

Abstract
Rationale:
The mitochondrial protein polymerase interacting protein 2 (Poldip2) is required for the activity of the tricarboxylic acid (TCA) cycle. As a consequence, Poldip2 deficiency induces metabolic reprograming with repressed mitochondrial respiration and increased glycolytic activity. Though homozygous deletion of Poldip2 is lethal, heterozygous mice are viable and show protection against aneurysm and injury-induced neointimal hyperplasia, diseases linked to loss of VSMC differentiation. Thus, we hypothesize that the metabolic reprograming induced by Poldip2 deficiency controls VSMC differentiation.
Objective:
To determine the role of Poldip2-mediated metabolic reprograming in phenotypic modulation of VSMC.
Methods and Results:
We show that Poldip2 deficiency in vascular smooth muscle in vitro and in vivo induces the expression of the serum response factor (SRF), Myocardin, and MRTFA and dramatically represses KLF4. Consequently, Poldip2-deficient VSMC and mouse aorta express high levels of contractile proteins and, more significantly, these cells do not dedifferentiate nor acquire macrophage-like characteristics when exposed to Cholesterol or PDGF. Regarding the mechanism, we found that Poldip2 deficiency upregulates the hexosamine biosynthetic pathway and OGT-mediated protein O-GlcNAcylation. Increased protein glycosylation causes the inhibition of a nuclear UPS responsible for SRF stabilization and KLF4 repression and is required for the establishment of the differentiated phenotype in Poldip2-deficient cells.
Conclusions:
Our data show that Poldip2 deficiency induces a highly differentiated phenotype in VSMCs through a mechanism that involves regulation of metabolism and proteostasis. Additionally, our study positions mitochondria-initiated signaling as key element of the VSMC differentiation programs that can be targeted to modulate VSMC phenotype during vascular diseases.
Keywords: VSMC differentiation, mitochondria, Poldip2, OGT, proteasome, metabolism, vascular smooth muscle
Subject Terms: Basic Science Research, Metabolism, Smooth Muscle Proliferation and Differentiation, Vascular Biology
Graphical Abstract
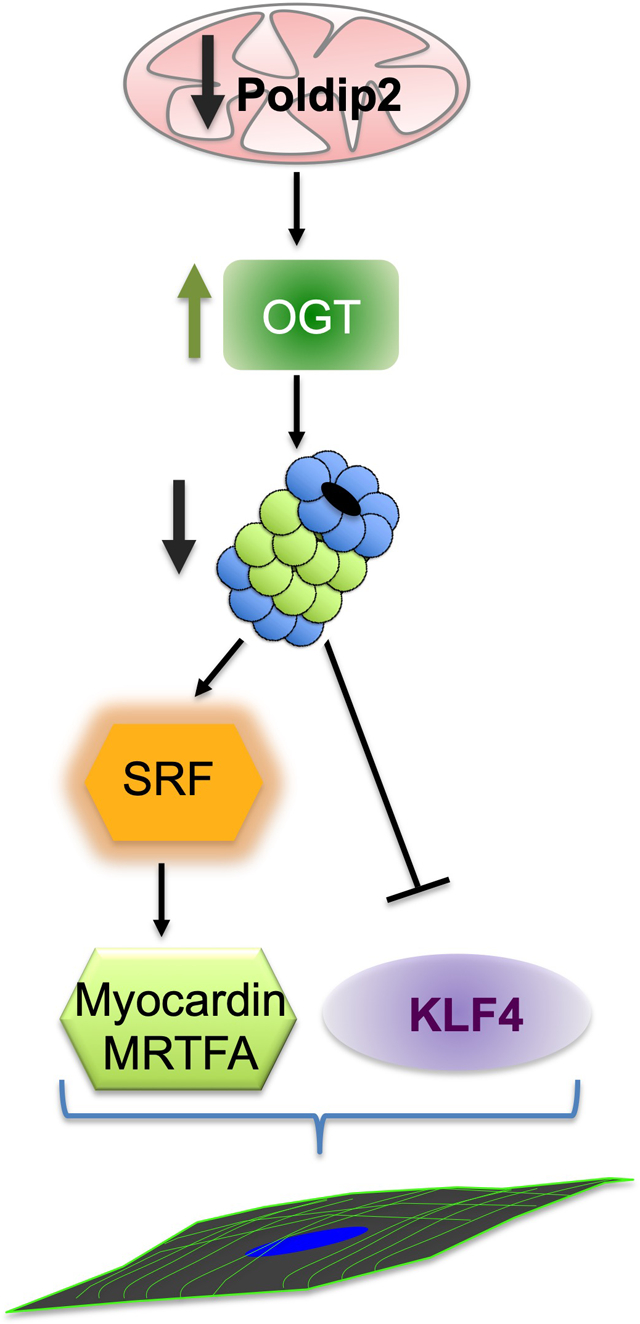
Preservation of VSMC differentiation disrupts inflammatory signaling and reduces the severity and size of the lesions in animal models of atherosclerosis. Thus, understanding the mechanisms that control the VSMC differentiation state is critically important. In this study, we show that deficiency of the mitochondrial protein Poldip2 induces a highly differentiated phenotype in VSMCs through a mechanism that involves regulation of metabolism and proteostasis. Our study positions mitochondria-initiated signaling as a key element of the VSMC differentiation program that can be targeted to modulate VSMC phenotype during vascular diseases.
INTRODUCTION
Vascular smooth muscle cells (VSMCs) comprise the majority of the cells in blood vessels. The contractile phenotype of fully differentiated VSMCs allows them to regulate blood flow and pressure, and thus they are essential for vascular function. The characterization of the VSMC phenotype is based on changes in cell morphology, response to agonists, and the expression of well-recognized SMC-specific genes from the contractile apparatus, such as myosin heavy chain (MYH11), Transgelin (TAGLN, aka SM22), Calponin1 (CNN1) and aortic smooth muscle actin (ACTA2, aka SMA). It is well appreciated that the Serum Response Factor (SRF), an ancient and evolutionarily conserved member of the MADS (MCM1, Agamous, Deficiens, SRF) box family, and its cofactors Myocardin (MYOCD) and Myocardin-related transcription factors (MRTFs) govern canonical programs of differentiation through their binding to highly conserved cis-regulatory elements termed CArG boxes.1–4
Additionally, the pluripotency and stemness transcription factor Krüppel-like factor (KLF)-4 activity negatively impacts the VSMC differentiation program by antagonizing Myocardin and expression of SMC-specific genes by a variety of mechanisms.5–8
A distinct characteristic of VSMCs is that they are not terminally differentiated and retain remarkable plasticity throughout life. In fact, in response to a variety of mechanical and chemical stimuli such as cholesterol or growth factors, VSMCs acquire proliferative and migratory capacity that is accompanied by loss of SMC markers and expression of macrophage antigens.
VSMC phenotypic plasticity plays key roles during vessel maturation or injury repair, but it is also linked to increased susceptibility to pathological vascular remodeling and vascular diseases. Consistently, preservation of VSMC differentiation, by increased Myocardin activity9 or reduced KLF4 expression10 in SMC-lineage, disrupts inflammatory signaling and reduces the severity and size of the lesions in animal models of atherosclerosis.
Recently, we reported that the nuclear-encoded mitochondrial protein polymerase delta interacting protein 2 (Poldip2) controls the lipoylation and activation of two critical catabolic enzymes: pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (αKGDH).11 As a result, Poldip2 deficiency represses the activity of the Krebs’s cycle resulting in metabolic reprograming with a lower rate of oxidative metabolism and higher glycolytic activity in a variety of cell types including VSMCs.11
Interestingly, though homozygous deletion of Poldip2 in mice results in perinatal lethality12 adult heterozygous deficient mice have no overt physiological phenotype. However, when subjected to physiological stress, Poldip2 heterozygote mice exhibit protection against aneurysm formation13 and injury-induced neointimal hyperplasia.14 These data suggested to us that the metabolic reprograming induced by loss of Poldip2 induces a more differentiated phenotype in VSMCs that seems to be preserved under pathological conditions, resulting in vascular protection.
The Hexosamine Biosynthetic Pathway (HBP) is a glucose dependent pathway that branches off glycolysis downstream of Hexokinase 2 (HK2). In addition to glucose, the HBP uses stoichiometric amounts of acetyl-CoA, uridine, and glutamine to yield its final product, Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is the sugar donor for the O-GlcNAc transferase (OGT)-catalyzed transfer of a single sugar onto specific serine and threonine residues of target proteins.15 This post-translational modification has been implicated in gene expression, stress responses, and nutrient sensing.16 Importantly, increased OGT-mediated glycosylation regulates proteostasis through the inhibition of the Ubiquitin Proteasome System (UPS).17, 18
Here, we report that downregulation of Poldip2 in human aortic smooth muscle cells (HASMCs) increases the activity of SRF, MRTFA and MYOCD while repressing KLF4, which translates into high expression of SMC-specific genes by a mechanism that involves OGT-mediated inhibition of a UPS. Most importantly, Poldip2-deficient HASMCs do not undergo phenotypic switching and do not express macrophage markers when exposed to cholesterol or platelet-derived growth factor (PDGF). Significantly, these results were corroborated in vivo, as Poldip2+/− mouse arteries exactly recapitulate the pattern of expression of differentiation markers and transcription factors and the metabolic reprogramming observed in vivo. These observations offer a mechanistic explanation for the vascular protection observed in Poldip2-deficient mice and position metabolic signaling as a key component of VSMC phenotypic modulation.
METHODS
The authors declare that all supporting data are available within the article and its online supplemental files. Analytical methods and materials will be made available upon request.
General reagents.
MG132, Azaserine and Osmi (Cayman, 13697, 14834 and 21894), Cycloheximide (Sigma, C1988), Bz-Val-Gly-Arg-AMC (Enzo Life Sciences, BML-BW9375–0005), Recombinant human PDGF-BB (R&D Systems Inc) and water-soluble Cholesterol (Sigma, C4951).
Mouse femoral artery injury model.
Poldip2+/− mice used were generated by a gene trap insertion in the first intron of Poldip2 (chromosome 11, NCBI Gene ID: 67811) at the Texas A&M Institute for Genomic Medicine (College Station, TX) and fully backcrossed with C57BL/6 as previously described13. Transluminal mechanical injury of the right femoral artery of 3 months old male mice was induced by introducing a guide wire as previously reported19–21. In brief, the right femoral artery was exposed by blunt dissection and was looped proximally and distally with 6–0 silk suture for temporary control of blood flow during the procedure. Arteriotomy was performed distal to the epigastric branch between the rectus femoris and vastus medialis muscles. A straight spring wire of 0.38 mm in diameter (Cook, Bloomington, IN) was inserted into the femoral artery through the epigastric artery toward the iliac artery and left in place for 1 min to denude and dilate the artery. Blood flow in the femoral artery was restored by releasing the proximal and distal femoral sutures. Animals were killed 2 weeks after surgery and arteries were pressure-perfused at 100 mm Hg with 0.9% sodium chloride, followed by pressure-fixation with 10% formalin. Femoral arteries were then carefully excised and embedded in paraffin. Mice were maintained on a standard chow diet. All procedures were approved by the Emory University Institutional Animal Care and Use Committee. Animal randomization and allocation concealment was performed for the analysis. Studies were performed in male mice to eliminate the possible confounding effects of reproductive hormones.
Cell culture.
Human aortic smooth muscle cells (HASMCs) were isolated from human aortic tissue by Lonza and cryopreserved at passage three. Cells used in this study originated from 27 to 32 year old male and female donors self-identified as Caucasian, Latino or African American. HASMCs were grown as recommended by the vendor in basal media 231 with the addition of growth supplements. After cells reached ~ 80% confluence, growth supplements were removed 48 h prior to experiments.
Cell transfection.
Cells were transfected using Lipofectamine RNAiMAX reagent (Thermo-Fisher) following the manufacturer’s suggested protocol. Sequences were: Poldip2 sense 5’-CGUGAGGUUUGAUCAGUAATT-3’; OGT sense 5’-TACGCGTGCCATCCAAATTAA-3’ and SRF sense 5’-CAAGARGGAGTTCATCGACAA-3’.
Western blots.
Total protein lysates were prepared in Hunter’s buffer. Nuclear and cytosolic fractions were prepared using the REAP method.22 Protein samples were separated on Express-Plus Page Gels (GenScript) with Tris-MOPS buffer containing SDS and transferred onto Immobilon-P membrane (Millipore, IPVH00010). All antibodies were incubated for 24h at 4°C. Protein bands were visualized using ECL Western Blotting Substrate (Pierce, 32106) and imaged on a Kodak Camera System.
Western blotting of aortic tissue.
Aortas from 3 months old wild type and Poldip2+/− male mice were harvested, cleaned of fat and connective tissue, and then flash frozen in liquid nitrogen. Frozen aortas were ground into a fine powder using a pre-chilled mortar and pestle. Proteins were extracted in RIPA buffer (25 mmol/L HEPES, 150 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 10 mmol/L Na-pyrophosphate, 10 mmol/L NaF, 1% Na deoxycholate, 1% Triton X 100, 0.1% SDS, 10% Glycerol, Na-orthovanadate and protease inhibitors) from their lysates, sonicated, and cleared at 14,000 × g for 20 minutes. Proteins were separated using SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore), blocked with 5% non-fat dairy milk, and incubated with primary antibodies.
Immunohistochemistry.
For immunofluorescent imaging of femoral arteries, sections were deparaffinized, rehydrated and then subjected to heat induced antigen retrieval with citrate buffer pH 6.0. Next, the sections were blocked for one hour at room temperature (4% BSA/0.4% Fish Gelatin in a 0.3% Triton containing TBS solution) and then incubated with transgelin (Abcam ab14106, 1:200 dilution), calponin 2 (CNN2) (Proteintech Cat# 13938–1-AP, 1:50 dilution), or myosin heavy chain 11 (MYH11) (Abcam ab53219, 1:100 dilution) primary antibodies overnight at 4°C. Again, the sections were washed and then incubated with Alexa Fluor® 647-AffiniPure Donkey Anti-Rabbit IgG (Jackson Immunoresearch Cat# 711-605-152) and counterstained with DAPI. Sections treated with secondary antibodies alone did not show specific staining. Confocal micrographs were acquired with a Zeiss LSM 800 Confocal Laser Scanning Microscope using a 63x oil objective lens and Zeiss ZEN acquisition software. When comparing sections from different experimental groups, all image threshold settings of the confocal microscope remained constant.
Immunocytochemistry
After siRNA transfection, HASMCs were plated on glass insert and serum deprived for 48 h. Cells were rinsed quickly in ice-cold PBS and fixed in 4% paraformaldehyde for 10 minutes at room temperature. They were then permeabilized in 0.01% Triton X-100 in PBS for 7.5 minutes and rinsed with PBS. Aldehyde groups were quenched with 50 mM NH4Cl for 10 minutes at room temperature. Cells were then blocked using 3% bovine serum albumin in PBS for 1 hour, incubated with anti-Ubiquitin antibody overnight, and then incubated with secondary antibodies for 1 hour at room temperature. Cells were mounted in Vectashield containing DAPI (Vector Laboratories). Images were acquired with a Zeiss LSM 510 META Laser Scanning Confocal Microscope.
List of antibodies used in this study:
| Protein | Company | Catalog number |
|---|---|---|
| Poldip2 | Abcam | ab172435 |
| ACTB | Sigma | A5441 |
| OGT | Abcam | ab96718 |
| Ubiquitin | Cell Signaling | 3936S |
| O-GlcNAc | Abcam | ab2739 |
| Hexokinase 2 | Cell Signaling | 2106S |
| PSMC1 | Bethyl Laboratories | A-303–821A |
| Cal 1/2/3 | Santa Cruz | SC-136987 |
| Calponin 1 | Sigma | 231R-14 |
| Calponin2 | Novus | NBP2–01325 |
| Calponin 3 | Genetex | GTX106174 |
| TAGLN | Abcam | ab14106 |
| ACTA2 | Sigma | A5228 |
| SRF | Santa Cruz | SC-335 |
| MYOCD | R&D Systems | MAB4828 |
| MRTFA | Novus | NBP1–88498 |
| KLF4 | R&D Systems | AF3640 |
| GFPT1 | Proteintech | 14132–1-AP |
| GFPT2 | Cell Signaling | 6917S |
| CD68 | Cell Signaling | 86985S |
| GAL3 | Cell Signaling | 12733S |
| Phospho-Thr/Pro | Cell Signaling | 9391S |
| Phospho-(Ser/Thr)/Phe | Cell Signaling | 9631S |
| Arginase 1 | BioLegend | 678802 |
| CCL2 | BD Biosciences | 551217 |
| SREBF1 | Santa Cruz | SC-366 |
Short-life reporter accumulation.
HASMCs were transfected with control or Poldip2 siRNA using RNAiMAX. The next day, cells were transfected with GFP-Ub (in an EGFP-C1 backbone, Addgene#11928) or YFP-CL1 (in an EGFP-C1 backbone, Addgene#1950) using Lipofectamine 3000 (Thermo Fisher) following the manufacturer’s recommendations. Total protein lysates were prepared after 24 hours and accumulation of fluorescent probe was quantified by western blots using an antibody against GFP.
Adenoviral expression of Poldip2 and PSMC1.
The human Poldip2 cDNA (Accession NP_056399) plus a C-terminal myc tag (EQKLISEEDL) was cloned into the pAdTrack-CMV vector, which allows expression of myc-tagged Poldip2 and GFP by two independent CMV promotors. The virus was packaged using the AdEasy Adenoviral Vector System (Agilent Technologies) as previously described.23, 24 The human PSMC1 cDNA (MGC24583) plus with C-terminal myc tag (EQKLISEEDL) and HIS tag was cloned into the pAdTrack-CMV vector, which allows expression of myc-tagged PSMC1 and His Tag by two independent CMV promoters. The adenovirus was obtained from Vigene Biosciences (VH827525).
Proteasome activity.
HASMCs were transfected with control siRNA or with siRNA against Poldip2. After 48h, protein lysates were prepared and 100ul were mixed with fusion protein for 48h. After collection, 100μl of lysates were transfer to a 96 well plate, mixed with Bz-Val-Gly-Arg-Amc to a final concentration of 50 mM and incubated at 37°C for 1h. Fluorescence readings were made using filters at specific wavelengths (360/380 excitation/460 emission). The results are presented as percentage of control 26S proteasome activity. In some samples, 50 μM MG132 was added 3h prior to the preparation of lysates as a control.
Phagocytosis of fluorescent particles.
This protocol was adapted from Rong et al, 2003. HASMCs were plated on glass coverslips and transfected with control or Poldip2 siRNA using Lipofectamine RNAiMAX (Thermo Fisher). After 24h, a water-soluble form of cholesterol (Sigma C-4951, which contains 40 mg of cholesterol per gram of compound and balance methyl-β-cyclodextrin) or vehicle were added to a final concentration of 10 μg/ml. After 48h, 1μM Sphero™-fluorescent particles (Spherotech, sky blue, average size of 0.78 μm) were added. After an additional 24h, cells were fixed with 4% paraformaldehyde in universal buffer and processed for nuclear (4′,6-diamidino-2-phenylindole (DAPI, for nuclei, blue) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (for membrane, red) was used. After the mounting, the digital images were captured by fluorescent microscope. ImageJ-win64 was used for quantification. The data is presented in number of fluorescent particles per nucleus.
Statistical analysis.
Images that better represent the average of the data set were selected as representative. Data are presented as mean ± SEM from a minimum of 4 independent experiments (biological replicates). Significance was determined using a t-test for unpaired samples, 1-way ANOVA or 2-way ANOVA followed by Dunnett post-hoc test. Prism6 or SPSS were used for statistical analysis. A threshold of P<0.05 was considered significant.
RESULTS
To characterize the phenotypic changes associated with the level of Poldip2 expression in VSMCs, we investigated the impact of Poldip2 downregulation on the expression of transcription factors known to control smooth muscle cell phenotype. As shown in Figure 1A, Poldip2-deficient human aortic smooth muscle cells (HASMCs) displayed a prominent upregulation of SRF, MRTFA and MYOCD. Interestingly, these changes were accompanied by a dramatic repression of KLF4 expression (Figure 1A). Consistent with this pattern of transcription factors expression, Poldip2-deficient HASMCs exhibited significant upregulation of proteins from the contractile apparatus, particularly from members of the Calponin family: TAGLN (aka SM22) and Calponin 2 (Figure 1A). These results were confirmed using an unrelated siRNA sequence against Poldip2 (Online Figure I)
Figure 1. Poldip2 loss-of-function increases vascular smooth muscle differentiation.
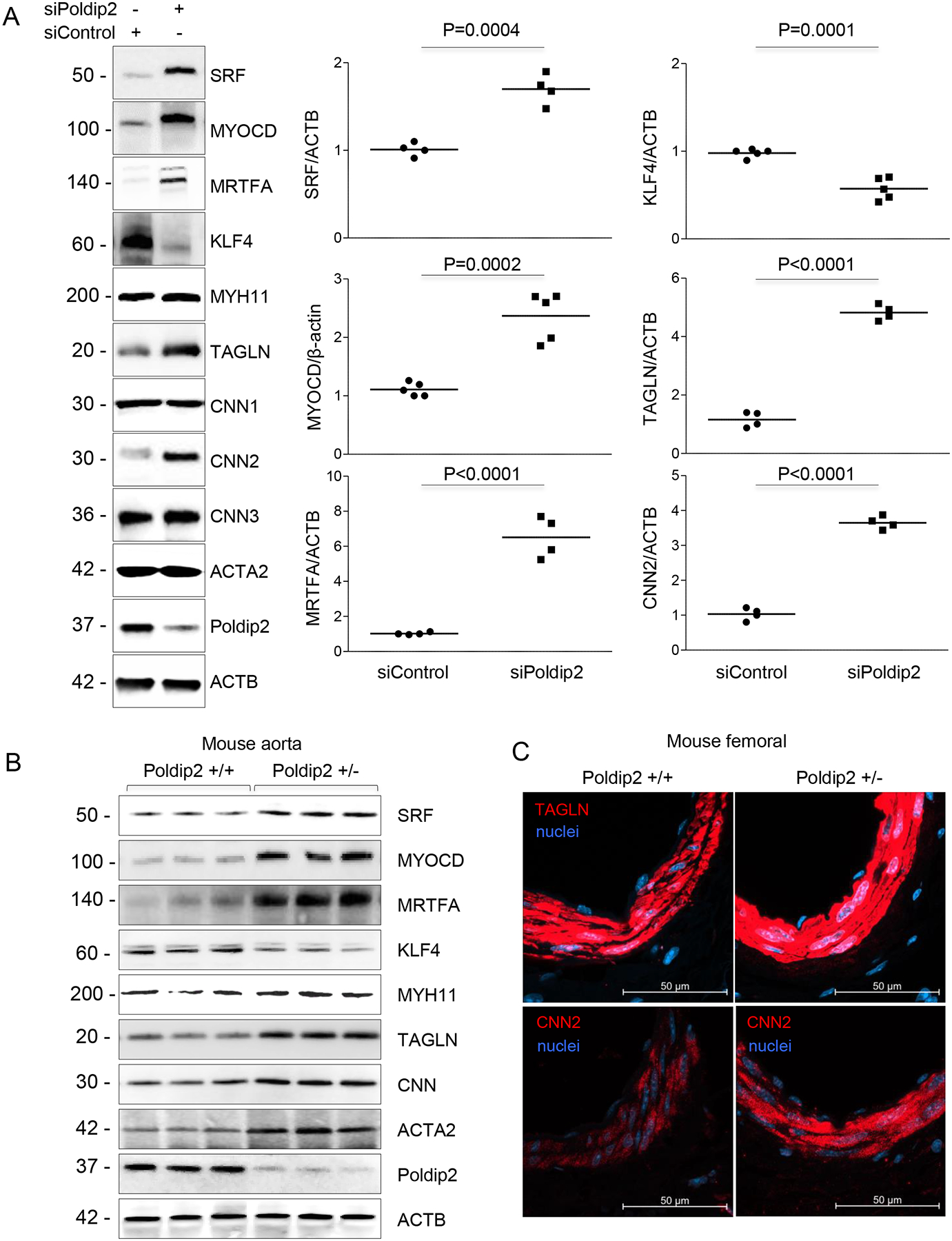
A. Representative western blots to evaluate the expression of VSMC differentiation markers and transcription factors when Poldip2 is downregulated. HASMCs were transfected with siRNA control or against Poldip2 for 48 h. Scatter plots and mean from 4–5 independent experiments are shown. P value indicated was calculated by independent t-test. B. Western blot to evaluate protein expression in aortas from wild type and Poldip2+/− mice. Protein lysate was prepared as described in methods section. Each lane corresponds to a different animal. C. Staining against TAGLN (SM22) and CNN2 in femoral tissue of wild type and Poldip2+/− mice.
Furthermore, increased differentiation in Poldip2-deficient HASMCs in vitro is recapitulated in the vasculature of Poldip2 heterozygous mice (Figure 1B–C). These results clearly underline the importance of Poldip2 in the regulation of VSMC differentiation in vivo.
It is well recognized that the exposure of VSMCs to PDGF-BB25, 26 or cholesterol27 in vitro induces phenotypic switching, and in vivo contributes to vascular disease.28, 29 Since previous reports indicated that the vasculature of Poldip2-deficient mice is protected against the development of vascular diseases under pathological conditions,13, 14 we tested whether Poldip2-deficient HASMCs are less permissive to undergo phenotypic switching when exposed to PDGF or cholesterol. As expected based on previous reports, exposure of control HASMCs to PDGF (Figure 2A) or to a form of water-soluble cholesterol (Figure 2B) significantly lowered the expression of most contractile proteins. On the other hand, after exposure to PDGF or cholesterol, Poldip2-deficient HASMCs preserved a highly differentiated phenotype with significantly higher levels of expression not only of members of the Calponin family but also from MYH11 and ACTA2 (Figure 2A–B). Furthermore, after incubation with cholesterol for 72 h and in contrast to control cells, Poldip2-deficient HASMCs did not express the macrophage-related genes CD68, LGALS3 (aka Galectin 3, Mac2), Arginase 1 (ARG1), SREBF130 or the macrophage-specific inflammatory cytokine CCL2, (Figure 3A) nor did they acquire phagocytic activity (Figure 3B).
Figure 2. Poldip2-deficient HASMCs remain differentiated after PDGF stimulation or cholesterol loading.
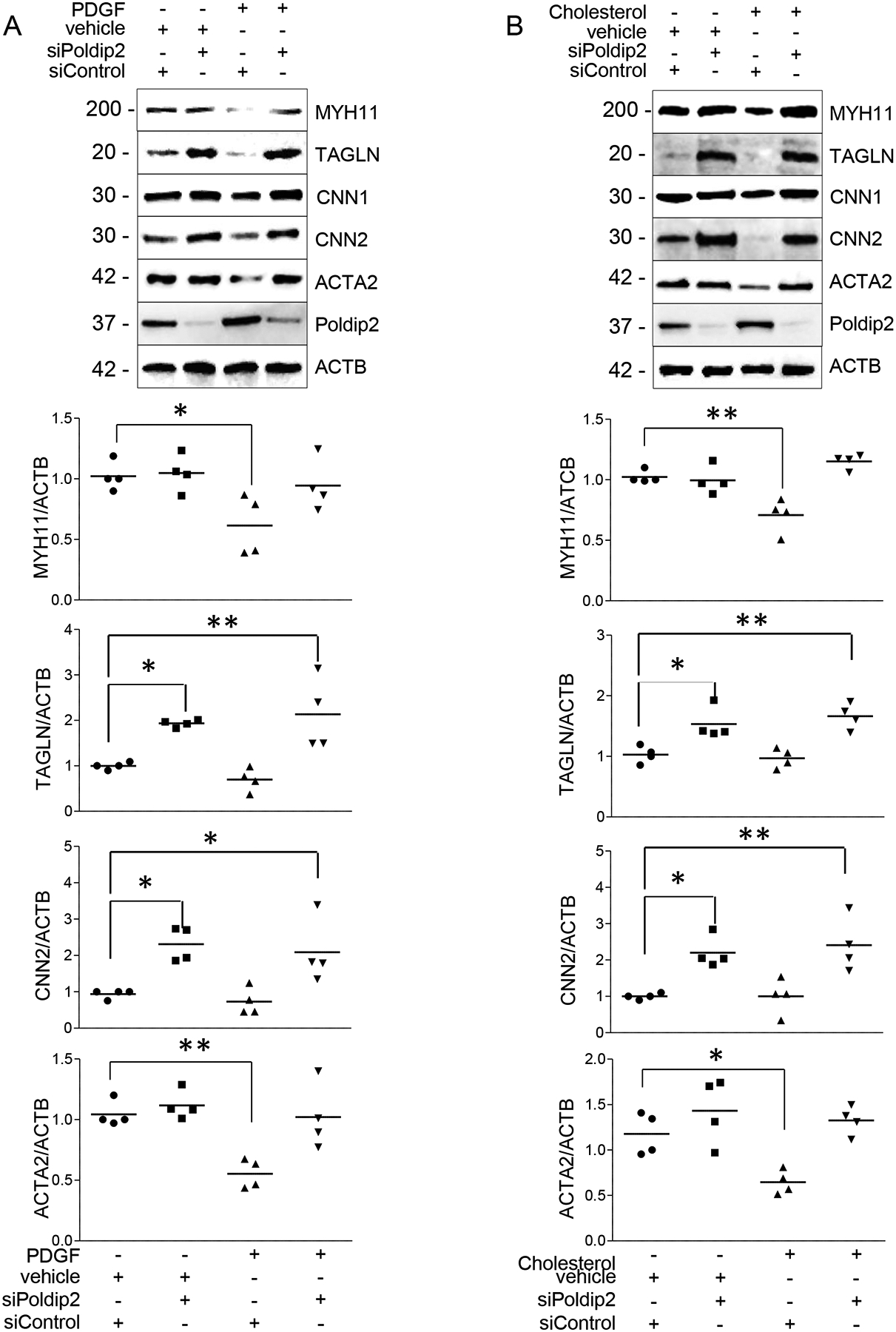
Expression of VSMC differentiation markers and transcription factors in HASMCs transfected with siRNA control or against Poldip2 were exposed to A) PDGF (10 ng/ml) or vehicle for 48h or B) Cholesterol (10 μg/ml) or vehicle for 72h. Scatter plots and mean from 4–5 independent experiments. P value indicated was calculated by two-way ANOVA, * < 0.05 ** <0.01 ***<0.001.
Figure 3. Poldip2-deficient HASMCs do not transdifferentiate to a macrophage-like phenotype.
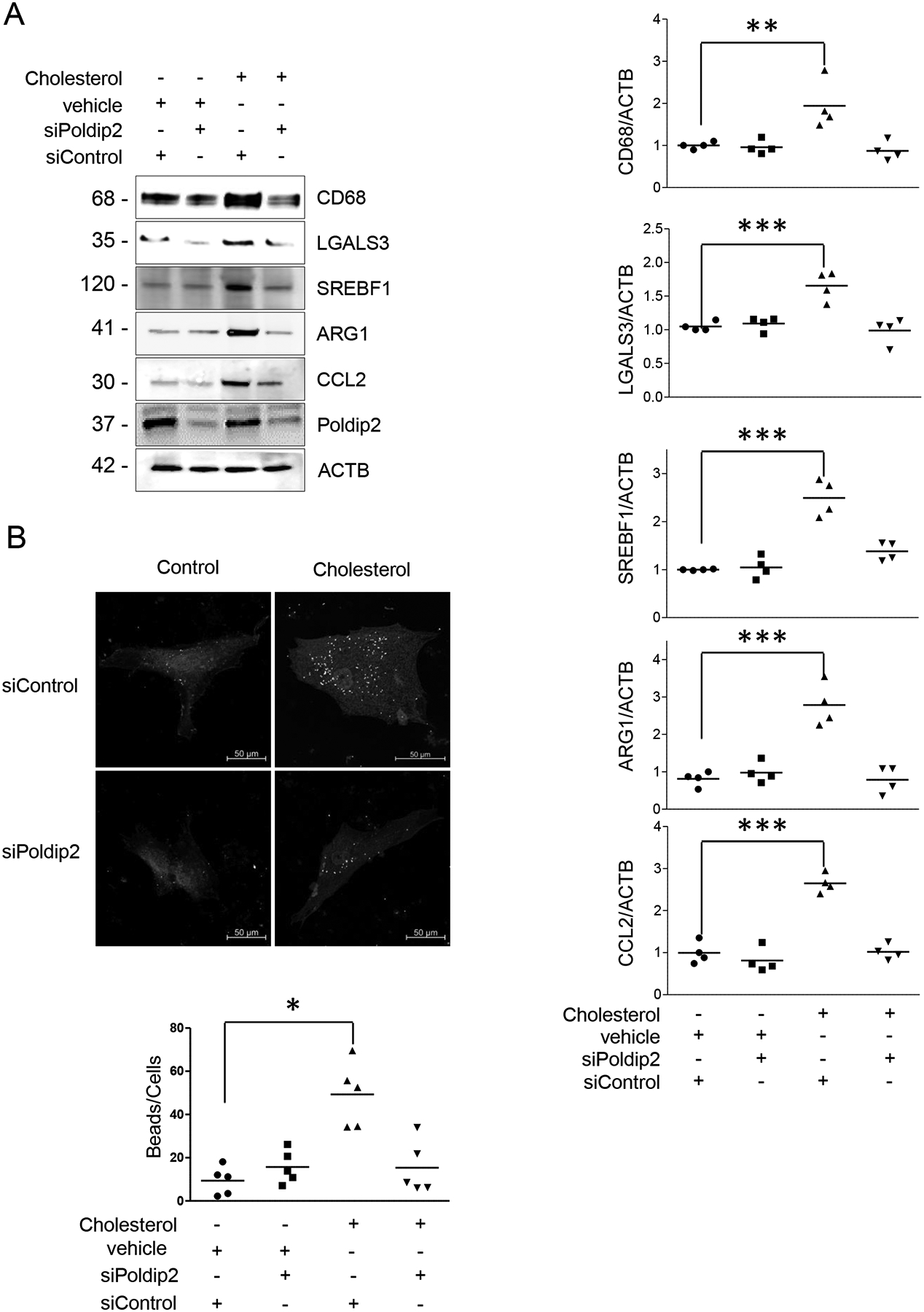
A) Expression of macrophage markers in control or Poldip2-deficient HASMCs exposed to cholesterol (of 10 μg/ml) for 72 h. B) Phagocytosis of fluorescent-beads in control or Poldip2-deficient HASMCs after Cholesterol exposure (72 h) was evaluated as described in the methods section. Scatter box plots and mean from 4–6 independent experiments are shown. P value indicated was calculated by two-way ANOVA, * < 0.05 ** <0.01 ***<0.001.
Femoral artery dilatation by intraluminal insertion of a large wire causes active proliferation and migration of dedifferentiated medial VSMCs at 2 weeks.21 Using this model, we tested whether VSMCs in the arteries of Poldip2+/− mice maintain a more differentiated phenotype than those in wild type animals at this time point. We found that in wild type animals, a significant population of cells in the media and nascent neointima was negative for TAGLN while arteries of Poldip2+/− mice retained the expression of this marker (Online Figure II). These results are consistent with the idea that preservation of the differentiated phenotype in Poldip2+/− mice inhibits neointima formation after injury.19
To begin to define the mechanisms by which reduced Poldip2 deficiency induces such a striking regulation of VSMC phenotype, we first investigated whether the regulation of SMC-specific transcription factors require de novo protein synthesis. As shown in the Online Figure III, SRF accumulation did not require de novo protein synthesis. In contrast, cycloheximide prevented the upregulation of MYOCD and MRTFA, suggesting that in Poldip2-deficient cells SRF is accumulated by stabilization of existing protein and that MRTFA and MYOCD might be upregulated at the transcriptional level by an SRF-dependent mechanism. The latter is supported by the ability of siSRF to block MRTFA and MYOCD accumulation in Poldip2-deficient cells (Online Figure IV) as well as the increase of TAGLN and CNN2. These data also show that SRF activity does not participate in the mechanism of KLF4 downregulation in Poldip2-deficient cells (Online Figure IV).
The fact that SRF was accumulating without de novo protein synthesis indicated to us that Poldip2 deficiency might affect VSMC proteostasis. The ubiquitin proteasome systems (UPS) are among the most important mechanisms of proteostasis, as they represent the primary site for degradation of short-lived regulatory protein and transcription factors.31 Previous reports have shown that stabilization of SRF by UPS inhibitors increases its transcriptional activity.32 Considering this and based on our results, we tested whether the increase of SRF half-life and activity was the result of the inhibition of a UPS in Poldip2-deficient cells.
Using a fluorogenic substrate, we confirmed that the overall UPS activity was significantly inhibited in Poldip2-deficient cells (Figure 4A). It is conceivable that Poldip2 deficiency inhibits UPS activity by regulating either the Ubiquitin ligase system or the proteolytic activity of the 26S complex. To explore these options, we used fluorescent UPS reporters, in which a short half-life UPS-degradation signal protein and a fluorescent tag that can be monitored by fluorometric detection are fused. 24 h after transfection, we observed the accumulation of both a Ubiquitin ligase dependent (Ub-GFP, Figure 4B), and a Ubiquitin ligase independent (CLP1-YFP, Figure 4C) reporter. The accumulation of both probes suggested that Poldip2 deficiency did not affect the Ubiquitin ligase system but rather the UPS proteolytic capacity directly. Consistently, poly-ubiquitinated proteins accumulated in Poldip2-deficient HASMCs (Figure 4D).
Figure 4. Proteasome activity is inhibited in Poldip2-deficient HASMCs.

A) Proteasome activity in control or Poldip2-deficient HASMCs were measured using a fluorogenic probe as described in the methods section. The UPS inhibitor MG132 was used as a negative control. B-C) The accumulation of the reporters Ub-GFP (B) and CL1-YFP (C) were evaluated by specific antibodies in control or Poldip2-deficient HASMCs. D-E) Representative western blot to evaluate the accumulation of poly-ubiquitinated proteins in total lysates (D) or cytosolic and nuclear fractions (E) of control or Poldip2-deficient HASMCs. F) Accumulation of Ubiquitin-associated proteins evaluated by immunocytochemistry in control or Poldip2-deficient HASMCs. Scatter plots and mean from 4–5 independent experiments are shown. P value indicated was calculated by independent t-test or two-way ANOVA, ***<0.001.
Interestingly, while poly-ubiquitinated proteins accumulated in both cytosolic and nuclear compartments of Poldip2-deficient HASMCs, the effect of Poldip2 downregulation seems to affect predominantly proteins from the nuclear compartment (Figure 4E–F). Thus, we sought to restore UPS activity by forced expression of the nuclear UPS activator PSMC1 (aka Rpt2).33 As shown in Figure 5, PSMC1 expression not only restored UPS activity (Online Figure V) but also impeded SRF accumulation and reversed the highly differentiated phenotype displayed by Poldip2-deficient HASMCs including KLF4 repression (Figure 5). These results clearly showed that the mechanism by which Poldip2 deficiency induces differentiation in vascular smooth muscle cells requires the inhibition of a nuclear UPS.
Figure 5. Increased differentiation in Poldip2-deficient HASMCs required the inhibition of a ubiquitin proteasome system.
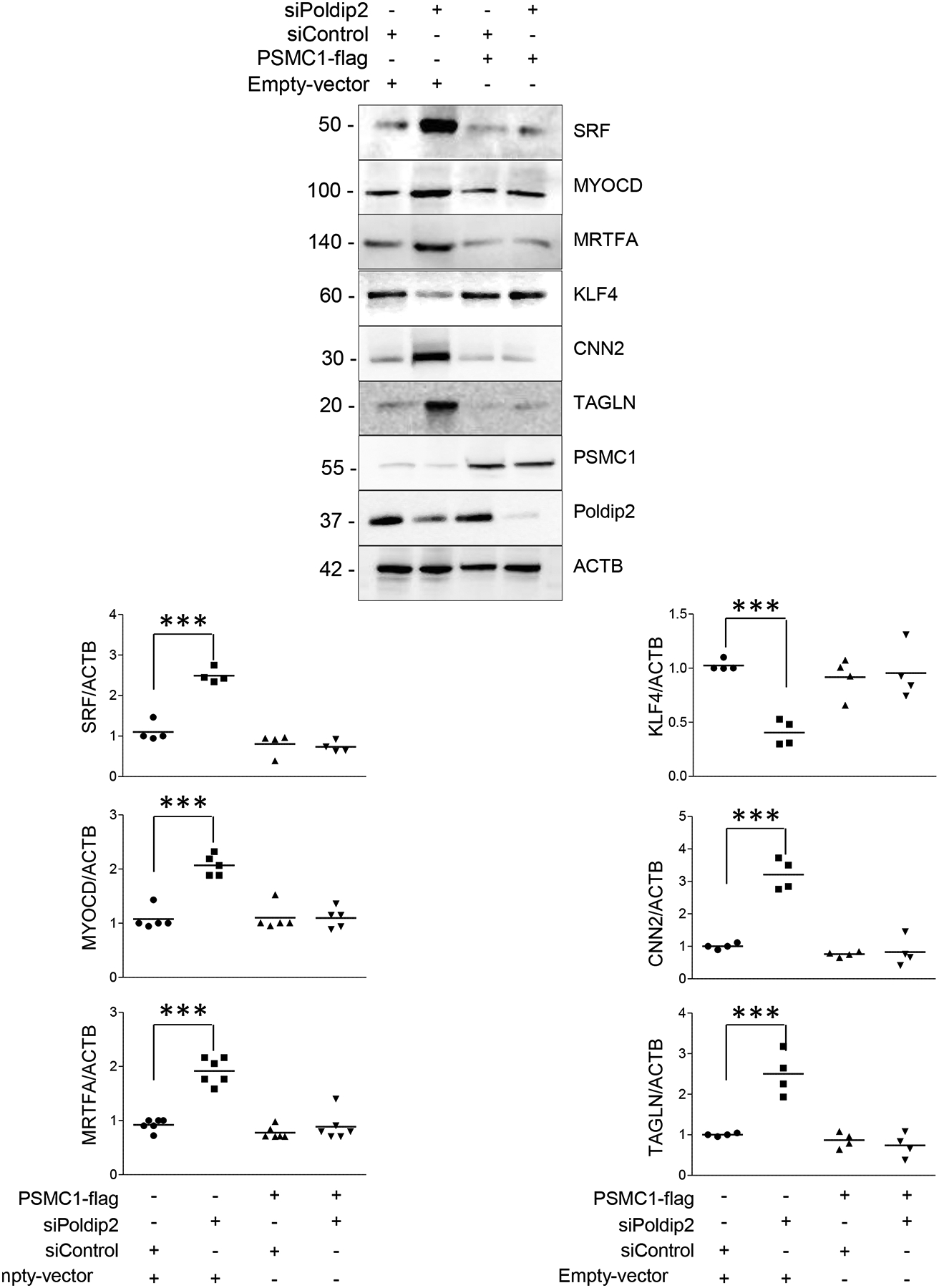
Representative western blots showing the expression of VSMC differentiation markers and transcription factors after UPS activity is rescued by PSMC1 expression in control and siPoldip2 treated cells. HASMCs were transfected as indicated in the methods section. Scatter plots and mean from 4–6 independent experiments are shown. P value indicated was calculated by two-way ANOVA, ***<0.001.
Recently, the inhibition of proteasome activity has been linked to cellular metabolic status through the hexosamine biosynthetic pathway (HBP). As mentioned before, the HBP is an alternative pathway of glucose catabolism downstream of Hexokinase 2 (HK2). In addition to glucose, the HBP uses stoichiometric amounts of acetyl-CoA, uridine, and glutamine to yield its final product, Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is the sugar donor for the O-GlcNAc transferase (OGT)-catalyzed transfer of a single sugar onto specific serine and threonine residues of target proteins.15 In particular, it has been shown that increased OGT-mediated glycosylation downstream of HBP inhibits the UPS activity.17, 18
Poldip2 deficiency directs the lipoic-activating enzyme (LAE) ACSM1 to degradation inhibiting the lipoylation of key enzymes of the TCA cycle, which results in repressed mitochondrial respiration and increased glycolytic activity.11 Of importance, forced expression of either the lipoyl transferase LIPT1 or the LAE ACSM1 reverses the expression of differentiation markers in Poldip2 deficient HASMCs (Online Figure VI A–B) confirming that increased differentiation caused by Poldip2 deficiency is downstream of impaired protein lipoylation. Thus, we posit that in Poldip2-deficient cells the UPS is inhibited by a glycolytic reprograming that might result in an increase in the flux through the HBP and the subsequent upregulation of OGT-mediated glycosylation.
Indeed, in HASMCs Poldip2 deficiency induced significant upregulation of HK2, the Glutamine-Fructose-6-Phosphate Transaminase 2 (GFPT2), one of the two isoforms that catalyzes the HBP’s rate-limiting reaction, and OGT (Figure 6A). These changes in expression were accompanied by a significant increase in the cell content of O-GlcNAc-modified proteins (Figure 6A).
Figure 6. Poldip2 deficiency increases flux through the hexosamine biosynthetic pathway and OGT-mediated protein glycosylation.

A) Representative western blot showing the expression of HK2, OGT and the accumulation of O-linked GlcNacylated proteins in control and Podip2-deficient HASMCs. Scatter plots and mean from 4–6 independent experiments are shown. P value indicated was calculated by independent t-test. B) Representative western blot to evaluate the OGT-mediated glycosylation in PSMC1. Control and Poldip2-deficient HASMCs were immunoprecipitated using a specific antibody against GlcNAc and blotted with a PSMC1 specific antibody. C) Representative western blot to evaluate phosphorylation of PSMC1 in control and siPoldip2 treated cells. PSMC1 separated by Immunoprecipitation and blotted with phospho specific antibodies. Some samples were treated with the OGT inhibitor OSMI (25 μM). Western blot is representative of 3 independent experiments.
The ability of the glutamine analog and HBP inhibitor Azaserine (40 μM)34 to reverse the accumulation of O-GlcNAc-modified proteins and the upregulation of OGT in Poldip2-deficient HASMCs suggests that the increase in HBP flux acts upstream and is responsible for the upregulation of OGT (Online Figure VII).
To validate that these changes are specific, physiologically relevant, and independent of agonist-stimulation, we recapped both: UPS inhibition and upregulation of the hexosamine pathway using a second unrelated siRNA against Poldip2 (Online Figure VIII), Poldip2+/− mouse aortas (Online Figure IX), and PDGF-stimulated cells (Online Figure X).
Consistent with the inhibition of a nuclear proteasome, the proteasome activator PSMC1 was among the GlcNAcylated proteins (Figure 6B). Considering that OGT-mediated glycosylation inhibits PSMC1 activation,18 and phosphorylation of 19S subunits are required for UPS activation,18, 35 we evaluated whether OGT-mediated glycosylation of Ser or Thr residues interferes with the phosphorylation of PSMC1. We found that while PSMC1 phosphorylation in Ser of Thr in the p-(Ser/Thr)-Phe motif was unaffected, the phosphorylation of threonine followed by a proline was dramatically reduced in Poldip2 deficient cells, which was recovered by inhibition of OGT with OSMI36 (25 μM) (Figure 6C).
Finally, we tested whether OGT mediates the inhibition of the UPS and the phenotypic changes observed in Poldip2-deficient HASMCs. As expected, downregulation of OGT completely prevented the accumulation of O-GlcNAc-modified proteins in Poldip2-deficient cells (Online Figure XI). Most significant, downregulation of OGT was able to restore UPS activity in Poldip2-deficient HAMSCs (Figure 7A) and impeded the accumulation of poly-Ubiquitinated proteins or UPS activity reporters (Figure 7B). These results demonstrate that the inhibition of UPS in Poldip2-deficient HASMCs is mediated by an OGT-dependent mechanism. Furthermore, and consistent with the role of UPS inhibition in the mechanism of phenotypic modulation initiated by Poldip2 deficiency (Figure 5), OGT downregulation prevented the increase in SRF, MYOCD and MRTFA and the downregulation of KLF4 observed in Poldip2-deficient HASMCs (Figure 8A). Consequently, OGT downregulation blocks the increased expression of genes from the contractile apparatus in Poldip2-deficient HASMCs (Figure 8A). A proposed model consistent with our findings is presented in Figure 8B.
Figure 7. Poldip2 deficiency induces inhibition of UPS by an OGT-dependent mechanism.

A) Proteasome activity in HASMCs transfected in control and Poldip2-deficient cells transfected with control siRNA or against OGT was measured using a fluorogenic substrate as described in the methods section. B) Immunoprecipitation to evaluate the accumulation of poly-ubiquitinated proteins, and the Ub-GFP reporter in Poldip2-deficient cells treated with siRNA control or against OGT. Scatter plots and mean from 4–5 independent experiments are shown. P value indicated was calculated by two-way ANOVA, * < 0.05, ***<0.001.
Figure 8. Poldip2 deficiency induces HASMC differentiation by an OGT-dependent mechanism.
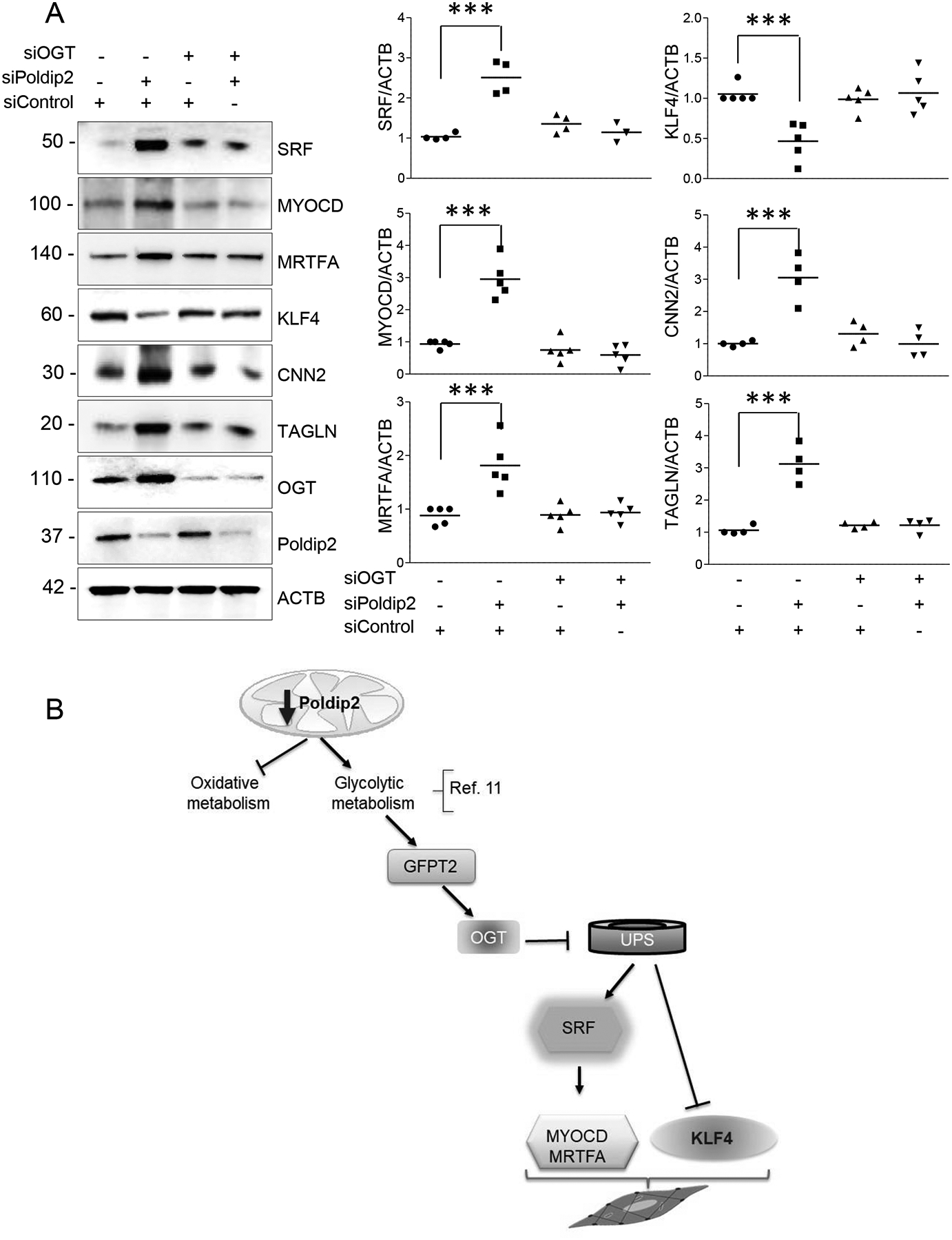
Representative western blot showing the expression of VSMC differentiation markers and transcription factors in Poldip2-deficient cells treated with siRNA control or against OGT. Scatter plots and mean from 4–5 independent experiments are shown. P value indicated was calculated by two-way ANOVA, ***<0.001. B. Model. Our data indicate that Poldip2 deficiency increases protein glycosylation by OGT downstream of HBP. OGT-mediated glycosylation induces the inhibition of UPS and subsequent stabilization of SRF and KLF4 repression. SRF, SRF-mediated upregulation of MRTFA and MYOCD and KLF4 repression enable the expression of SMC-specific genes.
DISCUSSION
Although early work in the field established a relationship between the rate of glycolysis and smooth muscle contractile capacity,37, 38 virtually nothing is known about the role of metabolism and mitochondria-initiated signaling in VSMC differentiation. In this study, we unveiled an unknown relationship between VSMC metabolic status and differentiation state. Furthermore, we recognize that proteostasis and UPS activity are regulated downstream of metabolic reprograming to control VSMC differentiation.
We recently described that Poldip2 is required for the lipoylation and activation of key enzymes of the TCA cycle and consequently for normal mitochondrial function.11 Consequently, Poldip2 deficiency impairs oxidative respiration and induces glycolytic metabolism.11 In the current study, we further characterized the metabolic reprogramming induced by Poldip2 deficiency and established that HK2 is highly upregulated and that a significant amount of glucose-6-phosphate (G6P) is being re-directed to the HBP. Since HBP uses stoichiometric amounts of G6P and acetyl-CoA to yield UDP-GlcNAc, the fact that Poldip2-deficient cells cannot oxidize Acetyl-CoA through the TCA cycle may explain why there is a preferential upregulation of HBP. Interestingly, while both GFPT isoforms are expressed in HASMCs, we only observed upregulation in the expression of GFPT2 an unrecognized isoform in smooth muscle.
Similarly, we observed the upregulation of OGT expression. Since OGT displays high affinity for UDP-GlcNAc (Km = 545 nM),39 in addition to increased HBP flux, the regulation of OGT itself may have a critical impact on the increase of protein O-GlcNAcylation. In our study, OGT upregulation is inhibited by Azaserine. This suggests that the upregulation of OGT expression occurs in response to the increased HBP flux, perhaps via accumulation of its substrate UDP-GlcNAc. Additional research is necessary to determine the exact mechanism of regulation of OGT by Poldip2 deficiency.
Since the metabolism of nitrogen, nucleotides, fatty acids, glucose and high energy phosphate groups all converge in the synthesis of its final product, UDP-GlcNAc, the flux through the HBP has been proposed as sensor of cell metabolism.40 Consequently, we now show that the HBP flux and its end point product O-GlcNAcylated proteins are capable of responding to and sensing mitochondrial metabolic status. Furthermore, our study shows that OGT-mediated protein O-GlcNAcylation tunes UPS activity to mitochondrial function.
Interestingly, our data indicate that Poldip2 deficiency preferentially impairs the degradation of nuclear proteins. Indeed, and consistent with previous reports,18 the nuclear 19S regulatory unit PSMC1 undergoes OGT-dependent O-GlcNAcylation. This condensation reaction with the hydroxyl groups of serine and threonine impedes PSMC1 phosphorylation at those sites and thus UPS activation.41 The fact that PSMC1 overexpression reverses the inhibition of UPS in Poldip2-deficient cells supports this notion.
While UPS inhibition may directly explain the increase in SRF activity, its role in KLF4 repression is probably more complex. Because Poldip2-deficient cells have higher levels of KLF4 messenger RNA (data not shown), we believe that KLF4 is regulated at the translational level and speculate that UPS may be responsible for the degradation of a factor that prevents KLF4 translation. Additional studies will be required to dissect the mechanism of KLF4 repression in Poldip2-deficient cells.
In our study, we found that MYOCD and MRTFA expression are SRF dependent. While it has been previously reported that MYOCD contains CArG box enhancer elements in its promoter,42 we are not aware that these elements have been found in MRTFA. Thus, we cannot rule out that the effect of SRF on MRTFA expression is indirect.
Generalizing the role of the VSMC phenotypic switch in vascular diseases is complex. In atherosclerosis, a more proliferative and secretory VSMC could be detrimental because it may cause a narrowing of the vessel. However, increased rates of proliferation and production of extracellular matrix have been shown to be beneficial as a result of plaque stabilization, decreasing the risk of eruption and embolism, which in fact are responsible for most of the mortality associated with atherosclerosis. Still, preservation of VSMC differentiation by increased MYOCD activity or KLF4 repression in SMC-lineage disrupts inflammatory signaling and reduces the severity and size of the lesions in animal models of atherosclerosis.9, 10
Tracking of SMC-lineage fate in animal models has evidenced the complexity and implications of VSMC phenotypic modulation. Indeed, lineage-tracing experiments in vivo have shown that under pathological circumstances they lose all VSMC-specific markers and transdifferentiate into foam and macrophage-like cells.43 Because the functional characteristics and transcriptome of cholesterol-loaded VSMC remain distinguishable from those of authentic macrophages,44 the exact role of these cells to atherosclerosis is still unclear. Nonetheless, data from mice and human studies indicate that they may directly contribute to disease progression.10, 28
In this study, we found that Poldip2-deficient cell differentiation is unaffected when they are exposed to growth factors or cholesterol, and in vivo they retain the expression of differentiation markers in the proliferative phase of an injury-induced neointima formation model. These results are consistent with previous reports showing that Poldip2-deficient animals are protected against vascular diseases. More importantly, our work shows that metabolic reprograming induced by Poldip2 downregulation modulates VSMC phenotype and restricts phenotypic switching under pathological stimuli. These data position Poldip2 as a possible pharmacological target to inhibit vascular diseases in which SMC phenotypic switching is involved
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Vascular smooth muscle cells (VSMCs) are not terminally differentiated and retain significant plasticity through life. VSMC phenotypic modulation is linked to vascular diseases.
It is increasingly appreciated that metabolic rewiring can inform cellular differentiation and phenotype. However, nothing is known about the role of mitochondria-initiated signaling and metabolism in VSMC differentiation.
Poldip2 is a nuclear-encoded mitochondrial protein required for oxidative metabolism. As a consequence, Poldip2 deficiency induces a metabolic reprogramming characterized by repressed mitochondrial respiration and increased glycolytic activity.
What New Information Does This Article Contribute?
Poldip2 deficient VSMC are highly differentiated in vitro and in vivo, and they display protection against agonist-induced phenotypic modulation.
Poldip2 deficiency re-directs glucose metabolism through the hexosamine pathway and induces glycosylation-mediated inhibition of a ubiquitin proteasome system (UPS) that results in stabilization of SRF and repression of KLF4, resulting in VSMC differentiation.
These results position metabolic signaling as a key component of VSMC phenotypic modulation.
ACKNOWLEDGEMENT
We thank Dr. Kathy Griendling for the generous gift of wild type and Poldip2 heterozygous mouse femoral specimens.
SOURCES OF FUNDING
This study was supported by The National Heart, Lung and Blood Institute of the National Institutes of Health under award HL095070 and by the American Heart Association grant AHA 19POST3438089.
Nonstandard Abbreviations and Acronyms:
- VSMC
Vascular smooth muscle
- SRF
Serum Response factor
- MYOCD
Myocardin
- MRTFA
Myocardin related transcription factor A
- KLF4
Krüppel-like factor
- OGT
O-Linked N-Acetylglucosamine Transferase
- GlcNAc
N-Acetylglucosamine
- UDP-GlcNAc
Uridine diphosphate N-acetylglucosamine
- UPS
Ubiquitin proteasome system
- Poldip2
Polymerase delta interacting protein 2
- MYH11
Myosin heavy chain
- CNN
Calponin
- ACTA2
Aortic smooth muscle actin
- TAGLN
Transgelin
- αKGDH
α-ketoglutarate dehydrogenase
- PDH
Pyruvate dehydrogenase
- HBP
Hexosamine Biosynthetic Pathway
- HK2
Hexokinase 2
- PDGF
Platelet-derived growth factor
- LGALS3
Lectin, Galactoside-Binding, Soluble, 3
- ARG1
Arginase 1
- SREBF1
Sterol Regulatory Element Binding Transcription Factor 1
- CCL2
C-C Motif Chemokine Ligand 2
- PSMC1
Proteasome 26S Subunit, ATPase 1
- GFPT2
Glutamine-Fructose-6-Phosphate Transaminase 2
Footnotes
DISCLOSURES
The authors declare no conflicts of interest with the contents of this article.
REFERENCES
- 1.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356 [DOI] [PubMed] [Google Scholar]
- 2.Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, Olson EN. Requirement of mads domain transcription factor d-mef2 for muscle formation in drosophila. Science. 1995;267:688–693 [DOI] [PubMed] [Google Scholar]
- 3.Shimizu RT, Blank RS, Jervis R, Lawrenz-Smith SC, Owens GK. The smooth muscle alpha-actin gene promoter is differentially regulated in smooth muscle versus non-smooth muscle cells. J Biol Chem. 1995;270:7631–7643 [DOI] [PubMed] [Google Scholar]
- 4.Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci U S A. 2002;99:14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of klf4, pelk-1, and hdac2 to a g/c repressor element in the sm22alpha promoter mediates transcriptional silencing during smc phenotypic switching in vivo. Circ Res. 2012;111:685–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, McDonald OG, Owens GK. A g/c element mediates repression of the sm22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res. 2004;95:981–988 [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem. 2005;280:9719–9727 [DOI] [PubMed] [Google Scholar]
- 9.Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. 2015;35:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paredes F, Sheldon K, Lassegue B, Williams HC, Faidley EA, Benavides GA, Torres G, Sanhueza-Olivares F, Yeligar SM, Griendling KK, Darley-Usmar V, San Martin A. Poldip2 is an oxygen-sensitive protein that controls pdh and alphakgdh lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc Natl Acad Sci U S A. 2018;115:1789–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown DI, Lassegue B, Lee M, Zafari R, Long JS, Saavedra HI, Griendling KK. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLoS One. 2014;9:e96657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, Gleason RL Jr, Taylor WR, Hart CM, Griendling KK, Lassegue B. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol. 2013;33:2154–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datla SR, L LH, Seidel-Rogol B, Dikalova AE, Harousseau M, Punkova L, Joseph G, Taylor WR, Lassegue B, Griendling KK. Poldip2 knockdown inhibits vascular smooth muscle proliferation and neointima formation by regulating the expression of pcna and p21. Lab Invest. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Love DC, Hanover JA. The hexosamine signaling pathway: Deciphering the “o-glcnac code”. Sci STKE. 2005;2005:re13. [DOI] [PubMed] [Google Scholar]
- 16.Aquino-Gil M, Pierce A, Perez-Cervera Y, Zenteno E, Lefebvre T. Ogt: A short overview of an enzyme standing out from usual glycosyltransferases. Biochem Soc Trans. 2017;45:365–370 [DOI] [PubMed] [Google Scholar]
- 17.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-glcnac modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–725 [DOI] [PubMed] [Google Scholar]
- 19.Datla SR, L LH, Seidel-Rogol B, Dikalova AE, Harousseau M, Punkova L, Joseph G, Taylor WR, Lassegue B, Griendling KK. Poldip2 knockdown inhibits vascular smooth muscle proliferation and neointima formation by regulating the expression of pcna and p21. Lab Invest. 2019;99:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, Griendling KK. Mechanisms of vascular smooth muscle nadph oxidase 1 (nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104 [DOI] [PubMed] [Google Scholar]
- 22.Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K. Reap: A two minute cell fractionation method. BMC Res Notes. 2010;3:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC. A protocol for rapid generation of recombinant adenoviruses using the adeasy system. Nat Protoc. 2007;2:1236–1247 [DOI] [PubMed] [Google Scholar]
- 25.Holycross BJ, Blank RS, Thompson MM, Peach MJ, Owens GK. Platelet-derived growth factor-bb-induced suppression of smooth muscle cell differentiation. Circ Res. 1992;71:1525–1532 [DOI] [PubMed] [Google Scholar]
- 26.Thyberg J, Palmberg L, Nilsson J, Ksiazek T, Sjolund M. Phenotype modulation in primary cultures of arterial smooth muscle cells. On the role of platelet-derived growth factor. Differentiation. 1983;25:156–167 [DOI] [PubMed] [Google Scholar]
- 27.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559 [DOI] [PubMed] [Google Scholar]
- 29.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. 1992;89:507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G. Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc Natl Acad Sci U S A. 2010;107:7817–7822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salomons FA, Verhoef LG, Dantuma NP. Fluorescent reporters for the ubiquitin-proteasome system. Essays in biochemistry. 2005;41:113–128 [DOI] [PubMed] [Google Scholar]
- 32.Sandbo N, Qin Y, Taurin S, Hogarth DK, Kreutz B, Dulin NO. Regulation of serum response factor-dependent gene expression by proteasome inhibitors. Mol Pharmacol. 2005;67:789–797 [DOI] [PubMed] [Google Scholar]
- 33.Kimura A, Kato Y, Hirano H. N-myristoylation of the rpt2 subunit regulates intracellular localization of the yeast 26s proteasome. Biochemistry. 2012;51:8856–8866 [DOI] [PubMed] [Google Scholar]
- 34.Rajapakse AG, Ming XF, Carvas JM, Yang Z. The hexosamine biosynthesis inhibitor azaserine prevents endothelial inflammation and dysfunction under hyperglycemic condition through antioxidant effects. Am J Physiol Heart Circ Physiol. 2009;296:H815–822 [DOI] [PubMed] [Google Scholar]
- 35.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic amp-dependent protein kinase through phosphorylation of rpt6. J Biol Chem. 2007;282:22460–22471 [DOI] [PubMed] [Google Scholar]
- 36.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits ogt activity in cells. ACS Chem Biol. 2015;10:1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paul RJ, Peterson JW. Relation between length, isometric force, and o2 consumption rate in vascular smooth muscle. Am J Physiol. 1975;228:915–922 [DOI] [PubMed] [Google Scholar]
- 38.Paul RJ. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am J Physiol. 1983;244:C399–409 [DOI] [PubMed] [Google Scholar]
- 39.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between o-glcnacylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer SP, Hart GW. Dynamic nuclear and cytoplasmic glycosylation: Enzymes of o-glcnac cycling. Biochemistry. 2003;42:2493–2499 [DOI] [PubMed] [Google Scholar]
- 41.Li N, Lerea KM, Etlinger JD. Phosphorylation of the proteasome activator pa28 is required for proteasome activation. Biochem Biophys Res Commun. 1996;225:855–860 [DOI] [PubMed] [Google Scholar]
- 42.Miano JM. Serum response factor: Toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593 [DOI] [PubMed] [Google Scholar]
- 43.Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, Fisher EA. Cholesterol loading reprograms the microrna-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.