

Abstract
Purpose of review
The purpose of this review is to provide an overview of recent advances in the development of histone deacetylase inhibitors (HDACi) for the treatment of cancer.
Recent findings
Recently, there has been a dramatic expansion of HDACi clinical investigation. There are now 11 HDACi in clinical trial, including inhibitors with a broad spectrum of HDAC isoform inhibitory activity as well as drugs with isoform selectivity. Over 70 combination therapy trials are in progress. Major areas of progress covered include the entry of new HDAC inhibitors into clinical development, recent progress in understanding of molecular mechanisms of HDACi anticancer activity, and a preclinical and clinical update on HDACi in combination.
Summary
In the period under review there have been advances in understanding of HDACi mechanisms of action, identification of rational combinations that address increased efficacy and overcoming resistance, and greatly expanded clinical development of pan-HDAC-inhibitory and isoform-selective inhibitors in monotherapy and combination therapy protocols.
Keywords: cancer therapy, clinical trial, combination therapy, histone deacetylase inhibitors
Introduction
Chromatin-templated activities including DNA transcription, replication, and repair are regulated by a variety of posttranslational modifications, among which histone acetylation plays a prominent role. Numerous nonhistone proteins are histone deacetylase (HDAC) substrates, including many of the most critical regulators of the malignant phenotype. Reciprocal acetylation/deacetylation has been shown to have profound effects on protein function, such as the recent demonstration that acetylation is absolutely required for p53 activation [1•], and small molecule HDAC inhibitors have shown promising anticancer activity preclinically and clinically [2–11].
Despite the central importance of HDAC substrates in the biology of cancer cells, at clinically achievable concentrations histone deacetylase inhibitors (HDACi) are rarely cytotoxic as single agents. Thus, from the onset of their development as oncology drugs, it has been commonly assumed that HDACi would ultimately be used in combination. As described here, in the past year progress has been made in moving toward the goal of effective combination therapy. There are now 11 HDACi in clinical trial, seven of which are undergoing testing in combination protocols. In addition to highlighting publications on monotherapy and combination therapy, we discuss advances in HDACi basic mechanisms of activity, HDACi pharmacodynamic assay development, and the impact of HDACi on systems level biological activities including angiogenesis, metabolism, and immune response. This article covers the time period from January 2007 to June 2008. Only a small fraction of preclinical and clinical articles published during this time period can be mentioned. The aim of this overview is to highlight major new findings and ideas related to the development of HDACi for the treatment of cancer.
Update on histone deacetylase inhibitors in clinical development
On 6 October 2006 the US Food and Drug Administration (USFDA) granted regular approval to vorinostat for the treatment of cutaneous manifestations of cutaneous T-cell lymphoma (CTCL) in patients with progressive, persistent, or recurrent disease on or following two systemic therapies. The pivotal study supporting approval was an open-label, single-arm, multicenter phase IIB trial of 74 patients with CTCL who had previously failed at least two prior systemic therapies, one of which must have contained bexarotene [12••]. There was a 30% objective response rate with a median response duration of 168 days in a heavily pretreated advanced CTCL patient population. In a supportive phase II single-institution trial of 33 CTCL patients there was a 31% response rate [13••].
In June 2008 Gloucester Pharmaceuticals (Cambridge, Massachusetts, USA) announced that a phase IIB registration trial of romidepsin in CTCL was completed and that the trial met its primary endpoint based on overall response rate. Gloucester Pharmaceuticals plans to submit a new drug application to the USFDA later in 2008 (http://www.gloucesterpharma.com/press/releases/pr060508.html).
New HDACi in clinical development include the hydroxamates CRA-024781 from Celera Genomics (South San Francisco, California, USA), ITF2357 from Italfarmaco (Milan, Italy), SB939 from S*BIO Pte Ltd (Singapore), and R306465 from Johnson & Johnson (Beerse, Belgium) (see Table 1 [14–17] for additional HDACi information). Novartis Pharmaceuticals (East Hanover, New Jersey, USA), which had two hydroxamate HDACi in clinical development, LAQ824 and LBH589 (panobinostat), has chosen to focus on development of panobinostat.
Table 1.
Histone deacetylase inhibitors in clinical trial
| Class | Compound | Structure | Isotype selectivitya,b | Company | Stage |
|---|---|---|---|---|---|
| Hydroxamic acid | Vorinostat/Zolinza (SAHA) | Class I, IIa, IIb, IV | Merck | USFDA approval for CTCL | |
| Belinostat/PXD101 | Class I, IIa, IIb, IV | TopoTarget | Phase II | ||
| Panobinostat/LBH589 | 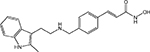 |
Class I, IIa, IIb, IV | Novartis | Phase III | |
| CRA-024781 (PCI 24781) | 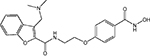 |
Class I, IIb | Celera Genomics | Phase I | |
| ITF2357 | Class I, II Class I, II | Italfarmaco | Phase II | ||
| SB939 | 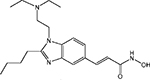 |
ND | S*BIO | Phase I | |
| R306465 (JNJ-16241199) | 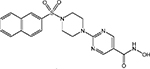 |
Class I | Johnson & Johnson | Phase I | |
| Cyclic peptide | Romidepsin/depsi peptide (FK 228) | 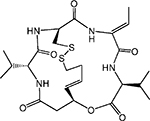 |
HDAC 1, 2, 4, 6 | Gloucester | Phase II |
| Aliphatic acid | VPA | 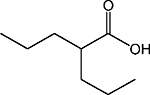 |
Class I, IIa | G2M | Phase II |
| Benzamide | SNDX-275/entinostat (MS-275) | 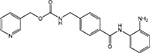 |
HDAC 1, 2, 3, 9 | Syndax/Bayer Schering Pharma AG | Phase II |
| MGCD0103 | 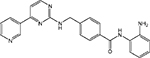 |
HDAC 1, 2, 3, 11 | MethylGene | Phase II |
CTCL, cutaneous T-cell lymphoma; HDAC, histone deacetylase; ND, isotype selectivity not known; USFDA, US Food and Drug Administration; VPA, valproic acid.
Class I: HDAC 1, 2, 3, 8; class IIa: HDAC 4, 5, 7, 9; class IIb: HDAC 6, 10; class IV: HDAC 11.
Interest in isoform-selective HDACi continues. The effects of the pan-HDACi hydroxamic acid derivatives are being compared with more isoform-selective compounds such as the benzamides SNDX-275 and MGCD0103 and the cyclic peptide romidepsin. In addition there is increasing interest in the class II enzyme HDAC6, which has been implicated in tumor cell motility and metastasis, in deacetylation of the molecular chaperone heat shock protein 90 (Hsp90), and in the regulation of tumor cell death via autophagy [18,19•]. The HDAC6 inhibitor tubacin has been a valuable probe of HDAC6 function [20], and novel small molecule inhibitors of HDAC6 have been synthesized that extend understanding of the structural requirements for selective HDAC6 inhibition [21].
Preclinical studies
Preclinical research in areas of particular interest published during the review period is summarized below.
Autophagy, the proteasome and apoptosis
Eukaryotic cells primarily use two regulated processes for large-scale catabolism, autophagy, and apoptosis. Recently, there has been intense interest in both processes as drug targets, and a variety of studies have demonstrated synergy of HDACi with autophagy and proteasome inhibitors.
Treatment with vorinostat induces both apoptosis and autophagy. To examine a potential role of autophagy in vorinostat resistance Carew et al. [22•] tested the effect of vorinostat on imatinib-resistant chronic myelogenous leukemia (CML), in the presence and absence of the autophagy inhibitor chloroquine. Chloroquine dramatically augmented the anticancer activity of vorinostat in CML cell lines and primary cells. Generation of reactive oxygen species (ROS) is a critical event in vorinostat-induced cell death, and chloroquine, which induces ROS in other systems, markedly augmented vorinostat-induced superoxide generation.
Dai et al. [23] in chronic lymphocytic leukemia cells and Duan et al. [24] in head and neck squamous cell carcinomas demonstrated that HDACi enhance nuclear factor-kappa B (NF-κB) activation resulting in HDACi resistance, and that this activation is inhibited by bortezomib. NPI-0052 (Nereus Pharmaceuticals, San Diego, California, USA), a second-generation proteasome inhibitor that unlike bortezomib, irreversibly binds to the 20S proteasome and inhibits all three proteasome catalytic activities, has entered clinical trial in refractory solid tumors and lymphoma. Miller et al. [25•] investigated the effect of combining SNDX-275 and valproic acid (VPA) with NPI-0052 in leukemia cells in vitro and reported synergistic apoptosis associated with superadditive increases in ROS, consistent with a role for oxidative challenge in the observed synergy.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) preferentially induces apoptosis in malignant cells. However, many cancers are resistant to TRAIL treatment. Various HDACi have been shown to synergize with TRAIL in inducing tumor cell death [26]. Lexatumumab (Human Genome Sciences, Rockville, Maryland, USA) is an agonistic monoclonal antibody to TRAIL-R2. Nawrocki et al. [27] demonstrated that vorinostat and trichostatin A (TSA) reduced lexatumumab resistance in colon cancer cells via induction of p21 expression, p21-dependent inhibition of cdc2, and subsequent instability and loss of expression of the antiapoptotic protein survivin.
Zhao et al. [28] demonstrated that apoptosis induced by the anti-CD20 antibody rituximab (Rituxan, Biogen Idec/Genentech, Cambridge, Massachusetts/San Francisco, California, USA), used in the treatment of B-cell non-Hodgkin’s lymphoma (NHL), is significantly enhanced by combination with vorinostat in vitro and in a lymphoma mouse model in vivo, in association with NF-κB inactivation and c-myc degradation.
Histone deacetylase inhibitors, translational inhibition, and heat shock protein 90 regulated proteolysis
In mantle cell lymphoma cells constitutively overexpressing cyclin D1, Kawamata et al. [29] demonstrated that vorinostat markedly downregulates cyclin D1 protein while having little effect on cyclin D1 gene expression or protein stability, and that the mechanism of downregulation is via inhibition of the PI3 kinase/Akt/mTOR/eIF4E-BP pathway, resulting in inhibition of cyclin D1 translation.
Acetylation of Hsp90 promotes proteasomal degradation of client proteins. Epidermal growth factor receptors (EGFRs) with activating mutations are dependent on Hsp90 for stability and function. Edwards et al. [30] showed that LBH589 induces Hsp90 acetylation and EGFR degradation in lung cancer cells with mutant EGFR, and that LBH589 and the EGFR tyrosine kinase inhibitor (TKI) erlotinib synergized in promoting apoptosis of nonsmall cell lung cancer (NSCLC) cells harboring EGFR mutations.
HDACs and DNA methyltransferase 1 (DNMT1) cooperate in epigenetic gene regulation. As shown by Zhou et al. [31•] DNMT1 forms a molecular chaperone complex with Hsp90, and LBH589 induces Hsp90 acetylation and DNMT1 degradation.
Histone deacetylase inhibitors, mammalian target of rapamycin inhibitors and angiogenesis
HDACi have been reported to inhibit angiogenesis through a variety of mechanisms, including HIF-1α downregulation and decreased expression of VEGF and other proangiogenic HIF-1α target genes. The mammalian target of rapamycin (mTOR) signaling pathway regulates angiogenesis as well as tumor survival and proliferation. Resistance to mTOR inhibitors has been associated with Akt phosphorylation and activation of survival mechanisms. Verheul et al. [32] tested the combination of LBH589 and the mTOR inhibitor rapamycin and reported that the combination abrogated constitutive and rapamycin-induced Akt phosphorylation, suppressed HIF-1α, and inhibited in-vivo tumor growth and angiogenesis.
Histone deacetylase inhibitors and nuclear receptor-targeted agents
Bigaku et al. [33] demonstrated potentiation of tamoxifen by HDACi in breast cancer cell lines, which was associated with downregulation of estrogen receptor and progesterone receptor protein and nuclear signaling. Fiskus et al. [34] reported that in estrogen receptorpositive breast cancer cells hydroxamic acid HDACi induced acetylation of Hsp90 and degradation of estrogen receptor-α with consequent attenuation of estrogen receptor signaling. Hsp90 acetylation was also associated with degradation of client proteins Her-2, phospho-ERK1/2, and phospho-Akt, which have been associated with intrinsic and acquired resistance to tamoxifen.
Progression of prostate cancer patients on hormonal therapy is thought to represent continued signaling of androgen receptor (AR) in the presence of low levels of androgen due to AR upregulation or activation [35]. Thus, agents that reduce AR expression or function are good candidates for therapy of castration-resistant prostate cancer. Marrocco et al. [36] demonstrated that vorinostat decreased AR mRNA and protein, inhibited androgen signaling, and acted synergistically with the antiandrogen bicalutamide to induce apoptosis in prostate cancer cells.
HDACi have been shown to reverse epigenetic repression of the tumor suppressor retinoic acid receptor beta (RARb) [37]. Kato et al. [38] demonstrated LAQ824 induced RARb expression and restored retinoid sensitivity in retinoic acid-resistant melanoma cell lines.
Reversal of epithelial-mesenchymal transition by histone deacetylase inhibitors
Epithelial-mesenchymal transition (EMT) is a process that occurs normally during embryonic development and is thought to play a critical role in tumor progression by promoting invasiveness and metastasis [39]. EMT has been associated with resistance to targeted TKI therapy. Both EMT and TKI resistance have been reported to be reversible by HDACi [40]. Transforming growth factor beta 1 (TGF-β1), when it is aberrantly expressed in the tumor microenvironment, is an important inducer of EMT. Shan et al. [41] studied the mechanism of TGF-β1-induced EMT in lung adenocarcinoma cells in vitro and reported that HDAC6 is a critical regulator of EMT and, thus, a therapeutic target in pathological EMT.
Histone deacetylase inhibitors, Wnt signaling and cancer stem cells
An area of considerable importance in anticancer therapeutics is the identification and targeting of cancer stem cells. Although much needs to be learned about cancer stem cell signaling, there is a documented role for the Wnt/β-catenin pathway in cancer stem/progenitor cell biology [42]. Jiang et al. [43•] reported that DACT3, which is transcriptionally repressed in colorectal cancer, profoundly suppresses Wnt/β-catenin signaling, and that DACT3 expression is repressed not by DNA methylation but by bivalent activating and repressive histone methylation marks at the DACT3 promoter. They observed pharmacological reactivation of DACT3 expression with the combination of TSA and the S-adenosylhomocysteine hydrolase inhibitor 3-deazaneplanocin A, and this combination induced dramatic apoptosis associated with inhibition of Wnt/β-catenin signaling. Their data demonstrating that DACT3 is a therapeutic target of histone modifications have implications for colorectal cancer treatment and potentially, for targeting of cancer stem/progenitor cells driven by Wnt/β-catenin signaling.
Molecular basis of histone deacetylase inhibitor efficacy and resistance
Fantin et al. [44] identified constitutive signal transducers and activators of transcription (STAT) activation as a biomarker predictive of resistance to vorinostat in patients with CTCL. Nuclear STAT1 and nuclear phosphotyrosine-STAT3 in CTCL patient skin biopsies correlated with lack of clinical response to vorinostat. The pan-Janus-activated kinase inhibitor JAK-I, used to block the JAK/STAT pathway, was synergistic with vorinostat in antiproliferative effect in CTCL cells in vitro. Epping et al. [45•] performed a functional genetic screen to identify HDACi targets and determined that retinoic acid signaling is a rate-limiting target determining HDACi therapeutic efficacy. Using the Eμ-myc transgenic mouse model of B-cell lymphoma to examine the molecular basis of vorinostat efficacy and resistance, Lindemann et al. [46] reported that overexpression of the antiapoptotic proteins Bcl-2 or Bcl-XL or deletion of the proapoptotic BH3-only proteins Bim and Bid resulted in resistance to vorinostat therapy.
Histone deacetylase inhibitors as radiation sensitizers
A number of investigators have established that HDACi act as radiation sensitizers and prolong the expression of gH2AX, consistent with inhibition of DNA double-strand break repair [47]. HDAC3 is a critical component of the N-CoR/SMRT corepressor complex, which is aberrantly recruited by mutant transcription factors. Bhaskara et al. [48] used a conditional knockout of Hdac3 to demonstrate that in interphase cells inactivation of HDAC3 results in apoptosis, preceded by DNA damage associated with defective DNA double-strand break repair. In contrast, quiescent Hdac3−/− mouse embryo fibroblasts were protected from DNA damage. Their data suggest that inhibition of HDAC3 results in DNA damage, impedes efficient DNA repair, and induces apoptosis with selectivity for cycling cells.
Histone deacetylase inhibitor effects on immune response
HDACi impact the immune system through a variety of mechanisms, including upregulation of tumor immune genes resulting in increased cytotoxic T-lymphocyte recognition, upregulation of major histocompatibility complex class I, class II and costimulatory molecules leading to increased CD4 and CD8 T-cell activation, induction of NKG2D ligand expression on tumor cells rendering tumor cells more sensitive to NK cytolysis, and upregulation of molecules including TAP1 and LMP2 that play a role in antigen processing and presentation [49,50]. In an examination of the role of LAQ824 in immunity, Brogdon et al. [51•] reported LAQ824 inhibited dendritic cell-regulated T helper 1 (Th1) effector and not Th2 effector cell activation and migration, and thus can alter the Th1 and Th2 balance in therapeutic settings. Using an immunocompetent orthotopic murine renal cell carcinoma model Kato et al. [52•] demonstrated synergistic antitumor activity of interleukin 2 and SNDX-275, accompanied by a decrease in immunosuppressive Foxp3+ regulatory T cells and evidence of enhanced splenocyte antitumor cytotoxicity.
Histone deacetylases and metabolism
Using a postnatal liver-specific mouse knockout model, to understand HDAC3 function, Knutson et al. [53] demonstrated that loss of HDAC3 disrupts metabolic transcriptional networks and results in significant imbalances in carbohydrate and lipid metabolism. Chung et al. [54] used 31P magnetic resonance spectroscopy to monitor tumor phospholipid metabolism in xenograft studies of the response to the pan-HDACi LAQ824. They report a correlation between increased phosphomonoester/total phosphorus ratio and tumor response, demonstrating HDACi-induced alterations in phospholipid metabolism and tumor bioenergetics.
Pharmacodynamic assay development
The great majority of HDACi pharmacodynamic studies have been an assessment of histone H3 and H4 acetylation in peripheral blood mononuclear cells. Although concentration-dependent modulation of this marker is frequently seen, this is rarely correlated with clinical benefit. A greater emphasis is now being placed on obtaining tumor cells, including tumor biopsies and circulating epithelial tumor cells, and in identifying noninvasive techniques that measure drug activity against tumor.
Using multiparameter flow cytometric analysis [55] of bone marrow aspirates from acute myeloid leukemia (AML) patients in a phase I trial of SNDX-275, Gojo et al. [56] demonstrated quantification of hyperacetylation in blast cells versus normal marrow elements and correlation of tumor cell hyperacetylation with apoptosis and with p21 expression on a single cell level. Kummar et al. [57] used this assay to quantify hyperacetylation in different cell lineages in an SNDX-275 phase I trial in solid tumor and lymphoma patients. A new assay of HDAC enzymatic activity in intact cells using a cell-permeant substrate with a fluorescent read-out has been developed and tested in clinical trials of MGCD0103 [58,59]. In a phase I study of belinostat Steele et al. [60] demonstrated that patients with stable disease, and not progressive disease, showed a significant increase in plasma M30 epitope, the product of caspase-dependent cleavage of cytokeratin-18, which is thought to represent apoptosis of epithelial tumor cells.
Update on histone deacetylase inhibitor clinical trials
HDACi clinical trials and American Society of Clinical Oncology (ASCO) annual meeting abstracts are summarized in Table 2 [61 –112,113•,114–117,118•,119]. Some of the published trials are highlighted below. In HDACi monotherapy trials the majority of objective responses have been reported in CTCL and, to a lesser extent, other hematological malignancies. Only occasional objective responses have been reported in solid tumor monotherapy trials. Encouragingly, combination therapy protocols are achieving objective responses in both hematological malignancies and solid tumors. Table 3 contains a list of drug combinations and indications in active HDACi combination therapy trials.
Table 2.
Published histone deacetylase inhibitors clinical trials January 2007–June 2008a
| HDACi | Combination drug(s) | Phase | Indication | References |
|---|---|---|---|---|
| Vorinostat | None | I | Advanced multiple myeloma | [61] |
| None | I | Advanced leukemias and MDS | [62] | |
| None | I | GI cancer (Japanese patients) | [63] | |
| None | I | Children with refractory solid tumors | [64] | |
| None | I | Solid tumors (Japanese patients) | [65] | |
| None | I | NHL (Japanese patients) | [66] | |
| None | I | Malignant gliomas | [67] | |
| None | II | Refractory CTCL | [13••] | |
| None | II | Persistent or recurrent epithelial ovarian or primary peritoneal carcinoma | [68] | |
| None | II | Relapsed or refractory breast, colorectal, or NSCLC | [69] | |
| None | II | Recurrent and/or metastatic head and neck cancer | [70] | |
| None | II | Recurrent/metastatic TCC failing first line platinum-based therapy | [71] | |
| None | II | Relapsed NSCLC | [72] | |
| None | II | Recurrent GBM | [73] | |
| None | II | HRPC | [74] | |
| None | II | Relapsed or refractory indolent NHL | [75] | |
| None | II | Relapsed DLBCL | [76] | |
| None | II | Metastatic breast cancer | [77] | |
| None | IIb | Persistent, progressive, or treatment refractory CTCL | [12••] | |
| Carboplatin and paclitaxel | I | Advanced solid malignancies | [78] | |
| 13cRA | I | Children with refractory neuroblastomas, medulloblastomas, PNETs, and atypical teratoid rhabdoid tumor | [79] | |
| Bortezomib | I | Relapsed/refractory multiple myeloma | [80] | |
| Bortezomib | I | Refractory solid tumors | [81] | |
| Bevacizumab | I | Kidney cancer | [82] | |
| Capecitabine | I | Advanced solid tumors | [83] | |
| Bexarotene | I | Advanced CTCL | [84] | |
| Gemcitabine and cisplatin/carboplatin | I | Advanced NSCLC | [85] | |
| Doxorubicin | I | Advanced solid tumors | [86] | |
| Idarubicin | I | Advanced acute leukemia | [87] | |
| Pemetrexed and cisplatin | I | Advanced cancer | [88] | |
| Azacitidine | I/II | MDS, AML | [89] | |
| Tamoxifen | II | Advanced breast cancer having failed prior aromatase inhibitor therapy | [90] | |
| PCI-24781 | None | I | Refractory advanced solid tumors | [91] |
| R306465 | None | I | Advanced tumors | [92] |
| LBH-589 | None | I | Advanced solid tumors or NHL | [93] |
| None | I | Advanced solid tumors or lymphoma | [94] | |
| W/wo docetaxel and prednisone | I | Castration-resistant prostate cancer | [95] | |
| SNDX-275 | None | I | Refractory and relapsed acute leukemias | [56] |
| None | I | Refractory solid tumors and lymphoid malignancies | [57] | |
| Azacitidine | I | Advanced NSCLC | [96] | |
| MGCD0103 | None | I | Advanced leukemia, MDS | [97] |
| None | I | Advanced solid tumors | [59] | |
| None | II | Relapsed or refractory NHL | [98] | |
| None | II | Relapsed or refractory NHL | [99] | |
| Gemcitabine | I/II | Refractory solid tumors | [100] | |
| Azacitidine | I/II | High-risk MDS or AML | [101] | |
| Belinostat | None | I | Advanced solid tumors | [60] |
| None | I/II | Unresectable hepatocellular carcinoma | [102] | |
| None | II | Metastatic or recurrent platinum-resistant (<6 months) EOC and LMP ovarian tumors | [103] | |
| Azacitidine | I | Advanced myeloid neoplasms | [104] | |
| Carboplatin and paclitaxel (BelCaP) | II | Relapsed EOC | [105] | |
| Romidepsin | None | I | Thyroid and other advanced cancers | [106] |
| None | II | CTCL, PTCL | [107] | |
| None | II | Metastatic HRPC | [108] | |
| None | II | Lung cancer | [109] | |
| Gemcitabine | I | Pancreatic and other advanced solid tumors | [110] | |
| ITF2357 | None | II | Relapsed/refractory Hodgkin’s lymphoma | [111] |
| VPA | None | I | Refractory advanced cancer | [112] |
| Epirubicin | I | Advanced solid tumors | [113•] | |
| Decitabine | I | AML | [114] | |
| Decitabine | I | NSCLC | [115] | |
| Azacytidine | I | Advanced solid cancers | [116] | |
| FEC100 | I/II | Locally advanced/metastatic breast cancer. | [117] | |
| Azacitidine, ATRA | I/II | AML, MDS | [118•] | |
| Hydralazine | II | Refractory solid tumors | [119] |
AML, acute myeloid leukemia; ATRA, all-trans retinoic acid; CTCL, cutaneous T-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; EOC, epithelial ovarian cancer; GBM, glioblastoma multiforme; GI, gastrointestinal; HDACi, histone deacetylase inhibitors; HRPC, hormone refractory prostate cancer; LMP, low malignant potential; MDS, myelodysplastic syndrome; NHL, non-Hodgkin’s lymphoma; NSCLC, nonsmall cell lung cancer; PNETs, primitive neuroectodermal tumors; PTCL, peripheral T-cell lymphoma; TCC, transitional cell urothelial cancer; VPA, valproic acid.
Summary of active HDACi clinical trials. References are to full-length papers or to abstracts from the 2007 and 2008 annual meetings of the American Society of Clinical Oncology.
Table 3.
Active histone deacetylase inhibitors clinical trialsa
| HDACi | Combination drug(s) | Indication |
|---|---|---|
| Vorinostat | Azacitidine, bortezomib, decitabine, docetaxel, 5-FU, doxorubicine, idarubicin, erlotinib, isotretinoin, gemcitabine, temozolomide, sorafenib, tamoxifen, flavopiridol, paclitaxel/bevacizumab, paclitaxel/carboplatin, gemcitabine/cisplatin or carboplatin, 5-FU/leucovorin calcium/oxaliplatin, isotretinoin/carboplatin, cytarabine/etoposide, irinotecan/5-FU/leucovorin, rituximab/ifosfamide/carboplatin/etoposide, radical prostatectomy, palliative radiotherapy | CTCL, GBM, RCC, MDS, AML, ALL, CML, breast cancer, NSCLC, melanoma, lymphoma, advanced malignant pleural mesothelioma, advanced solid tumors, gliomas, nasopharyngeal cancer or nasal natural killer T-cell lymphoma, multiple myeloma, NHL, leukemia, colorectal cancer, GI cancer, prostate cancer, pelvic malignancy |
| Romidepsin | Decitabine, flavopiridol, gemcitabine, bortezomib | CTCL, PTCL, squamous cell carcinoma of the head and neck, NHL, refractory thyroid cancer, pulmonary and pleural malignancies, pancreatic, advanced solid tumors, relapsed myeloma, advanced lung, esophageal or pleural cancer |
| Belinostat | Azacitidine, bortezomib, isotretinoin, carboplatin/paclitaxel | Hepatocellular carcinoma, primary peritoneal cancer, fallopian tube cancer, B-cell NHL, T-cell lymphomas, MDS, thymic carcinoma, advanced hematological cancers, advanced solid tumors, lymphomas, ovarian cancer |
| LBH589 | Ketoconazole, bortezomib, trastuzumab, dextromethorphan, gemcitabine, lenalidomide/dexamethasone, capecitabine/lapatinib, paclitaxel/carboplatin/bevacizumab | CTCL, CML, MDS, NSCLC, advanced hematological malignancies, clear cell renal carcinoma, advanced solid tumors, multiple myeloma, breast cancer, malignant pleural mesothelioma |
| SB939 | None | Advanced or metastatic solid tumors |
| ITF2357 | None | Relapsed/refractory Hodgkin’s lymphoma |
| PCI-24781 (CRA-024781) | None | Advanced cancer |
| SNDX-275 | Azacitidine, erlotinib, sargramostim (GM-CSF), exemestane | NSCLC, AML, CML, MDS, breast cancer |
| MGCD0103 | Azacitidine, docetaxel, gemicitabine | AML, MDS, CLL, lymphoma, solid tumors, Hodgkin’s lymphoma, NHL |
| VPA | Decitabine, bevacizumab, doxorubicin, etoposide, epirubicin, sunitinib, sorafenib, dasatinib, erlotinib, lapatinib, lenalidomide, karenitecin, azacitidine/carboplatin, azacitidine/ATRA, ATRA/theophyllamine, FEC 100 (5-FU, epirubicin, and cyclophosphamide), temozolomide/radiation therapy | CLL, AML, MDS, GBM, small lymphocytic lymphoma, advanced thyroid cancer, nasopharyngeal carcinoma, ovarian cancer, NHL, advanced solid tumors, melanoma, advanced breast cancer, malignant mesothelioma, neuronal tumors and brain metastases |
ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukemia; ATRA, all-trans retinoic acid; CML, chronic myelogenous leukemia; CTCL, cutaneous T-cell lymphoma; EOC, epithelial ovarian cancer; 5-FU, 5-fluorouracil; GBM, glioblastoma multiforme; GI, gastrointestinal; HDACi, histone deacetylase inhibitors; MDS, myelodysplastic syndrome; NHL, non-Hodgkin’s lymphoma; NSCLC, nonsmall cell lung cancer; PTCL, peripheral T-cell lymphoma; RCC, renal cell carcinoma; VPA, valproic acid.
Further information regarding these clinical trials can be found at (http://www.cancer.gov/search/SearchClinicalTrialsAdvanced.aspx).
Monotherapy trials
Modesitt et al. [68] in a Gynecologic Oncology Group study of vorinostat in patients with recurrent or persistent epithelial ovarian or primary peritoneal carcinoma who were platinum-resistant/refractory concluded that vorinostat was well tolerated but had minimal activity in this patient population. Richardson et al. [61] in a phase I trial of vorinostat in patients with relapsed/refractory multiple myeloma concluded that vorinostat was generally well tolerated and had modest single-agent activity, warranting investigation with other antimyeloma agents. Garcia-Manero et al. [62] studied vorinostat in patients with advanced leukemias and myelodysplastic syndromes (MDS) and observed seven of 41 patients, all with AML, had hematological improvement including two complete responses and two complete responses with incomplete blood count recovery. They observed that antioxidant gene expression may confer vorinostat resistance, and that further evaluation of vorinostat in AML/ MDS is warranted. Steele et al. [60] observed stable disease in 18 of 46 patients in a phase I trial of belinostat in patients with advanced solid tumors. Schrump et al. [109] studied romidepsin in a lung cancer phase II trial that included assessment of gene expression profiles pre-romidepsin and post-romidepsin treatment in laser-captured tumor cells and adjacent normal bronchial epithelial cells. Although no objective responses were observed, transient disease stabilization was noted in nine of 18 evaluable patients, and there was evidence of gene expression changes indicating a shift of the lung tumor cells toward the expression pattern of normal bronchial epithelia. In a phase I trial of MGCD0103 in solid tumors disease stabilization was observed in five of 32 evaluable patients [59], and in a MGCD0103 phase I in leukemia three of 39 patients achieved a complete bone marrow response including two patients who had been previously treated with chemotherapy for refractory AML [58].
Combination therapy trials
Sung and Waxman [120] reported a phase I trial of escalating doses of 5-fluorouracil in combination with a fixed dose of phenylbutyrate in patients with advanced colorectal cancer. They reported disease stabilization in three out of four patients who completed at least 8 weeks of treatment. Soriano et al. [118•] conducted a phase I/II study of VPA, 5-azacytidine, and all-trans retinoic acid (ATRA) in patients with AML or high-risk MDS. The overall response rate was 42%, and the response rate in previously untreated patients older than 60 years was 52%. Clinical activity was associated with global DNA hypomethylation and histone acetylation, but these epigenetic changes were not different between responders and nonresponders. Higher VPA levels were demonstrated in patients who responded versus patients who did not, suggesting the potential utility of targeting plasma levels of VPA rather than dosing based on weight.
Munster et al. [113•] have studied the potentiation of tumor cell sensitivity to topo II inhibitors by HDACi, which they have demonstrated is associated with HDACi-induced chromatin remodeling increasing access of topo II inhibitor to the DNA substrate. They have examined this combination in a phase I dose-escalation trial of VPA and epirubicin in advanced solid tumor patients. Partial responses were seen in 22% of patients and stable disease/minor responses were seen in 39% of patients. The authors concluded that noteworthy antitumor activity was observed in heavily pretreated patients and historically anthracycline-resistant tumors. Decitabine alone or in combination with VPA was studied by Blum et al. in AML [114]. The authors determined the optimal biological dose of decitabine based on gene reexpression and the maximum tolerated dose of VPA combined with decitabine. Of evaluable patients 52% responded. Reexpression of estrogen receptor was associated with clinical response. Low-dose decitabine showed promising activity. However, the addition of VPA at relatively low doses resulted in encephalopathy. The authors concluded that further studies of decitabine alone or with an alternate HDACi are warranted. Ramalingam et al. [78] studied vorinostat in combination with carboplatin and paclitaxel for advanced solid malignancies. Of 25 evaluable patients they observed partial responses in 11, including 10 of 19 NSCLC patients and one patient with head and neck cancer, all chemotherapy naive. They are currently conducting a randomized phase II of carboplatin and paclitaxel with either vorinostat or placebo in advanced NSCLC.
Conclusion
HDACi with broad isoform inhibitory activity or some degree of isoform selectivity are moving into clinical trial in a wide range of indications and combinations. Despite only modest evidence of single-agent activity, especially in solid tumors, the pace of clinical exploration of HDACi is markedly accelerating. There is reason to be encouraged by recent advances in understanding of HDACi mechanisms of action, selection of optimal indications, and identification of rational combinations and optimal trial design.
The clinical development of HDACi presents an instructive dichotomy. Histone acetyltransferases, HDACs, and the regulation of acetylation have been the subject of highly sophisticated molecular analyses, often at the level of a single gene locus, so that on a reductionist basis, this class of drug is exceptionally well understood. In contrast, to employ HDACi clinically, it is important to recognize that these drugs are highly pleiotropic and their impact should, therefore, be examined for impact on a systems level, including recognition of their activity as modulators of cell metabolism, the immune system and the vasculature. Recent events suggest we are entering a new, more mature phase of HDACi clinical development that will facilitate a comprehensive examination of this molecularly fascinating but imperfectly understood class of drug.
Acknowledgements
Research funding supporting this work was received from Bayer Schering Pharma AG, Syndax Pharmaceuticals, and the Intramural Research Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References and recommended reading
Papers of particular interest, published within the annual period of review, have
been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 724–726).
- 1.Tang Y, Zhao W, Chen Y, et al. Acetylation is indispensable for p53 activation. Cell 2008; 133:612–626.• Demonstration of the requirement for acetylation for p53 function.
- 2.Bishton M, Kenealy M, Johnstone R, et al. Epigenetic targets in hematological malignancies: combination therapies with HDAC is and demethylating agents. Expert Rev Anticancer Ther 2007; 7:1439–1449. [DOI] [PubMed] [Google Scholar]
- 3.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 2007; 5:981–989. [DOI] [PubMed] [Google Scholar]
- 4.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 2007; 74:659–671. [DOI] [PubMed] [Google Scholar]
- 5.Marchion D, Munster P. Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther 2007; 7:583–598. [DOI] [PubMed] [Google Scholar]
- 6.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25:84–90. [DOI] [PubMed] [Google Scholar]
- 7.Piekarz RL, Sackett DL, Bates SE. Histone deacetylase inhibitors and demethylating agents: clinical development of histone deacetylase inhibitors for cancer therapy. Cancer J 2007; 13:30–39. [DOI] [PubMed] [Google Scholar]
- 8.Rasheed W, Bishton M, Johnstone RW, Prince HM. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther 2008; 8:413–432. [DOI] [PubMed] [Google Scholar]
- 9.Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs 2007; 16:659–678. [DOI] [PubMed] [Google Scholar]
- 10.Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab 2007; 8:383–393. [DOI] [PubMed] [Google Scholar]
- 11.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541–5552. [DOI] [PubMed] [Google Scholar]
- 12.Olsen EA, Kim YH, Kuzel TM, et al. Phase Ilb multicenter trial of vorinostat in ·· patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 2007; 25:3109–3115.•• Clinical trial that led to USFDA approval of vorinostat for the treatment of CTCL.
- 13.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109:31–39.•• Clinical trial that led to USFDA approval of vorinostat for the treatment of CTCL.
- 14.Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 2008; 409:581–589. [DOI] [PubMed] [Google Scholar]
- 15.Buggy JJ, Cao ZA, Bass KE, et al. CRA-024781: a novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. Mol Cancer Ther 2006; 5:1309–1317. [DOI] [PubMed] [Google Scholar]
- 16.Golay J, Cuppini L, Leoni F, et al. The histone deacetylase inhibitor ITF2357 has antileukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia 2007; 21:1892–1900. [DOI] [PubMed] [Google Scholar]
- 17.Arts J, Angibaud P, Marien A, et al. R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br J Cancer 2007; 97:1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Gonzalez A, Lin T, Ikeda AK, et al. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res 2008; 68:2557–2560. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell 2007; 27:197–213.• Demonstration of the mechanism by which a class II HDAC regulates motility.
- 20.Haggarty SJ, Koeller KM, Wong JC, et al. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA 2003; 100:4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh Y, Suzuki T, Kouketsu A, et al. Design, synthesis, structure-selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J Med Chem 2007; 50:5425–5438. [DOI] [PubMed] [Google Scholar]
- 22.Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007; 110:313–322.• Demonstration that autophagy promotes HDACi resistance and potential for targeting autophagy in combination trials.
- 23.Dai Y, Chen S, Kramer LB, et al. Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clin Cancer Res 2008; 14:549–558. [DOI] [PubMed] [Google Scholar]
- 24.Duan J, Friedman J, Nottingham L, et al. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 2007; 6:37–50. [DOI] [PubMed] [Google Scholar]
- 25.Miller CP, Ban K, Dujka ME, et al. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007; 110:267–277.• Activity of an HDACi and a promising proteasome inhibitor.
- 26.Fulda S Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr Cancer Drug Targets 2008; 8:132–140. [DOI] [PubMed] [Google Scholar]
- 27.Nawrocki ST, Carew JS, Douglas L, et al. Histone deacetylase inhibitors enhance lexatumumab-induced apoptosis via a p21Cip1-dependent decrease in survivin levels. Cancer Res 2007; 67:6987–6994. [DOI] [PubMed] [Google Scholar]
- 28.Zhao WL, Wang L, Liu YH, et al. Combined effects of histone deacetylase inhibitor and rituximab on non-Hodgkin’s B-lymphoma cells apoptosis. Exp Hematol 2007; 35:1801–1811. [DOI] [PubMed] [Google Scholar]
- 29.Kawamata N, Chen J, Koeffler HP. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood 2007; 110:2667–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards A, Li J, Atadja P, et al. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol Cancer Ther 2007; 6:2515–2524. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Q, Agoston AT, Atadja P, et al. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res 2008; 6:873–883.• An HDACI induces posttranscriptional loss of DNMT1 expression.
- 32.Verheul HM, Salumbides B, Van Erp K, et al. Combination strategy targeting the hypoxia inducible factor-1{alpha} with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res 2008; 14:3589–3597. [DOI] [PubMed] [Google Scholar]
- 33.Biçaku E, Marchion DC, Schmitt ML, Munster PN. Selective inhibition of histone deacetylase 2 silences progesterone receptor-mediated signaling. Cancer Res 2008; 68:1513–1519. [DOI] [PubMed] [Google Scholar]
- 34.Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res 2007; 13:4882–4890. [DOI] [PubMed] [Google Scholar]
- 35.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol 2005; 23:8253–8261. [DOI] [PubMed] [Google Scholar]
- 36.Marrocco DL, Tilley WD, Bianco-Miotto T, et al. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol Cancer Ther 2007; 6:51–60. [DOI] [PubMed] [Google Scholar]
- 37.Pili R, Kruszewski MP, Hager BW, et al. Combination of phenylbutyrate and 13-cis retinoic acid inhibits prostate tumor growth and angiogenesis. Cancer Res 2001; 61:1477–1485. [PubMed] [Google Scholar]
- 38.Kato Y, Salumbides BC, Wang XF, et al. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther 2007; 6:70–81. [DOI] [PubMed] [Google Scholar]
- 39.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442–454. [DOI] [PubMed] [Google Scholar]
- 40.Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006; 66:944–950. [DOI] [PubMed] [Google Scholar]
- 41.Shan B, Yao TP, Nguyen HT, et al. Requirement of HDAC6 for TGF-beta 1-induced epithelial-mesenchymal transition. J Biol Chem 2008; 283: 21065–21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008; 26:2839–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang X, Tan J, Li J, et al. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell 2008; 13:529–541.• Epigenetic histone marks regulate β-catenin signaling.
- 44.Fantin VR, Loboda A, Paweletz CP, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res 2008; 68:3785–3794. [DOI] [PubMed] [Google Scholar]
- 45.Epping MT, Wang L, Plumb JA, et al. A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc Natl Acad Sci USA 2007; 104:17777–17782.• Demonstration that retinoic acid signaling is a rate-limiting target of HDACi.
- 46.Lindemann RK, Newbold A, Whitecross KF, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A 2007; 104:8071–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol 2007; 25:4051–4056. [DOI] [PubMed] [Google Scholar]
- 48.Bhaskara S, Chyla BJ, Amann JM, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 2008; 30:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diermayr S, Himmelreich H, Durovic B, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 2008; 111:1428–1436. [DOI] [PubMed] [Google Scholar]
- 50.Khan AN, Tomasi TB. Histone deacetylase regulation of immune gene expression in tumor cells. Immunol Res 2008; 40:164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brogdon JL, Xu Y, Szabo SJ, et al. Histone deacetylase activities are required for innate immune cell control of Th1 but notTh2 effector cell function. Blood 2007; 109:1123–1130.• HDACi can alter the Th1/Th2 balance.
- 52.Kato Y, Yoshimura K, Shin T, et al. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin Cancer Res 2007; 13:4538–4546.• Use of an immunocompetent mouse model allows observation of immune regulatory events.
- 53.Knutson SK, Chyla BJ, Amann JM, et al. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J 2008; 27:1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chung YL, Troy H, Kristeleit R, et al. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia 2008; 10:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung EJ, Lee S, Sausville EA, et al. Histone deacetylase inhibitor pharmacodynamic analysis by multiparameter flow cytometry. Ann Clin Lab Sci 2005; 35:397–406. [PubMed] [Google Scholar]
- 56.Gojo I, Jiemjit A, Trepel JB, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2007; 109:2781–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kummar S, Gutierrez M, Gardner ER, et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res 2007; 13:5411–5417. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Manero G, Assouline S, Cortes J, et al. Phase I study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 2008; 11:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siu LL, Pili R, Duran I, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol 2008; 26:1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steele NL, Plumb JA, Vidal L, et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res 2008; 14:804–810. [DOI] [PubMed] [Google Scholar]
- 61.Richardson P, Mitsiades C, Colson K, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma 2008; 49:502–507. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008; 111:1060–1066. [DOI] [PubMed] [Google Scholar]
- 63.Chin K, Hatake K, Hamaguchi T, et al. A phase I study of vorinostat (suberoylanilide hydroxamic acid, SAHA) in Japanese patients with gastrointestinal (GI) cancer [abstract]. J Clin Oncol 2008; 26:15656S. [Google Scholar]
- 64.Fouladi M, Park J, Sun J, et al. A phase I trial of vorinostat in children with refractory solid tumors: a Children’s Oncology Group Study [abstract]. J Clin Oncol 2007; 25:9569S. [Google Scholar]
- 65.Fujiwara Y, Yamamoto N, Yamada K, et al. A phase I and pharmacokinetic/pharmacodynamic study of vorinostat (suberoylanilide hydroxamic acid, SAHA) in Japanese patients with solid tumor [abstract]. J Clin Oncol 2007; 25:14015S. [DOI] [PubMed] [Google Scholar]
- 66.Tobinai K, Watanabe T, Kobayashi Y, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients (pts) with non-Hodgkin lymphoma (NHL) in Japan [abstract]. J Clin Oncol 2007; 25:18521S. [Google Scholar]
- 67.Wen PY, Puduvalli V, Kuhn J, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid) in combination with temozolomide (TMZ) in patients with malignant gliomas (NABTC 04–03) [abstract]. J Clin Oncol 2007; 25:2039S. [Google Scholar]
- 68.Modesitt SC, Sill M, Hoffman JS, Bender DP. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2008; 109:182–186. [DOI] [PubMed] [Google Scholar]
- 69.Vansteenkiste J, Van Cutsem E, Dumez H, et al. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or nonsmall cell lung cancer. Invest New Drugs 2008. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 70.Blumenschein GR Jr, Kies MS, Papadimitrakopoulou VA, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs 2008; 26:81–87. [DOI] [PubMed] [Google Scholar]
- 71.Cheung EM, Quinn DI, Tsao-Wei DD, et al. Phase II study of vorinostat (Suberoylanilide Hydroxamic Acid, SAHA) in patients with advanced transitional cell urothelial cancer (TCC) after platinum-based therapy - California Cancer Consortium/University of Pittsburgh NCI/CTEP-sponsored trial [abstract]. J Clin Oncol 2008; 26:16058S. [Google Scholar]
- 72.Traynor AM, Dubey S, Eickhoff J, et al. A phase II study of vorinostat (NSC 701852) in patients (pts) with relapsed nonsmall cell lung cancer (NSCLC) [abstract]. J Clin Oncol 2007; 25:18044S. [Google Scholar]
- 73.Galanis E, Jaeckle KA, Maurer MJ, et al. N047B: NCCTG phase II trial of vorinostat (suberoylanilide hydroxamic acid) in recurrent glioblastoma multi-forme (GBM) [abstract]. J Clin Oncol 2007; 25:2004S. [Google Scholar]
- 74.Hussain M, Dunn R, Rathkopf D, et al. Suberoylanilide hydroxamic acid (vorinostat) post chemotherapy in hormone refractory prostate cancer (HRPC) patients (pts): a phase II trial by the Prostate Cancer Clinical Trials Consortium (NCI 6862) [abstract]. J Clin Oncol 2007; 25:5132. [Google Scholar]
- 75.Kirschbaum M, Zain J, Popplewell L, et al. Phase 2 study of suberoylanilide hydroxamic acid (SAHA) in relapsed or refractory indolent non-Hodgkin lymphoma: a California Cancer Consortium study [abstract]. J Clin Oncol 2007; 25:18515S. [Google Scholar]
- 76.Crump M, Coiffier B, Jacobsen ED, et al. Oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in relapsed diffuse large B-cell lymphoma (DLBCL): final results of a phase II trial [abstract]. J Clin Oncol 2007; 25:18511S. [DOI] [PubMed] [Google Scholar]
- 77.Luu TH, Leong L, Morgan R, et al. Vorinostat (suberoylanilide hydroxamic acid) as salvage therapy in metastatic breast cancer (MBC): a California Cancer Consortium phase II study [abstract]. J Clin Oncol 2007; 25: 11502S. [Google Scholar]
- 78.Ramalingam SS, Parise RA, Ramanathan RK, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res 2007; 13:3605–3610. [DOI] [PubMed] [Google Scholar]
- 79.Fouladi M, Park JR, Sun J, et al. Aphase Itrial and pharmacokinetic (PK) study of vorinostat (SAHA) in combination with 13 cis-retinoic acid (13cRA) in children with refractory neuroblastomas, medulloblastomas, primitive neuroectodermal tumors (PNETs), and atypical teratoid rhabdoid tumor [abstract]. J Clin Oncol 2008; 26:10012S. [Google Scholar]
- 80.Badros AZ, Philip S, Niesvizk R, et al. Phase I trial of vorinostat plus bortezomib (bort) in relapsed/refractory multiple myeloma (mm) patients (pts) [abstract]. J Clin Oncol 2008; 26:8548S. [Google Scholar]
- 81.Schelman WR, Kolesar J, Schell K, et al. A phase I study of vorinostat in combination with bortezomib in refractory solid tumors [abstract]. J Clin Oncol 2007; 25:3573S. [Google Scholar]
- 82.Hammers HJ, Verheul H, Wilky B, et al. Phase I safety and pharmacokinetic/pharmacodynamic results of the histone deacetylase inhibitor vorinostat in combination with bevacizumab in patients with kidney cancer [abstract]. J Clin Oncol 2008; 26:16094S. [Google Scholar]
- 83.Townsley C, Oza AM, Tang P, et al. Expanded phase I study of vorinostat (VOR) in combination with capecitabine (CAP) in patients (pts) with advanced solid tumors [abstract]. J Clin Oncol 2008; 26:11096S. [Google Scholar]
- 84.Hymes K, Dummer R, Sterry W, et al. Phase I trial of oral vorinostat in combination with bexarotene in patients with advanced cutaneous T-cell lymphoma [abstract]. J Clin Oncol 2008; 26:8613S. [Google Scholar]
- 85.Misset J, Tredaniel J, Descourt R, et al. Vorinostat in combination with gemcitabine plus platinum in patients with advanced nonsmall-cell lung cancer: a phase I, dose-escalation study [abstract]. J Clin Oncol 2008; 26:14601S. [Google Scholar]
- 86.Daud A, Schmitt M, Marchion D, et al. Phase I trial of a sequence-specific combination of the HDAC inhibitor, vorinostat (SAHA) followed by doxor-ubicin in advanced solid tumor malignancies [abstract]. J Clin Oncol 2007; 25:3502S. [Google Scholar]
- 87.Kadia TM, Borthakur G, Wierda W, et al. A phase I study of the combination of a DNA topoisomerase inhibitor, idarubicin, with the histone deacetylase inhibitor vorinostat, in advanced acute leukemia [abstract]. J Clin Oncol 2007; 25:7066S. [Google Scholar]
- 88.Chen L, Vogelzang NJ, Blumenschein G, et al. Phase I trial of vorinostat (V) in combination with pemetrexed (Pem) and cisplatin (CDDP) in patients with advanced cancer [abstract]. J Clin Oncol 2007; 25:18088S. [Google Scholar]
- 89.Silverman LR, Verma A, Odchimar-Reissig R, et al. A phase I/II study of vorinostat, an oral histone deacetylase inhibitor, in combination with azacitidine in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Initial results of the phase I trial: a NewYork Cancer Consortium [abstract]. J Clin Oncol 2008; 26:7000S. [Google Scholar]
- 90.Munster PN, Lacevic M, Schmitt M, et al. Phase II trial of vorinostat, a histone deacetylase inhibitor to restore the hormone sensitivity to the antiestrogen tamoxifen in patients with advanced breast cancer having failed prior aromatase inhibitor therapy [abstract]. J Clin Oncol 2008; 26:3501S. [Google Scholar]
- 91.Undevia SD, Janisch L, Schilsky RL, et al. Phase I study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the histone deacetylase inhibitor (HDACi) PCI-24781 [abstract]. J Clin Oncol 2008; 26: 14514S. [Google Scholar]
- 92.Fong PC, Settatree S, Sinha R, et al. A first-in-man phase I study of R306465, a histone deacetylase (HDAC) inhibitor exploring pharmacokinetics (PK) and pharmacodynamics (PD) utilizing an electrochemiluminescent immunoassay in patients (p) with advanced tumours [abstract]. J Clin Oncol 2007; 25: 3578S. [Google Scholar]
- 93.Prince HM, George D, Patnaik A, et al. Phase I study of oral LBH589, a novel deacetylase (DAC) inhibitor in advanced solid tumors and nonhodgkin’s lymphoma [abstract]. J Clin Oncol 2007; 25:3500S. [Google Scholar]
- 94.Sharma S, Vogelzang NJ, Beck J, et al. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of LBH589, a novel deacetylase (DAC) inhibitor given intravenously on a new once weekly schedule [abstract]. J Clin Oncol 2007; 25:14019S. [Google Scholar]
- 95.Rathkopf DE, Wong BY, Ross RW, et al. A phase I study of oral panobinostat (LBH589) alone and in combination with docetaxel (Doc) and prednisone in castration-resistant prostate cancer (CRPC) [abstract]. J Clin Oncol 2008; 26:5152S. [Google Scholar]
- 96.Juergens RA, Vendetti F, Coleman B, et al. Phase I trial of 5-azacitidine (5AC) and SNDX-275 in advanced lung cancer (NSCLC) [abstract]. J Clin Oncol 2008; 26:19036S. [Google Scholar]
- 97.Lancet JE, Nichols G, Assouline S, et al. A phase I study of MGCD0103 given as a twice weekly oral dose in patients with advanced leukemias or myelodysplastic syndromes (MDS) [abstract]. J Clin Oncol 2007; 25:2516S. [Google Scholar]
- 98.Crump M, Andreadis C, Assouline S, et al. Treatment of relapsed or refractory nonhodgkin lymphoma with the oral isotype-selective histone deacetylase inhibitor MGCD0103: Interim results from a phase II study [abstract]. J Clin Oncol 2008; 26:8528S. [Google Scholar]
- 99.Bociek RG, Kuruvilla J, Pro B, et al. Isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 demonstrates clinical activity and safety in patients with relapsed/refractory classical Hodgkin lymphoma (HL) [abtract]. J Clin Oncol 2008; 26:8507S. [Google Scholar]
- 100.Hurwitz H, Nelson B, O’Dwyer PJ, et al. Phase I/II: The oral isotype-selective HDAC inhibitor MGCD0103 in combination with gemcitabine (Gem) in patients (pts) with refractory solid tumors [abtract]. J Clin Oncol 2008; 26:4625S. [Google Scholar]
- 101.Garcia-Manero G, Yang AS, Klimek V, et al. Phase I/II study of a novel oral isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 in combination with azacitidine in patients (pts) with high-risk myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML) [abstract]. J Clin Oncol 2007; 25:7062S. [Google Scholar]
- 102.Yeo W, Lim R, Ma BB, et al. A phase I/II study of belinostat (PXD101) in patients with unresectable hepatocellular carcinoma [abstract]. J Clin Oncol 2007; 25:15081S. [Google Scholar]
- 103.Mackay H, Hirte HW, Covens A, et al. A phase II trial of the histone deacetylase inhibitor belinostat (PXD101) in patients with platinum resistant epithelial ovarian tumors and micropapillary/borderline (LMP) ovarian tumors. A PMH phase II consortium trial [abtract]. J Clin Oncol 2008; 26:5518S. [Google Scholar]
- 104.Odenike O, Green M, Larson RA, et al. Phase I study of belinostat (PXD101) plus azacitidine (AZC) in patients with advanced myeloid neoplasms [abtract]. J Clin Oncol 2008; 26:7057S. [Google Scholar]
- 105.Finkler NJ, Dizon DS, Braly P, et al. Phase II multicenter trial of the histone deactylase inhibitor (HDACi) belinostat, carboplatin and paclitaxel (BelCaP) in patients (pts) with relapsed epithelial ovarian cancer (EOC) [abtract].J Clin Oncol 2008; 26:5519S. [Google Scholar]
- 106.Piekarz R, Luchenko V, Draper D, et al. Phase I trial of romidepsin, a histone deacetylase inhibitor, given on days one, three and five in patients with thyroid and other advanced cancers [abtract]. J Clin Oncol 2008; 26: 3571S. [Google Scholar]
- 107.Piekarz R, Frye R, Wright J, et al. Update of the NCI multiinstitutional phase II trial of romidepsin, FK228, for patients with cutaneous or peripheral T-cell lymphoma [abstract]. J Clin Oncol 2007; 25:8027S. [Google Scholar]
- 108.Parker C, Molife R, Karavasilis V, et al. Romidepsin (FK228), a histone deacetylase inhibitor: Final results of a phase II study in metastatic hormone refractory prostate cancer (HRPC) [abstract]. J Clin Oncol 2007; 25: 15507S. [Google Scholar]
- 109.Schrump DS, Fischette MR, Nguyen DM, et al. Clinical and molecular responses in lung cancer patients receiving romidepsin. Clin Cancer Res 2008; 14:188–198. [DOI] [PubMed] [Google Scholar]
- 110.Doss HH, Jones SF, Infante JR, et al. A phase I trial of romidepsin in combination with gemcitabine in patients with pancreatic and other advanced solid tumors [abstract]. J Clin Oncol 2008; 26:2567S. [Google Scholar]
- 111.Viviani S, Bonfante V, Fasola C, et al. Phase II study of the histone-deacetylase inhibitor ITF2357 in relapsed/refractory Hodgkin’s lymphoma patients [abstract]. J Clin Oncol 2008; 26:8532S. [Google Scholar]
- 112.Atmaca A, Al-Batran SE, Maurer A, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer 2007; 97:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Munster P, Marchion D, Bicaku E, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol 2007; 25:1979–1985.• Encouraging activity of a HDACi/cytotoxic drug combination therapy trial.
- 114.Blum W, Klisovic RB, Hackanson B, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol 2007; 25:3884–3891. [DOI] [PubMed] [Google Scholar]
- 115.Karpenko MJ, Liu Z, Aimiuwu J, et al. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in patients with NSCLC. [abstract]. J Clin Oncol 2008; 26:3502S. [Google Scholar]
- 116.Soriano AO, Braiteh F, Garcia-Manero G, et al. Combination of 5-azacytidine (5-AZA) and valproic acid (VPA) in advanced solid cancers: a phase I study [abstract]. J Clin Oncol 2007; 25:3547S. [Google Scholar]
- 117.Munster PN, Marchion DC, Schmitt M, et al. Phase I/II trial combining the HDAC inhibitor, valproic acid (VPA) and FEC100 (5-fluorouracil, epirubicin and cyclophosphamide) in locally advanced/metastatic breast cancer [abstract]. J Clin Oncol 2007; 25:1065S. [Google Scholar]
- 118.Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine,valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood 2007; 110:2302–2308.• Significant clinical activity in a three-drug HDACi trial in AML/MDS.
- 119.Candelaria M, Gallardo-Rincon D, Arce C, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol 2007; 18:1529–1538. [DOI] [PubMed] [Google Scholar]
- 120.Sung MW, Waxman S. Combination of cytotoxic-differentiation therapy with 5-fluorouracil and phenylbutyrate in patients with advanced colorectal cancer. Anticancer Res 2007; 27:995–1001. [PubMed] [Google Scholar]