

Summary
Epigenetic deregulation of gene transcription is central to cancer cell plasticity and malignant progression but remains poorly understood. We found that the uncharacterized epigenetic factor chromodomain on Y-like 2 (CDYL2) is commonly over-expressed in breast cancer, and that high CDYL2 levels correlate with poor prognosis. Supporting a functional role for CDYL2 in malignancy, it positively regulated breast cancer cell migration, invasion, stem-like phenotypes, and epithelial-to-mesenchymal transition. CDYL2 regulation of these plasticity-associated processes depended on signaling via p65/NF-κB and STAT3. This, in turn, was downstream of CDYL2 regulation of MIR124 gene transcription. CDYL2 co-immunoprecipitated with G9a/EHMT2 and GLP/EHMT1 and regulated the chromatin enrichment of G9a and EZH2 at MIR124 genes. We propose that CDYL2 contributes to poor prognosis in breast cancer by recruiting G9a and EZH2 to epigenetically repress MIR124 genes, thereby promoting NF-κB and STAT3 signaling, as well as downstream cancer cell plasticity and malignant progression.
Subject Areas: Molecular Mechanism of Gene Regulation, Stem Cells Research, Functional Aspects of Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
Up-regulation of CDYL2 is common in breast cancer and correlates with poor prognosis
-
•
CDYL2 regulates enrichment of methyltransferases G9a and EZH2 at MIR124 genes
-
•
microRNA-124 regulation by CDYL2 impacts STAT3 and NF-κB signaling
-
•
CDYL2 regulation of EMT, migration, invasion, and stemness is STAT3/NF-κB dependent
Molecular Mechanism of Gene Regulation; Stem Cells Research; Functional Aspects of Cell Biology; Cancer
Introduction
A key feature of epigenetic processes is their ability to establish and maintain the expression level of genes in a manner that is durable, yet can be altered when necessary. To this end, the deposition of histone lysine methylation marks on chromatin is tightly regulated. However, deregulation of epigenetic factors can cause pathologic changes in cell identity and function and is a near-universal feature of cancer cells. Such perturbations offer therapeutic opportunities, and several drugs targeting epigenetic regulators are in use or under investigation as cancer treatments. These include inhibitors of the histone methyltransferases EZH2 and G9a, which respectively impart the H3K27me3 and H3K9me2 marks on chromatin (Dawson and Kouzarides, 2012, Jambhekar et al., 2019, Simó-Riudalbas and Esteller, 2015). However, despite recent progress, the epigenetic regulation of cellular plasticity in cancer remains poorly understood, with several putative epigenetic factors still uncharacterized. Addressing these issues could uncover new epigenetic drug targets for cancer treatment.
Most tumors are of epithelial origin, but epithelial cells are inherently resistant to several key steps in malignant progression. Molecular and cellular changes that render carcinoma cells more mesenchymal-like are associated with increased propensity to migrate and invade the surrounding tissues (Puisieux et al., 2014, Shibue and Weinberg, 2017). This so-called epithelial-to-mesenchymal transition (EMT) is also linked to the emergence of cancer stem cells (CSC), a subset of cells within a tumor mass that are highly efficient at seeding new tumor growth and in the case of breast cancer, more efficient at forming cellular aggregates called mammospheres in vitro (Shibue and Weinberg, 2017). In breast cancer, different tumor subtypes and prognosis correlate with distinct EMT states. Tumors expressing the estrogen receptor alpha (ER), but not the human epidermal growth factor (EGF) receptor 2 (HER2), are more epithelial-like, less invasive, and have better prognosis, whereas those triple-negative (TN) for expression of ER, HER2, and the progesterone receptor (PR) are more mesenchymal-like, invasive, and have worse prognosis (Sarrio et al., 2008). However, the acquisition of EMT-like features in a subset of cells within the ER+/HER2- tumor could drive the malignant progression of these cancers.
The gene expression changes underlying EMT and stemness result from interconnected regulatory systems involving transcription factors, epigenetic factors, and non-coding RNAs. In breast cancer, active forms of the transcription factors p65/NF-κB and STAT3 promote EMT, migration, invasion, and stemness (Marotta et al., 2011, Yang et al., 2014, Zhou et al., 2008). Misregulation of EZH2 and G9a can also induce these cellular processes (Chang et al., 2011, Curry et al., 2015, Dong et al., 2012), as can aberrant silencing of the tumor suppressive microRNA-124 (miR-124) (Ji et al., 2019, Lv et al., 2011, Wang et al., 2016a), itself a regulator of p65/NF-κB and STAT3 signaling (Cao et al., 2018, Hatziapostolou et al., 2011, Mehta et al., 2017, Olarerin-George et al., 2013). Recently, EZH2 was implicated in miR-124 repression in renal carcinoma cells (Zhou et al., 2019), supporting an interplay between these pathways. However, by and large, epigenetic regulation of EMT and stemness in cancer remains poorly understood.
In this study, we investigated the molecular and cellular functions of the putative epigenetic factor chromodomain on Y-like 2 (CDYL2) in breast cancer. This is a member of the CDYL family of genes, which includes two autosomal homologs in humans, CDYL1/CDYL and CDYL2 (Dorus et al., 2003). The family is defined by the presence of an N-terminal chromodomain that binds to methylated histone H3 lysine 9 (H3K9) and H3K27 residues (Fischle et al., 2008, Franz et al., 2009) and a C-terminal domain homologous to enoyl coenzyme A hydratase/isomerase enzymes (Dorus et al., 2003). CDYL1 is implicated in cancer as a candidate oncogene or tumor suppressor, depending on the context (Mulligan et al., 2008, Wu et al., 2013), and its epigenetic mechanism involves its interaction with and regulation of several other epigenetic factors, notably the H3K9 methyltransferases G9a/EHMT2, GLP/EHMT1 and SETDB1/ESET (Mulligan et al., 2008), and EZH2 (Zhang et al., 2011). By contrast, very little is known about the roles of CDYL2 in physiology or disease or its putative epigenetic mechanism.
A potential role for CDYL2 in cancer was suggested by a genome-wide association study that identified an intronic SNP in CDYL2 associated with cancer risk (Michailidou et al., 2013). Here we show that CDYL2 expression is also frequently up-regulated in breast cancer, and that high expression correlates with poor outcome in the estrogen receptor-positive/human EGF receptor 2-negative (ER+/HER2−) and TN subtypes. We propose that high levels of CDYL2 expression promote epigenetic repression of MIR124 genes by increasing G9a and EZH2 recruitment and H3K9 and H3K27 methylation at upstream regulatory regions. This, in turn, contributes to CDYL2 induction of NF-κB and STAT3 signaling, consequent induction of EMT genes, and increased cell motility, invasiveness, and stemness. These findings implicate CDYL2 as candidate proto-oncogene and therapeutic target in breast cancer.
Results
High CDYL2 Expression Level in Breast Cancer Is Associated with Poor Prognosis
Datamining revealed that CDYL2 mRNA is up-regulated in four breast cancer cohorts within The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Network, 2012) (Figures 1A and S1A). Similarly, the NCBI GEO datasets GSE10780 (Chen et al., 2010) and GSE21422 (Kretschmer et al., 2011) identified CDYL2 up-regulation in invasive ductal breast carcinomas as well as ductal carcinoma in situ, compared with normal breast tissues (Figure 1A). Analysis of the paired Clinical Proteomic Tumor Analysis Consortium (CPTAC) (Ellis et al., 2013) and TCGA datasets revealed that CDYL2 protein expression correlated with mRNA levels (Figure 1B). We found that both TCGA mRNA and CPTAC protein levels for CDYL2 across breast cancer subtypes also showed similar patterns, being higher in the ER+ forms than TN forms (Figures S1B and S1C). We next asked if the expression level of CDYL2 correlates with clinical outcome. Patients were subdivided into three categories based on their expression of the ER, PR, and HER2, namely, ER+/HER2−, ER+/HER2+, and receptor TN. This revealed that high expression of CDYL2 correlated with worse survival in both ER+/HER2− and TN subtypes (Figures 1C and 1D), but not ER+/HER2+ (Figure 1E). We also analyzed the expression of CDYL2 in normal breast tissues over the course of breast cancer progression, across all breast cancer types, ER+/HER2− and TN. This showed up-regulation of CDYL2 from the earliest pre-metastatic stage (pN0) in all three patient cohorts (Figures S1D–S1F). To further probe a possible association between CDYL2 expression and breast cancer progression, we examined its correlation with cancer gene expression signatures in the Molecular Signature Database (MSigDB) (Subramanian et al., 2005). This uncovered a positive correlation between CDYL2 expression in both ER+/HER2− and TN breast cancer and the Rizki_tumor_invasiveness-2D-UP signature (Figures S1G and S1H), corresponding to genes up-regulated in an invasive breast cancer cell line relative to the non-invasive precursor cell line from which it was derived (Rizki et al., 2008). Finally, we extended our analysis to other cancer contexts, revealing an association between CDYL2 expression level and survival in colorectal carcinoma, rectal adenocarcinoma, lung squamous cell carcinoma, and lung adenocarcinoma (Figures S1I–S1L). These findings identify CDYL2 as a gene commonly up-regulated in breast cancer and a candidate modulator of cancer progression and patient survival in the ER+/HER2− and TN subtypes, and other cancer forms.
Figure 1.

High CDYL2 Expression Level in Breast Cancer Is Associated with Poor Prognosis
(A) CDYL2 mRNA expression in breast tumors compared with normal tissues, as derived from the Oncomine database and GEO2R analysis of the indicated GEO datasets.
(B) Paired analysis of CDYL2 mRNA (TCGA, RNA-seq) and protein levels (CPTAC, mass spectrometry) in individual tumor specimen.
(C–E) Kaplan-Meier overall survival (OS) analysis performed from TCGA breast cancer subtypes: ER+/HER2− (C), triple negative (TN) (D), and ER+/HER2+ (E) using best cutoff of CDYL2 expression (high and low). Significance using log rank p value and hazard ratio (CI).
CDYL2 Over-Expression in the Non-invasive Breast Cancer Cell Line MCF7 Induces Transcriptional Changes Associated with Malignant Progression
To ask if CDYL2 up-regulation could induce oncogenic transcriptional and cellular changes, we stably expressed a CDYL2 cDNA in the non-invasive breast cancer cell line MCF7 (MCF7-CDYL2) or empty vector (MCF7-Vector) (Figure 2A). CDYL2 over-expression did not affect cell growth (Figure S2A), whereas RNA sequencing (RNA-seq) revealed striking differences between MCF7-CDYL2 and MCF7-Vector cells, with 693 genes up-regulated and 174 genes down-regulated at least 2.5-fold (Figure 2B; Table S1). Gene set enrichment analysis (GSEA) of genes up- or down-regulated in MCF7-CDYL2 cells revealed positive associations with EMT, metastasis, invasive versus non-invasive ductal carcinoma, breast cancer relapse in bone, and atypical ductal hyperplasia compared with non-cancerous breast tissue (Figures 2C and 2D). A number of genes from these GSEA signatures were validated by qRT-PCR, focusing on genes that are individually associated with breast cancer cell plasticity and malignant progression. These include the proto-oncogenes SOX2, KLF4, MYC, MUC1, FOS, FOSL1/Fra-1, and JUN (Alam et al., 2014, Bakiri et al., 2015, Jiao et al., 2010, Kufe, 2013, Nair et al., 2014, Piva et al., 2014, Yu et al., 2011), and the secretory molecules LCN2, CTGF, CXCL8, INHBA, and IL6 (Rhodes et al., 2004, p. 8; Shimo et al., 2006, Singh et al., 2013, Singh et al., 2013, Sullivan et al., 2009, p. 8) (Figure 2E). Down-regulation of the tumor suppressor TP63, breast cancer metastasis suppressor BRMS1, and cytokine BMP5, which regulate EMT, metastasis, and stemness, among other processes (Gatti et al., 2019, Romagnoli et al., 2012), was also confirmed (Figure 2E). Together, these insights suggest that CDYL2 over-expression can induce transcriptional changes associated with malignant breast cancer, potentially by promoting EMT, invasiveness, and metastasis.
Figure 2.

CDYL2 Over-Expression in the Non-invasive Breast Cancer Line MCF7 Induces Transcriptional Changes Associated with Malignant Progression
(A) Western blot analysis of CDYL2 and beta-actin expression in MCF7-CDYL2 and MCF7-Vector cells.
(B) Volcano plot showing genes up- or down-regulated at least 2.5-fold at an adjusted p value less than 0.05 (t test).
(C) Selected molecular signatures over-represented in either the up-or down-regulated gene sets from (B).
(D) Heatmap showing expression of genes from molecular signatures in (C) in the triplicate RNA-seqs.
(E) qRT-PCR validation of selected differentially expressed genes from (C). Mean of three independent experiments ±S.D. All differential expressions were significant at p < 0.05 (t test).
CDYL2 Over-expression in MCF7 Cells Induces EMT-like Changes, Migration, Invasiveness, and Mammosphere Formation
Further probing if CDYL2 might induce EMT-like changes in MCF7 cells, we assessed the expression of a panel of established EMT markers. qRT-PCR analysis revealed CDYL2 up-regulation of mesenchymal markers TWIST1, SNAI1, FN1, VIM, CTNNB1, and SNAI2 (Figure 3A). Western blotting revealed down-regulation of epithelial marker E-Cadherin and up-regulation the mesenchymal markers Vimentin (VIM), TWIST1/Twist, and SNAI1/Snail (Figure 3B). However, CDYL2 over-expression did not alter the levels of ER-alpha (Figure 3B), down-regulation of which can induce EMT (Dhasarathy et al., 2007), suggesting an independent mechanism. Notably, 3 weeks after MCF7 cells were transduced with the CDYL2 over-expression construct a change in cell morphology occurred, with loss of the cobblestone-like morphology of monolayers, replaced by a more fibroblast-like morphology (Figure 3C), similar to previous descriptions of EMT in MCF7 (Lin et al., 2014, Yin et al., 2008).
Figure 3.

CDYL2 Over-Expression in MCF7 Cells Induces EMT-like Changes, Accompanied by Increased Migration, Invasiveness, and Mammosphere Formation
(A) qRT-PCR analysis of a panel of EMT marker genes, normalization to GAPDH. Shown is mean ± SD of three experiments. Differences significant at p < 0.05 (t test).
(B) Western blot analysis of a panel of EMT markers, ER-alpha, CDYL2, and beta-actin.
(C) 10× phase contrast micrograph of MCF7-CDYL2 and MCF7-Vector control. Scale bar, 200 μM.
(D) Schematic diagram of the xCELLigence quantitative, real-time migration and invasion assay system.
(E) xCELLigence assay comparing the relative migration efficiency (Cell Index, CI) of MCF7-Vector and MCF7-CDYL2 cells. Both (E) and (F) show technical quadruplicates ± SD. Experiments were repeated at least three times with similar results.
(F) Invasion assays were performed as in (E), except that the porous membrane separating the upper and lower chambers of the transwell was first overlaid with Matrigel.
(G) Zebrafish embryo cell invasion and migration assay. Shown are micrographs illustrating the metastasis of fluorescently labeled MCF7-CDYL2 or MCF7-Vector from the site of injection to the tail. Scale bar, 200 μM. Quantification of the percentage of embryo exhibiting tail metastases is shown below. Experiments were repeated three times.
(H) Mammosphere formation in MCF7-CDYL2 cells compared with MCF7-Vector controls. Cells were plated at the indicated seeding number per well in 96-well plates. Mammospheres with size >50 μm were counted after 8 days. Shown is a scatterplot of the results of a representative of three independent experiments indicating the median (black bar) and t test significance (∗∗∗∗p < 0.0001).
(I) Representative 4× phase microscopy images of mammospheres counted in (H). Scale bar, 700 μM.
(J) Mammosphere diameters were determined by image analysis. Shown is mean + SD of eight wells in which 1,000 cells were seeded. t test significance (∗p < 0.05).
(K and L) FACS analysis of antigenic profile associated with breast cancer stem cells (CD44+; CD24-low/negative). Shown are representative FACS scatterplots (K) and the mean of three independent experiments ± S.D. (L). t test significance (∗p < 0.05).
To further investigate the possibility that CDYL2 over-expression induced EMT in MCF7 cells, we analyzed the expression of the cell surface adhesion molecules EpCAM and CD49f. It was previously demonstrated that low expression levels of EpCAM are associated with both EMT and breast cancer stem cells. The EpCAM−/CD49f− subpopulation of transformed mammary epithelial cells was especially tumorigenic, confirming high tumor-initiating, stem-like capacity. On the other hand, the EpCAM+/CD49f− subpopulation was shown not to form tumors (Kim et al., 2015). We found that over-expression of CDYL2 in MCF7 cells results in the appearance of a substantial population of EpCAM−/CD49f− cells (Figure S2C, top panels). Notably, CDYL2 over-expression also induced an EpCAM−/CD49f− cell population in another ER+ breast cancer cell line, Cama-1 (Figure S2C, center panels).
Among the primary contributions of EMT to malignant progression is increased cancer cell migration and invasion. In vitro assays revealed that the MCF7-CDYL2 cells migrated more proficiently across a microporous membrane compared with controls (Figures 3D and 3E). Using an adaptation of this assay to test for invasive capacity, wherein the porous membrane was first overlaid with a Matrigel barrier, we found that MCF7-CDYL2 cells also had increased invasive capacity relative to controls (Figure 3F).
We then probed the effect of CDYL2 on MCF7 cell invasion and metastasis in vivo. Both MCF7-CDYL2 and control cells were fluorescently labeled and injected into the perivitelline space of zebrafish embryos. The presence of tail metastases was monitored by fluorescence microscopy 24 h later. MCF7-Vector cells rarely produced metastases (3.57% of fish), whereas MCF7-CDYL2 cells did so in 21.57% of cases (Figure 3G).
To test if CDYL2 over-expression additionally induced stem-like characteristics in MCF7 cells, we first performed a mammosphere assay. This is a functional assay to assess the enrichment of stem-like cells in a population. MCF7-CDYL2 cells yielded both more and larger mammospheres compared with controls (Figures 3H–3J). Consistent with this, flow cytometry analysis revealed that CDYL2 over-expression in MCF7 cells increased the fraction of cells bearing the stem-like antigenic profile of CD44-high/CD24-low cells compared with controls (Figures 3K and 3L). Taken together, this series of studies indicates that CDYL2 over-expression in MCF7 cells promotes a number of cellular phenotypes associated with cellular plasticity and malignant progression.
RNAi Knockdown of CDYL2 in the Invasive Breast Cancer Cell Line MDA-MB-231 Diminishes the Expression of EMT Markers and Inhibits Migration, Invasion, and Mammosphere Formation
We next analyzed the effect of CDYL2 loss of function in the highly invasive, cancer stem cell-enriched, mesenchymal-like breast cancer line MDA-MB-231. CDYL2 expression was inhibited by RNAi (Figures 4A and 4B) and high-throughput RNA-seq analysis performed. This revealed that compared with CDYL2 over-expression in MCF7 cells, the effects of its knock-down on MDA-MB-231 cell gene expression were more moderate, with no genes up- or down-regulated 2.5-fold or greater, except for CDYL2 itself (Table S2). However, using a fold-change cutoff of 1.25, we identified 204 genes up-regulated and 129 genes down-regulated (Figure 4C, Table S2). These gene lists were subjected to over-representation analysis using the GSEA molecular signatures database. This revealed that the down-regulated gene set was enriched in transcripts associated with EMT, metastasis, mammary stem cells, and invasive ductal carcinoma (Figures 4D and 4E). This suggests that CDYL2 knock-down might suppress EMT, metastasis, and stemness. To determine if this is the case, we performed essentially the same suite of assays used to probe the effect of CDYL2 over-expression on MCF7 cells (Figure 3).
Figure 4.

RNAi Knockdown of CDYL2 in the Invasive Breast Cancer Cell Line MDA-MB-231 Induces Transcriptional and Phenotypic Changes Associated with Inhibition of Malignancy
(A and B) CDYL2 knock-down validated by RT-qPCR (A) and western blotting (B).
(C and D) (C) Volcano plot showing genes up- or down-regulated at least 1.25-fold. (D) Selected molecular signatures from the MSigDB database that were over-represented in either the up- or down-regulated gene sets from (C).
(E) Heatmap showing expression of selected genes from (D) in the triplicate RNA-seq.
(F) qRT-PCR validation of differential expression of selected genes from (D). Expression normalized to GAPDH. Data are the mean ± SD of three independent experiments. All differential expressions significant at p < 0.05 (t test).
(G) Western blot analysis of a panel of EMT markers, CDYL2, and beta-actin.
(H) xCELLigence assay comparing the relative migration efficiency of MDA-MB-231 cells treated with esiLuc or esiCDYL2 (esiCD2). Both (H) and (I) show technical quadruplicates ± SD. Experiments were repeated at least three times with similar results.
(I) Invasion assays were performed as in (H), except that the porous membrane separating the upper and lower chambers of the transwell was first overlaid with Matrigel.
(J) Fluorescence microscopy analysis of with esiLuc or esiCDYL2 on the migration of MDA-MB-231 from the perivitelline space of zebrafish embryos to the tail. Representative images are shown. Scale bar, 700 μM. Quantification of the percentage of embryos exhibiting tail metastases (Mets) is shown to the right. Experiments were repeated three times with similar results.
(K) Mammosphere formation in MDA-MB-231 cells treated with esiLuc or esiCDYL2. Cells were plated at the indicated seeding number per well in 96-well plates. Mammospheres with size >50 μm were counted after 8 days. Shown is a scatterplot of the results of a representative of three independent experiments indicating the median (black bar) and t test significance (∗∗p < 0.01; ∗∗∗∗p < 0.0001).
(L) Representative 4× phase microscopy images of mammospheres counted in (K) are shown. Scale bar, 200 μM.
(M) The diameters of mammospheres from (K) were determined using image analysis software. Shown is the mean +SD of eight wells in which 1,000 cells were seeded. t test significance (∗∗p < 0.01).
(N and O) FACS analysis of antigenic profile associated with breast cancer stem cells (CD44+; CD24-low/negative). Shown are representative FACS scatterplots (N) and the mean of three independent experiments ± SD. (O). t test significance (∗∗p < 0.01).
We first confirmed by qRT-PCR that CDYL2 RNAi reduced the levels of a number of transcripts associated with EMT, namely, JUN, MYC, SNAI2, FOSL1, and TWIST1 (Figure 4F). Immunoblotting revealed induction of E-cadherin expression and diminished expression of Vimentin, Fibronectin, and Twist (Figure 4G). Fluorescence-activated cell sorting (FACS) analysis revealed that CDYL2 RNAi also induced the appearance of a subpopulation of EpCAM+/CD49− MDA-MB-231 cells (Figure S2C, lower panels), which was shown to be associated with loss of tumorigenicity (Kim et al., 2015). However, we did not observe morphological changes in these cells indicative of a mesenchymal-to-epithelial transition (MET), possibly because the duration of the RNAi treatment was not long enough for such phenotypes to emerge. Nonetheless, transduction of MDA-MB-231 cells with three independent small hairpin RNA (shRNA) sequences targeting CDYL2 (Figure S3A) did result in epithelial-like morphological changes after 2 weeks, with cells forming cobblestone-like monolayers (Figure S3B). These cells also exhibited down-regulation of a number genes associated with EMT induction that were strongly up-regulated in MCF7-CDYL2 cells (FOS, FOSB, JUNB, CXCL8, CTGF, LCN2, MUC1, ERBB4), but not down-regulated in MDA-MB-231 cells treated with transient CDYL2 RNAi (Figure S3C).
The effect of CDYL2 RNAi on motility and invasiveness of MDA-MB-231 cells was evaluated using in vitro assays, revealing that transient or stable RNAi of CDYL2 dramatically reduced the migratory and invasive ability of MDA-MB-231 cells, relative to controls (Figures 4H, 4I, S3D, and S3E). In vivo, CDYL2 RNAi significantly impaired the ability of MDA-MB-231 cells to metastasize from the perivitelline site of injection to the tail of zebrafish embryos (Figure 4J), indicating suppression of the invasive and/or migratory capacity. Knockdown of CDYL2 by transient or stable RNAi treatment also resulted in fewer and smaller mammospheres relative to negative controls (Figures 4K–4M and S3F). While FACS analysis revealed that the majority of control RNAi-treated MDA-MB-231 cells were CD44-high/CD24-low, CDYL2 RNAi induced a population of CD44-low/CD24-low cells, indicative of loss of stemness (Figures 4N, 4O, and S3G). Collectively, these assays indicate that CDYL2 is required for MDA-MB-231 cell migration, invasion, and stemness, as well as the full expression of its mesenchymal-like state.
Regulation of p65/NF-κB and STAT3 Signaling by CDYL2
Gene expression signature analysis of the effects of CDYL2 over-expression in MCF7 cells or knockdown in MDA-MB-231 revealed a potential role in regulating NF-κB/TNF-alpha and STAT3/interleukin-6 signaling (Figure 5A). Given the importance of these signaling pathways in controlling cancer cell EMT, stemness, motility, and invasiveness, we asked if their regulation by CDYL2 might contribute to its regulation of these cellular processes. Consistent with the transcriptomic analysis, over-expression of CDYL2 in MCF7 cells increased the levels of tyrosine 705-phosphorylated STAT3 (Figure 5B), the active form of this protein. It also increased the levels of serine 536 phosphorylation on the NF-κB TF p65 (Figure 5B), indicating an increase in canonical NF-κB pathway signaling. By contrast, the levels of both phosphoproteins were diminished in CDYL2 RNAi-treated MDA-MB-231 cells (Figure 5C). We also probed the levels of total p65 and STAT3 proteins, as well as beta-actin, as a loading control. The total p65 levels were not affected by either CDYL2 over-expression or RNAi, whereas total STAT3 levels were higher in MCF7-CDYL2 cells compared with controls (Figure 5B) and down-regulated after CDYL2 RNAi in MDA-MB-231 cells (Figure 5C).
Figure 5.

CDYL2 Regulation of NF-κB and STAT3 Signaling Contributes to Its Induction of Invasion and Mammosphere Formation
(A) Selected gene expression signatures enriched in the indicated RNA-seq datasets.
(B) Western blot of Ser536-phosphorylated p65 and Tyr705-phosphorylated STAT3 in MCF7-Vector versus MCF7-CDYL2. The levels of total p65, STAT3, and β-actin were also probed.
(C) As for (B), except comparing MDA-MB-231 cells treated with esiLuc or esiCDYL2.
(D and E) Western blot validation of RNAi knockdown of p65 (D) or STAT3 (E) in MCF7-Vector and MCF7-CDYL2 cells. β-actin is shown as loading control.
(F) qRT-PCR analysis of the effect of RNAi knockdown of p65 on the expression of a panel of NF-κB target genes that were up-regulated in MCF7-CDYL2 compared with MCF7-Vector cells. Data are represented as mean of three independent experiments ± SD. All differences were significant at p < 0.05 (t test).
(G) As in (F), except the effect of RNAi knockdown of STAT3 on the expression of a panel of its target genes up-regulated in MCF7-CDYL2 compared with MCF7-Vector cells was evaluated.
(H and I) xCELLigence invasion assays of MCF7-CDYL2 in MCF7 cells treated with either control RNAi or siRNA targeting p65 (H) or STAT3 (I). Graphs are representative of three independent experiments in quadruple runs per condition. Error bars represent the SD of quadruplicate readings at each time point.
(J and K) Mammosphere assay of MCF7-CDYL2 in MCF7 cells treated with either control RNAi or siRNA targeting p65 (J) or STAT3 (K). Mammospheres from 1,000 seeded cells with size >50 μm were counted after 8 days. Shown is a scatterplot of the results of a representative of three independent experiments indicating the median (black bar) and t test significance (∗∗∗p < 0.001; ∗∗∗∗p < 0,0001; ns, not significant).
We then asked if CDYL2 induction of genes associated with EMT, invasion, and stemness in MCF7 cells might be dependent on signaling via p65/NF-κB and STAT3. STAT3 and p65 were knocked down by transient transfection with small interfering RNA (siRNA) in both MCF7-Vector and MCF7-CDYL2 cells. A non-targeting siRNA was used as a control (Figures 5D and 5E). qRT-PCR analysis then revealed that p65 RNAi down-regulated several genes associated with NF-κB signaling, namely, CTGF, EGR1, FOS, IL6, CXCL8, INHBA, JUN, MYC, SNAI1, KLF4, SOX2, and TWIST1 (Figure 5F). Similarly, STAT3 RNAi down-regulated several genes associated with STAT3 signaling, including FOS, TWIST1, SOX2, JUN, MUC1, INHBA, IL6R, IL6ST, and TNF (Figure 5G). Strikingly, RNAi knockdown of either p65 or STAT3 potently suppressed both invasiveness (Figures 5H and 5I) and mammosphere induction by CDYL2 (Figures 5J and 5K). Taken together, these analyses indicate that p65/NF-κB and STAT3 signaling is regulated by CDYL2, and that both pathways are required for CDYL2 induction of invasion and mammosphere formation in MCF7 cells.
CDYL2 Binds Upstream of MIR124 Genes and Regulates miR-124 Expression
Consistent with the possibility that CDYL2 might be an epigenetic regulator of transcription, we found that it was enriched in the nucleus of both MCF7 and MDA-MB-231 cells, with a significant fraction present in the chromatin fraction (Figures S4A–S4C). To identify where on chromatin CDYL2 is bound, we performed CDYL2 chromatin immunoprecipitation (ChIP) in both MCF7-Vector and MCF7-CDYL2 cells followed by Illumina sequencing (ChIP-seq). This revealed several genomic loci that were more enriched in CDYL2 in the MCF7-CDYL2 cells compared with vector controls, including upstream of all three members of the MIR124 gene family (Figure 6A; Data S1, S2, and S3). Owing to their implication in tumor suppression, and the ability to regulate both p65/NF-κB and STAT3 signaling, EMT, invasion, and stemness (Cao et al., 2018, Hatziapostolou et al., 2011, Ji et al., 2019, Lv et al., 2011, Mehta et al., 2017, Olarerin-George et al., 2013, Wang et al., 2016a, Wang et al., 2016b), we decided to investigate CDYL2 regulation of the MIR124 genes further. We confirmed CDYL2 enrichment upstream of MIR124 genes using ChIP-qPCR (Figure 6B). A non-reactive IgG was used as negative ChIP control, whereas qPCR analysis did not detect enrichment of CDYL2 at an unrelated sequence (Figure 6B). We reasoned that CDYL2 repression of MIR124 genes might contribute to its regulation of STAT3 and NF-κB signaling in MCF7 and MDA-MB-231 cells. In agreement with this possibility, CDYL2 RNAi diminished its levels upstream of MIR124 genes in MDA-MB-231 (Figure 6C), with a corresponding increase in the expression of both the precursor (pri-mir-124) and mature (miR-124-3p) forms of microRNA-124 (Figure 6E). Stable knock-down of CDYL2 using three independent shRNAs also increased levels of both pre-mir-124 and miR-124-3p (Figure S5). In complementary analysis, both MIR124 transcripts were down-regulated by CDYL2 over-expression in MCF7 (Figure 6D). Supporting the notion that the alterations of miR-124 levels were sufficient to affect cell function, analysis of the MCF7-CDYL2 and MDA-MB-231 esiCDYL2 RNA-seq data showed that miR-124 target genes were commonly up-regulated in the former and down-regulated in the latter (Figure 6F). The miR-124 GSEA signature was also positively correlated with CDYL2 mRNA expression across all samples in the TCGA breast cohort, as well as in the ER+/HER2− and TN subtypes (Figures 6G–6I). Differential expression of several of the genes up-regulated in MCF7-CDYL2 cells or down-regulated in MDA-MB-231 cells treated with CDYL2 RNAi was validated by RT-qPCR (Figures 6J and 6K). In suppression assays, a miR-124-3p mimic strongly diminished the levels of the active, phosphorylated forms of both p65 and STAT3 (Figure 6L). miR-124-3p also suppressed the total levels of STAT3 protein (Figure 6L). In complementary experiments, a neutralizing anti-miR-124-3p oligonucleotide rescued esiCDYL2 suppression of phospho-p65 and phospho-STAT3 levels in MDA-MB-231 cells, compared with a control non-targeting anti-miR oligonucleotide (Figure 6M). The reduced total STAT3 levels observed upon esiCDYL2 treatment were also rescued by anti-miR-124-3p treatment (Figure 6M). These findings indicate that CDYL2 regulates miR-124 levels, possibly by its binding upstream of MIR124 genes, and that control of miR-124-3p levels by CDYL2 contributes to its regulation of NF-κB and STAT3 signaling.
Figure 6.

CDYL2 ChIP-Seq Analysis Identifies miR-124 as a Mediator of CDYL2 Regulation of STAT3 and NF-κB Signaling
(A) Relative enrichment of CDYL2 upstream of MIR124 genes in MCF7-Vector cells (blue) and MCF7-CDYL2 (red), as revealed by ChIP-seq analysis. Input control and gene positions relative to the peaks are shown below.
(B) ChIP-qPCR validation of data presented in (A). ChIP-qPCR signal at an unrelated negative control sequence is also shown. Shown is the mean enrichment as a percentage of input of three independent experiments, ± SD. (∗p < 0.05; ∗∗p < 0.01, t test)
(C) As in (B), except CDYL2 or IgG ChIP was performed using chromatin prepared from MDA-MB-231 cells treated with esiLuc or esiCDYL2.
(D) qRT-PCR analysis of pre-mir-124 and miR-124-3p levels in MCF7-CDYL2 and MCF7-Vector cells. Expression was normalized to an unrelated miRNA. Data represent the mean of three independent experiments ± SD. Significance determined by test (∗p < 0.05; ∗∗p < 0.01).
(E) As in (D), except qRT-PCR analysis was performed using microRNA (miRNA) prepared from MDA- MB-231 cells treated with esiLuc or esiCDYL2.
(F) Selected gene expression signatures enriched in the indicated RNA-seq datasets.
(G–I) Correlation between CDYL2 expression and the GSEA signature “TGCCTTA MIR124A” in the indicated TCGA breast cancer patient cohorts. The linear regression r and p value are indicated.
(J) qRT-PCR analysis of the expression of miR-124-3p target genes in MCF7-CDYL2 and MCF7-Vector cells. Data represented as mean of three independent experiments ± SD. (∗p < 0.05, by t test).
(K) As in (J), except qRT-PCR analysis was performed using RNA prepared from MDA-MB-231 cells treated with esiLuc or esiCDYL2.
(L and M) Western blot of phosphorylated p65 (Ser 536), total p65, phosphorylated STAT3 (Tyr 705), and total STAT3 in MCF7-CDYL2 cells treated with a miR124-3p mimic or miR control (L) or in MDA-MB-231 cells co-treated with esiCDYL2 and either an anti-miR-124-3p oligonucleotide or a control anti-miR (M). Data are representative of three independent experiments.
CDYL2 Interacts with G9a, GLP, and PRC2 Complex Components EZH2 and SUZ12
Because CDYL2 is enriched at MIR124 genes and negatively regulates miR-124 expression, we asked if it might promote an epigenetically repressive chromatin environment at these loci. However, the epigenetic mechanism of CDYL2 is not known. By analogy with CDYL1, we speculated that it may form a complex with the H3K9 di-methyltransferases G9a, GLP, or SETDB1 (Mulligan et al., 2008) and the Polycomb Repressive Complex 2 (PRC2) core components EZH2 and SUZ12 (Zhang et al., 2011). Using immunoprecipitation (IP) assays we found that anti-CDYL2, but not a control IgG, efficiently recovered endogenous CDYL2 from MCF7 lysates and co-immunoprecipitated (coIP) G9a and its heterodimeric partner (Tachibana et al., 2005), GLP (Figure 7A). After long exposure of the western blot membrane, we also detected the presence of small amounts of EZH2 and SUZ12 in the CDYL2 IP, but at a much lower percentage of input compared with G9a and GLP, suggesting a low abundance or labile interaction (Figure S6A). Reciprocal coIP assays confirmed G9a interaction with CDYL2, but did not identify CDYL2 association with EZH2 (Figures 7A and S6A). These data indicate that CDYL2 forms a complex with G9a and GLP and may interact marginally with EZH2 and SUZ12.
Figure 7.

CDYL2 Interaction with G9a, GLP, EZH2, and SUZ12 and Its Regulation of G9a, EZH2, H3K9me2, and H3K27me3 Levels Upstream of MIR124-2
(A) Immunoprecipitation (IP) of CDYL2, EZH2, and G9a was performed on MCF7 cell lysates and the presence of the indicated proteins in the resulting IP eluates determined by western blotting (WB). A non-specific IgG was used as negative control. Input lysate was used to assess the relative strength of each coIP signal. The experiment was repeated three times with similar results. (∗specific band; ∗∗H.C., IgG heavy chain).
(B) ChIP-qPCR analysis of the relative occupancy of CDYL2, EZH2, and G9a upstream of MIR124-2 in MCF7-CDYL2 compared with MCF7-Vector cells. IgG, negative control ChIP. qPCR analysis was also performed at an unrelated negative control sequence. Shown is the mean enrichment as a percentage of input of three independent experiments, ± SD. Significance was determined by t test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
(C) As in (B), except using antibodies specific to H3K9me2, H3K27me3, H3, and IgG. These ChIP analyses were conducted using the same lysates as in (B), so are paired analyses.
(D and E) Experiments were conducted as described in (B) and (C), except using chromatin lysates prepared from MDA-MB-231 cells treated with esiCDYL2 or esiLuc.
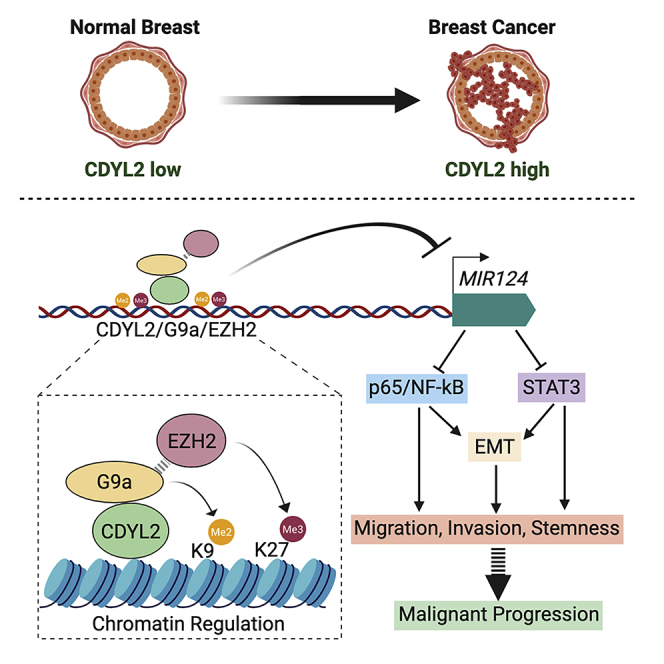
(F) Schematic model of the proposed contribution of CDYL2 to epigenetic regulation of MIR124, cell signaling, and malignancy-associated cellular processes.
CDYL2 Regulates the Enrichment of G9a and EZH2 Upstream of MIR124 Genes, as well as that of Their Cognate Methylation Marks H3K9me2 and H3K27me3
We next asked if CDYL2 might control the levels of G9a and EZH2 at a promoter-proximal region upstream of MIR124 genes. ChIP-qPCR assays indicated that CDYL2, G9a, and EZH2 were enriched upstream of all three MIR124 genes in both MCF7 and MDA-MB-231 cells (Figures 7B, 7C, S6B, and S6C). The enrichment of both methyltransferases was increased by CDYL2 over-expression in MCF7 cells (Figures 7B and S6B) and diminished by CDYL2 RNAi knockdown in MDA-MB-231 cells (Figures 7D and S6C). Increased levels of H3K9me2 and H3K27me3 were also observed upstream of MIR124 genes in MCF7-CDYL2 (Figures 7C and S6B), whereas levels of H3K9me2 and H3K27me3 at these loci were decreased upon CDYL2 RNAi in MDA-MB-231 (Figures 7E and S6C). The levels of total histone H3 at these loci were not affected by CDYL2 over-expression in MCF7 or its knockdown in MDA-MB-231 (Figures 7C and 7E, left panels). The same pattern of alterations was not observed at two independent control sequences (Figures 7C and 7E, right panels; Figures S6B and S6C). These findings indicate that in addition to interacting with G9a, and weakly so with EZH2, CDYL2 positively regulates the enrichment of both methyltransferases upstream of MIR124 genes, as well as those of the histone marks they regulate. To further probe the involvement of G9a and EZH2 in CDYL2 down-regulation of miR-124 levels, we treated MCF7-CDYL2 cells with the G9a/GLP inhibitor UNC0642 (Kim et al., 2017) or the EZH2 inhibitor CPI-169 (Bradley et al., 2014). We observed that UNC0642 treatment indeed resulted in increased levels of miR-124-3p (Figure S7A), relative to vehicle controls. However, treatment with CPI-169 at a range of doses did not affect miR-124-3p levels after 3, 4, or 5 days of treatment (Figure S7B and data not shown). This result further supports the importance of G9a/GLP methyltransferase activity in regulating miR-124 levels, but argues that inhibition of EZH2 enzymatic activity is not sufficient to antagonize CDYL2-mediated miR-124 down-regulation.
Discussion
Despite the emergence of epigenetic factors as important regulators of cancer cell plasticity and malignant progression, the underlying molecular mechanisms remain poorly understood. This is due in part to insufficient characterization of several putative epigenetic factors, including CDYL2. Our study shows that CDYL2 is frequently misexpressed in breast cancer and provides a proof of principle that this could promote cellular phenotypes associated with malignant progression. We present the first insights into the genes and cellular pathways of CDYL2 controls, and the epigenetic mechanisms it engages. Based on our findings, we propose that CDYL2 up-regulation contributes to poor prognosis in breast cancer by inducing epigenetic deregulation of genes and pathways important in tumorigenesis (MIR124, NF-κB, STAT3), resulting in cellular changes central to malignant progression (EMT, migration, invasion, stemness) (schematic diagram, Figure 7F).
Although we predicted CDYL2 to be an epigenetic repressor of transcription due to its homology to CDYL1 (Dorus et al., 2003, Fischle et al., 2008), this was not previously demonstrated. We have shown that CDYL2 is localized in the nuclear fraction of cells and binds to chromatin upstream of the MIR124 genes. Further implicating CDYL2 as an epigenetic repressor of transcription, CDYL2 over-expression in MCF-7 cells both increased its enrichment upstream of MIR124 genes and decreased the levels of MIR124 transcripts. Treatment of MCF7-CDYL2 cells with the G9a/GLP inhibitor UNC0642 increased expression of miR-124-3p, indicating that the catalytic activity of G9a and/or its heterodimeric partner GLP (Tachibana et al., 2005) is important for the CDYL2-mediated repression of MIR124 genes. Meanwhile, CDYL2 RNAi in MDA-MB-231 cells had the opposite effect. Our coIP data suggest that CDYL2 might regulate G9a levels at MIR124 genes via a mechanism involving physical association of the two factors, whereas they only weakly support this possibility in the case of EZH2. We speculate that an indirect mechanism could account for the strong effects of CDYL2 over-expression and RNAi on EZH2 enrichment upstream of MIR124 genes, such as the previously described regulation of EZH2 levels on chromatin by G9a methyltransferase activity (Mozzetta et al., 2014). However, the inhibition of the catalytic activity of EZH2 using the small molecule CPI-169 was not sufficient to de-repress miR-124 expression in MCF7-CDYL2 cells. This indicates that recruitment of the EZH2 methyltransferase activity to MIR124 genes by CDYL2 is not sufficient for their repression. Taken together, our findings are consistent with the idea that CDYL2 regulation of G9a enrichment at MIR124 genes promotes their transcriptional repression, likely via local increases in H3K9me2. Although CDYL2 also regulated the enrichment of EZH2 and H3K9me3 levels upstream of MIR124 genes, the weak interaction between CDYL2 and EZH2/SUZ12 and failure of CPI-169 to reverse CDYL2 repression of MIR124 genes in MCF7 cells suggests that these may be indirect and not sufficient for CDYL2 repression of miR-124.
MIR124 genes are emerging tumor suppressors commonly silenced in various cancers including breast (Conaco et al., 2006, Sullivan et al., 2009, p. 124; Wang et al., 2016a, p. 124; Wang et al., 2016b, p. 124). miR-124-3p directly targets STAT3 mRNA and antagonizes p65/NF-κB by inhibiting multiple components of its signaling pathway. It also regulates EMT, migration, invasion, and stemness (Ji et al., 2019, Lv et al., 2011, p. 124; Wang et al., 2016a, p. 124). Importantly, we showed that CDYL2 over-expression up-regulated the levels of active STAT3 and p65 in MCF7 cells, whereas CDYL2 RNAi down-regulated their levels in MDA-MB-231 cells. These observations were consistent with the effects of CDYL2 gain or loss of function on miR-124 levels, suggesting a functional association. Further supporting this possibility, CDYL2 up-regulation of STAT3 and NF-κB signaling in MCF7 cells was suppressed by a miR-124-3p mimic, whereas CDYL2 RNAi down-regulation of STAT3 and NF-κB signaling was rescued by a miR-124-3p inhibitor. Our data suggest that CDYL2 might regulate STAT3 levels via direct targeting by miR-124-3p, as the levels of total STAT3 were increased or diminished in accordance with CDYL2 gain or loss of function. Owing to the diversity of miR-124-3p targets implicated in NF-κB regulation across different cell types, it is challenging to determine which ones might connect CDYL2 with NF-κB regulation. Based on our studies, candidates include BIRC3, RELA, and STAT3 in MCF7 cells, all of which were up-regulated upon CDYL2 over-expression and are known to link miR-124 with NF-κB regulation (Cao et al., 2018, p. 3; Mehta et al., 2017, Wang et al., 2016a). In MDA-MB-231 cells, they include RELA and STAT3, which were down-regulated upon CDYL2 RNAi.
In accordance with the known effects of constitutive NF-κB and STAT3 signaling in cancer cells (Banerjee and Resat, 2016, Huber et al., 2004, p.; Wu et al., 2009), stable over-expression of CDYL2 induced migration and invasiveness in MCF7 cells in vitro. Likewise, whereas control MCF7 cells injected into zebrafish embryos seldom metastasized, cells over-expressing CDYL2 frequently did so. In complementary experiments, we showed that CDYL2 RNAi suppressed migration and invasion of MDA-MB-231 cells, which are normally highly invasive. CDYL2 RNAi also diminished the metastatic potential of MDA-MB-231 cells injected into zebrafish embryos. NF-κB and STAT3 signaling also promote the emergence of breast cancer stem cells, which are believed to play crucial roles in malignant progression (Marotta et al., 2011, Shostak and Chariot, 2011, Wang et al., 2016a, Zhou et al., 2008). We showed that CDYL2 over-expression augmented mammosphere formation in MCF7 cells, suggesting an increase in the proportion of stem-like cells in the culture. Consistent with this, we also observed an increase in the proportion of cells expressing the breast cancer stem cell marker profile CD44-high/CD24-low. In complementary assays, CDYL2 RNAi in MDA-MB-231 cells decreased both mammosphere formation and the fraction of CD44-high/CD24-low cells. These findings support the notion that CDYL2 not only promotes breast cancer cell migration and invasion but also stemness. As was the case for invasion, CDYL2 induction of both invasiveness and mammosphere formation was suppressed by RNAi knockdown of either NF-κB or STAT3, indicating key roles of these pathways in CDYL2 regulation of cancer cell biology.
It has been proposed that in certain malignancies, including breast, molecular and cellular changes that promote the emergence of mesenchymal-like cells constitute a key enabling step in the process of malignant progression (Huber et al., 2004, Puisieux et al., 2014, Sarrio et al., 2008, Shibue and Weinberg, 2017). We found that stable over-expression of CDYL2 induced morphological and molecular changes in the normally non-invasive, epithelioid, MCF7 cells, strongly indicative of an EMT. While transient transfection of MDA-MB-231 cells with siRNA targeting CDYL2 altered the expression of some genes consistent with a partial loss of mesenchymal cell identity, and impaired invasiveness and mammospheres formation in these cells, it did not induce any striking morphological changes. This is consistent with a partial reversal of the EMT state. However, when CDYL2 was knocked down for a longer duration using lentiviral RNAi vectors, we observed clusters of cells forming epithelial-like cobblestone monolayers after 2 to 3 weeks, suggesting a more advanced EMT reversal. These cells also exhibited down-regulation of several EMT-related genes (LCN2, CTGF, FOSB, JUNB, and MYC) that were over-expressed in MCF7-CDYL2 cells but not down-regulated by transient knock-down of CDYL2 in MDA-MB-231. This difference in gene expression may account for the observation of changes in cell morphology only after prolonged CDYL2 knock-down. In genetic suppression experiments, we found that the EMT-like gene expression program activated in MCF7-CDYL2 cells was antagonized by RNAi knock-down of either STAT3 or p65. These data argue that CDYL2 induction of migration, invasion, metastasis, and stemness in breast cancer cells might be due in part to its regulation of EMT states via up-regulation of NF-κB and STAT3 signaling.
Overall, our studies are consistent with an oncogenic effect of CDYL2 over-expression in breast cancer. This might contribute to the poor prognosis of the patients with ER+/HER2− and TN breast cancer whose cancers express high levels of CDYL2. Although not studied in depth here, we also observed a correlation between high CDYL2 expression and poor prognosis in lung and colorectal carcinomas, hinting at a wider role in cancer. Given the emergence of epigenetic factors as viable therapeutic targets in cancer, our study supports the further evaluation of CDYL2 as a candidate drug target in breast cancer, and potentially other malignancies.
Limitations of the Study
Our study provides several lines of molecular and cellular evidence supporting roles for CDYL2 in regulating EMT, stemness, and cancer cell migration and invasion. Future studies should further extend these findings using murine models of breast cancer cell growth, invasion, and stemness. Although we showed that both G9a and EZH2 were involved in the mechanism of CDYL2 regulation of MIR124 genes, several molecular details remain to be fully elucidated. For instance, given the weak coIP between CDYL2 and EZH2, it remains to be determined how over-expression or knock-down of CDYL2 exerts comparably strong effects on EZH2 chromatin levels at MIR124 genes. We speculate that this may be due to the previously described ability of G9a to regulate EZH2 enrichment at certain chromatin loci in a manner that depends on an intact G9a histone methyltransferase activity (Mozzetta et al., 2014), but this remains to be demonstrated in our models. An important aspect of this study is that it links the previously uncharacterized CDYL2 with pathways and genes of established importance in breast cancer, notably MIR124 genes and signaling via the STAT3 and NF-κB pathways. However, we do not exclude the possibility that CDYL2 regulation of other genes and pathways may also contribute to the cellular phenotypes we observed, and the role of CDYL2 in cancer.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Mulligan (peter.mulligan@inserm.fr).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. They are also available via NCBI GEO: GSE150320.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We gratefully acknowledge the technical assistance provided by Aurelie Jullien and Valery Attignon. We thank Christophe Ginestier, Toufic Renno, and Hichem Mertani for helpful technical discussions and sharing of protocols. This work was funded by grant number PLBIO2016-180 from l’Institut National du Cancer, grant number AJE20131128936 from La Fondation pour la recherche médicale, and financial support from le Centre Léon Bérard, and l’Institut national de la santé et de la recherche médicale, France (Inserm).
Author Contributions
M.S.: execution and analysis of majority of experiments; writing of original and updated manuscript; supervision and mentorship of other project participants; co-ordination with other co-authors. P.M.: conceptualization, funding acquisition, project administration, supervision and mentorship of other project members, investigation, formal analysis, writing of original and updated manuscript. P.S. and H.G.: conceptualization, project administration, funding acquisition, formal analysis; M.A.M.-P.: formal analysis; A.D.D.: investigation; L.B.B.: investigation; M.O.: conceptualization, investigation; G.I.: conceptualization and investigation; L.T., A.V., M.R., J.-P.F., V.L., S.N.M., A.P., J.B., and B.G.: formal analysis.
Declaration of Interests
The authors have no conflicts of interest to declare.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101141.
Supplemental Information
Illumina sequencing of input control for corresponding CDYL2 ChIP-seq in MCF7-CDYL2 and MCF7-Vector cells.
Illumina sequencing of CDYL2 ChIP in MCF7-CDYL2 cells.
Illumina sequencing of CDYL2 ChIP in MCF7-Vector cells.
References
- Alam M., Rajabi H., Ahmad R., Jin C., Kufe D. Targeting the MUC1-C oncoprotein inhibits self-renewal capacity of breast cancer cells. Oncotarget. 2014;5:2622–2634. doi: 10.18632/oncotarget.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri L., Macho-Maschler S., Custic I., Niemiec J., Guio-Carrion A., Hasenfuss S.C., Eger A., Muller M., Beug H., Wagner E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ. 2015;22:336–350. doi: 10.1038/cdd.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee K., Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int. J. Cancer. 2016;138:2570–2578. doi: 10.1002/ijc.29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley W.D., Arora S., Busby J., Balasubramanian S., Gehling V.S., Nasveschuk C.G., Vaswani R.G., Yuan C.-C., Hatton C., Zhao F. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Qiu J., Wang X., Lu Z., Wang D., Feng H., Li X., Liu Q., Pan H., Han X. Identification of microRNA-124 in regulation of Hepatocellular carcinoma through BIRC3 and the NF-κB pathway. J. Cancer. 2018;9:3006. doi: 10.7150/jca.25956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-J., Yang J.-Y., Xia W., Chen C.-T., Xie X., Chao C.-H., Woodward W.A., Hsu J.-M., Hortobagyi G.N., Hung M.-C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.-T., Nasir A., Culhane A., Venkataramu C., Fulp W., Rubio R., Wang T., Agrawal D., McCarthy S.M., Gruidl M. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res. Treat. 2010;119:335–346. doi: 10.1007/s10549-009-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaco C., Otto S., Han J.-J., Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. PNAS. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry E., Green I., Chapman-Rothe N., Shamsaei E., Kandil S., Cherblanc F.L., Payne L., Bell E., Ganesh T., Srimongkolpithak N. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenetics. 2015;7:84. doi: 10.1186/s13148-015-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Dhasarathy A., Kajita M., Wade P.A. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-α. Mol. Endocrinol. 2007;21:2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Wu Y., Yao J., Wang Y., Yu Y., Rychahou P.G., Evers B.M., Zhou B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Invest. 2012;122:1469–1486. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorus S., Gilbert S.L., Forster M.L., Barndt R.J., Lahn B.T. The CDY-related gene family: coordinated evolution in copy number, expression profile and protein sequence. Hum. Mol. Genet. 2003;12:1643–1650. doi: 10.1093/hmg/ddg185. [DOI] [PubMed] [Google Scholar]
- Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., Townsend R.R., Kinsinger C., Mesri M., Rodriguez H., Liebler D.C., Clinical Proteomic Tumor Analysis Consortium (CPTAC) Connecting genomic alterations to cancer biology with proteomics: the NCI clinical proteomic tumor analysis Consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W., Franz H., Jacobs S.A., Allis C.D., Khorasanizadeh S. Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J. Biol. Chem. 2008;283:19626–19635. doi: 10.1074/jbc.M802655200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz H., Mosch K., Soeroes S., Urlaub H., Fischle W. Multimerization and H3K9me3 binding are required for CDYL1b heterochromatin association. J. Biol. Chem. 2009;284:35049–35059. doi: 10.1074/jbc.M109.052332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti V., Bongiorno-Borbone L., Fierro C., Annicchiarico-Petruzzelli M., Melino G., Peschiaroli A. p63 at the crossroads between stemness and metastasis in breast cancer. Int. J. Mol. Sci. 2019;20:2683. doi: 10.3390/ijms20112683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziapostolou M., Polytarchou C., Aggelidou E., Drakaki A., Poultsides G.A., Jaeger S.A., Ogata H., Karin M., Struhl K., Hadzopoulou-Cladaras M., Iliopoulos D. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147:1233–1247. doi: 10.1016/j.cell.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M.A., Azoitei N., Baumann B., Grunert S., Sommer A., Pehamberger H., Kraut N., Beug H., Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambhekar A., Dhall A., Shi Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019;1 doi: 10.1038/s41580-019-0151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H., Sang M., Liu F., Ai N., Geng C. miR-124 regulates EMT based on ZEB2 target to inhibit invasion and metastasis in triple-negative breast cancer. Pathol. Res. Pract. 2019;215:697–704. doi: 10.1016/j.prp.2018.12.039. [DOI] [PubMed] [Google Scholar]
- Jiao X., Katiyar S., Willmarth N.E., Liu M., Ma X., Flomenberg N., Lisanti M.P., Pestell R.G. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J. Biol. Chem. 2010;285:8218–8226. doi: 10.1074/jbc.M110.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G., Ouzounova M., Quraishi A.A., Davis A., Tawakkol N., Clouthier S.G., Malik F., Paulson A.K., D’Angelo R.C., Korkaya S. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. 2015;34:671–680. doi: 10.1038/onc.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Lee H.-M., Xiong Y., Sciaky N., Hulbert S.W., Cao X., Everitt J.I., Jin J., Roth B.L., Jiang Y.-H. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 2017;23:213–222. doi: 10.1038/nm.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer C., Sterner-Kock A., Siedentopf F., Schoenegg W., Schlag P.M., Kemmner W. Identification of early molecular markers for breast cancer. Mol. Cancer. 2011;10:15. doi: 10.1186/1476-4598-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufe D.W. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2013;32:1073–1081. doi: 10.1038/onc.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-C., Lee Y.-C., Li L.-H., Cheng C.-J., Yang R.-B. Tumor suppressor SCUBE2 inhibits breast-cancer cell migration and invasion through the reversal of epithelial-mesenchymal transition. J. Cell. Sci. 2014;127:85–100. doi: 10.1242/jcs.132779. [DOI] [PubMed] [Google Scholar]
- Lv X.-B., Jiao Y., Qing Y., Hu H., Cui X., Lin T., Song E., Yu F. miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chin. J. Cancer. 2011;30:821. doi: 10.5732/cjc.011.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta L.L., Almendro V., Marusyk A., Shipitsin M., Schemme J., Walker S.R., Bloushtain-Qimron N., Kim J.J., Choudhury S.A., Maruyama R. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J. Clin. Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A.K., Hua K., Whipple W., Nguyen M.-T., Liu C.-T., Haybaeck J., Weidhaas J., Settleman J., Singh A. Regulation of autophagy, NF-κB signaling, and cell viability by miR-124 in KRAS mutant mesenchymal-like NSCLC cells. Sci. Signal. 2017;10:eaam6291. doi: 10.1126/scisignal.aam6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45:353–361. doi: 10.1038/ng.2563. 361.e1-e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzetta C., Pontis J., Fritsch L., Robin P., Portoso M., Proux C., Margueron R., Ait-Si-Ali S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell. 2014;53:277–289. doi: 10.1016/j.molcel.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Mulligan P., Westbrook T.F., Ottinger M., Pavlova N., Chang B., Macia E., Shi Y.J., Barretina J., Liu J., Howley P.M. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell. 2008;32:718–726. doi: 10.1016/j.molcel.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R., Roden D.L., Teo W.S., McFarland A., Junankar S., Ye S., Nguyen A., Yang J., Nikolic I., Hui M. c-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene. 2014;33:3992–4002. doi: 10.1038/onc.2013.368. [DOI] [PubMed] [Google Scholar]
- Olarerin-George A.O., Anton L., Hwang Y.-C., Elovitz M.A., Hogenesch J.B. A functional genomics screen for microRNA regulators of NF-kappaB signaling. BMC Biol. 2013;11:19. doi: 10.1186/1741-7007-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piva M., Domenici G., Iriondo O., Rabano M., Simoes B.M., Comaills V., Barredo I., Lopez-Ruiz J.A., Zabalza I., Kypta R., Vivanco M. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014;6:66–79. doi: 10.1002/emmm.201303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puisieux A., Brabletz T., Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- Rhodes D.R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A.M. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki A., Weaver V.M., Lee S.-Y., Rozenberg G.I., Chin K., Myers C.A., Bascom J.L., Mott J.D., Semeiks J.R., Grate L.R. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008;68:1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli M., Belguise K., Yu Z., Wang X., Landesman-Bollag E., Seldin D.C., Chalbos D., Barille-Nion S., Jezequel P., Seldin M.L., Sonenshein G.E. Epithelial-to-mesenchymal transition induced by TGF-beta1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 2012;72:6268–6278. doi: 10.1158/0008-5472.CAN-12-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D., Rodriguez-Pinilla S.M., Hardisson D., Cano A., Moreno-Bueno G., Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimo T., Kubota S., Yoshioka N., Ibaragi S., Isowa S., Eguchi T., Sasaki A., Takigawa M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J. Bone Miner Res. 2006;21:1045–1059. doi: 10.1359/jbmr.060416. [DOI] [PubMed] [Google Scholar]
- Shostak K., Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13:214. doi: 10.1186/bcr2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simó-Riudalbas L., Esteller M. Targeting the histone orthography of cancer: drugs for writers, erasers and readers. Br. J. Pharmacol. 2015;172:2716–2732. doi: 10.1111/bph.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.K., Simoes B.M., Howell S.J., Farnie G., Clarke R.B. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013;15:210. doi: 10.1186/bcr3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan N.J., Sasser A.K., Axel A.E., Vesuna F., Raman V., Ramirez N., Oberyszyn T.M., Hall B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–2947. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li Y., Dai Y., Liu Q., Ning S., Liu J., Shen Z., Zhu D., Jiang F., Zhang J., Li Z. Sulforaphane improves chemotherapy efficacy by targeting cancer stem cell-like properties via the miR-124/IL-6R/STAT3 axis. Sci. Rep. 2016;6:36796. doi: 10.1038/srep36796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Chen L., Wu Z., Wang M., Jin F., Wang N., Hu X., Liu Z., Zhang C.-Y., Zen K. miR-124-3p functions as a tumor suppressor in breast cancer by targeting CBL. BMC Cancer. 2016;16:826. doi: 10.1186/s12885-016-2862-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Zhang H., Wang P., Mao Z., Feng L., Wang Y., Liu C., Xia Q., Li B., Zhao H. Short-Form CDYLb but not long-form CDYLa functions cooperatively with histone methyltransferase G9a in hepatocellular carcinomas. Genes Chromosomes Cancer. 2013;52:644–655. doi: 10.1002/gcc.22060. [DOI] [PubMed] [Google Scholar]
- Wu Y., Deng J., Rychahou P.G., Qiu S., Evers B.M., Zhou B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Han S., Sun Y. An IL6-STAT3 loop mediates resistance to PI3K inhibitors by inducing epithelial-mesenchymal transition and cancer stem cell expansion in human breast cancer cells. Biochem. Biophys. Res. Commun. 2014;453:582–587. doi: 10.1016/j.bbrc.2014.09.129. [DOI] [PubMed] [Google Scholar]
- Yin L., Castagnino P., Assoian R.K. ABCG2 expression and side population abundance regulated by a transforming growth factor beta-directed epithelial-mesenchymal transition. Cancer Res. 2008;68:800–807. doi: 10.1158/0008-5472.CAN-07-2545. [DOI] [PubMed] [Google Scholar]
- Yu F., Li J., Chen H., Fu J., Ray S., Huang S., Zheng H., Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Yang X., Gui B., Xie G., Zhang D., Shang Y., Liang J. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J. Biol. Chem. 2011;286:42414–42425. doi: 10.1074/jbc.M111.271064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Tang K., Liu H., Zeng J., Li H., Yan L., Hu J., Guan W., Chen K., Xu H. Regulatory Network of two tumor-suppressive noncoding RNAs interferes with the growth and metastasis of renal cell carcinoma. Mol. Ther. Nucleic Acids. 2019;16:554–565. doi: 10.1016/j.omtn.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Zhang H., Gu P., Bai J., Margolick J.B., Zhang Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res. Treat. 2008;111:419–427. doi: 10.1007/s10549-007-9798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Illumina sequencing of input control for corresponding CDYL2 ChIP-seq in MCF7-CDYL2 and MCF7-Vector cells.
Illumina sequencing of CDYL2 ChIP in MCF7-CDYL2 cells.
Illumina sequencing of CDYL2 ChIP in MCF7-Vector cells.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. They are also available via NCBI GEO: GSE150320.