

Abstract
Methylation of cytosine residues in DNA influences chromatin structure and gene transcription, and its regulation is crucial for brain development. There is mounting evidence that DNA methylation can be modulated by hormone signaling. We analyzed genome-wide changes in DNA methylation and their relationship to gene regulation in the brain of Xenopus tadpoles during metamorphosis, a thyroid hormone-dependent developmental process. We studied the region of the tadpole brain containing neurosecretory neurons that control pituitary hormone secretion, a region that is highly responsive to thyroid hormone action. Using Methylated DNA Capture sequencing (MethylCap-seq) we discovered a diverse landscape of DNA methylation across the tadpole neural cell genome, and pairwise stage comparisons identified several thousand differentially methylated regions (DMRs). During the pre- to pro-metamorphic period, the number of DMRs was lowest (1,163), with demethylation predominating. From pre-metamorphosis to metamorphic climax DMRs nearly doubled (2,204), with methylation predominating. The largest changes in DNA methylation were seen from metamorphic climax to the completion of metamorphosis (2,960 DMRs), with 80% of the DMRs representing demethylation. Using RNA sequencing, we found negative correlations between differentially expressed genes and DMRs localized to gene bodies and regions upstream of transcription start sites. DNA demethylation at metamorphosis revealed by MethylCap-seq was corroborated by increased immunoreactivity for the DNA demethylation intermediates 5-hydroxymethylcytosine and 5-carboxymethylcytosine, and the cytosine dioxygenase ten eleven translocation 3 that catalyzes DNA demethylation. Our findings show that the genome of tadpole neural cells undergoes significant changes in DNA methylation during metamorphosis, and these changes likely influence chromatin architecture, and gene regulation programs occurring during this developmental period.
Keywords: DNA methylation, Xenopus, metamorphosis, transcription, TET enzymes, brain development
Introduction
Methylation of DNA is an important epigenetic modification that involves the addition of a methyl group to the carbon-5 position of cytosine (5-methylcytosine; 5-mC). In vertebrates, 5-mC is found predominantly in the context of cytosine-guanine (CpG) dinucleotides, located in intergenic regions, and within genes and transposable elements (Feng et al., 2010b; Suzuki and Bird, 2008; Zemach et al., 2010). Global changes in DNA methylation have been shown to be important for establishing tissue-specific gene expression patterns during embryogenesis in mammals (Goll and Bestor, 2005) and in Xenopus (Bogdanovic et al., 2011). Regions with large CpG content often found within gene promoters are known as ‘CpG islands’, and methylation of CpG islands often leads to gene repression, while demethylation leads to gene activation (Guo et al., 2011; Lister et al., 2013; Song et al., 2011a; Szulwach et al., 2011).
Methylation of DNA is catalyzed by DNA methyltransferases (DNMTs), of which there are three that have enzymatic activity in mammals. The de novo DNMTs, DNMT3a and DNMT3b, establish the pattern of DNA methylation during embyrogenesis, which is then preserved through subsequent rounds of cell division by DNMT1 (Rottach et al., 2009; Watanabe et al., 2002). DNA methylation imposes long-term, stable transcriptional silencing through physical blockade of transcription factor binding, and recruitment of methyl-CpG binding proteins, which in turn recruit corepressors or histone deacetylases to modify local chromatin into a transcriptionally silent state (Rottach et al., 2009). DNA methylation of gene regulatory regions typically leads to gene repression, and DNA demethylation gene activation (Maeder et al., 2013; Shin et al., 2014; Suzuki and Bird, 2008; Xu et al., 2012). DNA demethylation can be active or passive; active DNA demethylation is a DNA replication-independent enzymatic removal of 5-mC, while passive DNA demethylation occurs when the newly synthesized DNA does not inherit the pattern of DNA methylation during cell division, which may occur through exclusion of DNMT1 from the cell nucleus (Wu and Zhang, 2014).
Active DNA demethylation involves a complex set of enzymatic steps that depends in part on the ten-eleven translocation (TET) family of dioxygenases (Lu et al., 2015). These enzymes catalyze active DNA demethylation by oxidizing 5-mC to the intermediates 5-hydroxy methylcytosine (5-hmC), 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC) (He et al., 2011a; Ito et al., 2010; Ito et al., 2011; Lu et al., 2015; Maiti and Drohat, 2011). The isocitrate dehydrogenase family of enzymes (idh1/2/3) provide the substrate α-ketoglutarate for TET-mediated oxidation of 5-mC, and are necessary for TET-dependent 5-hmC production (Figueroa et al., 2010; Lian et al., 2012). The 5-hmC is removed by two different pathways. It can be further oxidized by TET enzymes to 5-fC and to 5-caC (He et al., 2011b; Ito et al., 2011), and then removed through the base-excision repair pathway catalyzed by thymine DNA glycosylase (TDG) (He et al., 2011b; Maiti and Drohat, 2011), or by nucleotide excision repair pathways catalyzed by growth arrest and DNA damage (GADD45 α/ β/ γ) enzymes (Schafer, 2013). Alternatively, 5-hmC may be deaminated to unmethylated cytosine by the combined actions of activity-induced cytidine deaminase (AID) and the apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) (Wu and Zhang, 2014).
The methylation state of DNA plays a critical role in animal development, particularly the nervous system. The DNMTs are essential for normal development; mice carrying mutations for these genes are embryonic lethal or die shortly after birth (Li et al., 1992; Okano et al., 1999). Rett syndrome in humans is caused by mutations in the X-linked gene that codes for methyl-CpG binding protein 2 (MeCP2), which binds to methylated cytosines through its methyl-CpG binding domain (Amir et al., 1999). Neural cell-specific deletion of DNMT genes in mice causes abnormal neurological development accompanied by defective cell differentiation, neurodegeneration and deficits in learning and memory (Fan et al., 2005; Feng et al., 2010a; Hutnick et al., 2009; Nguyen et al., 2007). In mouse and human there are dramatic changes in DNA methylation in the brain during the neonatal/early postnatal period. A recent, comprehensive genome-wide mapping of 5-mC revealed a dramatic increase in cytosine-adenine (CpA) methylation in developing mouse and human brain, coincident with the period of synaptogenesis and synaptic pruning (Lister et al., 2013).
While DNA methylation is recognized as an essential epigenetic modification regulating gene expression, how it is regulated is poorly understood. We previously showed that thyroid hormone (T3) can directly induce transcription of the dnmt3a gene in Xenopus and mouse neural cells (Kyono et al., 2016a; Kyono et al., 2016b). Thyroid hormone is essential for neurological development in mammals, and deficiency of T3 during the neonatal/early postnatal period in humans leads to a condition of severe mental and growth retardation known as cretinism (Bernal, 2009; Porterfield and Hendrich, 1993). Amphibian tadpole metamorphosis is entirely dependent on T3, and it is an important developmental model system for investigating T3 actions (Buchholz, 2017). Developmental processes that occur in the tadpole brain during metamorphosis mirror those in mammals, with T3 regulating cell proliferation, migration, myelination, synaptogenesis, and cell death (Denver, 1998b; Denver et al., 2009). The hormone binds to nuclear hormone receptors (thyroid hormone receptors - TRs) that modify chromatin structure and regulate gene transcription (Cheng et al., 2010). The gene regulation programs during amphibian metamorphosis underlie tissue morphogenesis (Shi, 2000), and correlate with histone modifications that are thought to be due primarily to T3 action (Shi, 2009). There are dynamic changes in DNA methylation during early Xenopus development (Bogdanovic et al., 2011; Bogdanovic et al., 2012), but to our knowledge, changes in DNA methylation during metamorphosis have not been investigated in any tissue. Such changes, if they occur, are likely to be important for gene regulation during metamorphosis, and for establishing stable, cell type-specific gene expression patterns in the juvenile adult.
In the current study, we used genome-wide Methylated DNA Capture sequencing (MethylCap-seq), and several biochemical and histochemical approaches, to investigate developmental changes in DNA methylation in the brain of Xenopus tadpoles during metamorphosis. We also conducted RNA sequencing (RNA-seq) to look for associated changes in gene transcription during metamorphosis. We focused on the brain region containing the preoptic area, thalamus and hypothalamus, which contains neurosecretory neurons that control hormone production during metamorphosis, and is highly responsive to T3 action (Denver, 1998a; Denver et al., 2009). Using MethylCap-seq, we identified several thousand differentially methylated regions (DMRs). We also found negative correlations between changes in DNA methylation and gene expression. The largest changes in DNA methylation occurred from metamorphic climax to the completion of metamorphosis, with DNA demethylation predominating. During this interval we found increases in mRNAs for genes that encode enzymes involved with DNA demethylation, and also immunoreactivity for the DNA demethylation intermediates 5-hydroxymethylcytosine (5-hmC) and 5-carboxymethylcytosine (5-caC), and the cytosine dioxygenase ten eleven translocation 3 (TET3).
Materials and Methods
Animal care and use
We obtained X. tropicalis tadpoles by in-house breeding or from Xenopus One (Dexter, MI), reared them in dechlorinated tap water (water temperature 25 °C, pH 7) and maintained them on a 13L:11D photoperiod. We fed tadpoles ad libitum with pulverized frog brittle (NASCO, Fort Atkinson, WI) or Sera Micron plankton food. Tadpoles were staged using the developmental staging system of Nieuwkoop and Faber (1994) (NF). We euthanized tadpoles before tissue collection by rapid decapitation. The animals were not anaesthetized before euthanasia because anaesthesia can cause changes in gene expression, and perhaps also DNA methylation. This method, and all procedures involving animals were conducted under an approved animal use protocol (PRO00006809) in accordance with the guidelines of the Institutional Animal Care and Use Committee at the University of Michigan.
Methylated DNA capture for deep sequencing and targeted analysis
To investigate genome-wide changes in DNA methylation in tadpole brain during metamorphosis, we conducted Methylated DNA Capture assay (MethylCap) using the MethylCap kit (Diagenode, Denville, NJ) following the manufacturer’s instructions. We microdissected the region of the tadpole brain containing the preoptic area/thalamus/hypothalamus (Supplemental Fig. 1A) at four stages of metamorphosis: NF stages 50 (pre-metamorphosis), 56 (pro-metamorphosis), 62 (metamorphic climax) and 66 (metamorphosis complete, juvenile adult). We included three biological replicates for each developmental stage, and for each biological replicate we pooled tissue from 15 animals for NF stage 50, 5 for stage 56, and 6 for stages 62 and 66.
We isolated genomic DNA using the DNeasy Blood and Tissue kit (Qiagen), and sonicated the DNA to an average size of 400 bp using a Covaris S2 sonicator (Covaris, Woburn, MA). To monitor DNA capture efficiency during the MethylCap procedure, we ‘spiked’ the MethylCap reaction with a cocktail of four lambda phage DNAs containing varying numbers of CG dinucleotides, and either methylated or not (described below), as described by Taiwo et al. (Taiwo et al., 2012) with minor modifications. Using PCR, we generated two unique DNA fragments that differed in CpG density using lambda phage DNA as template (one fragment with low, and one with high CpG density; see Supplemental Table 4 for oligonucleotide primer sequences). We conducted in vitro methylation of the two DNA fragments by incubating them with 10 U of M.SssI CpG methyltransferase (New England Biolabs [NEB], Ipsiwich, MA) and S-adenosylmethionine (SAM; 640 μM) for 8 hr (we replenished the SAM after 4 hr). We purified the DNA fragments by phenol/chloroform/isoamyl alcohol precipitation (Invitrogen Life Technologies, Carlsbad, CA), resuspended them in restriction enzyme buffer EB from Qiagen (Valencia, CA), and verified in vitro methylation by digestion of the fragments with 10 U of Hinp1I (NEB), a methylation-sensitive restriction enzyme; methylated DNA fragments were resistant to digestion, while unmethylated DNA fragments were not (data not shown). The two un-methylated, low and high CpG density DNA fragments served as negative controls. We combined the four DNA fragments into a cocktail (two methylated and two un-methylated) at a concentration of 0.2 pM for each DNA fragment, and added this to Buffer B of the MethylCap kit (Diagenode) to a final concentration of 0.0067 pM before proceeding with the capture reaction.
Using 1 μg of sonicated genomic DNA for each sample (each with the cocktail of lambda phage DNA described above added), we captured methylated DNA with magnetic beads, then eluted the captured DNA sequentially with three concentrations of NaCl; we retained DNA from the high salt elution buffer (900 mM NaCl) for deep sequencing. This salt concentration selects for DNA sequences with high CpG density, and therefore we may not have isolated regions of the genome with lower CpG density. However, Bogdanovic and colleagues (Bogdanovic et al., 2011) found no differences in overall distribution of DNA methylation in Xenopus embryos among different salt concentrations of elution buffer. We purified the eluates and the input DNA using the QIAquick PCR Purification kit (Qiagen). Real-time qPCR analysis targeting the three spiked-in DNA fragments showed that the capture efficiency was similar among samples (Supplemental Fig. 1B).
For MethylCap-seq, we sequenced captured DNA samples from tadpole brain at the four stages of metamorphosis, and we also selected three input DNA samples for sequencing, one from each of three NF stages 50, 56 and 62. The sequencing libraries were constructed on the Apollo instrument (an automated system) using IntegenX reagents (now Wafergen, Fremont, CA) at the University of Michigan DNA sequencing core. We sequenced the libraries on Illumina HiSeq 2000 using High Output mode (50 bp, single-end).
Bioinformatics Analysis of MethylCap-seq Data
We conducted quality processing on raw reads (fastq) files from each sample to remove duplicate reads, poor quality bases, and adapter sequences using dedupe.sh and bbduk.sh from the BBTools suite (v37.90). Quality assessments were done on all raw and processed data using FastQC (v0.11.7) and MultiQC (v1.5). We aligned the quality control-processed reads to the X. tropicalis genome (v4.1) using hisat2 (v2.1.0) and the -- dta option with all other defaults. Uniquely mapped reads were selected for further analysis. We sorted aligned reads and indexed them using the samtools sort and samtools index commands, respectively (samtools v1.8). Read depth-normalized bigwig files were created from sorted alignment files for ease of viewing in genome browsers using the bamCoverage command from deepTools (v3.0.2).
We called methylation peaks using PePr (v1.1.18) (Zhang et al., 2014) with default parameters except for --shiftsize and --windowsize, which were each set to 200. For each stage, peaks were called with both the --broad and --sharp flags. We then combined broad and sharp peaks (at each stage) using the bedops --not-element-of 1 command (v2.4.34) to add all sharp peaks that were not partially contained within broad peaks and combine them with the broad peaks.
We called differentially methylated regions (DMRs) using PePr (v1.1.18) as above with the exception that we used the --normalization flag, which was set inter-group, and --diff flag. As with peak calling, DMRs were called with the --broad and --sharp flags set, and combined in the same manner. We filtered the DMRs to accept only those with a p-value ≤0.0005.
For each stage, we calculated the GC content % of each peak using the bedtools nuc command from the bedtools suite (v2.27.1). From these values, we calculated the mean GC content % of peaks at each stage. For CpG and CpA enrichment calculations, two different enrichment scores were calculated: Cp(G∣A) frequency enrichment and Cp(G∣A) observed/expected ratio enrichment. We used the bedtools nuc command to calculate the number of occurrences of each nucleotide (A,T,C,G,N), and the number of occurrences of CpG and CpA, within each peak. From these values, we calculated Cp(G∣A) frequency for each stage by normalizing the number of bases in a Cp(G∣A) context within peaks to the total length of the peaks using a formula recreated based on that developed in the MEDIPS package (Lienhard et al., 2014). See formula 1a and 1b below.
| (1a) |
| (1b) |
We calculated a Cp(G∣A) observed/expected value for each stage by multiplying the number of Cp(G∣A) occurrences in peaks by the total length of the peaks, and dividing that by the product of the total number of C and the total number of (G∣A) in the peaks. See formula 2a and 2b below.
| (2a) |
| (2b) |
These same Cp(G∣A) frequency and Cp(G∣A) observed/expected ratios were calculated for the genomic region not containing peaks. We then calculated enrichment scores by dividing the Cp(G∣A) frequency of peaks by the Cp(G∣A) frequency of the genomic region not containing peaks using a formula recreated based on that developed in the MEDIPS package (Lienhard et al., 2014). This was done for each stage. See formulas 3 and 4 below.
| (3a) |
| (3b) |
| (4a) |
| (4b) |
To investigate peak and DMR distributions relative to genomic features, we used the bedmap command from the bedops suite (v2.4.34) with the --prec 3 and --fraction-either 0.50 flag values set to identify peaks or DMRs as overlapping a genomic feature (intron, exon, tss, promoter, TSS, gene body, etc.) if more than 50% of the peak overlapped the genomic feature, or vice versa. Genomic features correspond to the MNHN gene model annotation published by Buisine and colleagues (Buisine et al., 2015).
With the exception of a few steps, we implemented most of the workflow in Snakemake v4.7.0.
RNA-sequencing library preparation
To investigate the relationship between changes in gene expression and DNA methylation, we conducted RNA sequencing (RNA-seq) on RNA isolated from X. tropicalis tadpole brain during metamorphosis. We micro-dissected the preoptic area/thalamus/hypothalamus of the tadpole brain (Supplemental Fig. 1A) at the same four stages of metamorphosis as for MethylCap-seq. We included three biological replicates for each stage of metamorphosis. After extraction of total RNA using the TRIZOL reagent (Invitrogen Life Technologies, Carlsbad, CA), we further purified the total RNA using the RNeasy Mini kit (Qiagen), then submitted samples to the University of Michigan DNA sequencing core for sequencing library preparation and deep sequencing. All samples had RNA Integrity Numbers (RIN) of 8 or greater as analyzed using the TapeStation (Agilent). Each library was prepared from 130 ng of RNA using the Illumina TruSeq mRNA Sample Prep v2 kit (Illumina Inc., San Diego, CA), where total RNA was poly-A selected, fragmented and reverse transcribed with random primers. The 3’ ends of the cDNA libraries were adenylated, ligated with adapters that contain hexanucleotide barcodes, and PCR-amplified to construct the final libraries. The quality of the final libraries were assessed using the TapeStation (Agilent) and quantified by qPCR using a commercial kit (Kapa Biosystems,Wilmington MA). All libraries were pooled and sequenced to produce single-end 50 bp reads in two lanes on the Illumina HiSeq 2000 platform with High Output mode using version 3 reagents.
Bioinformatics Analysis of RNA-seq Data
We conducted quality processing on raw reads (fastq) files from each sample to remove poor quality bases and adapter sequences using cutadapt (v1.11). We did quality assessments on all raw and processed data using FastQC (v0.11.7) and MultiQC (v0.8). The quality control processed reads were aligned to the Xenopus tropicalis genome (v4.1) and quantified using bowtie2/RSEM (v2.1.0) with default parameters. We conducted gene-level quantification using a GTF file containing each of 22,820 custom-annotated X. tropicalis genes of a custom annotation (the MNHN gene model) built by Nicolas Buisine and Laurent Sachs based on high-throughput RNA paired-end tag sequencing that identified the 5’ and 3’ ends of transcripts expressed in different X. tropicalis tissues, including the brain (Buisine et al., 2015). A gene-level abundance matrix was generated using the abundance_estimates_to_matrix.pl script from the Trinity suite (v2.2.0).
We sorted and indexed aligned reads using the samtools sort and samtools index commands from samtools (v1.8), respectively. Read depth-normalized bigwig files were created from sorted alignment files for ease of viewing in genome browsers using the bamCoverage command from deepTools (v3.0.2). We conducted differential expression analysis using the R package DESeq2 (v1.22.0) to identify differentially expressed genes with a false discovery rate ≤ 0.01.
With the exception of a few steps, we implemented most of the workflow in Snakemake v4.7.0.
Bioinformatics Analysis of Relationship between DMRs and DEGs
To investigate possible relationships between DMRs and DEGs, we used the bedmap command from the bedops suite (v2.4.34) with --echo and --echo-map flags set to identify DMRs within 50 kb (--range 50000) of DEGs or non-DEGs. Similarly, we used bedmap to identify DMRs within the gene-body of DEGs or non- DEGs. Using these values, we ran a Fisher’s Exact Test to test 1) whether there is a difference in the proportion of DMRs within 50 kb (associated with) of DEGs and non- DEGs and 2) whether there is a difference in the proportion of DMRs within the gene-body of DEGs and non- DEGs.
We also analyzed possible correlations between DMRs and DEGs considering their direction of change (i.e, we tested the hypothesis that DNA demethylation is correlated with gene activation, and vice versa). For each comparison, we used the bedmap command from the bedops suite (v2.4.34) with --echo and --echo-map flags set to intersect DMRs with the following regions from DEGs for the same comparison: 5 kb upstream of the DEG, the DEG gene-body, and 5 kb upstream + the DEG gene-body. Pearson correlation coefficients and p-values were then calculated for each comparison between the DMR height (log2 A/B) and the DEG fold-change (log2 A/B) value for the intersecting region. We plotted the values and fit a regression line to the data. We also calculated Pearson correlation coefficients and p-values for each comparison between the DMR direction (1 for positive DMR height [log2 A/B value], −1 for negative DMR height [log2 A/B value]) and the intersecting DEG region fold-change direction (1 for positive DEG log2 fold-change A/B, −1 for negative log2 fold-change A/B). We plotted the values were plotted and fit a regression line to the data.
Targeted analysis of DNA methylation using bisulfite sequencing
We bisulfite-converted 400 ng of genomic DNA extracted from X. tropicalis tadpole brain using the BisulFlash DNA modification kit (Epigentek, Farmingdale, NY) (three biological replicates per developmental stage). We used the Methprimer software (Li and Dahiya, 2002) to design oligonucleotide primers to specifically amplify DNA that had been converted by bisulfite treatment (oligonucleotide primers sequences are given in Supplemental Table 4), and conducted PCR using GoTaq DNA polymerase (Promega) with the following PCR conditions: (94°C for 2 min; 49 cycles of 94°C for 30 sec, 55 or 60°C 30 sec, 72°C 45 sec). We purified the PCR products using the Qiagen PCR purification kit (Qiagen) and subcloned them into pGEM-Teasy vector (Promega) following the manufacturer's instructions. After transformation of E. coli with the resulting plasmids, we conducted colony PCR on bacterial colonies using SP6 and T7 olignoucletide primers, purified the PCR products and conducted Sanger sequencing using SP6 or T7 primers. We sequenced 5-7 colonies for each biological replicate.
Targeted analysis of DNA methylation by 5-mC-sensitive restriction digest
We used 5-mC-sensitive restriction digest followed by qPCR (5-mC chop-qPCR) to analyze changes in DNA methylation in tadpole brain at discrete genomic regions. We incubated 1 μg of genomic DNA in a 50 μl reaction with 10 units of Hinp1I or HpyCH4IV (NEB) at 37 °C overnight, purified the DNA using phenol/chloroform/isoamyl alcohol (Invitrogen, Life Technologies, Carlsbad, CA) and resuspended in 40 uL of buffer AE (Qiagen, 10mM Tris, 0.5mM EDTA, pH 9.0). To measure the extent of restriction digest, we conducted genomic DNA qPCR using primers flanking the restriction sites (see Supplemental Table 4 for oligonucleotide primer sequences), which was normalized to the quantity of the DNA amplicon at an adjacent genomic region that did not contain restriction sites.
Reverse transcriptase real-time quantitative PCR (RTqPCR)
We used RTqPCR to validate gene expression changes identified by RNA-seq, and also for targeted analysis of developmental changes in genes that code for enzymes involved with DNA demethylation. We isolated RNA from the preoptic area/thalamus/hypothalamus of pools of 2-5 brains for each biological replicate; the number pooled depended on the stage/size of the animal, and was held constant within a developmental stage. We treated 1 μg total RNA with 20 units of DNAse I (Promega) to remove genomic DNA, then synthesized cDNA using the High Capacity cDNA Synthesis Kit (Applied Biosystems Inc. [ABI], Foster City, CA). We conducted real-time qPCR using ABsolute Blue qPCR SYBR Low ROX Mix (ABgene Thermo Scientific, Surrey, UK) and the Fast 7500 Real-Time PCR System (ABI) or the StepOne Real Time PCR Systems (Life Technologies). We designed oligonucleotide primers to span exon/exon boundaries (Supplemental Table 4). We used a relative quantification method (Crespi and Denver, 2006; Yao et al., 2007) to compare mRNA levels by generating standard curves for each gene using pooled cDNA. We normalized mRNA levels to the reference gene ef1a, which did not change during metamorphosis (data not shown).
Immunohistochemistry (IHC)
We obtained brains for IHC by dissecting the heads of X. tropicalis tadpoles at six different NF stages of metamorphosis (50, 54, 56, 58, 62 and 66). We fixed tadpole heads in 4% paraformaldehyde made in 0.6X phosphate buffered saline (PBS) overnight at 4°C, then dissected the brains out of the skull and further fixed them in 4 % PFA for 2-3 hr at 4°C. We transferred the brains to 30% sucrose in 0.6X PBS, incubated at 4°C overnight followed by saturation in a 2:1 solution of sucrose:Tissue-Tek Cryo-OCT compound (Fisher Scientific) overnight at 4°C. We embedded the brains in a mold in OCT compound, snap froze and stored brains at −80°C until cryosectioning. We made 16 μM transverse cryosections on Superfrost slides (ThermoFisher Scientific) and stored the slides at −80°C until processing for IHC.
For IHC, we air-dried the slides for 30 minutes, rehydrated them in 0.6X PBS and subjected them to antigen retrieval by immersing slides in 0.01 M sodium citrate (pH 6) at 95 °C for ten minutes. We let the slides cool to room-temperature, then blocked with Superblock (Pierce Chemical Co.) plus 5% normal goat serum and 0.3% Tween-20 in 0.6X PBS for 1-2 hr at room temperature. After blocking, we incubated slides overnight at 4°C with one of the following: rabbit polyclonal antiserum to 5-hmC (1:500; Diagenode C15410205-20), rabbit polyclonal antiserum to 5-caC (1:500; Diagenode C15410204-20) or protein A-purified IgG from a rabbit polyclonal antiserum generated against Xenopus TET3 (xTET3; 0.7 μg/ml; see Supplemental Methods for information on xTET3 antiserum generation and validation). We used DyLight 550-conjugated goat anti-rabbit IgG secondary antibody (#84541, Thermo Scientific, Grand Island, NY, USA) and mounted the slides using ProLong™ Diamond Antifade Mountant (Thermo Fisher Scientific, catalog # P36965). We captured micrographic images using an Olympus IX81 inverted fluorescence microscope (Olympus, Tokyo, Japan) with a Retiga camera and carefully matched sections for anatomical level following the Xenopus brain atlas developed by Tuinhof and colleagues (Tuinhof et al., 1998). We captured digital micrographic images and uniformly adjusted the brightness, contrast and evenness of illumination using Adobe Photoshop CS6 (Adobe Systems, Inc., San Jose, CA).
Data analysis and statistics
We analyzed data using the computer programs SigmaPlot (v. 13.0) or SYSTAT (v. 13; both from Systat Software, San Jose, CA). We used one-way ANOVA followed by Fisher’s least significant difference (Fisher’s LSD) post hoc test, or Student’s independent sample t-test (p=0.05). Derived values were Log10-transformed before statistical analysis if the variances were found to be heterogeneous.
Information regarding reagents used and deep sequencing data generated during the course of this study is given in the Key Resource Table associated with this manuscript.
Results
MethylCap-seq revealed a dense and diverse DNA methylation landscape across the tadpole neural cell genome
We analyzed the region of the tadpole brain containing the preoptic area, thalamus and hypothalamus (Supplemental Fig. 1A). The MethylCap assay enriches methylated cytosines using the recombinant methyl-CpG-binding domain of human MeCP2. Capture efficiency did not differ among developmental stages (Supplemental Fig. 1B). Genome-wide profiling of DNA methylation by MethylCap-seq during metamorphosis revealed a dense and diverse DNA methylation landscape across the tadpole neural cell genome. High quality reads were mapped on the reference genome with stringent parameters to minimize noise (see Materials and Methods). Independent profiles derived from three biological replicates per stage were highly correlated (0.9781 < r < 0.9879, Spearman correlation coefficient). The MethylCap-seq data reported in this study have been deposited in the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI), accession number GSE139267.
The GC content of peaks was not different among developmental stages and averaged 44.52% (Supplemental Table 1). Within the peak sequences, CpG dinucleotides were present at a higher frequency relative to the genome at each developmental stage, while CpA dinucleotides were not (Fig. 1A,B). The CpG and CpA enrichment statistics are given in Supplemental Table 2. These data support that the MethylCap-seq technique achieved the purification of methylated regions of the genome containing high CG content. The mean number of peaks declined with development, although this was not statistically significant (Nieuwkoop-Faber [NF] 50 – 111,789; NF 56 – 111,962; NF 62 – 110,835; NF 66 – 106,016; Fig. 1B).
Fig. 1. Genome-wide patterns of DNA methylation in tadpole brain during metamorphosis analyzed by MethylCap-seq.

We conducted MethylCap-seq on DNA isolated from X. tropicalis tadpole brain (preoptic area/thalamus/hypothalamus) at four stages of metamorphosis (n=3 per stage). A. CpG dinucleotides (left panel) are enriched at MethylCap-seq peaks at each stage of development, while CpA dinucleotides (right panel) are not. Shown are plots of the peak CpG (left panel) and CpA (right panel ) frequencies for each stage of metamorphosis (NF - Nieuwkoop-Faber). The CpG and CpA frequency values were binned (50 bins) prior to plotting. B. The mean number of total MethylCap-seq peaks across the genome, and peaks found within genes (presented by gene region or feature; 50% or more of the peak had to occur within the gene region or feature to be counted) at four stages of metamorphosis. C. The number of MethylCap-seq peaks and their distribution among different regions of the genome did not differ among developmental stages. Gene refers to the gene-body, which we defined as the region from the first exon through the last exon. The promoter region was defined as 5 kb upstream from the end of the first exon (i.e., it included the first exon). Intergenic refers to regions not within any gene-body, and non-intergenic is regions within a gene body. CpG islands are regions identified by the UCSC genome browser.
The mean number of peaks was equally distributed among genes and intergenic regions, and this distribution did not change with development (Fig. 1B). Within genes (defined as the beginning of the first exon through the end of the last exon), the majority of peaks were found within introns. Approximately 20% of the peaks were in proximal promoter regions (defined as ≤ 1 kb upstream of the transcription start site – TSS), 7% in more distal promoter regions (defined as between 1 and 5 kb upstream of the TSS), and about 20% were in CpG islands (regions identified by the University of California, Santa Cruz Genome Browser; Fig. 1C; Supplemental Fig. 2). The length and height of the peaks did not differ between developmental stages (Supplemental Fig. 3). We found four general patterns of DNA methylation across the tadpole neural cell genome: within gene bodies localized to exons (Supplemental Fig. 4A), within gene bodies localized to introns (Supplemental Fig. 4B), in intergenic regions (see rightmost peaks, Supplemental Fig. 4A&B) and little or no methylation (Supplemental Fig. 4C).
Changes in DNA methylation in tadpole brain during metamorphosis analyzed by MethylCap-seq
Our analysis revealed >10,000 distinct genomic regions that displayed statistically significant (p≤0.0005) variation in the level of DNA methylation. The number of differentially methylated regions (DMRs) increased with the progression of metamorphosis, with the largest number of DMRs (2,960) appearing between metamorphic climax (NF stage 62) and the completion of metamorphosis (NF stage 66; Fig. 2A). At the beginning of metamorphosis (NF stage 50 to 56) DNA demethylation predominated; of 1,163 DMRs, 738 became demethylated and 425 became methylated (Fig. 2B). Between pro-metamorphosis (NF stage 56) and metamorphic climax (NF stage 62), this pattern shifted, where DNA methylation predominated; of 2,904 DMRs, 694 became demethylated and 2,210 methylated. The DNA methylation pattern changed during the completion of metamorphosis (NF stage 62 to 66), where DNA demethylation predominated; of 2,960 DMRs, 2,425 became demethylated and 535 methylated. If we compare pre-metamorphosis (NF stage 50) with metamorphic climax (NF stage 62), we find that DNA methylation predominated (923 demethylated vs. 1,281 methylated). If we compare pre-metamorphosis (NF stage 50) with the completion of metamorphosis (NF stage 66), we find that DNA demethylation predominated (1,633 demethylated vs. 697 methylated). Several examples of changes in peak heights for MethylCap-seq and RNA-seq reads mapped to the reference genome are shown in Supplemental Figs. 5 and 6.
Fig. 2. Changes in DNA methylation in tadpole brain during metamorphosis analyzed by MethylCap-seq.
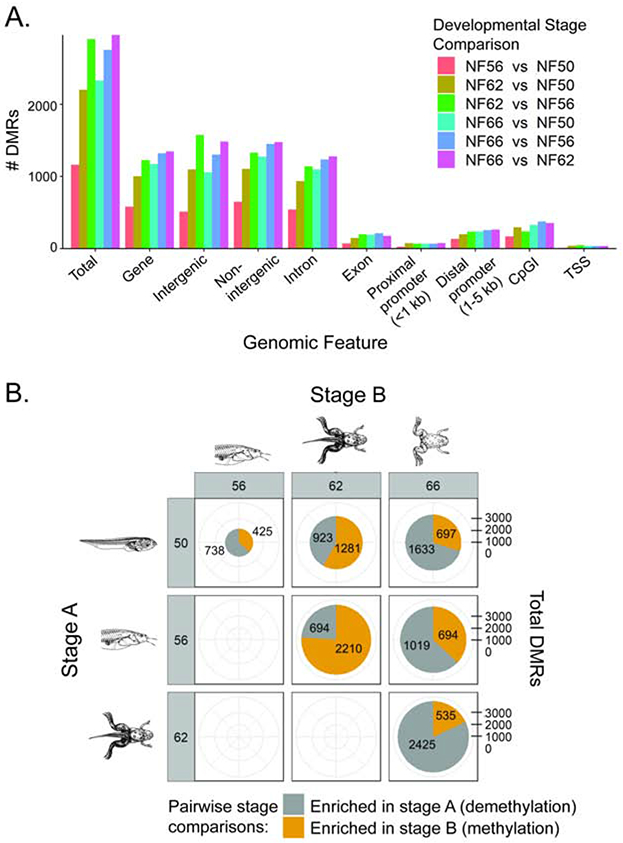
We conducted MethylCap-seq on DNA isolated from X. tropicalis tadpole brain (preoptic area/thalamus/hypothalamus) at four stages of metamorphosis (n=3 per stage). A. Shown is the distribution of DMRs among different genomic features during metamorphosis. Gene refers to the gene-body, which we defined as the region from the first to the last exon. The promoter region was defined as 5 kb upstream of the first exon through the first exon. Intergenic refers to regions not within a gene or gene promoter region. CpG islands are regions identified by the UCSC genome browser. Approximately half of the DMRs occurred within or proximal to genes, the other half were intergenic. Most of the DMRs found within genes were in introns. There were no differences among developmental stages in the distribution of DMRs across the genome. B. Shown in the pie charts are the numbers of differentially methylated regions (DMRs) and the direction of change in DNA methylation (increase is gray, decrease is yellow) within each pairwise comparison (stage A vs. stage B). The number of DMRs in each category is also given. The total number of DMRs increased during metamorphosis. In the first comparison (NF stage 50 vs. 56; pre-metamorphosis to early pro-metamorphosis) there was greater demethylation, in the second (NF stage 56 vs. 62; late pro-metamorphosis to metamorphic climax) greater methylation, and in the third (NF stage 62 vs. 66; metamorphic climax to the completion of metamorphosis) greater demethylation. Comparison of methylation changes from pre-metamorphosis (NF stage 50) to the completion of metamorphosis (NF stage 66) showed that DNA demethylation predominated.
The shift toward demethylation at the completion of metamorphosis was also seen in our bulk genome biochemical analyses, where we saw a decrease in 5-mC content between pre-metamorphosis (NF stage 50-52) and immediately after metamorphic climax (NF stage 63-64) measured by 5-mC enzyme immunoassay (EIA), luminometric assay (LUMA) and cytosine extension assay (Supplemental Fig. 7).
We also saw a decrease in the total height, but not the length of the DMRs during metamorphosis (shown in the small bar graphs at the top of the graph panels in Supplemental Fig. 8), supporting greater DNA demethylation from pro-metamorphosis to the completion of metamorphosis. Similar to the total methylation peaks, we found that the DMRs were distributed roughly equally among genes and intergenic regions, and there were no differences in the relative distribution among these regions when comparing stages of metamorphosis (Fig. 2A; Supplemental Fig. 9). There was no change in the distance from TSSs of intergenic DMRs in any of the comparisons (Supplemental Fig. 10).
Gene expression changes in the tadpole preoptic area/thalamus/hypothalamus during metamorphosis analyzed by RNA sequencing
To investigate relationships between DNA methylation and gene expression we conducted RNA-sequencing (RNA-seq) on X. tropicalis tadpole brain (preoptic area/thalamus/hypothalamus) at four stages of metamorphosis (NF stages 50, 56, 62 and 66). For RNA-seq we generated an average of 27.4 million reads per sample, which we then mapped to the X. tropicalis genome. Using DESeq software (Anders and Huber, 2010) we found that 5,220 of 22,820 annotated genes showed changes in expression in tadpole brain during metamorphosis. Pairwise comparisons by developmental stage of the number of genes up or downregulated are shown in Fig. 3A, and all data are presented in Supplemental Table 3. This analysis showed that, of all differentially expressed genes (DEGs), approximately half were upregulated, and half were downregulated. We conducted reverse transcriptase real-time quantitative PCR (RTqPCR) for 14 DEGs (6 up-, 6 down- and 2 non-regulated) chosen based on their magnitude change (low, medium and high). All DEGs tested by RTqPCR showed the expected developmental changes as found by RNA-seq (Supplemental Fig. 11; gene annotation and primer sequences in Supplemental Table 4). The RNA-seq data reported in this study have been deposited in GEO at NCBI, accession number GSE140120. A full analysis of gene expression changes, gene ontology and pathways will be published elsewhere.
Fig. 3. Gene expression changes in X. tropicalis tadpole brain during metamorphosis analyzed by RNA-seq and their relationship to changes in DNA methylation analyzed by MethylCap-seq.
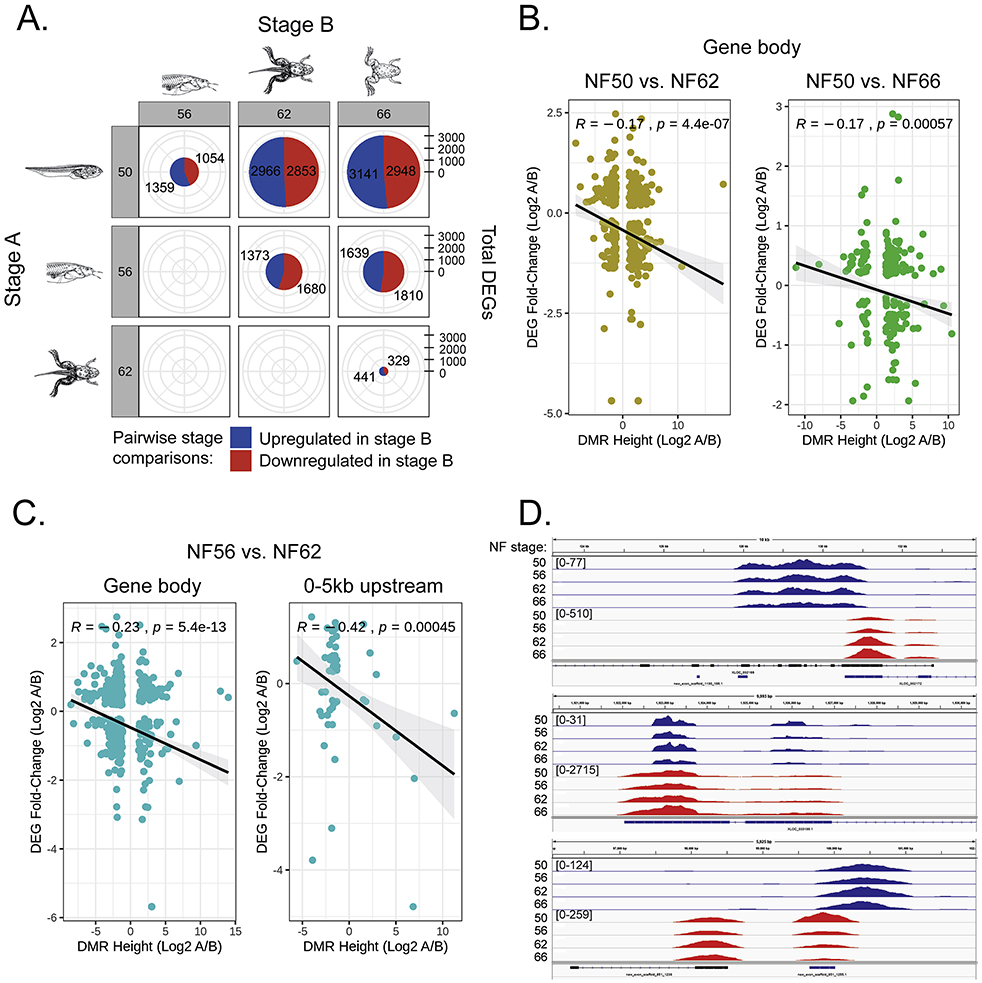
We conducted RNA-seq on total RNA isolated from X. tropicalis tadpole brain (preoptic area/thalamus/hypothalamus) at four stages of metamorphosis (n=3 per stage). We used DESeq software to identify genes whose mRNA levels changed during metamorphosis (differentially expressed genes - DEGs). A. Shown are pie charts with pairwise comparisons among the four stages of metamorphosis of DEGs. The numbers in the red areas of the pie charts represent genes upregulated in Stage B, while the numbers in the blue areas of the pie charts represent genes downregulated in Stage B. Gene regulations changes increased as metamorphosis proceeded, with roughly half the genes upregulated and half downregulated. B. Correlation between overlapping DMRs and DEGs by direction and magnitude of change within gene bodies comparing pre-metamorphosis (NF 50) with metamorphic climax (NF 62) and pre-metamorphosis and the completion of metamorphosis (NF 66). Shown are scatter plots, Pearson correlation coefficients, and p-values for each comparison, with DEG fold-change on the y-axis and differentially methylated region (DMR) height on the x-axis (Log2A/B, where A is NF 62 or 66 and B is NF 50). The values were plotted and a regression line was fit (black line). A 95% Confidence Interval (light-grey region) is also shown around the regression line. There were no statistically significant correlations for these comparisons in the region 5 kb upstream of the transcription start site (TSS; data not shown). C. Correlation between overlapping DMRs and DEGs by direction and magnitude of change within gene bodies and at 5 kb upstream of TSSs comparing pro-metamorphosis (NF 56) with metamorphic climax (NF 62). D. Three examples of a negative correlation between DNA methylation and gene transcription in tadpole brain during metamorphosis (top and middle panels: DNA demethylation; bottom panel: DNA methylation). Shown are screenshots of MethylCap-seq (blue) and RNA-seq (red) reads at discrete genomic regions at four stages of metamorphosis. Reads were mapped to the reference genome using the UCSD Genome Browser. NF stage – Nieuwkoop-Faber developmental stage. Numbers in brackets represent the peak height range. Gene IDs and symbols: (top) XLOC_002166; unknown, (middle) XLOC_030199.1; avpr1a, (bottom) new_exon_scaffold_851 1255.1; unknown.
DNA Methylation and Gene Transcription
We analyzed the relationship between DMRs and DEGs by calculating the Odds Ratios and 95% confidence intervals for DMRs’ association with DEGs versus non-DEGs, at each of the pairwise stage comparisons (Supplemental Fig. 12). We looked at the region 50 kb upstream of the TSS, or within the gene body. An Odds Ratio of 1.5 means that a DMR is 1.5 times more likely to be associated with a DEG than a non-DEG. For most stage comparisons, DMRs were enriched within 50 kb upstream of the TSS of DEGs vs non-DEGs, and this association was even greater if the DMR was located within the gene-body (Supplemental Fig. 12).
To test the hypothesis that increased DNA methylation is correlated with downregulation of gene expression, and vice versa, we examined correlations between the direction and magnitude of change in DNA methylation and gene expression. For this analysis we binned data based on DMR location, looking at DMRs within DEG gene bodies, or DMRs at 0-5 kb upstream of DEG TSSs. We found statistically significant negative correlations between DMRs and DEGs within gene bodies when comparing pre-metamorphosis (NF stage 50) and metamorphic climax (NF stage 62; p=4.4e-7), and pre-metamorphosis and the completion of metamorphosis (NF stage 66; p=0.00057) (Fig. 3B). There were no statistically significant correlations among DMRs and DEGs in the 0-5 kb upstream region in these comparisons. When we looked at the period from pro-metamorphosis (NF stage 56) to metamorphic climax (NF stage 62) we found statistically significant negative correlations between DMRs and DEGs, for DMRs within gene bodies (p=5.4e-13) and also within the 0-5 kb upstream regions (p=0.00045; Fig. 3C). Examples of screenshots of MethlCap-seq and RNA-seq reads mapped to the reference genome for genomic regions showing a negative correlation between DNA methylation and gene transcription are given in Fig. 3D and Supplemental Fig. 5 (Supplemental Fig. 6 shows was an example of a positive correlation, and two examples of no correlation between DNA methylation and gene transcription).
Analysis of changes in DNA methylation using targeted, biochemical assays
We looked at changes in DNA methylation using several biochemical assays. We chose two genes that are known T3 response genes, DNA methyltransferase 3a (dnmt3a) and Krüppel-like factor 9 (klf9). The frog dnmt3a gene has two T3 response elements (TREs), one located −7.1 kb from the TSS, designated TRE-A, and one located +5.1 kb from the TSS, designated TRE-B (22) (Fig. 4A). The TRE in the frog klf9 gene is located −5.9 kb upstream of the TSS within a 180 bp, ultraconserved enhancer module called the klf9 synergy module (KSM) (Bagamasbad et al., 2015).
Fig. 4. DNA demethylation during metamorphosis at the dnmt3a locus correlates increased dnmt3a mRNA.
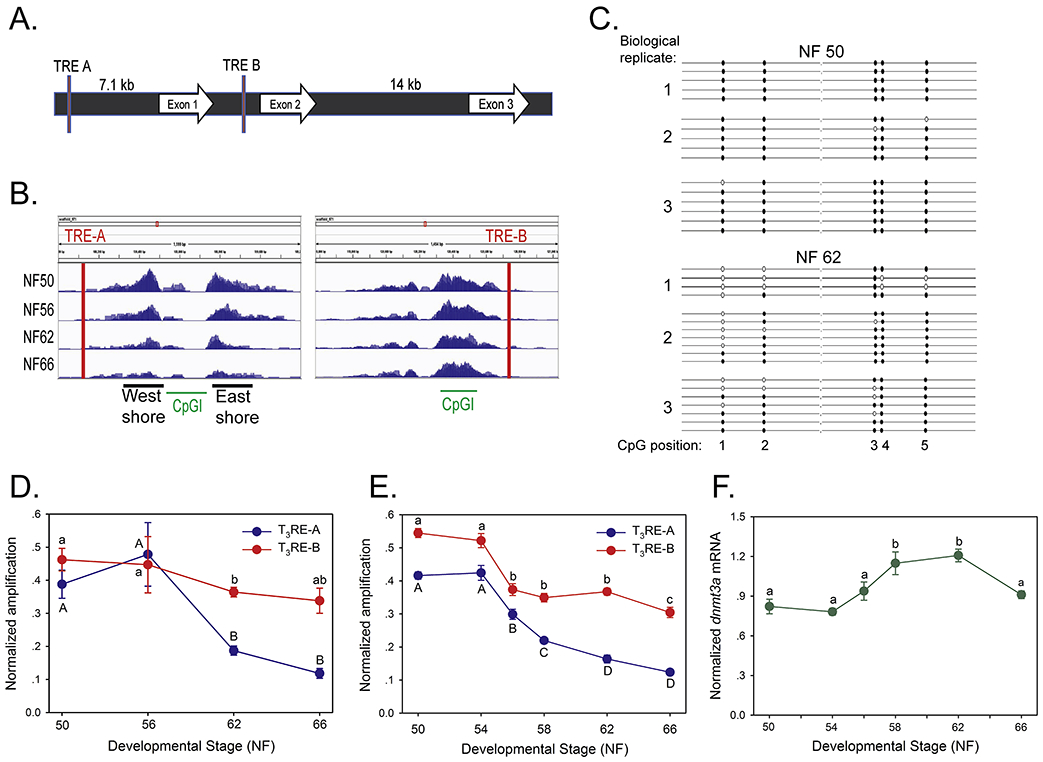
We isolated genomic DNA from X. tropicalis tadpole brain (preoptic area/thalamus/hypothalamus) at different stages of metamorphosis and analyzed DNA methylation using several approaches. A. Schematic depiction of the X. tropicalis dnmt3a locus. The locations of the two TREs that we identified previously are shown as red bars (Kyono et al., 2016a). B. IGV genome browser tracks for MethylCap-seq reads at the X. tropicalis dnmt3a locus at four stages of metamorphosis (NF – Nieuwkoop-Faber). CpGI – CpG island, with the CpG island ‘shores’ indicated. The locations of genomic fragments of the dnmt3a gene that we amplified by PCR for each of the analyses described below are given in Supplemental Fig. 13. C. Lollipop diagram depicting results from bisulfite sequencing of the region of the ‘west shore’ proximal to the dnmt3a TRE-A. We extracted DNA from tadpole brain at NF stages 50 and 62 (pre-metamorphosis and metamorphic climax), conducted bisulfite sequencing, and subcloned PCR-amplified DNA into the pGEM-Teasy vector for direct DNA sequencing. We sequenced 4-7 clones for each biological replicate (n=3/developmental stage). Closed circles show methylated cytosines, open circles unmethylated cytosines. D. We conducted MethylCap assay that targeted the regions of the dnmt3a gene proximal to the two TREs that we identified previously (primer sequences in Supplemental Table 4). Points represent the mean±SEM (n=4/developmental stage) of the normalized PCR amplification signal (normalized to the input). Means with the same letter are not significantly different (p<0.001, ANOVA; lower case TRE-A, upper case TRE-B). E. We conducted methylation-sensitive restriction digest/qPCR (chop-qPCR) assays that targeted the regions of the dnmt3a gene proximal to the two TREs that we identified previously (primer sequences in Supplemental Table 4). Points represent the mean±SEM (n=5/developmental stage) of the normalized PCR amplification signal (normalized to the quantity of the amplicons from neighboring genomic regions that did not contain restriction sites). Means with the same letter are not significantly different (p<0.0001, ANOVA; lower case TRE-A, upper case TRE-B). F. We measured dnmt3a mRNA by RTqPCR in the region of the preoptic area/thalamus/hypothalamus of X. tropicalis tadpole brain during metamorphosis. The dnmt3a mRNA was normalized to the reference gene alpha-actinin mRNA which did not change during development (data not shown). Points represent the means ± SEM (n=5/developmental stage), and means with the same letter are not significantly different (p < 0.001; ANOVA).
Our MethylCap-seq analysis showed that the dnmt3a gene was heavily methylated in pre-metamorphic tadpole brain (NF stage 50), but exhibited a progressive decline in methylation during metamorphosis (Fig. 4B). The largest demethylation occurred at regions flanking the CpG island (“CpG island shores”) located near dnmt3a TRE-A; methylation at the dnmt3a TRE-B also decreased with metamorphosis, but the change was less than at TRE-A. We confirmed demethylation occurring proximal to the TRE-A using targeted bisulfite sequencing (Fig. 4C). Of the five CpG dinucleotides located in the upstream (‘west’) shore, the first three CpGs (CpG-1, CpG-2, and CpG-3) became demethylated between pre-metamorphosis (NF stage 50) and metamorphic climax (NF stage 62). In pre-metamorphic tadpoles 93.3% of CpG1, 100% of CpG2, and 93.3% of CpG3 were methylated; whereas, at metamorphic climax 33.3% of CpG1, 66.7% of CpG2 and 77.8% of CpG3 were methylated. The other two CpGs in this region (CpG4 and CpG5) did not show changes in methylation between the two stages.
We also confirmed demethylation at dnmt3a using targeted MethylCap-qPCR analysis (the locations of oligonucleotide primers used for this and the assays described below are shown in Supplemental Fig. 13). Regions proximal to the two dnmt3a TREs became demethylated between pro-metamorphosis and metamorphic climax (NF stages 56 and 62) (Fig. 4D). The reduction in the MethylCap-qPCR signal was greater at TRE-A than at TRE-B, which is consistent with the MethylCap-seq data. We also confirmed DNA demethylation during metamorphosis at regions proximal to the two dnmt3a TREs using 5-mC-sensitive restriction digest followed by qPCR (5-mC chop-qPCR; Fig. 4E). DNA methylation at both TRE-A and TRE-B showed progressive declines from NF stage 54 through the completion of metamorphosis (NF stage 66), and as with the MethylCap assays, the decrease in methylation was greater at TRE-A. The DNA demethylation at this locus correlated with a developmental increase in dnmt3a mRNA analyzed by RTqPCR (Fig. 4F).
Unlike dnmt3a, the entire klf9 gene, including the KSM, exhibited very low MethylCap-seq signal that was not different from background (input) and did not change during metamorphosis (Supplemental Fig. 14A). Targeted MethylCap-qPCR analysis also found no developmental changes at the klf9 locus (Supplemental Fig. 14B). Using 5-mC chop-qPCR, we found a small, but statistically significant decrease in DNA methylation at the KSM during metamorphosis (Supplemental Fig. 14C). The klf9 mRNA level increased strongly (~20 fold) during metamorphosis (Supplemental Fig. 14D). Analysis of the homologous regions (dnmt3a and the KSM) in the genome of the related species X. laevis using targeted MethylCap-qPCR showed similar changes to those observed in X. tropicalis (Supplemental Fig. 15).
The mRNAs for genes that encode enzymes that catalyze active DNA demethylation increase in tadpole brain during metamorphosis
We used RTqPCR to analyze the mRNA levels for genes that encode enzymes that catalyze DNA demethylation (tet2, tet3, idh1/2/3, gadd45α/β/γ, tdg, idax, aid and apobec2) at five stages of metamorphosis (NF stages 50, 54, 56, 62 and 66). We found that the mRNA levels for all genes, except aid and apobec2 which did not change, were lowest at NF stage 50, then showed progressive increases during metamorphosis, and were maximal at metamorphic climax (NF stage 62; ~2 to 3 fold increases relative to NF stage 50) when circulating T3 concentration is maximal (Leloup, 1977) (Fig. 5). Of the 10 genes that increased during metamorphosis, mRNAs for 8 genes remained at similar elevated levels in the post-metamorphic frog (NF stage 66), with gadd45γ and idh2 showing statistically significant declines (Fig. 5).
Fig. 5. The mRNAs for genes that code for enzymes involved with active DNA demethylation increase during metamorphosis in tadpole brain.
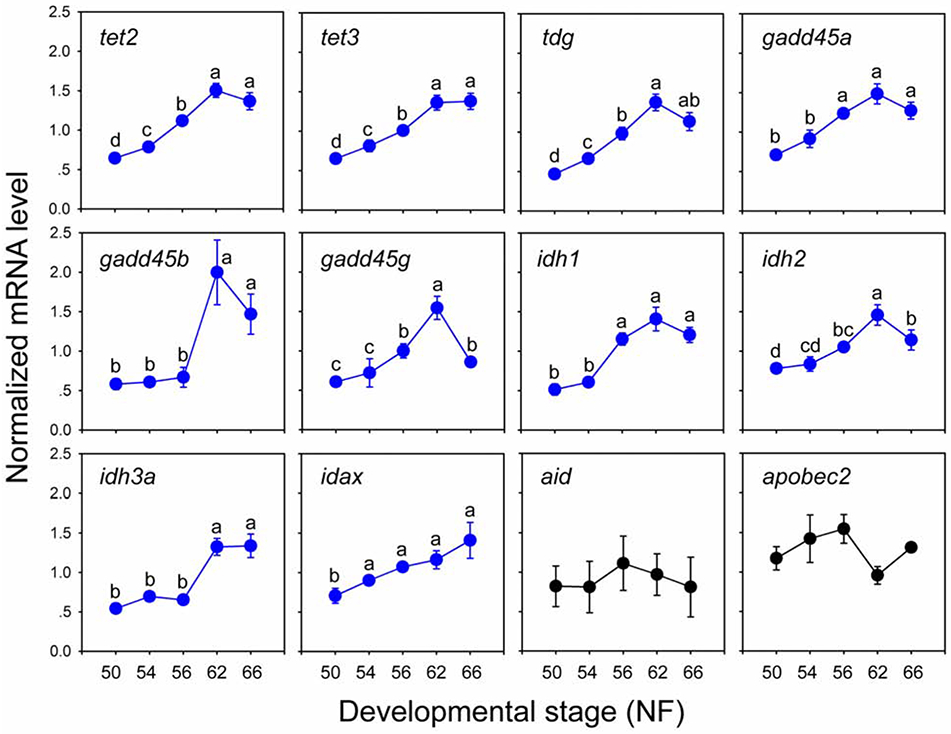
We analyzed mRNA levels using RTqPCR in the region of the preoptic area/thalamus/hypothalamus of X. tropicalis tadpole brain at different stages. All mRNAs were normalized to the reference gene alpha-actinin mRNA which did not change during development (data not shown). Points represent the means ± SEM (n=5/developmental stage). All genes, except aid and apobec2 showed statistically significant increases during metamorphosis (p<0.05, ANOVA).
Immunoreactivity for 5-hmC, 5-caC and TET3 increased in tadpole brain during spontaneous metamorphosis
We used immunohistochemistry (IHC) to analyze changes in immunoreactivity (ir) of the DNA demethylation intermediates 5-hmC and 5-caC, and the cytosine dioxygenase TET3 in tadpole brain during metamorphosis. We generated rabbit antiserums to Xenopus TET3 (see Supplemental Methods) and we verified the titer and specificity by EIA, dot blot, immunocytochemistry, IHC and ChIP assays conducted on chromatin isolated from X. laevis embryos (Supplemental Fig. 16).
We first analyzed the distribution in tadpole brain of 5-hmC-ir, 5-caC-ir and TET3-ir by conducting IHC on transverse sections from NF stage 62 tadpoles (following the brain atlas of Tuinhof and colleagues (Tuinhof et al., 1998), with modifications by Yao and colleagues (Yao et al., 2004); Supplemental Fig. 17). We chose this stage to map these antigens in the brain because it is when the mRNA levels for genes that encode enzymes involved with DNA demethylation are highest (Fig. 5). We saw no 5-hmC-ir or 5-caC-ir in the most rostral regions of the telencephalon (data not shown). There was pronounced 5-hmC-ir and 5-caC-ir in several brain regions including the region of the pallium, amygdala and bed nucleus of the stria terminalis, anterior preoptic area (location of neurosecretory neuron cell bodies; Supplemental Figs. 18&19, region E), the suprachiasmatic nucleus (Supplemental Figs. 18&19, region H) thalamic nuclei and the ventral hypothalamic nucleus (Supplemental Figs. 18&19, regions I and K).
The TET3-ir showed a similar distribution pattern in the tadpole brain as 5-hmC-ir and 5-caC-ir (Supplemental Fig. 20). It was undetectable in the most rostral regions of the telencephalon (brain regions A,B & C in Supplemental Fig. 17; data not shown) and was first detected in the most rostral pallium. We saw TET3-ir throughout the pallium, the anterior preoptic area (Supplemental Fig. 20, region E) and the succeeding brain regions (Supplemental Fig. 20, regions H, I, K, M), with pronounced signal in the thalamic nuclei (anterior, ventromedial, posterior, lateral), the ventral hypothalamic nucleus and the tegmentum. The TET3-ir was low or absent in the hindbrain and spinal cord (data not shown).
We chose the tadpole brain region containing the thalamic nuclei and ventral hypothalamic nucleus (Supplementary Fig. 17, region K) for IHC analysis during spontaneous metamorphosis, as this region showed high 5-hmC-ir, 5-caC-ir and TET3-ir in metamorphic climax stage animals (Supplemental Figs. 18, 19 & 20). This region contains neurosecretory neuron cell bodies and axons that project to the median eminence to control pituitary hormone secretion, and it is highly responsive to T3 action (Denver, 1998b).
The 5-hmC-ir in tadpole brain was low or nondetectable during pro-metamorphosis (NF stages 54 and 56; Fig. 6), then increased at NF stage 58 and reached a maximum at metamorphic climax (NF stage 62). The intensity of 5-hmC-ir was highest in the thalamic nuclei, with lower signal in the ventral hypothalamus. We detected 5-caC-ir at earlier developmental stages than 5-hmC-ir (Fig. 6; or TET3-ir, see below). At NF stage 50, we saw low but distinct 5-caC-ir that was localized to the thalamic nuclei. The signal increased during metamorphosis and reached a maximum at metamorphic climax. Throughout metamorphosis the 5-caC-ir increased in the thalamic nuclei and appeared in the ventral hypothalamus.
Fig. 6. Immunoreactivity for 5-hmC, 5-caC and TET3 increases in tadpole brain during spontaneous metamorphosis.
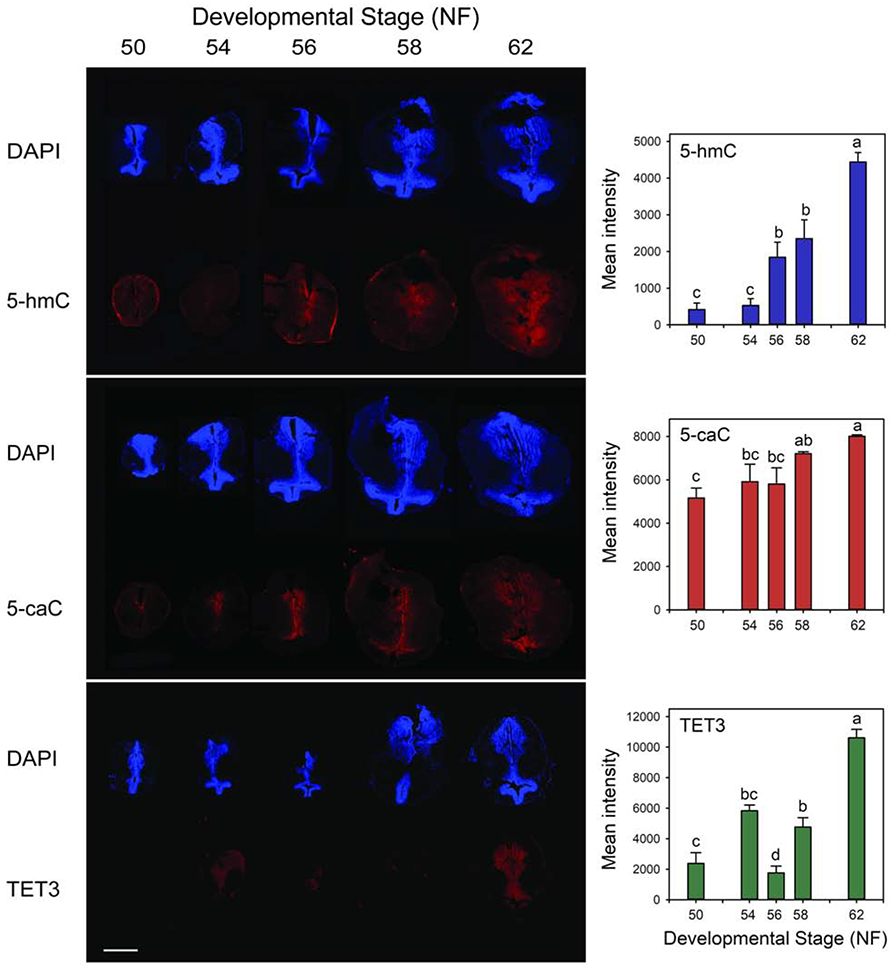
We conducted immunohistochemistry on the region of the tadpole brain containing the thalamic nuclei and ventral hypothalamus (section K in Supplemental Fig. 17; abbreviations are defined in Supplemental Table 5) at different stages of metamorphosis. Shown are representative brain sections stained with DAPI and with antibodies to the three different antigens, captured at 4X magnification. Scale bar = 0.5 mm. Graphs to the right of the brain images provide quantification of the immunoreactivity. Bars represent the means ± SEM (n=4/developmental stage). Data were analyzed by one-way ANOVA (p < 0.001). Letters indicate results of the post-hoc test (Fishers’s LSD); means with the same letter are not significantly different (p<0.05).
Similar to 5-hmC-ir, there was faint TET3-ir in early pro-metamorphic tadpole brain (NF stages 54 and 56), which then increased at NF stage 58 and reached a maximum at NF stage 62 (Fig. 6). These changes in TET3-ir parallel changes in tet3 mRNA (Fig. 5). The TET3 signal was highest in the thalamic nuclei, with lower signal in the ventral hypothalamus. Due to technical problems we only had n=2 for NF stage 66; the 5-hmC-ir and 5-caC-ir were similar to NF stage 62 in these samples, while the TET3-ir was lower.
Discussion
The regulation of cytosine methylation in DNA is crucial for normal neural development and function (Bogdanovic et al., 2011; Feng et al., 2005; Guo et al., 2011; Lister et al., 2013; Shin et al., 2014; Song et al., 2011b; Szulwach et al., 2011). Here we present the first genome-wide analysis of changes in DNA methylation, paired with analysis of changes in gene expression in the brain of a nonmammalian vertebrate during postembryonic development. This developmental period in Xenopus, metamorphosis, is characterized by profound changes in gene regulation (Brown et al., 1995; Shi, 2000; Wen et al., 2019), further evidenced here by our RNA-seq analysis, and it shares many similarities with the late fetal/neonatal developmental period of humans (Sachs and Buchholz, 2017; Tata, 2006). We focused on the part of the tadpole brain that houses the neurosecretory neurons that control pituitary hormone secretion, a brain region that is highly dependent on T3 for its development and maturation (Denver, 1998a; Etkin, 1965; Etkin, 1968; Kikuyama et al., 1979). Using MethylCap-seq, we discovered changes in DNA methylation at discrete regions of the tadpole neural cell genome during metamorphosis, with the largest changes occurring during the period from metamorphic climax to the completion of metamorphosis, when DNA demethylation accounted for over 80% of the methylation changes. The changes in DNA methylation were negatively correlated with changes in gene expression, with the strongest relationship seen for methylation changes occurring within gene bodies of DEGs. In agreement with our genome-wide analyses, biochemical and immunohistochemical analyses showed a decrease in 5-mC, but increases in the DNA demethylation intermediates 5-hmC and 5-caC, and also TET3 during metamorphosis. Taken together, these findings provide additional evidence for predominant DNA demethylation during metamorphosis. The DNA demethylation that we observed is likely dependent, at least in part, on increased expression of genes that encode enzymes that catalyze DNA demethylation.
MethylCap-seq revealed a dense and diverse DNA methylation landscape across the genome of tadpole neural cells
We found that CpG, but not CpA dinucleotides, are enriched within the DNA sequences covered by the MethylCap-seq peaks. This is in contrast to mouse and human, where investigators found pronounced CpA methylation in the frontal cortex that increased during the early postnatal period (Lister et al., 2009); the significance of non-CpG methylation in mammalian neurons and embryonic stem cells is not understood (Shin et al., 2014). In vertebrate genomes, CpG dinucleotides are often present in clusters (termed ‘CpG islands’), and many CpG islands overlap with promoters of constitutively active (i.e., housekeeping) genes; these regions are generally free of DNA methylation, which is thought to maintain the promoters active (Deaton and Bird, 2011). In Xenopus tadpole brain during metamorphosis we found that gene promoters and CpG islands had low methylation compared with other regions of the genome, which is consistent with earlier findings in Xenopus, zebrafish and mammals (Bogdanovic et al., 2011; Bogdanovic et al., 2016). The MethylCap-seq peaks in tadpole brain were approximately equally distributed between genes and intergenic regions.
MethylCap-seq showed changes in DNA methylation in tadpole brain during metamorphosis, with predominant DNA demethylation from climax to completion
Our MethylCap-seq analysis discovered significant changes in DNA methylation at thousands of loci in tadpole brain during metamorphosis. During the pre- to pro-metamorphic transition there was more demethylation, which may represent active DNA demethylation supported by TETs and other enzymes in the demethylation pathway; these genes are expressed during this developmental period, albeit at relatively low levels. On the other hand, these changes could be due to passive DNA demethylation, since neural cell proliferation peaks during this period (Denver et al., 2009). From pro-metamorphosis to metamorphic climax methylation predominated, which may be driven by the activation of dnmt3a transcription during pro-metamorphosis and metamorphic climax, when circulating [T3] is maximal; Xenopus dnmt3a is directly regulated by T3 via two TREs (Kyono et al., 2016a).
From metamorphic climax to the completion of metamorphosis we saw predominant DNA demethylation, which is supported by MethylCap-seq analysis, biochemical analyses of 5-mC (directly by EIA, and indirectly by LUMA and cytosine extension assay), and increased levels of the DNA demethylation intermediates 5-hmC and 5-caC analyzed by IHC. These changes during late metamorphosis are likely dependent on the increased expression of enzymes that catalyze DNA demethylation, evidenced by increases in mRNAs for these genes, and also increased TET3 protein.
We also found, using targeted analyses, that genomic regions in proximity to known TREs of T3-regulated genes (klf9 and dnmt3a) (Denver and Williamson, 2009; Kyono et al., 2016a) became demethylated during metamorphosis. Thyroid hormone response elements are critical regulatory regions mediating T3 action. The most common TRE found in vertebrate genomes consists of two contiguous 6-bp half sites (AGGTCA) separated by a 4-bp spacer (DR+4). DNA demethylation near TREs, as occurred at the dnmt3 and klf9 TREs in this study, could modulate chromatin accessibility, and perhaps the function of TRs (it should not influence TR binding to DNA, since DR+4 TREs do not contain CG dinucleotides.) On the other hand, DNA demethylation at these sites may be induced by liganded TR action in chromatin (S. Raj, Y. Kyono and R.J. Denver, unpublished data). Studies in HEK293T cells support that TRα can directly interact with TET3, and that this may influence TR function in chromatin (Guan et al., 2017). Our preliminary findings support that T3 can promote DNA demethylation at discrete loci in Xenopus tadpole brain (S. Raj, Y. Kyono and R.J. Denver, unpublished data; but see Kasai et al. (Kasai et al., 2015)).
Roles for DNA methylation in animal development
Our findings support that changes in DNA methylation are linked to the gene regulation programs during tadpole metamorphosis, which underlie the dramatic morphogenetic changes that occur during this developmental period. Critical roles for DNA methylation in development have been demonstrated in several vertebrate species. For example, DNA methylation plays a pivotal role in the long-term transcriptional silencing seen with genomic imprinting, X chromosome inactivation, and the silencing of repetitive elements (Bird, 2002; Weber and Schubeler, 2007). The depletion of enzymes that catalyze DNA methylation or DNA demethylation leads to a diversity of developmental disorders, including severe neurological defects (Edwards et al., 2017; Fedotova and Illarioshkin, 2019; Kumar et al., 2018; Ross and Bogdanovic, 2019; Wu and Zhang, 2017; Wu et al., 2018). Many studies support that DNA methylation is a central regulator of cell identity (Bogdanovic and Lister, 2017; Wu et al., 2018; Zeng and Chen, 2019). This is hypothesized to be directly related to the modulation of the chromatin environment, and consequently gene transcription (Bogdanovic and Lister, 2017). Bogdanovic and colleagues (Bogdanovic et al., 2016) found that transcriptional activation of genes involved with body plan formation in zebrafish, X. tropicalis and the mouse depends on active DNA demethylation of functionally conserved enhancer elements.
Role for DNA methylation in the regulation of gene transcription
Our findings show that during the metamorphic transition, decreased DNA methylation correlates with upregulation of gene expression; whereas, increased DNA methylation at a locus correlates with downregulation of gene expression. We found negative correlations within gene bodies and the 0-5 kb upstream region (proximal and distal promoter), and this relationship was stronger for gene bodies. Our findings provide compelling support, using a vertebrate developmental model system, for the hypothesis that DNA demethylation promotes gene activation in vivo, while DNA methylation is associated with gene repression.
Mounting evidence supports that changes in DNA methylation at gene promoters and enhancers can influence transcription (Bogdanovic et al., 2016; Hackett et al., 2013; Wu and Zhang, 2017; Wu et al., 2018). DNA methylation at gene regulatory regions follow this general rule, as there is an inverse correlation between expression of nearby genes and DNA methylation at cell-type specific enhancers, promoters and within gene bodies (Guo et al., 2011; Hon et al., 2013; Lister et al., 2013). The DNA demethylation intermediate 5-hmC is enriched at enhancers, promoters and bodies of actively transcribed genes, which is consistent with DNA demethylation being associated with transcriptional activation (Nestor et al., 2016; Taylor et al., 2016; Tsagaratou et al., 2014). There is evidence from zebrafish and mouse that TET proteins, which catalyze conversion of 5-mC to 5-hmC and other DNA demethylation intermediates, play pivotal roles in enhancer activation through modulation of chromatin accessibility (Bogdanovic et al., 2016; Li et al., 2016). In vitro methylation of target DNA sequences represses transcription in Xenopus embryos (but not in oocytes) (Bogdanovic et al., 2011). Whole-genome bisulfite sequencing found that the genome of Xenopus embryos undergoes DNA demethylation during development, and that these differentially methylated regions were enriched in epigenomic marks indicative of enhancers (H3K4me1, p300) (Bogdanovic et al., 2016).
There are two mechanisms that have been proposed by which DNA methylation at gene regulatory regions can influence transcription. First, nuclear proteins that bind 5-mC (methyl-CpG binding proteins: MBDs) (Hendrich and Bird, 1998) are known to interact with protein complexes that contain the corepressor Sin3A, which in turn recruits histone deacetylases to generate a repressive chromatin state (Nan et al., 1998). Second, transcription factor binding to DNA may be impaired by cytosine methylation when the recognition motif contains CpG dinucleotides (Bogdanovic and Lister, 2017).
Genes that are activated during tadpole metamorphosis display different kinetic profiles, that may be related in part to DNA methylation at the locus. That is, there may be a correlation between the level of DNA methylation and how early the gene is activated during metamorphosis. This may be illustrated by the low methylation level at klf9, which shows very early developmental activation and rapid kinetics after T3 treatment (Bagamasbad et al., 2015; Bagamasbad et al., 2008; Furlow and Kanamori, 2002; Hu et al., 2016). This is in contrast to dnmt3a, which is heavily methylated, and exhibits late activation during metamorphosis and slow kinetics after T3 treatment (Kyono et al., 2016a). This hypothesis requires further investigation.
Our RNA-seq experiment documents the dramatic changes in gene expression that underlie development of the Xenopus tadpole preoptic area/thalamus/hypothalamus. Between pre-metamorphosis and metamorphic climax more than 5000 genes change their expression level, and this number is nearly equally divided between up and downregulated genes. This is the first analysis of its kind in any tadpole tissue (other studies have looked only at gene regulation responses to exogenous T3 in pre-metamorphic tadpoles); a full analysis of the RNA-seq data will be published elsewhere.
Molecular mechanisms for DNA demethylation in tadpole brain during metamorphosis
We found that mRNA levels for genes that encode enzymes involved in the active DNA demethylation pathway increased in tadpole brain during metamorphosis, with highest levels during metamorphic climax, when circulating T3 is highest (Leloup, 1977). We also show that for one of the genes, tet3, the increase in its mRNA level coincided with increased TET3 protein in tadpole brain. TET3 catalyzes the oxidation of 5-mC to the active DNA demethylation intermediates 5-hmC and 5-caC (Ross and Bogdanovic, 2019; Wu and Zhang, 2017). Consistent with the increase in tet3 mRNA and TET3 protein during metamorphosis, and the role of TET3 in direct catalysis of DNA demethylation, we found increases in the DNA demethylation intermediates 5-hmC and 5-caC in tadpole brain during metamorphosis.
To our knowledge, this is the first demonstration, in a vertebrate developmental model system, of a developmental increase in the expression of enzymes involved with DNA demethylation, that is coincident with an increase in DNA demethylation intermediates, and consequently DNA demethylation. It is noteworthy that immunoreactivity for TET3, 5-hmC and 5-caC was minimal in rostral regions of the telencephalon, the hindbrain and spinal cord in tadpole brain at metamorphic climax, but was high in the part of the brain that houses the thalamic nuclei, the ventral hypothalamus and the tegmentum, regions of the tadpole brain that are known to be highly responsive to T3 (Denver, 1998a; Etkin, 1968; Etkin et al., 1965). Diotel and colleagues (Diotel et al., 2017) used IHC to detect 5-hmC in neurons of pre-metamorphic X. laevis tadpoles (NF stage 50) and newly metamorphosed frogs (NF stage 66) in the same brain regions as the current study; this marker of DNA demethylation was excluded from proliferating cells. The 5-hmC level in mouse brain was highest in neurons, and it strongly increased postnatally when neurons were maturing and forming synapses (Cadena-del-Castilloa et al., 2014; Lister et al., 2009; Song et al., 2011b; Szulwach et al., 2011). Furthermore, 5-hmC is enriched within gene bodies, and exhibited a positive correlation with gene transcription (Hahn et al., 2013; Hahn et al., 2014; Song et al., 2011b; Szulwach et al., 2011). The increase that we saw in TET3 in tadpole brain supports that this enzyme is responsible, at least in part, for the increase in 5-hmC and 5-caC, and DNA demethylation that we saw after metamorphic climax.
In mammals there are three Tet genes, and all are expressed in the brain, primarily in neurons, with Tet3 being the most abundant (Kaas et al., 2013; Li et al., 2014; Mi et al., 2015; Szwagierczak et al., 2010); these enzymes support the high levels of 5-hmC found in neuronal cells (Szulwach et al., 2011; Szwagierczak et al., 2010). In Xenopus, only tet2 and tet3 genes have been identified. The TET proteins play critical roles in modulating DNA methylation, and have profound influence on animal development (Kaas et al., 2013; Li et al., 2014; Mi et al., 2015; Tan and Shi, 2012). In mouse embryonic stem cells in which Tet 1, 2 & 3 were knocked out, and in tetl, 2 & 3 triple morphant zebrafish embryos, the levels of 5-mC at promoters, enhancers and DNaseI-hypersensitive sites were significantly higher compared to wild type embryos (Bogdanovic et al., 2016; Dawlaty et al., 2014; Lu et al., 2014). Inhibition of TET enzymes in the brain leads to dysregulation of methylation at, and transcription of genes involved with synaptogenesis and synaptic function (Campbell and Wood, 2019). TET3 has been shown to play a critical role in the maintenance of neural progenitors and the terminal differentiation of neurons (Antunes et al., 2019; Li et al., 2015; Lv et al., 2014). This enzyme has also been shown to function as a synaptic activity sensor, and is required for homeostatic synaptic plasticity (Campbell and Wood, 2019; Yu et al., 2015). In Xenopus, Xu and colleagues (Xu et al., 2012) demonstrated an essential role for TET3 in eye and neural development.
Conclusions
The genome of neural cells located in the region of the preoptic area/thalamus/hypothalamus of X. tropicalis tadpole brain undergoes changes in DNA methylation during metamorphosis that are negatively correlated with changes in gene expression. DNA methylation predominated during the pro-metamorphic period, possibly mediated by increased expression of dnmt3a, which is activated by T3 during these stages of metamorphosis. DNA demethylation predominated from metamorphic climax to the completion of metamorphosis, and this change may depend, in part, on the upregulation of genes that encode enzymes involved with active DNA demethylation. The activation of these genes, for example tet3, is likely responsible for the increased levels of TET3 protein, and the generation of the DNA demethylation intermediates 5-hmC and 5-caC. Taken together, our data support that changes in DNA methylation are likely crucial for the coordination of gene regulation programs that underlie tissue morphogenesis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal antiserum to 5-hmC | Diagenode | C15410205-20 |
| Rabbit polyclonal antiserum to 5-caC | Diagenode | C15410205-20 |
| Rabbit polyclonal antiserum to xTET3 | This paper | N/A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| MethylCap kit | Diagenode, Denville, NJ | C02020010 (AF-100-0048) |
| TruSeq mRNA Sample Prep v2 kit | Illumina Inc., San Diego, CA | RS-122-2001 |
| DNA modification kit | Epigentek, Farmingdale, NY | P-1026-050 |
| Deposited Data | ||
| Raw and analyzed MethylCap-seq data | This paper | GSE139267 |
| Raw and analyzed RNA-seq data | This paper | GSE140120 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Xenopus tropicalis, Nigerian strain | Xenopus One (Dexter, MI) | |
| Xenopus laevis | Xenopus One (Dexter, MI) | |
| Oligonucleotides | ||
| Primers for RTqPCR – see Supplemental Table 4 | This paper | N/A |
| Primers for synthesizing spike-in controls using lambda DNA as template – see Supplemental Table 4 | This paper | N/A |
| Primers for monitoring spike-in controls after methylated DNA capture using RTqPCR – see Supplemental Table 4 | This paper | N/A |
| Primers for methylation-sensitive restriction digest/qPCR assays – see Supplemental Table 4 | This paper | N/A |
| Primers for MethylCap-qPCR assay – see Supplemental Table 4 | This paper | N/A |
| Primers for bisulfite sequencing – see Supplemental Table 4 | This paper | N/A |
| Primers for subcloning xtet3 cDNA – see Supplemental Table 4 | This paper | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| BBTools suite (v37.90) | BBMap – Bushnell B. | https://sourceforge.net/proiects/bbmap/ |
| Samtools v1.8 | Li et al., 2009 | https://sourceforge.net/proiects/samtools/ |
| PePr (v1.1.18) | Zhang et al., 2014 | https://github.com/shawnzhangyx/PePr/ |
| bowtie2/RSEM (v2.1.0) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Methprimer software | Li and Dahiya, 2002 | https://www.urogene.org/methprimer/ |
| Other | ||
Highlights for Kyono et al.
Using Methylated DNA Capture-sequencing, we discovered changes in DNA methylation at discrete regions of the tadpole neural cell genome during metamorphosis, with the largest changes occurring during the period from metamorphic climax to the completion of metamorphosis when 80% of the changes represented demethylation.
Changes in DNA methylation were negatively correlated with changes in gene expression analyzed by RNA sequencing, with the strongest relationship seen for methylation changes occurring within gene bodies of differentially expressed genes.
We provide evidence that the DNA demethylation is driven by increased expression of genes that encode enzymes that catalyze DNA demethylation, especially members of the ten eleven translocation (TET) family of cytosine dioxygenases.
Acknowledgements:
We thank Jessica Head and Nil Basu for conducting the LUMA assay, and Ling Huang, Arasakumar Subramani, Pamela Navarette Ramirez and Samantha Fontana for technical assistance. We are grateful to Yujiang Geno Shi for providing the Xenopus tet3 expression vector pCMV-flag-xTET3.
Funding: This work was supported by grants from the National Science Foundation (IOS 0922583 and IOS 1456115) to R.J.D. Y. Kyono was supported by a Ruth L Kirschstein National Research Service Award (NS07329401) from the Nationals Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
Literature Cited
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY, 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W, 2010. Differential expression analysis for sequence count data. Genome Biology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes C, Sousa N, Pinto L, Marques CJ, 2019. TET enzymes in neurophysiology and brain function. Neurosci. Biobehav. Rev 102, 337–344. [DOI] [PubMed] [Google Scholar]
- Bagamasbad P, Bonett R, Sachs L, Buisine N, Raj S, Knoedler J, Kyono Y, Ruan Y, Ruan X, Denver R, 2015. Deciphering the regulatory logic of an ancient, ultraconserved nuclear receptor enhancer module. Molecular Endocrinology 29, 856–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagamasbad P, Howdeshell KL, Sachs LM, Demeneix BA, Denver RJ, 2008. A role for basic transcription element-binding protein 1 (BTEB1) in the autoinduction of thyroid hormone receptor beta. J. Biol. Chem. 283, 2275–2285. [DOI] [PubMed] [Google Scholar]
- Bernal J, 2009. Thyroid Hormones and Brain Development.
- Bird A, 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Bogdanovic O, Lister R, 2017. DNA methylation and the preservation of cell identity. Curr Opin Genet Dev 46, 9–14. [DOI] [PubMed] [Google Scholar]
- Bogdanovic O, Long SW, van Heeringen SJ, Brinkman AB, Gomez-Skarmeta JL, Stunnenberg HG, Jones PL, Veenstra GJC, 2011. Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis. Genome Research 21, 1313–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O, Smits AH, de la Calle Mustienes E, Tena JJ, Ford E, Williams R, Senanayake U, Schultz MD, Hontelez S, van Kruijsbergen I, Rayon T, Gnerlich F, Carell T, Veenstra GJ, Manzanares M, Sauka-Spengler T, Ecker JR, Vermeulen M, Gomez-Skarmeta JL, Lister R, 2016. Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat Genet 48, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O, van Heeringen SJ, Veenstra GJC, 2012. The epigenome in early vertebrate development. Genesis 50, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DD, Wang Z, Kanamori A, Eliceiri B, Furlow JD, Schwartzman R, 1995. Amphibian metamorphosis: a complex program of gene expression changes controlled by the thyroid hormone. Recent Prog Horm Res 50, 309–315. [DOI] [PubMed] [Google Scholar]
- Buchholz DR, 2017. Xenopus metamorphosis as a model to study thyroid hormone receptor function during vertebrate developmental transitions. Mol. Cell. Endocrinol 459, 64–70. [DOI] [PubMed] [Google Scholar]
- Buisine N, Ruan X, Bilesimo P, Grimaldi A, Alfama G, Ariyaratne P, Mulawadi F, Chen J, Sung W-K, Liu ET, Demeneix BA, Ruan Y, Sachs LM, 2015. Xenopus tropicalis genome re-scaffolding and re-annotation reach the resolution required for in vivo ChIA-PET analysis. Plos One DOI: 10.1371/journal.pone.0137526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena-del-Castilloa C, Valdes-Quezada C, Carmona-Aldana F, Arias C, Bermudez-Rattoni F, Recillas-Targa F, 2014. Age-Dependent Increment of Hydroxymethylation in the Brain Cortex in the Triple-Transgenic Mouse Model of Alzheimer's Disease. Journal of Alzheimers Disease 41, 845–854. [DOI] [PubMed] [Google Scholar]
- Campbell RR, Wood MA, 2019. How the epigenome integrates information and reshapes the synapse. Nat. Rev. Neurosci 20, 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SY, Leonard JL, Davis PJ, 2010. Molecular aspects of thyroid hormone actions. Endocr Rev 31, 139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi E, Denver R, 2006. Leptin (ob gene) of the South African clawed frog Xenopus laevis. Proc. Natl. Acad. Sci. U. S. A 103, 10092–10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, Powell BE, Cheng AW, Faull KF, Lyko F, Jaenisch R, 2014. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 29, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A, 2011. CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver RJ, 1998a. The molecular basis of thyroid hormone-dependent central nervous system remodeling during amphibian metamorphosis. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol 119, 219–228. [DOI] [PubMed] [Google Scholar]
- Denver RJ, 1998b. The molecular basis of thyroid hormone-dependent central nervous system remodeling during amphibian metamorphosis. Comp. Biochem. Physiol. C- Pharmacol. Toxicol. Endocrinol 119, 219–228. [DOI] [PubMed] [Google Scholar]
- Denver RJ, Hu F, Scanlan TS, Furlow JD, 2009. Thyroid hormone receptor subtype specificity for hormone-dependent neurogenesis in Xenopus laevis. Dev Biol 326, 155–168. [DOI] [PubMed] [Google Scholar]
- Denver RJ, Williamson KE, 2009. Identification of a thyroid hormone response element in the mouse Kruppel-like factor 9 gene to explain its postnatal expression in the brain. Endocrinology 150, 3935–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diotel N, Mérot Y, Coumailleau P, Gueguen M-M, Sérandour AA, Salbert G, Kah O, 2017. 5-hydroxymethylcytosine marks postmitotic neural cells in the adult and developing vertebrate central nervous system. J. Comp. Neurol 525, 478–497. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Yarychkivska O, Boulard M, Bestor TH, 2017. DNA methylation and DNA methyltransferases. Epigenetics & Chromatin 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin W, 1965. The phenomena of amphibian metamorphosis. IV. The development of the median eminence. J. Morphol 116, 371–378. [DOI] [PubMed] [Google Scholar]
- Etkin W, 1968. Hormonal control of amphibian metamorphosis, in: Etkin W, Gilbert LI (Eds.), Metamorphosis: a problem in developmental biology. Appleton-Century- Crofts, New York, pp. 313–348. [Google Scholar]
- Etkin W, Kikuyama S, Rosenblu J, 1965. Thyroid feedback to hypothalamic neurosecretory system in frog larvae. Neuroendocrinology 1, 45-&. [Google Scholar]
- Fan GP, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge WH, Shen Y, Hao W, ten Hoeve J, Shuai K, Sun YE, 2005. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 132, 3345–3356. [DOI] [PubMed] [Google Scholar]
- Fedotova EY, Illarioshkin SN, 2019. DNA Methylation in Neurodegenerative Diseases. Russian Journal of Genetics 55, 271–277. [Google Scholar]
- Feng J, Chang H, Li E, Fan G, 2005. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res 79, 734–746. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan GP, 2010a. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng SH, Cokus SJ, Zhang XY, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE, 2010b. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U. S. A 107, 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJM, Thompson CB, Levine RL, Melnick A, 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlow JD, Kanamori A, 2002. The transcription factor basic transcription element-binding protein 1 is a direct thyroid hormone response gene in the frog Xenopus laevis. Endocrinology 143, 3295–3305. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH, 2005. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74, 481–514. [DOI] [PubMed] [Google Scholar]
- Guan W, Guyot R, Samarut J, Flamant F, Wong J, Gauthier K, 2017. Methylcytosine dioxygenase TET3 interacts with thyroid hormone nuclear receptors and stabilizes their association to chromatin. Proc. Natl. Acad. Sci. U. S. A 114, 8229–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JJU, Ma DKK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song HJ, 2011. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci 14, 1345–U1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA, 2013. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Qiu RX, Wu XW, Li AX, Zhang HY, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, Lu Q, 2013. Dynamics of 5-Hydroxymethylcytosine and Chromatin Marks in Mammalian Neurogenesis. Cell Reports 3, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Szabó PE, Pfeifer GP, 2014. 5-Hydroxymethylcytosine: a stable or transient DNA modification? Genomics 104, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X-J, Chen T, Zhu J-K, 2011a. Regulation and function of DNA methylation in plants and animals. Cell Res. 21, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song C-X, Zhang K, He C, Xu G-L, 2011b. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Bird A, 1998. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B, 2013. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nature Genet. 45, 1198–U1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Knoedler JR, Denver RJ, 2016. A mechanism to enhance cellular responsivity to hormone action: Kruppel-like factor 9 promotes thyroid hormone receptor-beta autoinduction during postembryonic brain development. Endocrinology 157, 1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, Silva AJ, Schweizer FE, Fan G, 2009. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet 18, 2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y, 2010. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y, 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GI, King JR, Song HJ, Sweatt JD, 2013. TET1 Controls CNS 5-Methylcytosine Hydroxylation, Active DNA Demethylation, Gene Transcription, and Memory Formation. Neuron 79, 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai K, Nishiyama N, Izumi Y, Otsuka S, Ishihara A, Yamauchi K, 2015. Exposure to 3,3 ',5-triiodothyronine affects histone and RNA polymerase II modifications, but not DNA methylation status, in the regulatory region of the Xenopus laevis thyroid hormone receptor beta A gene. Biochem. Biophys. Res. Commun 467, 33–38. [DOI] [PubMed] [Google Scholar]
- Kikuyama S, Miyakawa M, Arai Y, 1979. Influence of thyroid hormone on the development of preoptic-hypothalamic monoaminergic neurons in tadpoles of Bufo bufo japonicus. Cell Tissue Res. 198, 27–33. [DOI] [PubMed] [Google Scholar]
- Kumar S, Chinnusamy V, Mohapatra T, 2018. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Frontiers in Genetics 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyono Y, Sachs LM, Bilesimo P, Wen L, Denver RJ, 2016a. Developmental and thyroid hormone regulation of the DNA methyltransferase 3a gene in Xenopus tadpoles. Endocrinology 157, 4961–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyono Y, Subramani A, Ramadoss P, Hollenberg AN, Bonett RM, Denver RJ, 2016b. Liganded thyroid hormone receptors transactivate the DNA methyltransferase 3a gene in mouse neuronal cells. Endocrinology 157, 3647–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup J, 1977. La triiodothyronine, hormone de la metamorphose des amphibiens. CR Acad. Sci. Paris, D 284, 2261–2263. [Google Scholar]
- Li E, Bestor TH, Jaenisch R, 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R, 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431. [DOI] [PubMed] [Google Scholar]
- Li T, Yang D, Li J, Tang Y, Yang J, Le W, 2015. Critical role of Tet3 in neural progenitor cell maintenance and terminal differentiation. Mol. Neurobiol 51, 142–154. [DOI] [PubMed] [Google Scholar]
- Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y, Bredy TW, 2014. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. U. S. A 111, 7120–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yue XJ, Pastor WA, Lin LZ, Georges R, Chavez L, Evans SM, Rao A, 2016. Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc. Natl. Acad. Sci. U. S. A 113, E8267–E8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee C-W, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC Jr., Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG, 2012. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L, 2014. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR, 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR, 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu FL, Liu YT, Jiang L, Yamaguchi S, Zhang Y, 2014. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 28, 2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhao BS, He C, 2015. TET family proteins: oxidation activity, interacting molecules, and functions in diseases. Chem. Rev 115, 2225–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv XH, Jiang HH, Liu YL, Lei XP, Jiao JW, 2014. MicroRNA-15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET3 during early neocortical development. Embo Reports 15, 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK, 2013. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol 31, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC, 2011. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi YJ, Gao XC, Dai JX, Ma Y, Xu LX, Jin WL, 2015. A Novel Function of TET2 in CNS: Sustaining Neuronal Survival. International Journal of Molecular Sciences 16, 21846–21857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan XS, Cross S, Bird A, 1998. Gene silencing by methyl-CpG-binding proteins, in: Chadwick DJ, Cardew G (Eds.), Epigenetics, pp. 6–16. [DOI] [PubMed] [Google Scholar]
- Nestor CE, Lentini A, Nilsson CH, Gawel DR, Gustafsson M, Mattson L, Wang H, Rundquist O, Meehan RR, Klocke B, Seifert M, Hauck SM, Laumen H, Zhang H, Benson M, 2016. 5-Hydroxymethylcytosine Remodeling Precedes Lineage Specification during Differentiation of Human CD4(+) T Cells. Cell Reports 16, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Meletis K, Fu DD, Jhaveri S, Jaenisch R, 2007. Ablation of de novo DNA methyltransferase dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Developmental Dynamics 236, 1663–1676. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E, 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Porterfield SP, Hendrich CE, 1993. The role of thyroid hormones in prenatal and neonatal neurological development-current perspectives. Endocr. Rev 14, 94–106. [DOI] [PubMed] [Google Scholar]
- Ross SE, Bogdanovic O, 2019. TET enzymes, DNA demethylation and pluripotency. Biochemical Society Transactions 47, 875–885. [DOI] [PubMed] [Google Scholar]
- Rottach A, Leonhardt H, Spada F, 2009. DNA methylation-mediated epigenetic control. J Cell Biochem 108, 43–51. [DOI] [PubMed] [Google Scholar]
- Sachs LM, Buchholz DR, 2017. Frogs model man: In vivo thyroid hormone signaling during development. Genesis 55, 1–10. [DOI] [PubMed] [Google Scholar]
- Schafer A, 2013. Gadd45 proteins: key players of repair-mediated DNA demethylation. Advances in experimental medicine and biology 793, 35–50. [DOI] [PubMed] [Google Scholar]
- Shi Y-B, 2000. Amphibian metamorphosis: from morphology to molecular biology. Wiley-Liss, New York. [Google Scholar]
- Shi Y-B, 2009. Dual functions of thyroid hormone receptors in vertebrate development: the roles of histone-modifying cofactor complexes. Thyroid 19, 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Ming GL, Song H, 2014. DNA modifications in the mammalian brain. Philosophical Transactions of the Royal Society B-Biological Sciences 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C-X, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen C-H, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C, 2011a. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C, 2011b. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A, 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9, 465–476. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, Vasanthakumar A, Godley LA, Chang Q, Cheng X, He C, Jin P, 2011. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H, 2010. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 38, e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, Beck S, Butcher LM, 2012. Methylome analysis using MeDIP-seq with low DNA concentrations. Nature Protocols 7, 617–636. [DOI] [PubMed] [Google Scholar]
- Tan L, Shi YG, 2012. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 139, 1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata JR, 2006. Amphibian metamorphosis as a model for the developmental actions of thyroid hormone. Mol. Cell. Endocrinol 246, 10–20. [DOI] [PubMed] [Google Scholar]
- Taylor SEB, Li YH, Smeriglio P, Rath M, Wong WH, Bhutani N, 2016. Stable 5-Hydroxymethylcytosine (5hmC) Acquisition Marks Gene Activation During Chondrogenic Differentiation. J. Bone Miner. Res 31, 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, Aijo T, Lio CWJ, Yue XJ, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A, 2014. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc. Natl. Acad. Sci. U. S. A 111, E3306–E3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinhof R, Ubink R, Tanaka S, Atzori C, van Strien FJC, Roubos EW, 1998. Distribution of pro-opiomelanocortin and its peptide end products in the brain and hypophysis of the aquatic toad, Xenopus laevis. Cell And Tissue Research 292, 251–265. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Suetake I, Tada T, Tajima S, 2002. Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis. Mechanisms of Development 118, 187–190. [DOI] [PubMed] [Google Scholar]
- Weber M, Schubeler D, 2007. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Current Opinion in Cell Biology 19, 273–280. [DOI] [PubMed] [Google Scholar]
- Wen L, He C, Sifuentes CJ, Denver RJ, 2019. Thyroid Hormone Receptor Alpha Is Required for Thyroid Hormone-Dependent Neural Cell Proliferation During Tadpole Metamorphosis. Frontiers in Endocrinology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y, 2014. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 156, 45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XJ, Zhang Y, 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nature Reviews Genetics 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Wu XW, Li G, Xie RY, 2018. Decoding the role of TET family dioxygenases in lineage specification. Epigenetics & Chromatin 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Xu C, Kato A, Tempel W, Abreu JG, Bian CB, Hu YG, Hu D, Zhao B, Cerovina T, Diao JB, Wu FZ, He HH, Cui QY, Clark E, Ma C, Barbara A, Veenstra GJC, Xu GL, Kaiser UB, Liu XS, Sugrue SP, He X, Min JR, Kato Y, Shi YG, 2012. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 151, 1200–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Stenzel-Poore M, Denver RJ, 2007. Structural and functional conservation of vertebrate corticotropin-releasing factor genes: evidence for a critical role for a conserved cyclic AMP response element. Endocrinology 148, 2518–2531. [DOI] [PubMed] [Google Scholar]
- Yao M, Westphal N, Denver R, 2004. Distribution and acute stressor-induced activation of corticotrophin-releasing hormone neurones in the central nervous system of Xenopus laevis. Journal of Neuroendocrinology 16, 880–893. [DOI] [PubMed] [Google Scholar]
- Yu HM, Su YJ, Shin J, Zhong C, Guo JJU, Weng YL, Gao FY, Geschwind DH, Coppola G, Ming GL, Song HJ, 2015. Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat. Neurosci 18, 836–U265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D, 2010. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 328, 916–919. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Chen TP, 2019. DNA Methylation Reprogramming during Mammalian Development. Genes 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lin Y-H, Johnson TD, Rozek LS, Sartor MA, 2014. PePr: a peak-calling prioritization pipeline to identify consistent or differential peaks from replicated ChIP-Seq data. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.