

Abstract
Objective:
To select optimal therapies based on the detection of actionable genomic alterations in tumor samples is a major challenge in precision medicine.
Methods:
We describe an effective process (opened December 1, 2017) that combines comprehensive genomic and transcriptomic tumor profiling, custom algorithms and visualization software for data integration, and preclinical 3-dimensiona ex vivo models for drug screening to assess response to therapeutic agents targeting specific genomic alterations. The process was applied to a patient with widely metastatic, weakly hormone receptor positive, HER2 nonamplified, infiltrating lobular breast cancer refractory to standard therapy.
Results:
Clinical testing of liver metastasis identified BRIP1, NF1, CDH1, RB1, and TP53 mutations pointing to potential therapies including PARP, MEK/RAF, and CDK inhibitors. The comprehensive genomic analysis identified 395 mutations and several structural rearrangements that resulted in loss of function of 36 genes. Meta-analysis revealed biallelic inactivation of TP53, CDH1, FOXA1, and NIN, whereas only one allele of NF1 and BRIP1 was mutated. A novel ERBB2 somatic mutation of undetermined significance (P702L), high expression of both mutated and wild-type ERBB2 transcripts, high expression of ERBB3, and a LITAF-BCAR4 fusion resulting in BCAR4 overexpression pointed toward ERBB-related therapies. Ex vivo analysis validated the ERBB-related therapies and invalidated therapies targeting mutations in BRIP1 and NF1. Systemic patient therapy with afatinib, a HER1/HER2/HER4 small molecule inhibitor, resulted in a near complete radiographic response by 3 months.
Conclusion:
Unlike clinical testing, the combination of tumor profiling, data integration, and functional validation accurately assessed driver alterations and predicted effective treatment.
Comprehensive genomic studies reveal that eachtumor harbors unique alterations. The observed tumor individuality argues for a personalized care model with therapeutic interventions tailored to the patient’s specific tumor profile. However, the success of targeted therapies depends on the accurate identification of actionable oncogenic driver mutations.
Several DNA alterations, including large genomic rearrangements, point mutations, and InDels, have emerged as potential sources of oncogenic driver mutations. Classical examples include EML4-ALK fusion,1,2 and oncogenic EGFR mutations in lung adenocarcinoma,3,4 and amplification of the ERBB2 gene in HER2-positive breast cancer.5,6 Importantly, targeting driver mutations, including ALK and EGFR in lung cancer, has resulted in significant increases in survival.7 Conversely, DNA alterations can render tumors resistant to targeted therapies (eg, EGFR T790M,8 ESR19). The diversity of acquired molecular alterations from patient to patient requires robust and precise comprehensive molecular diagnostic analysis to profile the molecular landscape of individual tumors.
Extensive molecular profiling increases the number of actionable alterations and the need for prioritization and validation of potential targeted therapies. A recent advance in precision medicine is the use of 3-dimensional (3D) culture models for drug testing. Multiple studies argue that freshly extracted human cancer cells will grow in 3D culture in a superior manner to growth in mice or in 2-dimensional culture for drug sensitivity studies.10,11 The major advantage of 3D model systems is time-to-reporting results (approximately 2 weeks), unlike animal models, which can require many months before drug testing can be performed. Other considerations are the low take-rate of patient-derived xenografts (PDXs), the need to use immune-compromised mice for PDX studies, overall cost, problems of scalability to the clinical laboratory, and the incompatibility of several key signaling pathways between murine and human cells.
Herein, we describe a genomically driven personalized therapeutic strategy that includes preclinical validation. This strategy uses comprehensive tools to analyze the genomic landscape of a patient’s cancer, integrates this information with pathway analysis, and employs a 3D microcancer model to assess drug sensitivity before patient treatment. Genomic profiling and drug sensitivity data are communicated to the treating physician. This multifaceted approach is described in the context of a patient with widely metastatic, chemotherapy refractory breast cancer who achieved a sustained therapeutic response to treatment selected through participation on this study.
PATIENTS AND METHODS
Eligible patients provide written informed consent to participate on protocols approved by the Mayo Clinic Institutional Review Board (IRB#13–000942 and IRB#14–004094). Tumor tissue is collected in conjunction with a clinically directed biopsy procedure to minimize patient morbidity. Integrated analysis is performed after compilation of the genomic data in collaboration with a multidisciplinary molecular tumor board to select targetable pathways of interest for the microcancer drug sensitivity assays (Figure 1A). Research-related genomic profiling and drug sensitivity data are communicated to the tumor board and clinical investigator for consideration and discussion with the treating physician. Findings of potential clinical relevance are verified in a College of American Pathologists (CAP)/Clinical Laboratory Improvement Amendments (CLIA) certified clinical laboratory as appropriate and used to direct therapy at the discretion of the health care provider.
FIGURE 1.
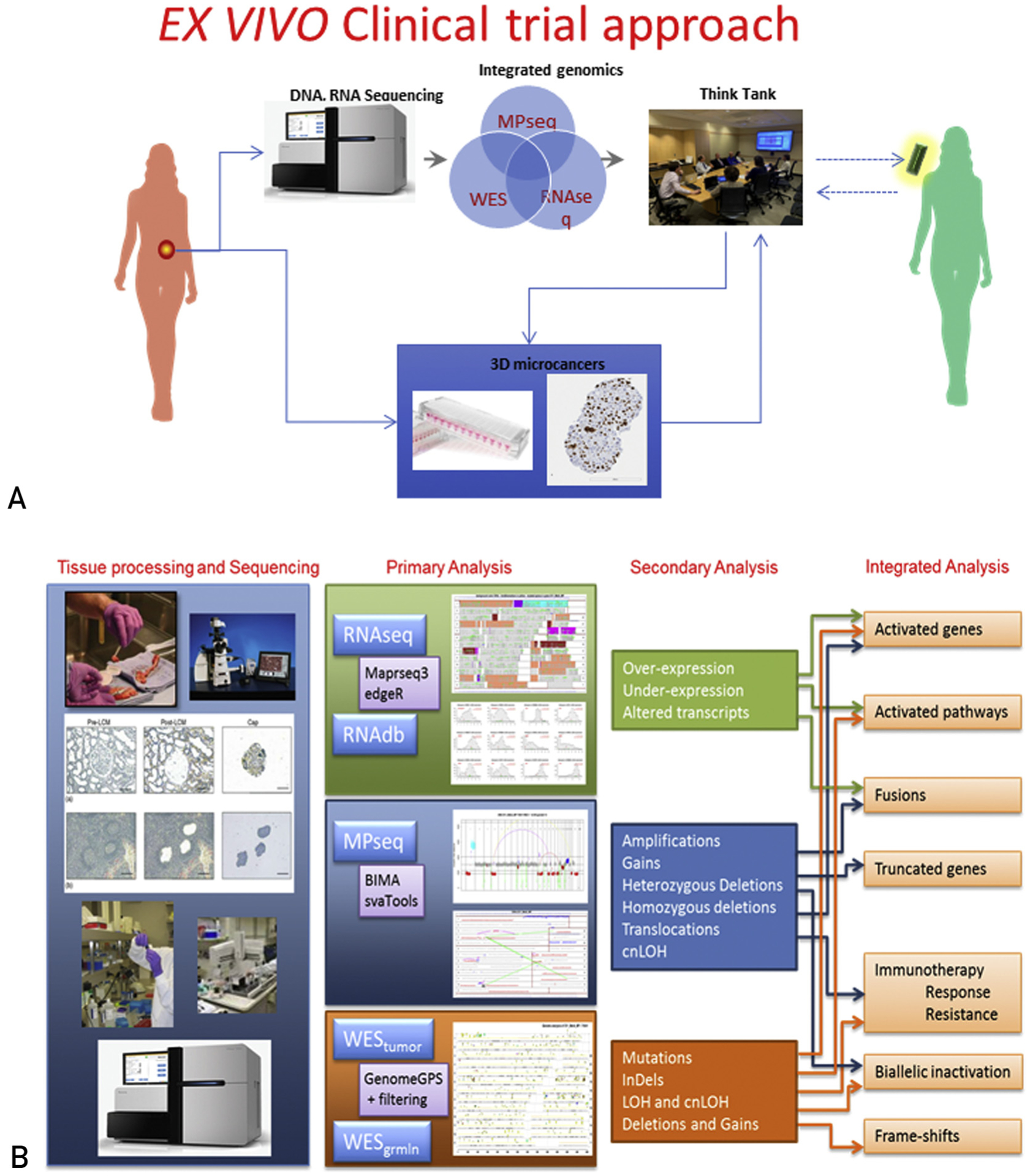
(A) Protocol schema. Tumor tissue of sufficient cellularity is split in two, and is either (1) flash frozen for subsequent pathology review, isolation of genomic material, and DNA/RNA sequencing, or (2) suspended in tissue culture media, minced, and cryopreserved for subsequent 3-dimensional microcancer analysis. Integrated genomic data are reviewed by a molecular tumor board to inform agent selection for screening in the 3-dimensional models. The molecular tumor board reconvenes to review drug sensitivity data in combination with the clinical and genomic data to generate an informed list of treatment regimens. Findings of potential clinical relevance are verified in a CAP/CLIA certified clinical laboratory as appropriate and used to direct therapy at the discretion of the health care provider. (B) Overall schematic illustrating the flow of the integrated Genomics pipeline. CAP = College of American Pathologists; CLIA = Clinical Laboratory Improvement Amendments; cnLOH = copy neutral loss of heterozygosity; MPseq = mate-pair sequencing; RNAseq = RNA sequencing; WES = whole-exome sequencing.
Integrated Genomic Analysis
A schematic describing the integrated genomics pipeline is shown in Figure 1B. Tumor alterations at the DNA level such as rearrangements, amplifications, copy number variants, copy neutral loss of heterozygosity, and mutations are investigated using mate-pair whole-genome DNA sequencing (MPseq) and whole-exome sequencing (WES). Gene fusions are investigated both by MPseq and RNA sequencing (RNAseq), whereas abnormal gene expression is investigated by RNAseq alone. Selected genes are investigated at the protein level by immunoblotting to confirm functional status by phosphorylation.
Tissue Handling at the Time of Collection
One portion of the tumor tissue is flash frozen in liquid nitrogen and maintained at −80°C for subsequent pathology review and isolation of genomic material. A second portion is immediately suspended in tissue culture media for subsequent 3D microcancer analysis.
Comprehensive Genomics, Next Generation Sequencing, and Bioinformatics
Pathologists review hematoxylin and eosinestained sections from the flash frozen specimen to guide tumor tissue macrodissection before DNA and RNA isolation. To reduce expense and increase robustness and sensitivity, we combine state-of-the-art techniques, including MPseq to detect global rearrangements, WES of both germline DNA, and tumor DNA to detect somatic mutations, and RNAseq to detect fusions and transcriptomic expression. MPseq was developed at Mayo Clinic following our previously published protocols12–15 as a novel cytogenetics tool to detect the chromosomal breakpoints involved in chromosome rearrangements, and to pinpoint the genes involved. The combination of MPseq analysis with WES and RNAseq data provides unprecedented accuracy in profiling genomic rearrangements, deletions, amplifications, aneuploidy, loss of heterozygosity, point mutations, gene phasing, and overall gene expression. MPseq, RNAseq, and WES are performed and analyzed as described in the Supplemental Methods (available online at http://www.mayoclinicproceedings.org).
Functional Analysis
Genomic findings are validated by phosphorylation assays using immunoblotting techniques (see Supplemental Methods), and potential drug sensitivities are tested with 3D microcancer models (see Supplemental Methods).
RESULTS
Patient and Tumor Characteristics
A 48-year-old woman presented at month 0 with stage IV, estrogen receptorepositive (25%), progesterone receptorenegative (0%), HER2-negative (IHC 2 +, fluorescence in situ hybridization [FISH] nonamplified), infiltrating lobular breast cancer. Initial sites of metastasis included the brain, bone, liver, adrenal gland, and regional lymph nodes. She had an excellent radiographic response to paclitaxel, completed palliative radio-therapy to the brain lesions, and progressed through letrozole. Biopsy results of the liver metastasis (month 7) were consistent with hormone receptor–negative (0%), HER2-negative (IHC 2+, FISH nonamplified), metastatic adenocarcinoma. She started vinorelbine shortly thereafter with stable disease followed by extracranial progression by month 10; radiographic imaging of the central nervous system was stable at that time. Tissue from the liver metastasis biopsy at month 7 was retrieved for (1) FoundationOne genomic testing, which identified mutations in NF1 (Q1399*), BRIP1 (Y1131fs*18), CDH1 (P30fs*3), RB1 (Q62*, splice site 2490–1G>A), and TP53 (I195fs*52); and (2) comprehensive genomics and preclinical functional validation as described below.
Structural Variant Analysis
Structural variant analysis of the tumor at the DNA level, assessed with MPseq, revealed an aneuploid genome (Figure 2A, 2B) with deletions of chromosomes 1p, 4p, 12p, 15, 16q, 17p, 22, and Xq; a double gain of 1q; large deletions on 4q, 12q, 14q, and 18p; and a large single gain on 12. The tumor also exhibited a few subclonal events (20% to 30% of the tumor cells) comprising a large deletion on 8q, a complex rearrangement on 8p (resulting in a possible ANK1-FGFR1 fusion), a rearrangement on 16p (resulting in a LITAF-BCAR4 fusion), a large deletion on 18, a gain on 20, and a small deletion on 3. No amplification was observed on chromosome 17q, where ERBB2 resides, consistent with the clinical FISH result. Considering the level of the large clonal deletions, the tumor percentage was calculated to be 80%, which is consistent with the pathology review (estimated tumor cellularity >70%). The ANK1-FGFR1 potential fusion detected by MPseq was not detected with RNAseq; however, the LITAF-BCAR4 fusion was detected and resulted in BCAR4 overexpression. BCAR4 is not normally expressed in adult tissues and is associated with antiestrogen resistance in breast cancer,16 reportedly through an ERBB2/3 signaling pathway.17–19 Detailed description of the junctions (magenta lines in Figure 2B) is provided in Supplemental Table 1 (available online at http://www.mayoclinicproceedings.org).
FIGURE 2.

Summary of integrated genomics data (MPseq, RNAseq, WES) generated by sequencing the breast cancer liver metastasis. (A) Whole-genome linear plot depicting structural variants and aneuploidy. (B) Whole-genome U-plot depicting mutations (circles) and structural variants. Gray segments depict the diploid areas. Red segments depict deleted areas. Blue segments depict gained areas. Magenta lines depict junctions. Cyan lines depict predicted gene fusions confirmed by RNAseq. (C) Summary table of genomic alterations. (D) Altered genes (24) overlapping with COSMIC’s Cancer Gene Census list. (E) Altered genes involved in breast cancer related pathways and potential targeted therapies. cnLOH = copy neutral loss of heterozygosity; MPseq = mate-pair sequencing; RNAseq = RNA sequencing; WES = whole exome sequencing.
Mutational Analysis
Differential analysis of tumor WES versus germline WES revealed 395 point mutations and InDels in 343 genes (Supplemental Table 2, available online at http://www.mayoclinicproceedings.org). Figure 2B depicts an integrated whole-genome map of the liver metastasis, including rearrangements and point mutations (lines and circles; numbers in the Y-axis denote human chromosomes). Figure 2C depicts a summary table of genomic alterations. Of these altered genes, 24 overlap with the Catalogue of Somatic Mutations in Cancer’s Cancer Gene Census list20 (Figure 2D). Integrated KEGG-pathway analysis of all affected genes indicated “bladder cancer” as the top cancer-related pathway, with alterations in ERBB2, TP53, RB1, CDH1, and RPS6KB1. Numerous alterations were identified in key pathways involved in breast cancer,21 including DNA repair (ATM, TP53, BRIP1, XPA), RAS/MAPK signaling (NF1, RASA2, CACNB4, IL1R2, PPP5C, SRF), cell cycle (RB1, ATM, TP53, WEE1), and GFR signaling (ERBB2, SHC3, RPS6KB1; Figure 2E).
MPseq and WES information were over-laid to identify genes with potential biallelic loss of function. Thirty-sex such genes were identified, including TP53, CDH1, FOXA1, and NIN (Supplemental Table 3, available online at http://www.mayoclinicproceedings.org). For example, biallelic involvement of CDH1 was due to a frame-shift mutation that rendered one allele dysfunctional, whereas the other allele was missing because of 16q deletion. This finding was supported in RNAseq with almost 100% alternative allele expression of the mutated CDH1 transcript (59 of 63 reads). Biallelic loss of CDH1 is a diagnostic marker associated with lobular breast cancer, consistent with the patient’s histologic diagnosis. Though not directly targetable, the loss of E-cadherin (the protein product of CDH1) expression results in the activation of numerous receptor tyrosine kinases, including EGFR and HER2.22
Potential treatments are available for tumors with inactivated ATM, RB1, NF1, SMARC4, and TP53 according to the TARGET list of 135 genes with known targeted therapies from the Broad Institute (http://www.tumorportal.org). These genes require biallelic inactivation to promote cancer. Only TP53 had a potential biallelic inactivation owing to complete deletion of one allele of 17p and a deleterious frame shift single nucleotide deletion on the second allele. The TP53 single nucleotide deletion was seen in 15 of 42 reads in RNAseq, presumably because of wild type TP53 expression in contaminating normal cells. Potential therapies for tumors lacking p53 expression include Chk123 and Wee1 inhibitors.24,25 The potential deregulation of both p53 and Wee1 also suggest the use of CDK inhibitors.26 A BRIP1 frame-shift InDel was also noted that would potentially lead to a functionally inactive transcript. BRIP1 associates with BRCA1 to repair damaged DNA, and mutations of BRIP1 have been linked to increased PARP inhibitor sensitivity.27
An ERBB2 (C2105T) somatic mutation was noted. The resulting proline-to-leucine substitution on position 702 of the NM_004448 transcript corresponds to a position just after the transmembrane domain in the intracellular region of the HER2 protein. This variant of unknown significance was observed (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5461196/) in another lobular carcinoma. The ERBB2 mutation is expressed as expected in the RNAseq at a fairly high level (249/516 reads showed the mutation) and in all tumor cells in the sample, suggesting that the ERBB2 mutation was an early event in tumor progression.
RNAseq indicated that the overall expression of ERBB2 was relatively high compared with a large cohort of human tumors, and toward the low end of tumor samples obtained from HER2-positive patients (Figure 3A). RNAseq also revealed a high overall expression of ERBB3 but not EGFR, suggesting that HER3 can act as a partner to HER2 in this tumor. Consistent with this finding, RNAseq suggested an increased overall expression of NRG2, but not NRG1 or EGF, a known HER3/4 ligand. Finally, immunoblotting analysis showed expression of EGFR, HER2, and HER3, but low levels of HER4 in the tumor sample, whereas tyrosine phosphorylation analysis confirmed that HER2, HER3, and HER4 were phosphorylated and therefore active, whereas EGFR was not (Figure 3B).
FIGURE 3.
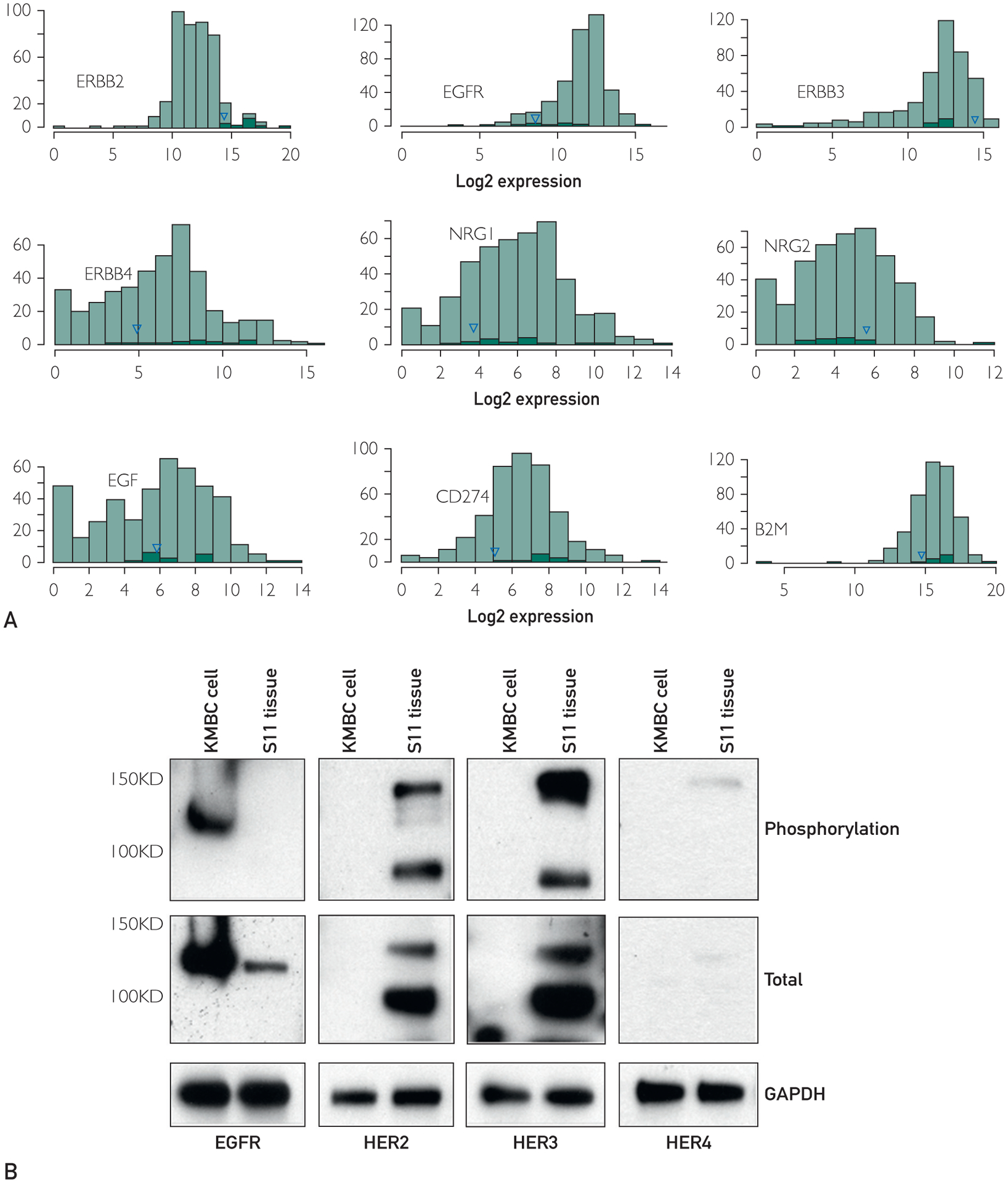
Relative gene expression and HER2/ErbB activation data. (A) Histograms contrasting the gene expression of selected genes (ERBB2, EGFR, ERBB3, ERBB4, NRG1, NRG2, EGF, CD274 [PDL1], and B2M) in the breast cancer liver metastasis to genes in a database of various other cancers. Black bars correspond to Her2-positive patients. The blue triangle denotes the study patient described herein. (B) Immunoblotting results for total EGFR, HER2, HER3, and HER4 protein expression and their tyrosine phosphorylation levels (activated state). KMCH1 cholangiocarcinoma cells serve as positive control for EGFR, HER2, and HER3 expression
Mutational burden was estimated at 8 mutations per megabase, which is considered high for breast cancer28 and may be an indication for immunotherapy benefit in some cancers.29,30 Therefore, we also investigated PD-L1 involvement by first comparing the expression of CD274 (the gene that codes PD-L1) in our sample with other tumor samples (Figure 3A). The expression was at the lower end. This result was consistent with PD-L1 IHC, performed as a clinical test, which showed no PD-L1 staining in the liver metastasis. B2M also appeared to be lower (one of the alleles would be deleted because of the deletion on chromosome 15).
Preclinical Validation Using Ex Vivo Tumor Models
An integral part of our genomically driven personalized cancer therapeutic strategy is the preclinical validation of potential nodes of vulnerability. To this end, we use a modi-fied hanging-drop 3D culture model established from live tumor cells (Figure 4A). The 3D microcancer model was extensively validated in PDXs and patient tumor tissue for accuracy, reproducibility, cellular heterogeneity, morphology, and drug sensitivity (data not shown). To confirm that our microcancer model faithfully recapitulates the original liver metastasis, we performed MPseq. As seen in Figure 4B, the 3D model faithfully recapitulates the genomic profile of the liver metastasis (see Figure 2B).
FIGURE 4.
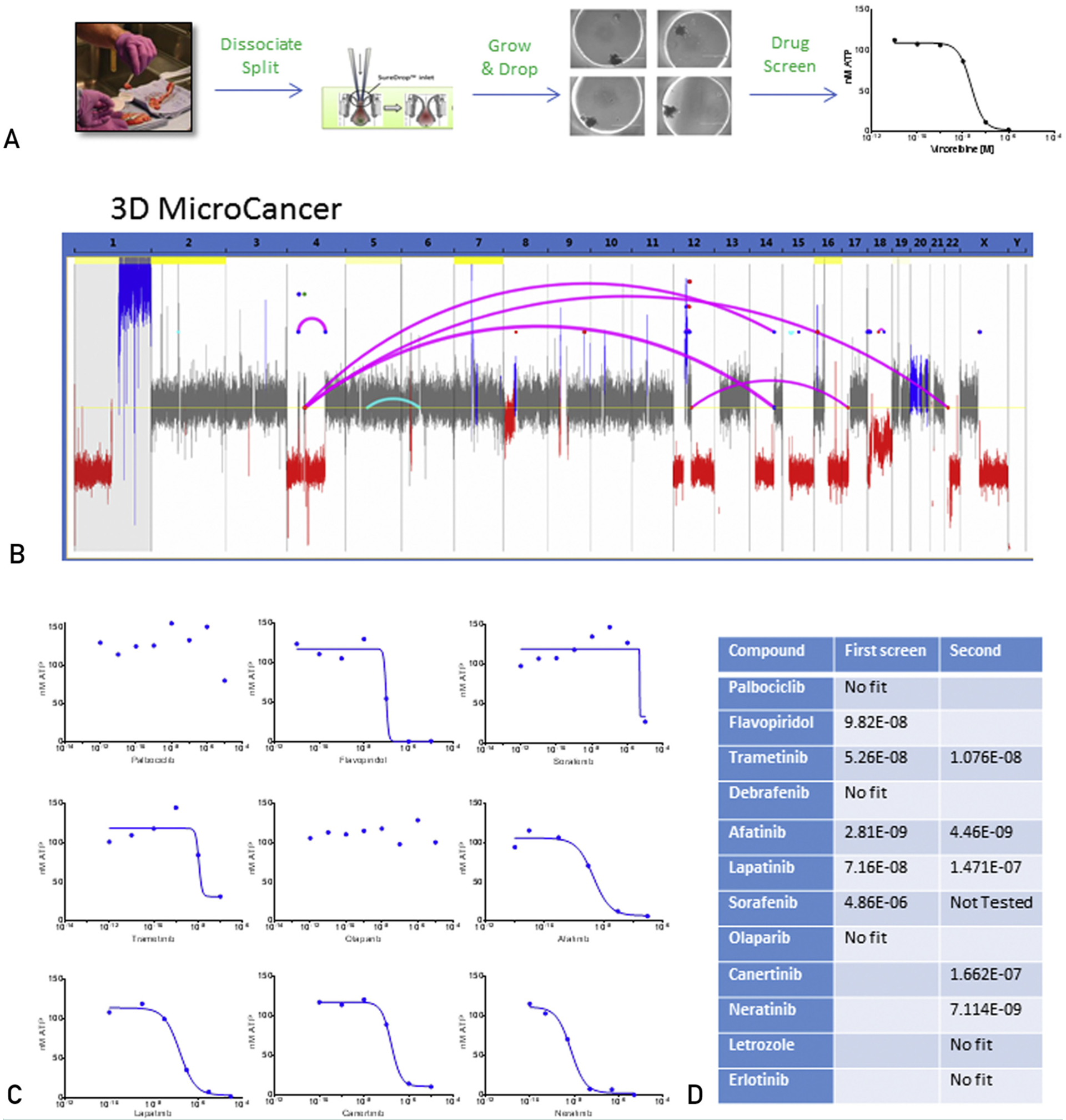
Summary of 3-dimensional (3D) microcancer data. (A) Schematic of the overall microcancer experimental process. (B) Whole-genome linear plot representation depicting structural variants and aneuploidy in the 3D microcancer of the breast cancer liver metastasis. (C) Dose response curves of the breast cancer liver metastasis 3D microcancer to indicated therapies. Y axis is adenosine triphosphate in nM, a measure of cell viability, whereas x axis is drug concentration (M). (D) Table of tested compounds and the related inhibitory concentrations at 50% obtained after testing the breast cancer liver metastasis 3D microcancers.
An initial drug screen was based on the potential deregulation of genes involved in the cell cycle, DNA repair, and ERBB signaling. Three-dimensional microcancers derived from the liver metastasis were grown in 96-well plates and treated for 6 days with select targeted compounds, chemotherapeutics, and combination treatments. Dose-response curves and the associated IC50 values were calculated after measuring cellular adenosine triphosphate. The assay indicated no sensitivity to: letrozole, a therapy that the patient progressed on; the PARP inhibitor olaparib; the RAF inhibitors sorafenib and dabrafenib; or the CDK4/6 inhibitor palbociclib (Figure 4C). A strong but partial response was observed with the MEK inhibitor trametinib. Strong antitumor responses were observed with the nonspecific CDK inhibitor, flavopiridol, the relatively nonselective ERBB2 inhibitor lapatinib and especially the EGFR/HER2/HER4 inhibitor afatinib, which inhibits all active ErbB homodimers and heterodimers. The strong sensitivity to HER2 targeted therapy was consistent with the increased expression and mutation of HER2 in this tumor, the increased expression of BCAR4,19 and the loss of E-cadherin, which was previously reported to confer increased sensitivity to HER2 targeted therapy in lobular breast carcinoma.31
To confirm this conclusion, we repeated the microcancer drug screen focusing on ERBB pathway inhibitors. The data confirmed sensitivity to lapatinib and afatinib with nearly identical IC50 values as in the first screen (Figure 4C, 4D); they also indicated sensitivity to other HER2 targeted therapies, including canertinib, neratinib, and trastuzumab/pertuzumab. No sensitivity was indicated for the selective EGFR inhibitor erlotinib.
Clinical Outcomes
Genomic profiling and drug sensitivity data were conveyed to the treating provider at month 10. In retrospect, clinical sequencing had identified ERBB2 P702L as a variant of unknown significance, providing further rationale for the use of HER2 targeted therapy. Given these findings and the documentation of disease progression on standard therapies, the patient’s treatment was changed to afatinib (40 mg oral daily). A follow-up PET scan after 3 months revealed near complete resolution of the extracranial metastatic lesions (Figure 5A, 5B). Subsequent positron emission tomography imaging 2 months later (ie, at month 5 of afatinib) was consistent with a durable response. The radiographic findings were supported by corresponding changes in CA27.29 (Figure 5C).
FIGURE 5.
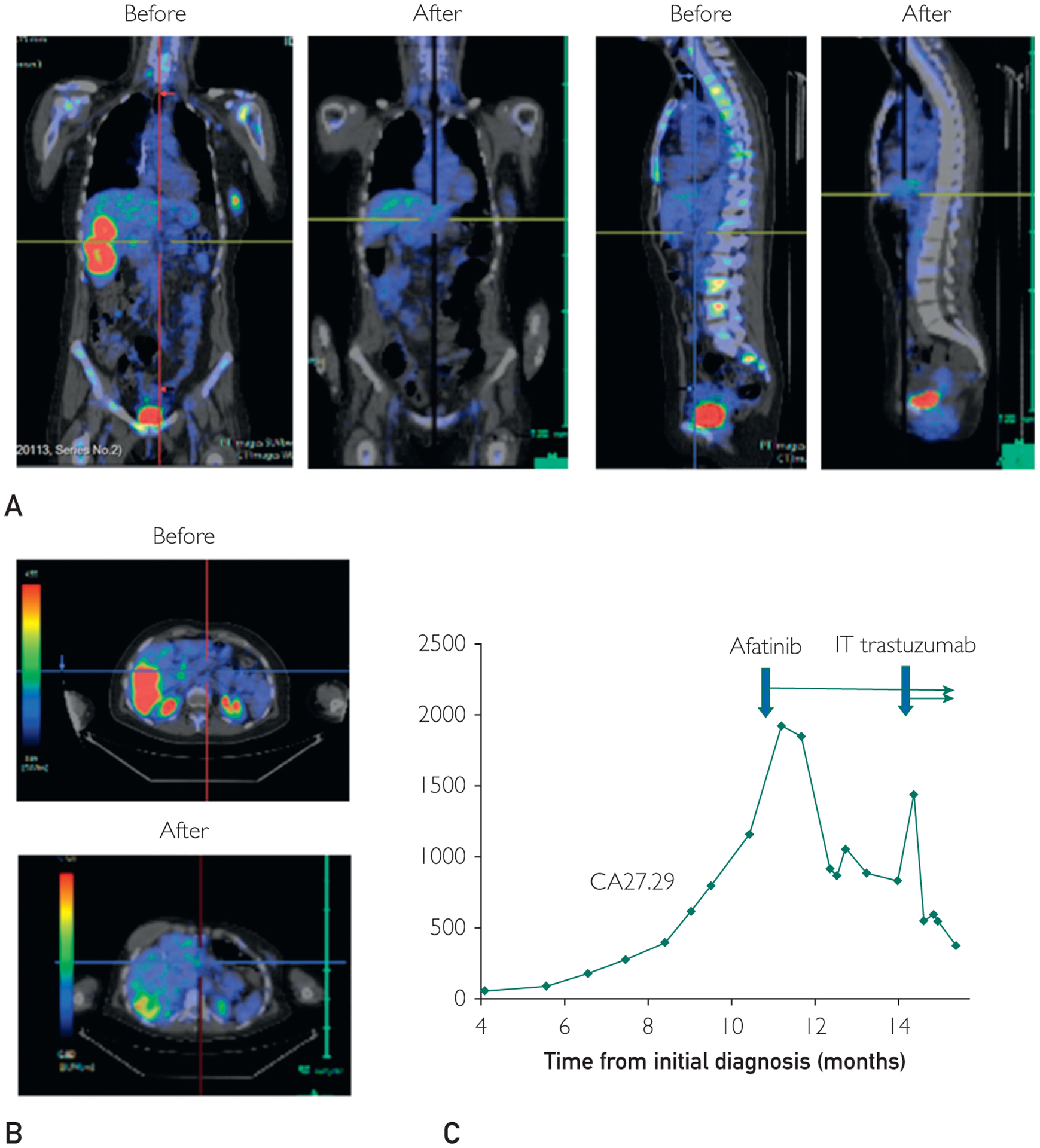
Summary of clinical results during treatment relevant to the study. (A, B) Representative postitron emission tromograph images before treatment relevant to the study and restaging scans 3 months after treatment relevant to the study revealing a near complete extracranial response. (C) Serial CA27.29 values and time points of treatment initiation relevant to the study.
Despite the robust extracranial response, the patient ultimately experienced disease progression in the brain and leptomeninges. Intrathecal trastuzumab was added to her treatment regimen. She received an 80-mg dose twice weekly for 4 weeks, with a partial magnetic resonance imaging response in both the parenchymal brain lesions and the leptomeningeal disease. Intrathecal trastuzumab was administered weekly thereafter. Infections and medical complications led to multiple treatment interruptions and ultimately the discontinuation of all therapy in favor of best supportive care.
DISCUSSION
Despite the promise of precision medicine, the degree of benefit remains to be fully realized in clinical practice.32–36 This might be explained by the fact that the success of targeted therapies depends on both the accurate identification of actionable oncogenic molecular alterations and a reliable means by which to determine which of these alterations are most relevant biologically. The data presented here support the combination of integrated genomics with an informed ex vivo functional drug screen to aid oncologists in the selection of individualized targeted therapies when standard of care options are limited.
A comprehensive approach is feasible and essential to uncover the genomic landscape of each tumor and identify actionable somatic alterations. Our approach integrates MPseq, RNAseq, and WES followed by targeted protein analysis. However, our approach does not incorporate comprehensive proteomics, epigenomics, and metabolomics that could also provide additional information. On the other hand, it is more cost effective and more sensitive than paired-end whole-genome sequencing and more comprehensive, but less sensitive than current targeted NGS panels. Using this approach, we can identify almost all altered genes because of mutations or structural alterations, including chromosomal rearrangements, deletions, and gains. Pathway analysis of altered genes points to critical roles in the cell cycle, DNA repair, and cell signaling leading to various potential therapies. Integrated genomics analysis allows for interrogation of the biallelic inactivation of numerous tumor suppressor genes and identification of oncogenic alterations. In most cases, however, this is not sufficient to inform cancer therapy.
This is evident in the current case where potential therapies were suggested by molecular changes in tumor suppressor genes that were often not biallelic. In addition, HER2 was not amplified, and the ERBB2 mutation was a variant of unknown significance. Therefore, a crucial component of our genomically driven personalized therapy strategy is the ability to assess drug sensitivity in our 3D microcancer model, before patient treatment. Three-dimensional culture models are gaining traction as preclinical testbeds. A key attribute of our 3D microcancer model is that it does not rely on the expansion of single cancer stem cells or of populations of tumor cells, and therefore is not subject to selective pressure and tumor evolution, which often limit the utility of preclinical models for drug testing studies.37,38 Our model also largely maintains the cellular tumor microenvironment and faithfully represents the genomic diversity of the patient’s original tumor. Combined with accuracy, reproducibility, and clinically relevant turnover of less than 2 weeks, the microcancer model is an excellent assay for the functional validation of driver mutations in individual patient tumors, as suggested by extensive genomic profiling. In the absence of obviously clinically actionable mutations, this approach can identify the most effective systemic treatment approach.
Using the microcancer model in our clinical example, we prescreened potential drug mono and combination therapies. Clearly, obtaining biopsy specimens from multiple affected sites could provide a more complete picture of genomic tumor heterogeneity and response to treatment. However, clinical considerations and cost often constrain the number of sites tested clinically. The results validated one of the potential targets as an oncogenic driver (ERBB2) and invalidated others (BRIP1 and NF1). Importantly, the strategy suggested a change in patient management that resulted in a significant and sustained therapeutic response.
Several studies show that ERBB2 mutations are enriched in E-cadherin mutated high-grade infiltrating lobular carcinoma, and are associated with a worse prognosis.31,39–41 The data argue for a more careful interrogation of the ERBB2 status in these patients (eg, amplification, mutation, fusion). Combining integrated genomics with functional drug testing provides both in depth genomic information on both genes, and drug sensitivity data to justify the use of anti-HER2 targeted therapy.
The integrated approach to precision medicine presented here has a turnaround time of four weeks and can be accomplished both in surgical tissue and in tumor biopsy specimens. This is clinically feasible. An additional advantage is the generation of crucial data that link diverse genomic alterations to drug sensitivity. As an example, a number of patients with HER2-positive early-stage breast cancer have a recurrence despite optimal HER2-directed (neo)adjuvant therapy.42–45 Functional validation in a 3D microcancer setting could identify genomic alterations that can be associated with reduced efficacy of HER2-targeted therapy.
CONCLUSION
Unlike available clinical tests, an integrated genomic approach can provide a comprehensive picture of the genomic landscape of each tumor and identify actionable somatic alterations. Although actionable means that there is a drug available to target that alteration, the relative effectiveness of the drug in each patient’s tumor cannot be assessed by genomics. We provide evidence that functional validation of targeted therapies is feasible, is timely, and can be performed ex vivo in 3D cultures of the patient’s own tumor. The combination of tumor profiling, data integration, and functional validation, accurately assessed driver alterations and predicted effective treatment.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients and their family members for generously participating in the related studies. We also thank the Mayo Clinic investigators, laboratory collaborators, and clinical research coordinators for their efforts on behalf of the patients and protocol.
Grant Support: This work was supported by funding from the Mayo Clinic Center of Individualized Medicine.
Abbreviations and Acronyms:
- 3D
3-dimensional
- FISH
fluorescence in situ hybridization
- MPseq
mate-pair whole-genome DNA sequencing
- PDX
patient-derived xenografts
- RNAseq
RNA sequencing
- WES
whole-exome sequencing
Footnotes
SUPPLEMENTAL ONLINE MATERIAL Supplemental material can be found online at http://www.mayoclinicproceedings.org. Supplemental material attached to journal articles has not been edited, and the authors take responsibility for the accuracy of all data.
Potential Competing Interests: Dr Vasmatzis is the owner of WholeGenome, LLC. The other authors report no competing interests.
REFERENCES
- 1.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. [DOI] [PubMed] [Google Scholar]
- 2.Boland JM, Erdogan S, Vasmatzis G, et al. Anaplastic lymphoma kinase immunoreactivity correlates with ALK gene rearrangement and transcriptional up-regulation in non-small cell lung carcinomas. Hum Pathol. 2009;40(8):1152–1158. [DOI] [PubMed] [Google Scholar]
- 3.Oberndorfer F, Mullauer L. Molecular pathology of lung cancer: current status and perspectives. Curr Opin Oncol. 2018;30(2):69–76. [DOI] [PubMed] [Google Scholar]
- 4.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. [DOI] [PubMed] [Google Scholar]
- 5.Press MF, Jones LA, Godolphin W, Edwards CL, Slamon DJ. HER-2/neu oncogene amplification and expression in breast and ovarian cancers. Prog Clin Biol Res. 1990;354A:209–221. [PubMed] [Google Scholar]
- 6.Pritchard KI, Shepherd LE, O’Malley FP, et al. HER2 and responsiveness of breast cancer to adjuvant chemotherapy. N Engl J Med. 2006;354(20):2103–2111. [DOI] [PubMed] [Google Scholar]
- 7.Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat Rev Clin Oncol. 2018;15(11):694–708. [DOI] [PubMed] [Google Scholar]
- 8.Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018; 29(suppl 1):i10–i19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013; 45(12):1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sachs N, Clevers H. Organoid cultures for the analysis of cancer phenotypes. Curr Opin Genet Dev. 2014;24:68–73. [DOI] [PubMed] [Google Scholar]
- 11.Shroyer NF. Tumor organoids fill the niche. Cell Stem Cell. 2016;18(6):686–687. [DOI] [PubMed] [Google Scholar]
- 12.Vasmatzis G, Johnson SH, Knudson RA. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012; 120(11):2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy SJ, Wigle DA, Lima JF, et al. Genomic rearrangements define lineage relationships between adjacent lepidic and invasive components in lung adenocarcinoma. Cancer Res. 2014; 74(11):3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson S, Smadbeck J, Smoley S, et al. SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq). Cancer Genet. 2018;(221):1–18. [DOI] [PubMed] [Google Scholar]
- 15.Smadbeck J, Johnson S, Smoley S, et al. Copy number variant analysis using genome-wide mate-pair sequencing. Genes Chromosomes Cancer. 2018;57(9):459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meijer D, van Agthoven T, Bosma PT, Nooter K, Dorssers LC. Functional screen for genes responsible for tamoxifen resistance in human breast cancer cells. Mol Cancer Res. 2006; 4(6):379–386. [DOI] [PubMed] [Google Scholar]
- 17.Godinho M, Meijer D, Setyono-Han B, Dorssers LC, van Agthoven T. Characterization of BCAR4, a novel oncogene causing endocrine resistance in human breast cancer cells. J Cell Physiol. 2011;226(7):1741–1749. [DOI] [PubMed] [Google Scholar]
- 18.Godinho MF, Sieuwerts AM, Look MP, et al. Relevance of BCAR4 in tamoxifen resistance and tumour aggressiveness of human breast cancer. Br J Cancer. 2010;103(8):1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godinho MF, Wulfkuhle JD, Look MP, et al. BCAR4 induces antioestrogen resistance but sensitises breast cancer to lapatinib. Br J Cancer. 2012;107(6):947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balko JM, Giltnane JM, Wang K, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23(8):1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142(1):1–10. [DOI] [PubMed] [Google Scholar]
- 24.Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36): 4354–4361. [DOI] [PubMed] [Google Scholar]
- 25.Leijen S, van Geel RM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36): 4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt M, Rohe A, Platzer C, Najjar A, Erdmann F, Sippl W. Regulation of G2/M transition by inhibition of WEE1 and PKMYT1 kinases. Molecules. 2017;22(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11): 1104–1107. [DOI] [PubMed] [Google Scholar]
- 28.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. [DOI] [PubMed] [Google Scholar]
- 30.Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria JC, Postel-Vinay S. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. 2016;22(17):4309–4321. [DOI] [PubMed] [Google Scholar]
- 31.Ross JS, Wang K, Sheehan CE, et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19(10):2668–2676. [DOI] [PubMed] [Google Scholar]
- 32.Hilal T, Nakazawa M, Hodskins J, et al. Comprehensive genomic profiling in routine clinical practice leads to a low rate of benefit from genotype-directed therapy. BMC Cancer. 2017;17(1):602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 2016;76(13):3690–3701. [DOI] [PubMed] [Google Scholar]
- 34.Schwaederle M, Parker BA, Schwab RB, et al. Precision oncology: the UC San Diego Moores Cancer Center PREDICT Experience. Mol Cancer Ther. 2016;15(4):743–752. [DOI] [PubMed] [Google Scholar]
- 35.Stockley TL, Oza AM, Berman HK, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. [DOI] [PubMed] [Google Scholar]
- 37.Ben-David U, Siranosian B, Ha G, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560(7718):325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ben-David U, Ha G, Tseng YY, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017; 49(11):1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deniziaut G, Tille JC, Bidard FC, et al. ERBB2 mutations associated with solid variant of high-grade invasive lobular breast carcinomas. Oncotarget. 2016;7(45):73337–73346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ping Z, Siegal GP, Harada S, et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget. 2016;7(49):80655–80663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross JS, Gay LM, Wang K, et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti-HER2 targeted therapies. Cancer. 2016;122(17):2654–2662. [DOI] [PubMed] [Google Scholar]
- 42.Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet. 2017;389(10075):1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurvitz SA, Martin M, Symmans WF, et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018;19(1):115–126. [DOI] [PubMed] [Google Scholar]
- 45.Gianni L, Pienkowski T, Im YH, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016;17(6):791–800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.