transporters play critical roles in cellular and physiological functions, and have been linked to Notch biology, in flies26. NVS-ZP7–4 is the first chemical tool to probe this important yet poorly understood biology and further investigate ZIP7 as a druggable node in the Notch pathway.
Results
Identification of small-molecule Notch inhibitors.
We screened a subset of the Novartis chemical library (~1.2 × 106 compounds) to identify inhibitors of Notch signaling using U2OS cells containing a HES-Luciferase (HES-Luc) Notch reporter gene, and a tetracycline (Tet)-inducible Notch ICD. Treatment of cells with doxycycline led to the production of the ICD and activation of HES-Luc. Compounds with known mechanisms unrelated to Notch and possessing highly reactive or promiscuous moieties were removed. Hits were then tested in a Notch co-culture assay, where human cervical cancer C33A cells containing a HES-Luc reporter were co-cultured with L cells expressing the Notch ligand Jagged 127. The screen resulted in the selection of NVS-ZP7–1 (1), which blocked Notch signaling in a concentration dependent manner (Supplementary Fig. 1a). The enantiomer NVS-ZP7–2 (2) was prepared and found to be inactive; confirming that the biological activity of this scaffold is stereospecific (Supplementary Fig. 1b). Further studies of the structure activity relationship of the scaffold showed that replacement of the benzoxazine of NVS-ZP7–1 by a quinazolinone (NVS-ZP7–3) (3) offered a modest improvement in activity, and subsequent fluorination of the aminobenzothiazole (NVS-ZP7–4) (4) (Fig. 1a) or replacement with a 3,4-difluorobenzamide moiety further improves activity (NVS-ZP7–5) (5). Diazirine-containing analogs (for example, NVS-ZP7–6 (6)) were generated for proteomics. Loss of the 2-hydroxyl group (NVS-ZP7–7) (7) had little impact on activity, while heterocyclic benzamides were found to be inactive (NVS-ZP7–8) (8) (Supplementary Fig. 1).
Fig. 1 |. Identification and characterization of molecules that inhibit Notch signaling.
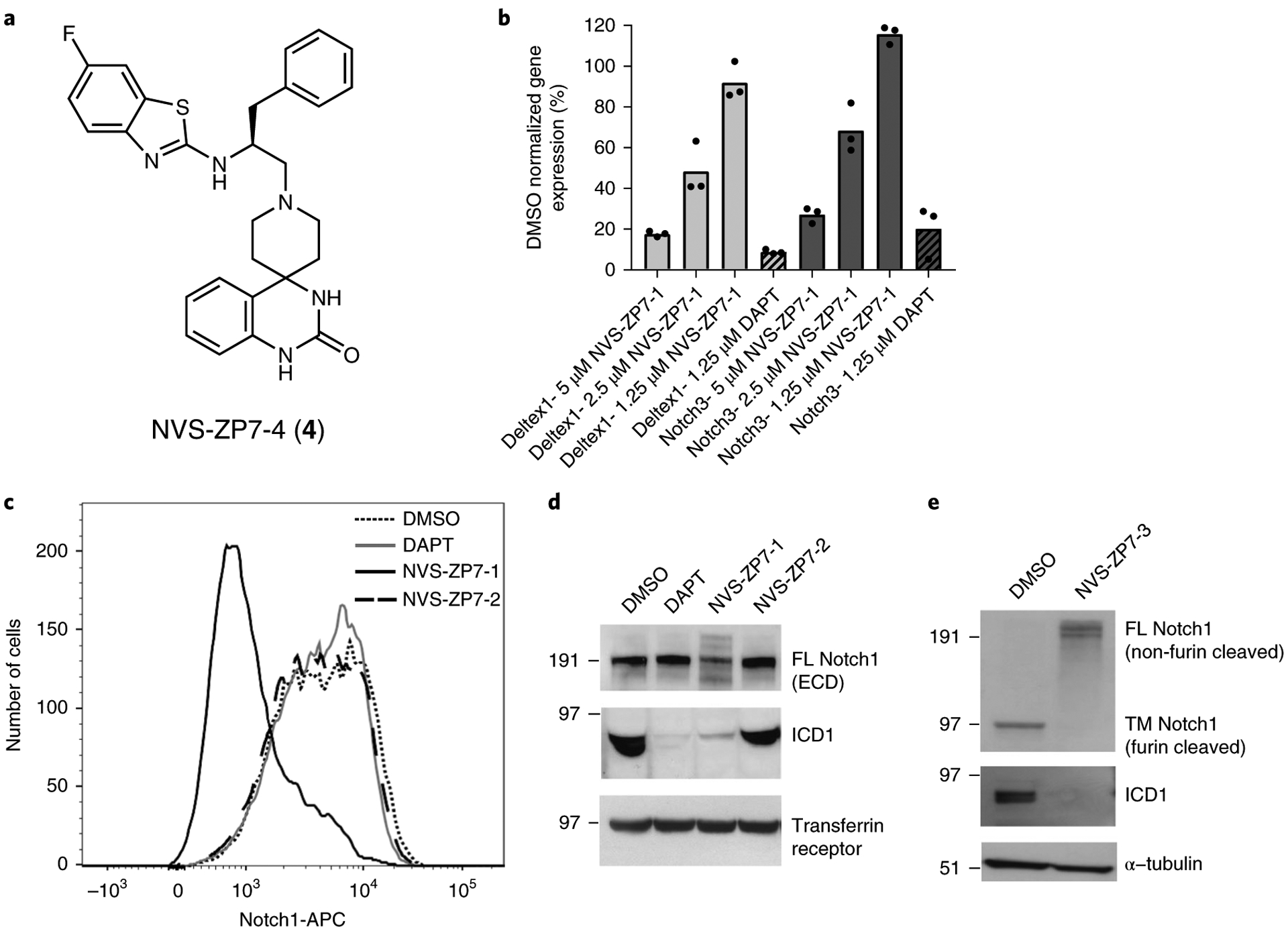
a, Structure of representative hit compound NVS-ZP7–4. b, Notch pathway target gene mRNA expression in a Notch1 mutant T-ALL cell line. HPB-ALL cells were treated with a single dose of the gamma-secretase inhibitor DAPT or three doses of NVS-ZP7–1 for 48 h. Percentage of DMSO normalized gene expression is plotted for DTX1 and Notch3 and represents the data for the technical replicates of the compound-treated samples from one individual experiment. The average readout value for these samples is represented by the dot-plot bar graph. Each experiment was performed three independent times. c, Cell surface expression of Notch1 in HPB-ALL cells. Cells were treated with 10 μM of NVS-ZP7–1 (black line), NVS-ZP7–2 (dashed line), DAPT (gray line), and DMSO (dotted line) for 48 h. Results from one biological replicate shown. Experiment was performed three independent times. d. Full length Notch1 extracellular domain (ECD) and Notch1 intracellular domain protein (ICD1) expression in HBP-ALL cells treated with 10 μM of compounds for 48 h. Full length gels are shown in Supplementary Fig. 11, and this experiment was repeated two independent times with representative data shown. e, Full length and Notch1 intracellular domain protein (ICD1) expression in MT-3 cells treated with 2 μM of compounds for 48 h. Notch1 western blot uses an antibody that has a C-terminal epitope that can detect full length non-furin-cleaved Notch1 (FL Notch1) as well as the furin-cleaved transmembrane domain/intracellular domain of Notch1 (TM Notch1). Full length gels are shown in Supplementary Fig. 12 and this experiment was repeated two independent times with representative western blot data shown.
Notch signaling is constitutively active in T-ALL cell lines, such as HPB-ALL, with activating mutations in the HD and PEST domains5. NVS-ZP7–1 treatment of HPB-ALL cells, dose dependently inhibited mRNA expression of the well-characterized Notch target genes, Deltex1 and Notch3 (Fig. 1b). NVS-ZP7–1 was less potent than DAPT, a gamma-secretase inhibitor, and known Notch signaling modulator. In contrast to DAPT, treatment of HPB-ALL cells with NVS-ZP7–1 resulted in decreased levels of Notch1 on the cell surface as monitored by flow cytometry (Fig. 1c). To further understand the effects of NVS-ZP7–1 on Notch signaling we monitored the various forms of the Notch receptor by western blotting. NVS-ZP7–1, but not its enantiomer, NVS-ZP7–2, reduced the levels of the Notch ICD similarly to DAPT (Fig. 1d). Notch is synthesized in the ER and is cleaved by a furin-like convertase in the trans-Golgi network generating an extracellular domain that can be detected by an antibody to the amino (N) terminus of the receptor8. Treatment of HPB-ALL cells with NVS-ZP7–1 resulted in detection of multiple altered mobility forms of full length (FL) Notch1 that were not observed on DAPT treatment (Fig. 1d). Use of a Notch1 antibody that detects an epitope in the carboxy (C) terminus, allowed us to monitor effects of compound treatment on FL non-furin-cleaved Notch1 (FL Notch1) as well as the transmembrane ICD form of Notch1 generated on furin cleavage (TM Notch1). Interestingly, treatment of Notch mutant MT-3 cells28 with NVS-ZP7–3 resulted in a clear increase in levels of FL Notch1 (Fig. 1e) suggesting furin processing of Notch1 in the Golgi was inhibited by compound treatment.
NVS-ZP7 selectively induces apoptosis and ER stress.
To characterize antiproliferative effects of the NVS-ZP7 compounds in T-ALL cell lines we first assessed NVS-ZP7–1 and NVS-ZP7–2 in a series of orthogonal assays. Induction of cleaved PARP (Supplementary Fig. 2a,b) and caspase 3/7 activity (Supplementary Fig. 2c) was observed in the Notch3 mutant TALL-1 cells29 but not in the Notch wild type SUPT11 cells5. We also demonstrated that NVS-ZP7–1 induced apoptosis and cell death in a propidium iodide and annexin V fluorescence-activated cell sorting (FACS) assay (Supplementary Fig. 2d,e). To extend these observations we tested a more potent compound, NVS-ZP7–3, in a panel of T-ALL cell lines that differed in their Notch mutation status. Compound treatment resulted in significant increases in dead and apoptotic cells in the annexin V/PI FACS assay in three of four Notch mutant cell lines but in none of the Notch wild type lines (Fig. 2a). P12-Ichikawa cells have a unique activating mutation in Notch1 with a 12-amino acid insertion between the negative regulatory region and the transmembrane domain5. Interestingly, treatment of P12-Ichikawa cells with NVS-ZP7–3 did not result in an increase in dead/apoptotic cells and this lack of sensitivity may be due to their unique mutation.
Fig. 2 |. NVS-ZP7–3 treatment induces apoptosis and ER stress in Notch pathway-active T-ALL cell lines.
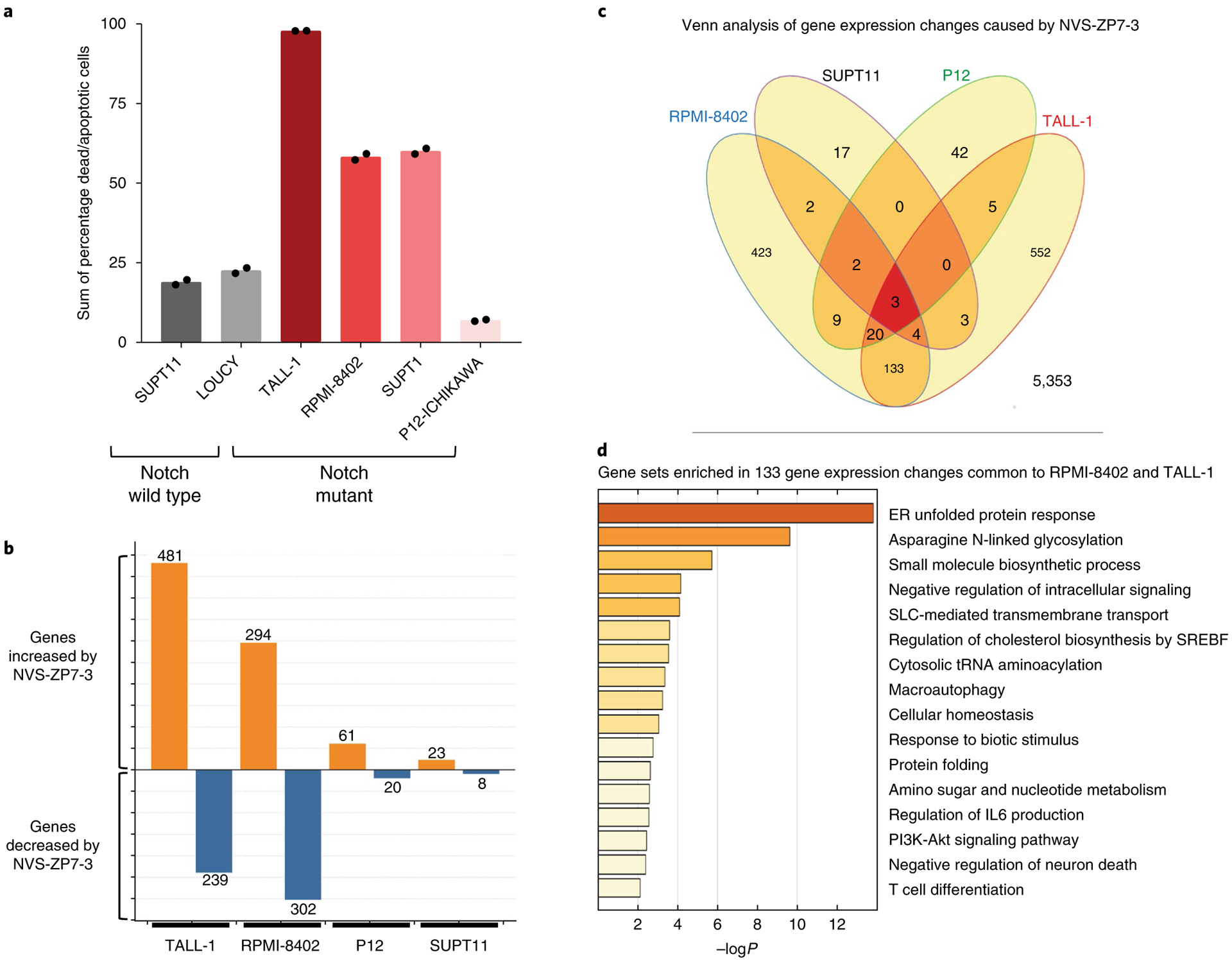
a, Increase in percentage of dead and apoptotic cells by annexin V/PI staining after 72 h of 2 μM NVS-ZP7–3. Each bar graph represents the data from one individual experiment in which two independent samples are treated with compound. The average readout value for these samples is represented by the dot-plot bar graph. Each experiment was performed three independent times. Basal levels of apoptotic/dead cells from DMSO-treated cells are used to normalize the data. Notch pathway-active T-ALL cell lines are indicted in shades of red, while Notch pathway-inactive cell lines are shown in black/gray. b, Number of statistically significant gene expression level changes observed by microarray analysis. Both increased (orange) and decreased (blue) gene counts were greater in TALL-1 and RPMI-8402 cells. c, Venn analysis of gene expression changes associated with NVS-ZP7–3 treatment identifies three gene expression changes common to all four cell lines and 133 gene expression changes common to TALL-1 and RPMI-8402. d, Gene sets enriched in 133 genes common to TALL-1 and RPMI-8402 cells. List is sorted by P values for biological process with unfolded protein response and asparagine N-linked glycosylation processes highlighted.
To further characterize the mechanism of action of these compounds, microarray analysis was used to compare gene expression profiles of mutant and wild type Notch T-ALL cell lines treated with NVS-ZP7–3. The number of significantly changing gene probe sets (adjusted P < 0.001 and a fold change greater than two) was higher in T-ALL cell lines that undergo apoptosis/cell death (RPMI-8402 and TALL-1) following compound treatment (Fig. 2b and Supplementary Fig. 3). Comparison of expression changes identified 133 genes common to TALL-1 and RPMI-8402 and gene-set enrichment analysis revealed effects on ER unfolded protein response (UPR) and N-linked glycosylation (Fig. 2c,d and Supplementary Dataset 1). To confirm the microarray profile and demonstrate induction of UPR in the NOTCH1-mutant RPMI-8402 cell line, we monitored mRNA and protein readouts of this pathway detecting increased levels of HSPA5 (BIP) and DDIT3 (CHOP) mRNA as well as spliced XBP1 and phosphorylation of EIF2 (Supplementary Fig. 4). As positive controls for ER stress, we treated cells with thapsigargin, a specific inhibitor of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and/or tunicamycin, an inhibitor of N-glycosylation, and observed similar induction of UPR mRNA and protein markers (Supplementary Fig. 4b,d). However, not all ER stress-inducing compounds inhibited Notch signaling (Supplementary Fig. 4a) nor gave the same pattern of antiproliferative effects in the T-ALL panel as NVS-ZP7–3 (Supplementary Fig. 4e). In summary, gene expression analysis provided insight into the mechanism of action and revealed that our NVS-ZP7 compounds induce UPR in Notch mutant T-ALL cells lines.
Characterization of a NVS-ZP7–4 compound-resistant cell line.
The different profile of gamma-secretase inhibitors (DAPT) and NVS-ZP7 compounds indicated that the NVS-ZP7 chemotype inhibits Notch signaling through a unique mechanism, and we therefore sought to identify the efficacy target(s). We generated and characterized a compound-resistant cell line and, in parallel, performed a genome-wide siRNA screen in the presence and absence of compound. Our goal was to intersect the hits from these two approaches to prioritize targets for further validation. We selected a compound-resistant TALL-1 cell line (referred to as TLR1) by increasing the dose of NVS-ZP7–4 over a six-month time frame, eventually exceeding the initial half-maximum inhibitory concentration (IC50) tenfold. In contrast to the parent cell line, the resistant cell line did not undergo apoptosis/death following NVS-ZP7–4 compound treatment (Fig. 3a). To verify that resistance was not due to upregulation of multidrug resistance proteins, we tested cytotoxic compounds such as vinblastine and taxol and observed no differences in antiproliferative effects in the parental and resistant line (data not shown). In addition, treatment with NVS-ZP7–4 no longer reduced the levels of Notch ICD in the resistant cell line. However, DAPT was able to block Notch signaling and reduce ICD in both the parental and resistant lines (TLR1), indicating that Notch signaling was not generally suppressed in the resistant cell line (Fig. 3b). To identify genomic changes in the resistant cell line, whole-exome sequencing was performed and 33 genes with single nucleotide polymorphisms (SNPs) were identified in the resistant line (Fig. 3c and Supplementary Dataset 2). Quantitation of RNA expression of the 33 genes in RPMI-8402 and TALL-1 cells revealed that 13 altered genes were expressed in both TALL-1 and RPMI-8402 cells (Supplementary Dataset 2).
Fig. 3 |. Generation and characterization of a NVS-ZP7–4 compound-resistant cell line.
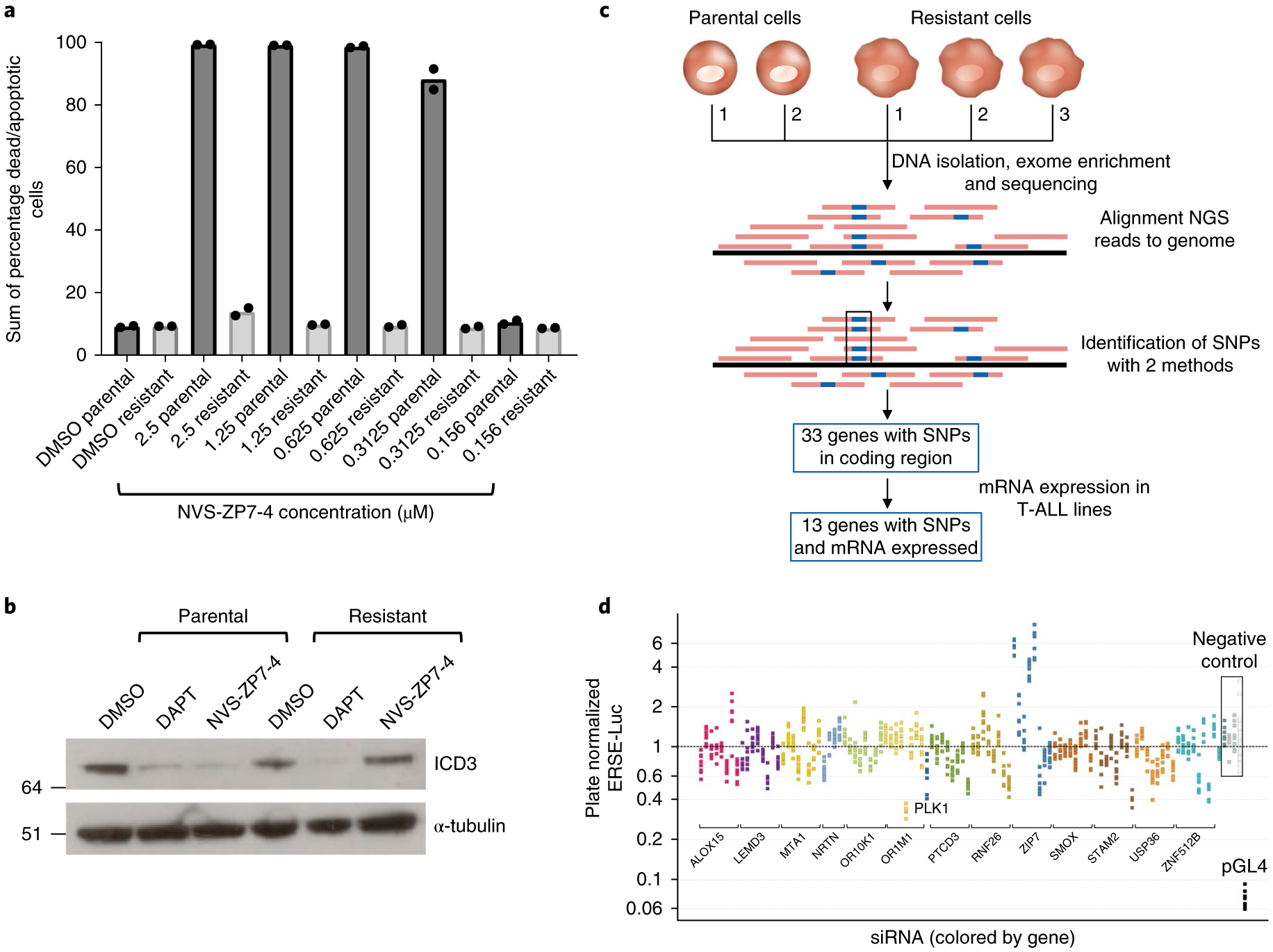
a, Comparison of effects of NVS-ZP7–4 on apoptosis/cell death in the parental TALL-1 and resistant TALL-1 cell line (TLR1). Dose response of NVS-ZP7–4 following 72 h of compound treatment. Data shown are the sum of percentage dead and apoptotic cells by annexin V/propidium iodide staining after compound treatment. Each bar graph represents the data from one individual experiment in which two independent samples are treated with compound. The average readout value for these samples is represented by the dot-plot bar graph. Each experiment was performed three independent times. b, Levels of Notch3 intracellular domain (ICD3) in the parental (TALL-1) and compound-resistant TALL-1 (TLR1) cell line following 20 h of 1 μM NVS-ZP7–4 or 10 μM DAPT treatment. Full length gels are shown in Supplementary Fig. 13, and the experiment was performed two independent times with representative western blot data shown. c, Schematic of sequencing and mutation detection in compound-resistant cell line. NGS, next generation sequencing. d, Effect of siRNA knockdown of genes identified from resistant cell line sequencing in combination with 20 nM NVS-ZP7–4 in ERSE reporter gene assay. Plate median normalized ERSE-Luc data are shown on the y axis.
Phenotypic effects due to RNAi knockdown of ZIP7.
We investigated the effects of short interfering RNA knockdown of our 13 prioritized genes, identified from exome sequencing. We leveraged our prior insight into UPR induction on compound treatment (Fig. 2d and Supplementary Fig. 4) and generated a cell line expressing an ER stress reporter (ERSE-Luc). From cell line profiling30, we determined that the adherent cell line HSC-3 was sensitive to compound treatment and was amenable to well-by-well screening in a high-throughput format. In addition, the ‘gain of signal’ HSC-3 ERSE-Luc assay was able to differentiate between general antiproliferative effects of siRNAs and siRNAs that functionally interacted with compound. Of the 13 genes identified from the resistance cell line sequencing only siRNAs to SLC39A7 (referred to as ZIP7 here), in combination with a low dose of NVS-ZP7–4 (20 nM) resulted in enhanced induction of the ERSE-Luc relative to negative-control siRNAs (Fig. 3d). Prior to embarking on additional experiments to validate ZIP7, we performed a genome-wide siRNA screen using the ERSE-Luc assay in the presence and absence of compound. The screen was performed at an IC20 concentration of compound, providing the opportunity to identify genes for which reduced protein levels sensitize cells to compound treatment. Similar to haplo-insufficiency profiling31, hits in this assay may include the efficacy target as well as related pathway components. Validation of hits from the screen revealed that only a few genes (ZIP7, ARRDC2, GSC2 and PLRG1) showed a differential effect relative to the DMSO control (Fig. 4a and Supplementary Fig. 5a,b). Four out of eight ZIP7 siR-NAs, induced a stronger increase in ERSE-Luc activity at 20 nM NVS-ZP7–4 treatment than in the DMSO treatment, resulting in a log2(fold change) > 1 (Fig. 4b). In contrast, ARRDC2, GSC2 and PLRG1 failed to show significant gene-level activity (Supplementary Fig. 5c–e). In addition, investigation of sources of off-target effects in the screen highlighted ZIP7 3′ untranslated regions (UTR) as the major off-target of siRNAs that showed the differential effect (Supplementary Fig. 6a–c). We focused our efforts on ZIP7 and quantitated the level of mRNA knockdown by four ZIP7 siRNAs (Fig. 4c) and tested the two most effective siRNAs in combination with a full dose–response of NVS-ZP7–4 in the ER stress and Notch signaling reporter gene assays. We detected modest IC50 shifts in NVS-ZP7–4 dose responses in the ERSE-Luc (Fig. 4d) and HES-Luc (Fig. 4e) assays with both ZIP7 siRNAs. From these data, ZIP7, an ER-localized zinc transporter, emerged as our lead target for the NVS-ZP7 compound series.
Fig. 4 |. Genetic screens reveal increased ER stress and decreased Notch signaling following ZIP7 siRNA knockdown.
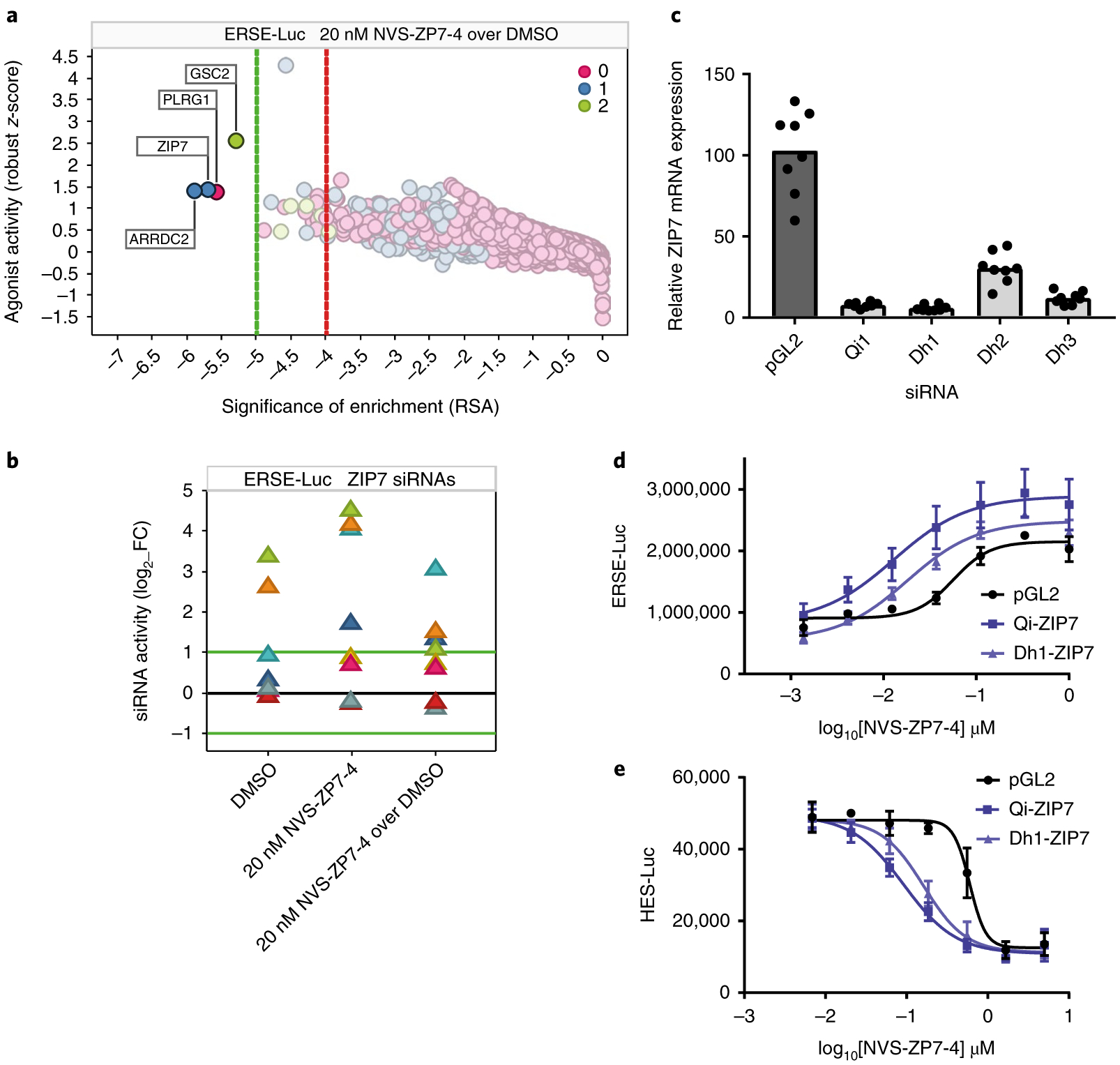
a, Gene on-target activity in the ERSE-Luc assay agonist side (increased signaling) was plotted as robust z-score (activity of the 75th percentile siRNA per gene) as a function of RSA (significance of enrichment) for NVS-ZP7–4 over DMSO control. Color was set to depict the number of siRNAs per gene with significant activity (robust z-score > + 2). Dotted lines represent stringent (green) and loose (red) thresholds of significance of enrichment, determined on the basis of randomized dataset analysis. b, The impact of the individual ZIP7 siRNAs (log2 fold change; luminescence for test siRNA over median luminescence for all siRNAs) in the ERSE-Luc assay is shown for each of the three screening conditions (DMSO, 20 nM NVS-ZP7–4, 20 nM NVS-ZP7–4 over DMSO). c, Quantitation of ZIP7 mRNA expression after treatment of HSC-3 cells with control (pGL2) or four independent ZIP7 siRNAs for 24 h. Data are representative of eight technical replicates from one biological sample represented in a box-plot graph. d, NVS-ZP7–4 dose response in ERSE-Luc assay in combination with siRNA knockdown with two independent ZIP7 siRNAs or a control siRNA (pGL2). Error bars represent s.d. of the mean from six biological replicates (n = 6) in an individual experiment. Each experiment was performed three independent times. e, NVS-ZP7–4 dose response in Notch signaling HES-luciferase assay in combination with siRNA knockdown with two independent ZIP7 siRNAs or a control siRNA (pGL2). Error bars represent the s.d. of the mean from six biological replicates (n = 6) in an individual experiment. Each experiment was performed three independent times.
Genetic and proteomic validation of ZIP7.
To confirm that ZIP7 is the physiological target of NVS-ZP7–4, we re-introduced the point mutation (V430E) identified in the compound-resistant TALL-1 cell line into the endogenous ZIP7 gene using CRISPR gene editing. The design for CRISPR gene editing used a repair single-stranded oligodeoxyribonucleotide containing the V430E sequence, as well as a mCherry Cas9 expression vector, to allow for cell sorting and enrichment (Fig. 5a and Supplementary Fig. 7a). After expansion of the mCherry-positive cells, we selected cells by addition of NVS-ZP7–4 or DMSO for 10 d. NVS-ZP7–4- and DMSO-selected cells were sequenced and a fivefold enrichment of V430E mutation was detected in NVS-ZP7–4-treated cells (Supplementary Fig. 7b). Introduction of the ZIP7 V430E mutation, shifted the dose–response of NVS-ZP7–4 tenfold in the annexin V apoptosis assay such that the response was similar to the original compound-resistant cell line (TLR1) (Fig. 5b). We used an identical approach to introduce the ZIP7 V430E mutation in a second Notch mutant T-ALL line (RPMI-8402) with a similar result (Supplementary Fig. 7c,d).
Fig. 5 |. Genetic validation of ZIP7 as the target of NVS-ZP7–4.
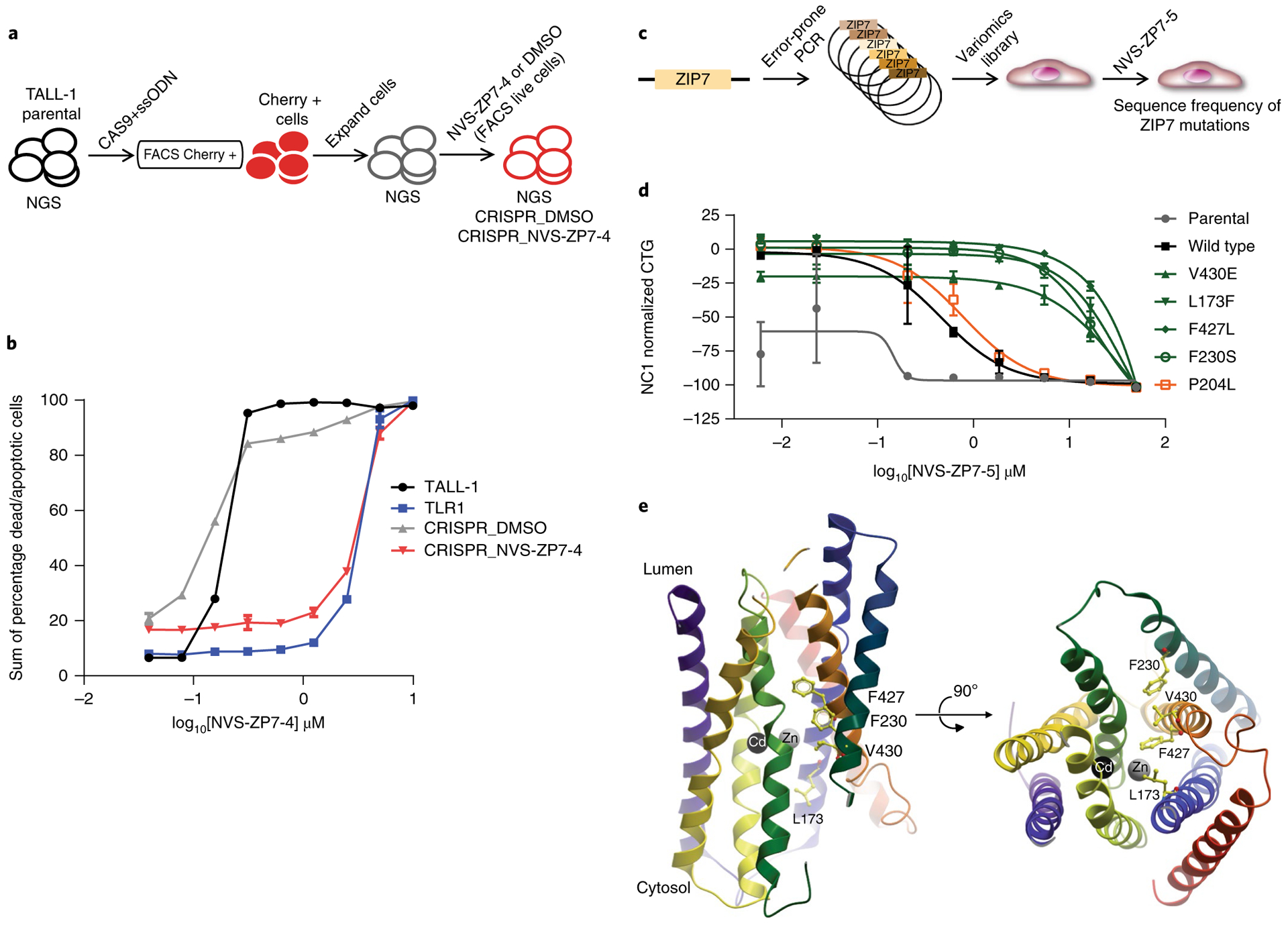
a, Schematic of the process to generate the ZIP7 V430E CRISPR knock-in cell line in the TALL-1 cellular background. b, Effect of 72 h of NVS-ZP7–4 treatment on apoptosis/cell death as measured by annexin V/propidium iodide staining in TALL-1 cells, spontaneous resistant TLR1 cells, as well as TALL-1 cells, following introduction of the ZIP7 V430E mutation with CRISPR and short-term selection with either DMSO (CRISPR_DMSO) or NVS-ZP7–4 (CRISPR_NVS-ZP7–4). Data shown are the sum of percentage dead and apoptotic cells by annexin V/propidium iodide staining after compound treatment. The x–y line graph represents the data from one individual experiment in which two independent samples are treated with compound. The average readout value for these samples is represented by the points and connecting lines with standard error bars. This exact experiment was performed two independent times. c, Schematic of process to generate ZIP7 variomics library and identify mutations that confer resistance to NVS-ZP7 compounds. d, Effect of 72 h of NVS-ZP7–4 treatment on proliferation (CTG) of parental HSC-3 cells or HSC-3 cells expressing wild type or mutant ZIP7 (V430E, L173F, F427L, F230S, P204L). Error bars represent s.d. of the mean from three biological replicates (n = 3) in an individual experiment. Each experiment was performed two independent times. e, Homology model of hZIP7 on the basis of the crystal structure of bacterial zinc transporter ortholog BdZIP (PDB code 5TSA) with transmembrane helices shown as ribbons. Zinc and cadmium ions at the binuclear center of the transporter are shown as gray and black spheres, respectively. Mutations that confer resistance to NVS-ZP7 compounds are shown in yellow ball-and-stick representation. Rotated view (right panel) is also shown.
To further characterize ZIP7 mutations that modulate NVS-ZP7–5 binding, we conducted saturating mutagenesis (variomics) of ZIP7 followed by identification of mutations that confer resistance (Fig. 5c)32,33. Resistant clones were sequenced and four mutations (L173F, F230S, F427L and V430E) were identified that occurred with high frequency (Supplementary Table 1). Expression constructs for all four ZIP7 mutants were generated and introduced into HSC-3 cells resulting in shifts in IC50 of NVS-ZP7–5 in proliferation assays of 15–25-fold relative to expression of wild type ZIP7 (Fig. 5d). A control mutation, P204L, showed no difference in IC50 relative to wild type ZIP7. To predict the location of the mutations within the ZIP7 structure, we prepared a homology model of human ZIP7 on the basis of the crystal structure of a bacterial ZIP transporter ortholog34, and subsequently mapped the positions of the four mutants in this model along a narrow cleft (Fig. 5e). The co-localization of the mutations strongly suggests that NVS-ZP7–5 and analogs bind in close proximity and further validates ZIP7 as the target of the NVS-ZP7 compounds.
To test whether the NVS-ZP7 compounds engage ZIP7 protein directly, we performed photoaffinity labeling with a chemical probe, NVS-ZP7–6, derived from NVS-ZP7–4 by appending a linker bearing a photoreactive diazirine group and terminal alkyne to allow introduction of a biotin enrichment handle with click chemistry. RPMI-8402 cells were incubated with varying concentrations of the photoaffinity labeling probe, NVS-ZP7–6, to identify proteins with saturable binding, and NVS-ZP7–4 was used as free competitor (Fig. 6a). The samples were analyzed by quantitative mass spectrometry and ZIP7 was identified as one of only a limited number of proteins that was enriched by NVS-ZP7–6 and where this labeling was competed by NVS-ZP7–4 (Fig. 6b). We further validated that ZIP7 as well as TRAM1 are enriched with the photoaffinity probe and competed by pre-incubation with NVS-ZP7–7 with western blotting (Supplementary Fig. 8a–c). The convergence of data from the genetic and proteomic target ID approaches indicates that the NVS-ZP7 scaffold indeed targets ZIP7.
Fig. 6 |. NVS-ZP7–4 interacts with ZIP7, increases ER Zn2+ levels in the ER and modulates Notch signaling.
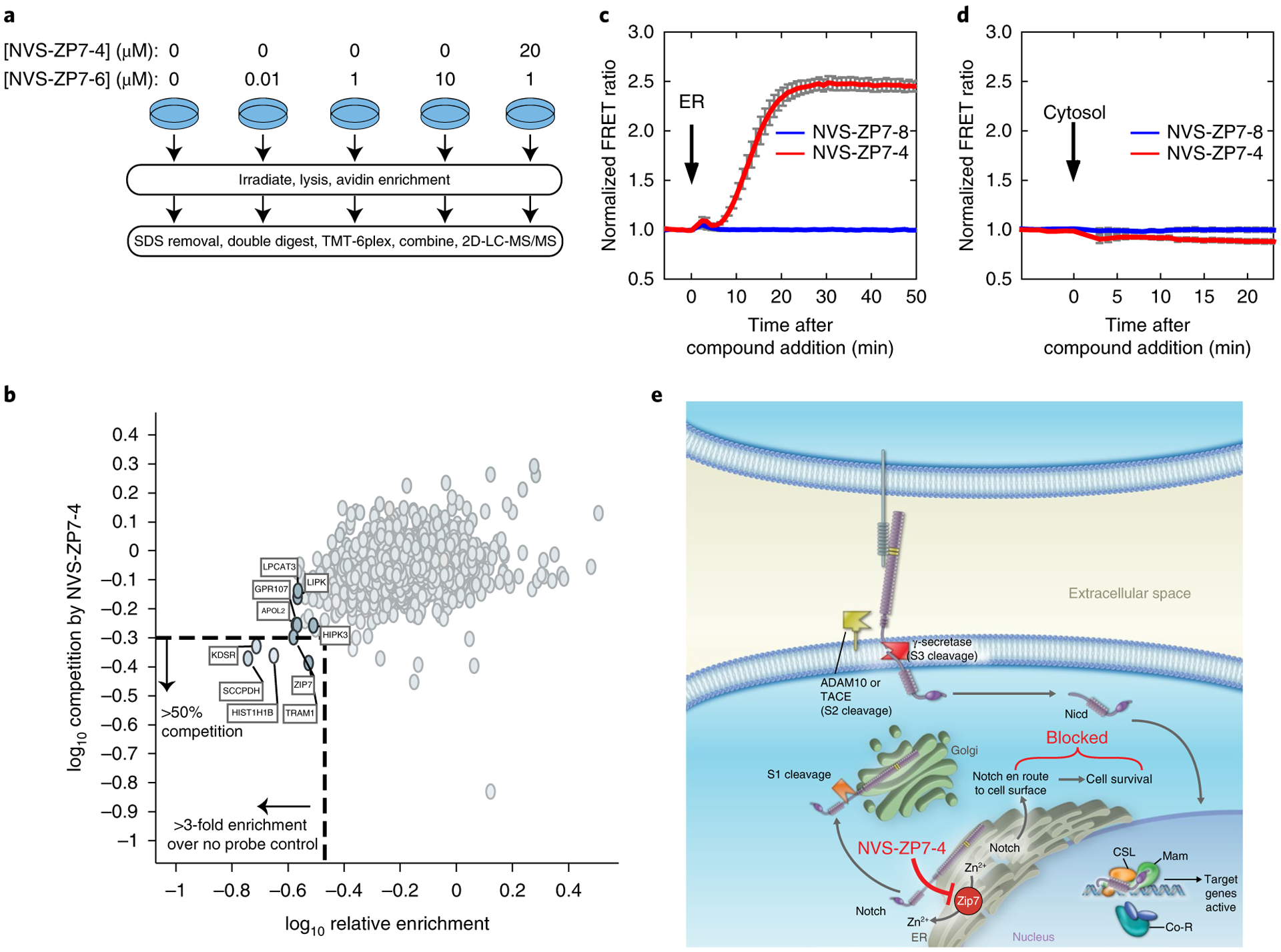
a, Schematic overview of photoaffinity labeling probe titration strategy. Cells were treated with varying concentrations of the NVS-ZP7–6 to identify proteins with saturatable binding. Additionally, cells were pretreated with NVS-ZP7–4 to identify that proteins specifically bound the pharmacophore. NGS, next generation sequencing. b, Identification of proteins that are enriched by NVS-ZP7–6 and where this labeling can be competed by NVS-ZP7–4. The x axis denotes the enrichment observed in the presence of 1 μM NVS-ZP7–6 relative to 0 μM NVS-ZP7–6 (log10 (0 μM NVS-ZP7–6/1 μM NVS-ZP7–6)). Proteins to the left of the vertical dashed lined are enriched by more than threefold over no probe control. Plotted on the y axis is the magnitude of competition observed in the presence of pre-incubated free competitor compound NVS-ZP7–4. Proteins below the dashed horizontal line show ≥ 50% reduction in enrichment in the presence of 20 μM NVS-ZP7–4 (log10 ((20 μM NVS-ZP7–4 + 1 μM NVS-ZP7–6)/1 μM NVS-ZP7–6). c, Quantitation of Zn2+ levels in the ER following NVS-ZP7–4 treatment. U2OS cells were transfected with ER-ZapCY1 for 24 h before imaging. Cells were treated with 1 μM NVS-ZP7–8 (blue, n = 7 cells) or 1 μM NVS-ZP7–4 (red, n = 6 cells) at the time indicated by the arrow. FRET ratios were normalized to the average FRET ratio of each cell before compound treatment. Error bars are the s.e.m. d, Quantitation of cytosolic Zn2+ levels following NVS-ZP7–4 treatment. U2OS cells were transfected with NES-ZapCV2 24 h before imaging. Cells were treated with 1 μM NVS-ZP7–8 (blue, n = 4 cells) or 1 μM NVS-ZP7–4 (red, n = 4 cells) at the time indicated by the arrow. FRET ratios were normalized to the average FRET ratio of each cell before compound treatment. Error bars are the s.e.m. e, Model of ZIP7 modulation by NVS-ZP7–4 and effects on Notch signaling.
ER and cytosolic zinc quantitation using Förster resonance energy transfer (FRET) sensors.
Mammalian ZIP7 proteins localize to the ER and Golgi compartments, where they contribute to the release of Zn2+ cations into the cytosol from ER/Golgi stores22,23. We wanted to determine whether modulation of cellular zinc levels would impact the response of NVS-ZP7–4 in the Notch and ER stress reporter gene assays. Treatment of cells with zinc ionophores such as pyrithione significantly rescued the effects of NVS-ZP7–4 in the Notch and ER stress reporter gene assays, suggesting that modulation of zinc levels was important for the cellular phenotypes (Supplementary Fig. 9). To quantify the effects of NVS-ZP7–4 on ER zinc, we used a genetically encoded FRET sensor35,36. The ER zinc FRET sensor (ER-ZapCY1) was expressed in U2OS cells and calibrated with a Zn2+-specific chelator followed by addition of excess Zn2+ and pyrithione (Supplementary Fig. 10) and as described previously35,37. A robust increase in normalized FRET ratio of ER-ZapCY1 was seen in cells within a few minutes following NVS-ZP7–4 addition, and the response was maintained up to 3 h or until the experiment ended (Fig. 6c). The magnitude of the effect of NVS-ZP7–4 was similar to that seen with addition of excess Zn2+ and pyrithione in the sensor calibration experiments (Supplementary Fig. 10). To investigate if this response was ER specific, we used the cytosolic zinc FRET sensor NES-ZapCV238. No changes in normalized FRET ratio on NVS-ZP7–4 in cells expressing the cytosolic zinc sensor were detected, suggesting that NVS-ZP7–4 is not altering cytosolic zinc levels (Fig. 6d). No effect on FRET ratio with either the ER or cytosolic sensor was detected with the inactive analog NVS-ZP7–8 (Fig. 6c,d).
Taken together, the data for the NVS-ZP7 series suggest that the compounds inhibit ZIP7, which increases ER zinc levels, and this leads to induction of UPR and inhibition of Notch signaling. We propose a model where the increased ER zinc affects Notch receptor trafficking and maturation resulting in selective apoptotic induction in Notch mutant T-ALL lines (Fig. 6e).
Discussion
Notch receptors have been implicated as oncogenic drivers in a variety of different human cancers, most notably Notch1 in T-ALL5. In this article, we describe a cell-based high-throughput screen that led to the identification of novel small-molecule inhibitors of the Notch pathway. A compound series, exemplified by NVS-ZP7–4, was shown to inhibit Notch signaling in T-ALL cell lines and induce ER stress, accompanied by apoptosis. A comprehensive approach to mechanism of action and target identification, combining genetics, cell biology and chemical proteomics, led to the discovery of ZIP7 as the target of this compound series. This work represents a description of a small-molecule ZIP7 modulator.
Notch receptors are large transmembrane proteins whose cellular trafficking through the Golgi and ER has long been appreciated as a key mode of regulation39,40. Cellular homeostasis and survival depend on correct ER/Golgi function and one of the quality control mechanisms that prevent the accumulation of damaged proteins is the UPR. Cells enter apoptosis if ER stress is protracted or if they are unable to restore homeostasis41. Treatment of T-ALL cell lines with NVS-ZP7–4 resulted in decreased levels of Notch receptors on the cell surface and apoptosis in Notch mutant T-ALL lines (Figs. 1c and 2). Transcriptional analysis of T-ALL cell lines that differed in terms of Notch pathway activity and Notch receptor mutation status revealed ER stress upregulation as a key feature of the NVS-ZP7 mechanism of action (Fig. 2b–d and Supplementary Fig. 4). This connection between Notch trafficking, ZIP7 and ER stress is consistent with data from a recently published forward genetic screen in Drosophila26. Interestingly, the authors demonstrated that mutations in Catsup, the Drosophila ortholog of ZIP7, resulted in defects in Notch trafficking and signaling in tissues that require a high level of Notch signaling. In addition, characterization of other membrane-bound proteins in Catsup mutant clones in Drosophila did not show effects on Notch ligand Delta, DE-Cadherin or α -Spectrin26. Small-molecule and cDNA enhancers of SERCA, an ER Ca2+ ATPase, were also identified in high-throughput screens, and were shown to target leukemia-associated Notch1 alleles42. We confirmed that thapsigargin, an inhibitor of SERCA, does inhibit Notch signaling; however, the antiproliferative profile of thapsigargin, as well as a second ER stress-inducing compound (tunicamycin), is distinct from NVS-ZP7 compounds in a T-ALL cell panel (Supplementary Fig. 4). A complete understanding of the impact of ZIP7 on protein trafficking awaits detailed global proteomics studies. Our data suggest that there may be a unique sensitivity of Notch mutant T-ALL, with constitutively active Notch signaling, to modulation of ZIP7 activity that is distinct from other compounds that modulate proper ER/Golgi function (thapsigargin and tunicamycin).
Identification of biological targets of active small molecules from phenotypic screening is challenging. As described above, we focused on extensive characterization of the mechanism of action of the NVS-ZP7–4 compounds before attempting target identification. Screening for compound-resistant variants has been used extensively in organisms such as yeast31 and the use of chemotype-specific resistance in mammalian cells is emerging as a valuable target identification approach43. We generated a compound-resistant T-ALL cell line and a combination of exome sequencing of the resistance cell line, as well as a full genome siRNA screen, led to a focus on ZIP7 as the physiological target of the NVS-ZP7 series. Others have suggested that the ‘gold standard’ evidence of the direct target of a chemical inhibitor is obtained on introduction of a resistance-conferring point mutation in both cellular and biochemical contexts43. We introduced the V430E mutation into the endogenous ZIP7 gene using CRISPR (Fig. 5b and Supplementary Fig. 7d) and were able to shift the dose–response of NVS-ZP7–4 in an apoptosis assay by tenfold. In addition, we conducted saturating mutagenesis (variomics) of ZIP7, identified resistance-conferring mutations and overexpressed these mutants in cells, resulting in shifts in IC50 of NVS-ZP7–5 in proliferation assays (Fig. 5d). When we mapped the positions of the four mutants in this ZIP7 homology model (Fig. 5e) we noted that the mutations are co-localized and speculate that NVS-ZP7 compounds bind in close proximity to this site. Although we were unable to determine the effects of the V430E mutation or other mutations in a biochemical assay, due to lack of any protein reagents or biochemical assays for ZIP proteins, we did show through photoaffinity labeling18,44 that the NVS-ZP7 compounds physically engage the ZIP7 protein (Fig. 6b and Supplementary Fig. 8). All of the genetic data, as well as the published connections between Notch, ZIP7/Catsup and ER stress, are consistent with ZIP7 as the target of NVS-ZP7–4 and provide a chemical tool to further investigate ZIP7 biology.
Elucidation of the functional roles of proteins often relies on the intersection of insights from genetic modulation of the protein as well as low molecular weight tools to acutely affect protein function. Zinc is an essential metal ion that plays a critical role in development, and its deficiency contributes to human disease45. Despite the evidence that zinc acts as a signaling factor and regulation of zinc levels is critical, no specific modulators of zinc transporters exist to acutely alter zinc levels in specific organelles. Mammalian ZIP7 proteins contribute to the release of Zn2+ cations into the cytosol from ER/Golgi stores22,23. Two recent reports using tissue-specific knock-out of ZIP7 demonstrate a role for ZIP7 in dermal development46 and intestinal epithelial self-renewal47. Both reports highlight ER stress induction and effects on ER function in mice lacking ZIP7. Using genetically encoded zinc FRET sensors, we demonstrate increased zinc levels in the ER within 10 min after treatment with NVS-ZP7–4 (Fig. 6c). These data, combined with published reports23,48 are consistent with a model where NVS-ZP7–4 is an inhibitor of ZIP7 activity. Further studies will be required to determine the cellular effects of NVS-ZP7 compounds on protein function, as well as the exact ZIP7-binding mode of these compounds. NVS-ZP7–4 represents a unique tool to modulate ER zinc levels and elucidate the roles of ZIP7 on cellular processes. Finally, we believe that the target identification and validation workflow described here, intersecting both genome- and proteome-wide techniques, provides a roadmap for future phenotypic discovery efforts.
Methods
Chemistry.
Chemical synthesis and characterization are provided as Supplementary Note 1 and spectra as Supplementary Note 2
Cell culture.
HPB-ALL, TALL-1, RPMI-8402, PF382, Loucy, SUP-T1, SUP-T11, P12-Ichikawa, MT-3 (DSMZ), C33a-Hes-Luc clone38 (Parental ATCC), HSC-3 ERSE-Luc (Parental JCRB), U2OS ICD Inducible (Parental ATCC), U2OS (ATCC), SN3T9, L cells. Cell lines were acquired from the sources indicated and authenticated by SNP fingerprinting. Cell lines were routinely maintained in growth media (indicated by supplier specifications) supplemented with 10% FBS and 1% penicillin–streptomycin. SN3T9 and L cells were obtained from G. Weinmaster (UCLA) and maintained in DMEM media (Thermo Fisher) with 10% FBS and 1% penicillin–streptomycin. HSC-3 ERSE-Luc, C33a-Hes-Luc and U2OS ICD4A48 cell lines were generated at Novartis and are described below. The growth media for the generated lines were further supplemented with 2 μg ml−1 puromycin (HSC-3 ERSE-Luc), 0.8 μg ml−1 puromycin (C33a-Hes-Luc) and 200 μg ml−1 G418 and 0.8 μg ml−1 puromycin (U2OS ICD4 A48). The Notch mutation status of the T-ALL cell lines is noted in Supplementary Table 2.
Generation of C33a-Hes-Luc cells.
The 14 × CSL-hHESR-Luciferase construct contains a portion of the native human HES1 promoter with two CSL binding sites (bold) as well 12 additional CSL binding sites (indicated in bold capital letters) upstream of the pGL4 luciferase (Promega). The 14 × CSL-hHESR-Luciferase construct was transiently transfected into C33a cells using Fugene6 reagent (Roche) and transfected cells were selected with 0.8 μg ml−1 puromycin. Stable clonal lines were screened for activity by co-culture with either Jagged-expressing SN3T9 cells or control L cells. At 48 h after co-culture, luciferase activity was assessed with Bright-Glo reagent (Promega). A single clonal line was used for all experiments.
14 × CSL-hHESR-luciferase:
g gt ac cG TG GG AA CG TG GG AA CG TG GG AA CG TG GG AA CG TG GG AA C G TG GG AA GC GG CC GC TG GG AA CG TG GG AA CGTGGGAACGTGGGA ACGTGGGAACGTGGGAAgcttgggaaagaaagtttgggaagtttcacacgagcc gttcgcgtgcagtcccagaTATATATAgaggccgccagggcctgcggatcacacaggatctgga gctggtgctgataacagcggaatcccctgtctacctctctccttggtcctggaaAAGCTT
Generation of HSC-3 ERSE-Luc.
HSC-3 cells were transduced with ERSE-Luciferase Reporter (catalog no. 336851, SA Biosciences) according to manufacturer’s instructions. Transduced cells were selected with 2 μg ml−1 of puromycin and stable clonal lines were selected. ERSE-Luciferase activity was quantitated with Bright-Glo (Promega), 6–7 h following compound treatment. A single clonal line was used for all experiments.
Generation of U2OS ICD4 A48.
A DNA construct containing the intracellular domain of Notch 4 (ICD4) was obtained from J. Griffin at Dana Farber Cancer Institute and used as a template for PCR and cloning into the pTRE-tight vector (catalog no. PT3720–5, Clontech). U2OS Tet-on cells (catalog no. 631143, Clontech) were maintained with Tet-approved FBS (catalog no. 631105, Clontech) for all experiments. U2OS Tet-on cells were transfected with 500 ng 14 × CSL-hHESR-luciferase and 250 ng pTRE_tightICD4 using Fugene6 reagent (Roche). At 24 h after transfection, cells were selected with 200 μg ml−1 of G418 and 1.2 μg ml−1 of puromycin. Stable clonal lines were selected by single-cell cloning and screened for Notch activation of the 14 × CSL-hHESR-luciferase with Bright-Glo (Promega). After plating cells, 2 μg ml−1 of doxycycline was added to induce production of the ICD4 protein and activation of Notch signaling. Luciferase activity was quantitated 48 h following doxycycline addition. A single clonal line was used for all experiments.
Notch target gene and ER stress induction mRNA assay.
To quantitate mRNA levels of both Notch target genes and ER stress-induced target gene expression, 4 × 105 cells per well were seeded into 96-well tissue culture plates (Corning) and compound dilutions were prepared in DMSO and added to each well. DAPT (Calbiochem) and DMSO (ATCC) were the controls used for this assay. Cells were incubated with compound at 37 °C and 5% CO2 for specified time points. RNA was isolated using the Qiagen RNeasy 96 kit. cDNA was made using the TaqMan Reverse Transcription reagents (Life Technologies) and the MJ Research PTC-225 Thermal cycler. TaqMan gene expression assays were run using either the TaqMan Universal PCR Master Mix or the TaqMan Fast Advanced Master Mix along with gene expression probes for Deltex1, Notch3, CHOP and BiP, as well as the housekeeping gene PP1A. Details of the probes used are described in Supplementary Table 3. TaqMan gene expression assays were run on the ABI Prism 7900HT Fast Real-Time PCR system or the ViiA 7 Real-Time PCR system (Thermo Fisher). All TaqMan reagents were purchased from Thermo Fisher (Applied Biosystems). Samples are normalized to the DMSO control target gene values. To quantitate the levels of each target gene, 2–[delta][delta]Ct method49 was calculated and then converted to percentage normalized relative gene expression using a power calculation.
FACS assay to assess cell surface levels of Notch1 receptor.
HPB-ALL cells were diluted to 1 × 106 cells ml−1 in FACS buffer (PBS; 0.1% BSA; 0.01% NaN3). A total of 2.5 × 105 cells per well were added to each well of a 96-well plate and centrifuged at 1,500 r.p.m. for 5 min at 4 °C before removing the supernatant. Anti-Notch1-APC (Supplementary Table 4) or mouse IgG isotype control labeled with APC (R&D Systems) was added to the cell pellets, 10 μl in 100 μl of FACS buffer and incubated for 1 h at 4 °C. The cells were washed and pelleted twice with 100 μl FACS buffer. Finally, cells were resuspended in 200 μl FACS buffer and fluorescence values were measured with a BD FACSCanto II cytometer (BD Biosciences). The amount of cell surface-bound anti-Notch1-APC was assessed by measuring the mean channel fluorescence of 10 K collected cell events. Cells were initially gated using forward and side scatter properties to isolate single cells. Data for each sample were then plotted on a histogram with APC-positive cells having values above the negative isotype control sample (not shown). Data were analyzed using FlowJo7.5.5 (Tree Star).
Vybrant apoptosis assay.
Staining for FITC annexin V and propidium iodide binding was performed in T-ALL cell lines as follows in 96-well v-bottom plates. 0.1 × 106 − 0.2 × 106 cells were treated with compounds for 72 h as indicated in the figures. Cells were washed with 1 × PBS and resuspended in 1 × annexin binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2) containing 5 μl FITC annexin V (Thermo Fisher) and 1 μg ml−1 propidium iodide (Thermo Fisher). Cells were spun down at 1,500 r.p.m. for 4–5 min between washes and staining. Cells were incubated at room temperature for 15 min in the dark and then fluorescence values were measured with a BD FACSCanto II cytometer (BD Biosciences). Cells were initially gated using forward and side scatter properties to isolate single cells. Scatter plots of propidium iodide versus FITC annexin V staining were gated into quadrants. FITC annexin V and propidium iodide-negative cells were assigned as the viable cell population. FITC annexin V-positive and propidium iodide-negative cells are undergoing apoptosis. Propidium iodide and FITC annexin V-positive constitute dead cells and propidium iodide-positive and FITC annexin V-negative are considered cell debris. BD FACS Diva Software (version 6) and Flow Jo (Tree Star) were used for analysis. To generate graphs the sum of the apoptotic and dead quadrants were plotted. Plots can also be shown as persentage dead and apoptotic or the total sum of dead and apoptotic cells. An example FACS gating strategy is shown in Supplementary Fig. 15.
Cleaved PARP ELISA.
To assess if NVS-ZP7–1 was stimulating the cleavage of PARP, an indicator of apoptosis, the Path Scan Cleaved PARP (Asp 214) sandwich ELISA kit (Cell Signaling) was used. Included in this assay were control compounds including staurosporine (Calbiochem), which is known to induce apoptosis along with negative controls that included DMSO and NVS-ZP7–2 (inactive version of NVS-ZP7–1). The T-ALL cell lines TALL-1 and SUPT11 were used for this assay. Cells (4 × 106) were plated into six-well assay plates and treated with a 10 μM final concentration of compound. Samples were collected at 24 and 48 h after compound addition. Samples were prepared and analyzed according to the conditions outlined in the protocol provided by Cell Signaling for this particular ELISA kit. The SpectraMax microplate reader (Molecular Devices) was used to read absorbance at 450 nm of each sample. Bar charts were then generated using the absorbance reads in GraphPad Prism.
Caspase-Glo 3/7 assay.
To assess if NVS-ZP7–1 was stimulating the activity of caspase 3/7, an indicator of apoptosis, the Caspase-Glo 3/7 assay system (Promega) was used. Included in this assay were control compounds including staurosporine (Calbiochem), which is known to induce apoptosis, along with negative controls that included DMSO and NVS-ZP7–2 (inactive version of NVS-ZP7–1). The T-ALL cell lines TALL-1 and SUPT11 were used for this assay. Cells (1 × 103) were plated into 384-well assay plates and treated with a 10 μM final concentration of compound. Caspase-Glo 3/7 reagent was prepared according to the Promega protocol and added to the cells 48 h after compound addition. Each sample was incubated with the Caspase-Glo 3/7 reagent for 45 min and then luminescence (relative luminescence units) was read out on the EnVision multilabel plate reader (PerkinElmer). Bar charts were then generated using the luminescence reads in GraphPad Prism.
Western blotting.
For HPB-ALL western blots, 4 × 106 cells were cultured in six-well plates. For MT-3 western blots, 10 × 106 cells were cultured in 10-cm plates. For RPMI-8402, 2.5 × 106 cells were cultured in six-well plates. The cells were treated with compounds for time points indicated in Figs. 1d,e and 3b and Supplementary Fig. 4d. Cells were lysed in 1× Cell Lysis buffer (Cell Signaling Technologies) containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg ml−1 leupeptin, supplemented with the HALT protease and phosphatase inhibitor cocktail (Thermo Fisher). Total protein (30 μg) was electrophoresed per lane on a 4–12% Bis–Tris SDS–PAGE gel. For high molecular weight proteins, electrophoresed gels were soaked in 2× NuPage Transfer buffer (Thermo Fisher) and 10% methanol for 15–20 min with gentle shaking. Proteins were transferred to nitrocellulose membranes (Thermo Fisher) using the iBlot detection and transfer system (Thermo Fisher). Membranes were incubated for 1 h at room temperature in 4% milk dissolved in 1× TBST. Membranes were incubated with primary antibodies according to supplier instructions. After incubation, membranes were washed with 1× TBST for 3 × 10 min at room temperature, and probed with a 1:10,000 dilution of either donkey anti-rabbit IgG or sheep anti-mouse IgG conjugated to horseradish peroxidase (Amersham Life Science) in 1× TBST for 1 h at room temperature. Following 3 × 10 min wash in 1× TBST at room temperature the blots were developed using enhanced chemiluminescence (Amersham Life Science). Antibodies are described in Supplementary Table 4.
siRNA transfection optimization and assay development.
Transfection conditions for human cervical C33a-Hes-Luc or the oral carcinoma HSC-3 ERSE-Luc cell lines were individually optimized using commercially available cationic lipids. The best transfection conditions were those that produced the least reduction in cell viability with negative controls and greatest reduction in luciferase readout with positive controls.
Cells were reverse transfected with siRNAs using RNAiMax lipofectamine (Thermo Fisher) transfection reagent. siRNA sequences are described in Supplementary Table 5. RNAiMax lipofectamine (180 μl) was diluted in 30 ml OptiMEM. This mix (10 μl) was added to 0.3 μl of a 2-μM siRNA in a 384-well plate. The plate was incubated at room temperature for 30 min. Cells were trypsinized using standard protocol for TrypLE (Thermo Fisher). Cell suspension at 3,750 cells per well in 10 μl 20% FBS media was added to wells containing siRNA. Plates were incubated at 37 °C and 5% CO2 for 48 h.
After 48 h 10 μl of a dose–response of compounds was added to the transfected cells. For C33a-Hes-Luc assay, 10 μl of 4 × SN3T9 or L cells were added to activate the Notch pathway. Cells were incubated for the time indicated in Fig. 4d,e. Readout was performed using Bright-Glo reagent to measure luminescence or CellTiterGlo reagent, as a toxicity counter screen (Roche) using the EnVision (PerkinElmer). Six biological replicates and three independent experiments were performed for each condition.
siRNA screen for NVS-ZP7–4 targets.
Genome-wide coverage of siRNA library or siRNA against human candidate target genes with 4–8 siRNAs per gene coverage (Dharmacon and Qiagen) were screened in 1,536-well plates. For each siRNA, 60 nl at 2 μM stock was transferred to assay plates using acoustic liquid dispenser. A 2 μl amount of diluted transfection reagent containing 12 nl of RNAiMax lipofectamine and 2 μl of OptiMEM was dispensed into each well of the assay plates and incubated for 30 min at room temperature. A 2 μl amount of HSC-3-ERSE-Luc cells suspension in PRMI/10% FCS/PS/2 μg ml−1 puromycin were added into assay plates at 750 cells per well and incubated for 48 h at 37 °C. To each well 2 μl NVS-ZP7–4 in RPMI/10% FCS/PS/2 μg ml−1 puromycin were added in assay plates at a final concentration of 20 nM and incubated for 9 h at 37 °C. The assay plates were equilibrated to room temperature and 3 μl per well of Bright-Glo reagent (Promega) was added. Luminescence level was measured after 5 min of incubation at room temperature. The activity readout (relative luminescence units) was normalized by dividing the median readout of siRNA-transfected wells per plate to give a fold change (FC) of activity. The FC was log2-tranformed (log2(FC)). For the individual conditions (DMSO and 20 nM NVS-ZP7–4), the log2(FC) was further transformed with the robust z-score calculation per condition: each siRNA FC was subtracted from the condition median siRNA FC and then divided by the median absolute deviation of siRNAs FC in the condition. For the differential siRNA activity calculations, the log2(FC) in DMSO was subtracted from the log2(FC) in the treated condition (20 nM NVS-ZP7–4) and further transformed by the robust z-score calculation.
On-target analysis.
To summarize the activity of approximately eight siRNAs for each gene, compared with all siRNAs/genes tested, the redundant iRNA activity (RSA) method50 was used for each individual or differential condition. The RSA is a directional test and was employed on the assay agonist (RSA up) and antagonist (RSA down) sides. In addition, gene-level Q1 and Q3 quartiles of activity, representing the activity (robust z-score) of the siRNAs at the 25th and 75th percentiles out of eight siRNAs activity per gene, respectively, were calculated and the quartiles were graphed as a function of the corresponding RSA log10P. The RSA log1010 P thresholds depicted by the green and red lines represent the thresholds of − 5 (seldom attained in a randomized dataset) and − 4 (some false positives expected), respectively, which were informed by performing RSA/quartile determination from the randomized dataset.
Off-target analysis.
A major source of off-target effects in siRNA screens result from the siRNAs acting as microRNA (miRNA)-like molecules and off-targeting transcript through targeting RNA-induced silencing complexes to their 3′ UTRs. Methods concentrating in identifying the 3′ UTRs commonly bound by siRNAs with a certain activity in a screening assay are available. GESS51,52 was employed to identify potential sources of off-target effects in the siRNA screen. The input data were prepared as indicated in the original publication. GESS results were annotated with the most current NCBI Entrez gene annotation database gene_info table (current Entrez gene_id, symbol and description; downloaded on 2017/04/04 from: ftp://ftp.ncbi.nih.gov/gene/DATA/GENE_INFO/). The GESS model tests the statistical significance of enrichment of siRNAs for off-target miRNA-like seed match binding to each human transcriptome mRNA 3′ UTR between two categories of siRNAs measurement in the screen (Tail A: 2,500 most active siRNAs on the down/antagonist assay side; Tail C: 2,500 most active siRNAs on the up/agonist assay side; siRNAs in between considered as the B category are not considered in the GESS analysis), The 3′ UTR sequence for the human transcriptome were derived from Ref-seq. The off-target and on-target analyses were cross-compared by plotting the 3′ UTR transcript-level off-target statistic (Chi-squared log10P) as a function of the gene-level on-target statistic (RSA up/agonist log10P) per gene for each individual and differential condition (Supplementary Fig. 5a–c). ZIP7 was observed as the common strongest significant outlier in the two orthogonal analyses, reinforcing ZIP7 as the top hit in the screen.
Microarray.
Ten million cells (TALL-1, RPMI-8402, SUPT11 and P12-ICHIKAWA) were plated in triplicate for each condition and compound was added on the same day. The DMSO final concentration for all compound treatment conditions was 0.5%. NVS-ZP7–3 was tested at a final concentration of 1 μM with DMSO-treated cells serving as the negative control. Cells were treated with compound for 16 h and then collected for RNA isolation using the Qiagen RNeasy Plus Mini Kit. RNA quality and concentration were measured using the Nanodrop ND-1000 spectrophotometer (Thermo Scientific). Gel electrophoresis was run (E-Gel Precast Agarose Gel, Thermo Scientific) to ensure that the RNA samples were not degraded. The GeneChip IVT Express kit (Affymetrix) was used for cDNA synthesis and in vitro transcription before hybridization to Affymetrix GeneChip Human Genome 133 Plus 2.0 arrays. Open source microarray analysis software from the R/Bioconductor consortium was used to analyze the microarray data (http://www.bioconductor.org/). arrayQualityMetrics was used for microarray technical quality assessment53, and data were background corrected and quantile normalized using the gcrma algorithm54. The raw data passed quality assessment (data not shown). Differentially expressed genes were calculated using a moderated t-statistic in limma applying a simple regression model comparing compound-treated to vehicle-treated biological triplicates (two-sided t-test). A Benjamini–Hochberg multiple hypothesis testing correction was applied and used to threshold probe sets for subsequent meta-analysis55. Probe sets were considered significantly changed if they had an adjusted P < 0.001 and a fold change greater than two (|log2FC| > 1). Genes with at least one probe set passing these criteria were considered significantly changed between groups, and for cases where multiple probe sets correspond to one gene, the probe set with the best F-statistic was selected to represent the gene in subsequent steps. The publicly available Metascape (http://metascape.org) resource was sed for gene-set enrichment analysis56.
Exome sequencing.
Exome sequencing was performed for two parental and three NVS-ZP7–4-resistant TALL-1 lines. Genomic DNA was extracted using DNeasy Blood and Tissue kit (Qiagen). Library preparation and exome capture were performed using the SureSelectXT Human All Exon V4 + UTR (71 megabase) capture baits as described in Agilent SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library Protocol without modification. The resulting sequencing libraries were then multiplexed and sequenced on an Illumina HiSeq 2000 instrument using TruSeq chemistry with a read length of 2 × 91 base pairs.
The raw sequence reads were aligned to the human genome (hg19) using BWA version 0.5.957. SNPs were called in two different ways. First, GATK version 1.6–1158 was used to call SNPs for each of the resistant samples as well as for two samples of the unmutagenized reference strain TALL-1. The SNPs of the two reference strain samples were then subtracted from the SNPs of the resistant samples. Second, we used a slightly modified version of the SNP calling method described previously59 to obtain SNPs at positions where the resistant mutant differs from the parental strain. SNPs were only kept if called against both of the reference strain samples. The combined set of SNPs from both methods was annotated using VEP for Ensembl v6960.
Next generation sequencing and variant analysis CRISPR.
Illumina sequencing libraries were generated from PCR amplicons using the Nextera DNA Library Preparation Kit (Illumina, catalog no. FC-121–1031) following the manufacturer’s recommendations. Libraries were sequenced on a MiSeq sequencer (Illumina) using 101 base paired-end reads to minimum of 1,000-fold coverage, following the manufacturer’s recommendations. Variants were called using standard methods.
ZIP7 variomics Library generation.
The 1,410-nucleotide open reading frame encoding the C-terminal 347 amino acid of isoform 1 (NP_008910.2) of ZIP7/SLS39A7 was cloned into vector pXP1510 (lentiviral vector with G418 selection marker and EF1α promoter driving open reading frame expression). Schematic of pXP1510 cloning vector is shown in Supplementary Fig. 16.
Error-prone PCR-mediated mutagenesis was performed using the Ex Taq hot start enzyme (TaKaRa Hot Start Ex Taq Kit, Clontech) in the presence of 100 μM manganese chloride to induce mutations in the ZIP7 open reading frame. Briefly, 100 ng of plasmid DNA (pXP1510-ZIP7) was mutagenized by PCR according to the manufacturer’s protocol using the following primers: XPO2011-F, 5′ -CAGATCCAAGCTGTGACC-3′, and XPO2010-R, 5′ -GTAACGTCTACGTGTCTG −3′. PCR products were separated by gel electrophoresis and purified using a Qiaquick PCR purification kit (Qiagen). After restriction enzyme digest (NotI-HF, AscI, DpnI), the DNA was purified with the PCR purification kit (Qiagen) mentioned above and cloned into pXP1510. Mutagenized DNA was transformed into Escherichia coli XL10-Gold ultracompetent cells (Agilent). A total of 48 colonies were chosen for plasmid sequencing to infer the mutation rate of the library, which was 0.7 non-silent mutations per kilobase.
Generation of lentiviral ZIP7 variomics library, infection of HSC-3 cells and selection of NVS-ZP7 resistant cells.
For virus packaging, pXP1510-ZIP7WT and pXP1510-ZIP7–100 μM MnCl2 variomics libraries were transfected into HEK293TN cells with TransIT-293T transfection reagent (Mirus) and Cellecta packaging mix (Cellecta) according to the manufacturer’s protocol. HSC-3 cells were infected with lentivirus in the presence of 8 μg ml−1 polybrene (Applied Bioanalytical). Cells stably expressing ZIP7 were selected with 400 μg ml−1 G418 creating stable cell pools. The cell pools expressing the ZIP7 variomics library were treated with 10 μM NVP-ZP7–5 for 12 d in six-well format to identify compound-resistant mutants. DNA was collected from pools of cells that survived compound treatment and then sequenced. ZIP7 PCR products were amplified, combined and cloned into pXP1510 using NotI and AscI. Frequency and location of 45 mutations in ZIP7 are shown in Supplementary Table 3. Expression constructs for wild type ZIP7 as well as five ZIP7 mutants were re-introduced into HSC-3 cells and proliferation examined following NVS-ZP7–5 treatment for 72 h.
ZIP7 homology modeling.
Homology modeling was conducted with MOLSOFT ICM61. The crystal structure of the bacterial zinc transporter ortholog BdZIP (PDB code 5TSA) served as a structural template to model the transmembrane (TM) helices of human ZIP7 (residues 129–469) following the strategy described to model human ZIP434. Predicted unstructured loops connecting TM3 and TM4 and between TM7 and TM8 were omitted.
Photoaffinity labeling experiments.
In brief, 40 million RPMI-8402 cells were added to individual wells of a six-well plate in OptiMEM Reduced Serum Media with 0% FBS. DMSO was added to cells in four wells, while cells in the remaining well were treated with 20 μM NVS-ZP7–4 for 1 h at 37 °C with 5% CO2. DMSO-treated cells were then treated with 0, 0.01, 1, or 10 μM NVS-ZP7–6, while NVS-ZP7–4 -reated cells were treated with 1 μM NVS-ZP7–6 for 1 h at 37 °C with 5% CO2. Irradiation, cell lysis, enrichment, sample processing and data acquisition were performed as previously described44, except that the samples were first digested with chymotrypsin before digestion with trypsin. Mass spectrometry data are shown in Supplementary Dataset 3.
FRET sensor cell culture and transfection.
U2OS cells were cultured in DMEM medium (Invitrogen) supplemented with 10% FBS and penicillin–streptomycin at 37 °C with 5% CO2. All sensor constructs were transiently transfected with Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions. The zinc ER FRET sensor, ER-ZapCY135, and zinc cytosol FRET sensor, NES-ZapCV238, constructs have been described previously. Cells were imaged or collected 24–48 h after transfection.
Live cell FRET sensor fluorescence imaging.
Imaging experiments were performed on a Nikon Ti-E wide-field fluorescence microscope equipped with Nikon elements software, Ti-E perfect focus system, an iXon3 EMCCD camera (Andor), mercury arc lamp, and YFP FRET (434/16 excitation, 458 dichroic, 535/20 emission) and CFP (434/16 excitation, 458 dichroic, 470/24 emission) filter sets. External excitation and emission filter wheels were controlled by a Lambda 10–3 filter changer (Sutter Instruments), while dichroic mirrors were placed on cubes in the dichroic turret. Images were collected using a ×60 oil objective (numerical aperture 1.40), 100 ms exposure time, electron mutiplying gain 1 MHz at 16-bit readout mode with an electron mutiplying gain multiplier of 200, and a neutral density filter with 25% light transmission. Cells were maintained at 37 °C and 5% CO2 in a LiveCell environment chamber (Pathology Devices) during the experiments. Images were collected every minute.
FRET sensor data analysis.
All imaging data were analyzed in MATLAB (Mathworks). Images were background corrected by drawing a region of interest in a blank area of the image and subtracting the average fluorescence intensity of the background region of interest from the average intensity of each cell. FRET ratios for each cell were calculated by dividing the background-corrected YFP FRET intensity by the background-corrected CFP intensity ((Icellular FRET – Ibackground FRET)/(Icellular CFP – Ibackground CFP)). Unless otherwise indicated, error bars are s.e.m.
Statistical analysis.
All statistical tests were done using Prism7 software (GraphPad) unless specifically stated in the Methods.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
The JAGGED1 and DLL1 expressing cell lines were kindly provide by G. Weinmaster (UCLA). HPB-ALL cells were kindly provided by A. Stasser (Walter and Eliza Hall Institute for Medical Research). The authors thank J. Paulk for his insights on the manuscript and A. Abrams for his artwork in schematic diagrams. This work was financially supported in part by NIH Director’s Pioneer Award GM114863 (to A.E.P.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41589-018-0200-7.
Competing interests
E.N., S.G., L.L., S.B., S.M. Brittain, P.B.-E., J.J.L., J.R.T., M.S., Y.Y., N.G., G.R., S.S., M.B., A.L., F.S., A.C., K.X.X., S.H., J.R.-H., W.A.W., K.T., D.H., R.I.M., N.G., A.D., H.G., J.S., J.D., S.M.C., G.B., E.L.G., Z.B.K., C.A., J.A.P., O.W., J.A.T., J.L.J., R.K.J., S.M. Bushell, and C.J.F. are (or were at the time the research was conducted) employees of Novartis.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-018-0200-7.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
The microarray data has been deposited in GEO (GSE115690). Other datasets that were generated during the current study are provided as Supplementary Information or are available from the corresponding author upon reasonable request.
References
- 1.Imming P, Sinning C & Meyer A Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov 5, 821–834 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B & Hopkins AL How many drug targets are there? Nat. Rev. Drug Discov 5, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Whitfield JR, Beaulieu ME & Soucek L Strategies to inhibit Myc and their clinical applicability. Front. Cell Dev. Biol 5, 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hori K, Sen A & Artavanis-Tsakonas S Notch signaling at a glance. J. Cell Sci 126, 2135–2140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng AP et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Espinoza I, Pochampally R, Xing F, Watabe K & Miele L Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. 6, 1249–1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aster JC, Pear WS & Blacklow SC The varied roles of Notch in cancer. Annu. Rev. Pathol 12, 245–275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopan R & Ilagan MX The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guruharsha KG, Kankel MW & Artavanis-Tsakonas S The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet 13, 654–666 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson ER, Sandberg R & Lendahl U Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Shih Ie. M.& Wang TL Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 67, 1879–1882 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Aster JC & Blacklow SC Targeting the Notch pathway: twists and turns on the road to rational therapeutics. J. Clin. Oncol 30, 2418–2420 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Imbimbo BP Therapeutic potential of gamma-secretase inhibitors and modulators. Curr. Top. Med. Chem 8, 54–61 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Ran Y et al. γ -Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol. Med 9, 950–966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner BK & Schreiber SL The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem. Biol 23, 3–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moffat JG, Vincent F, Lee JA, Eder J & Prunotto M Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov 16, 531–543 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Schenone M, Dančík V, Wagner BK & Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol 9, 232–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schirle M & Jenkins JL Identifying compound efficacy targets in phenotypic drug discovery. Drug Discov. Today 21, 82–89 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Palacino J et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 11, 511–517 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Huang SM et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Garbaccio RM & Parmee ER The impact of chemical probes in drug discovery: a pharmaceutical industry perspective. Cell Chem. Biol 23, 10–17 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Huang L, Kirschke CP, Zhang Y & Yu YY The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. J. Biol. Chem 280, 15456–15463 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Taylor KM, Morgan HE, Johnson A & Nicholson RI Structure-function analysis of HKE4, a member of the new LIV-1 subfamily of zinc transporters. Biochem. J 377, 131–139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hojyo S & Fukada T Zinc transporters and signaling in physiology and pathogenesis. Arch. Biochem. Biophys 611, 43–50 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Jeong J & Eide DJ The SLC39 family of zinc transporters. Mol. Aspects Med 34, 612–619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groth C, Sasamura T, Khanna MR, Whitley M & Fortini ME Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 zinc transporter Catsup. Development 140, 3018–3027 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindsell CE, Shawber CJ, Boulter J & Weinmaster G Jagged: a mammalian ligand that activates Notch1. Cell 80, 909–917 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Wu Y et al. Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052–1057 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Bernasconi-Elias P et al. Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene 35, 6077–6086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoepfner D et al. High-resolution chemical dissection of a model eukaryote reveals targets, pathways and gene functions. Microbiol. Res 169, 107–120 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Huang Z et al. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep. 3, 577–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bill A et al. Variomics screen identifies the re-entrant loop of the calcium-activated chloride channel ANO1 that facilitates channel activation. J. Biol. Chem 290, 889–903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T et al. Crystal structures of a ZIP zinc transporter reveal a binuclear metal center in the transport pathway. Sci. Adv 3, e1700344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Y, Dittmer PJ, Park JG, Jansen KB & Palmer AE Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+with genetically encoded sensors. Proc. Natl Acad. Sci. USA 108, 7351–7356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park JG & Palmer AE Quantitative measurement of Ca2+ and Zn2+ in mammalian cells using genetically encoded fluorescent biosensors. Methods Mol. Biol 1071, 29–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter KP, Carpenter MC, Fiedler B, Jimenez R & Palmer AE Critical comparison of FRET-sensor functionality in the cytosol and endoplasmic reticulum and implications for quantification of ions. Anal. Chem 89, 9601–9608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiedler BL et al. Droplet microfluidic flow cytometer for sorting on transient cellular responses of genetically-encoded sensors. Anal. Chem 89, 711–719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortini ME Notch signaling: the core pathway and its posttranslational regulation. Dev. Cell 16, 633–647 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi H & Haltiwanger RS Significance of glycosylation in Notch signaling. Biochem. Biophys. Res. Commun 453, 235–242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walter P & Ron D The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Roti G et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 23, 390–405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapoor TM & Miller RM Leveraging chemotype-specific resistance for drug target identification and chemical biology. Trends Pharmacol. Sci 38, 1100–1109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas JR et al. A photoaffinity labeling-based chemoproteomics strategy for unbiased target deconvolution of small molecule drug candidates. Methods Mol. Biol 1647, 1–18 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Hara T et al. Physiological roles of zinc transporters: molecular and genetic importance in zinc homeostasis. J. Physiol. Sci 67, 283–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bin BH et al. Requirement of zinc transporter SLC39A7/ZIP7 for dermal development to fine-tune endoplasmic reticulum function by regulating protein disulfide isomerase. J. Invest. Dermatol 137, 1682–1691 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Ohashi W et al. Zinc transporter SLC39A7/ZIP7 promotes intestinal epithelial self-renewal by resolving ER stress. PLoS Genet. 12, e1006349 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodruff G et al. The zinc transporter SLC39A7 (ZIP7) is essential for regulation of cytosolic zinc levels. Mol. Pharmacol 94, 1092–1100 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 50.König R et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 4, 847–849 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Sigoillot FD et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 9, 363–366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yilmazel B et al. Online GESS: prediction of miRNA-like off-target effects in large-scale RNAi screen data by seed region analysis. BMC Bioinformatics 15, 192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kauffmann A, Gentleman R & Huber W arrayQualityMetrics–a Bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F & Spencer F A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc 99, 909–917 (2004). [Google Scholar]
- 55.Smyth G in Bioinformatics and Computational Biology Solutions Bioinformatics and Computational Biology Solutions Using R and Bioconductor (eds. Gentleman R et al.) ch. 23 (Springer, 2005). [Google Scholar]
- 56.Tripathi S et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wacker SA, Houghtaling BR, Elemento O & Kapoor TM Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol 8, 235–237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McLaren W et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abagyan R et al. ICM-A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem 15, 488–506 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.